

Abstract
Mutations in fibroblast growth factor receptors (Fgfrs) 1–3 cause skeletal disease syndromes in humans. Although these Fgfrs are expressed at various stages of chondrocyte and osteoblast development, their function in specific skeletal cell types is poorly understood. Using conditional inactivation of Fgfr1 in osteo-chondrocyte progenitor cells and in differentiated osteoblasts, we provide evidence that FGFR1 signaling is important for different stages of osteoblast maturation. Examination of osteogenic markers showed that inactivation of FGFR1 in osteo-chondro-progenitor cells delayed osteoblast differentiation, but that inactivation of FGFR1 in differentiated osteoblasts accelerated differentiation. In vitro osteoblast cultures recapitulated the in vivo effect of FGFR1 on stage-specific osteoblast maturation. In immature osteoblasts, FGFR1 deficiency increased proliferation and delayed differentiation and matrix mineralization, whereas in differentiated osteoblasts, FGFR1 deficiency enhanced mineralization. Furthermore, FGFR1 deficiency in differentiated osteoblasts resulted in increased expression of Fgfr3, a molecule that regulates the activity of differentiated osteoblasts. Mice lacking Fgfr1, either in progenitor cells or in differentiated osteoblasts, showed increased bone mass as adults. These data demonstrate that signaling through FGFR1 in osteoblasts is necessary to maintain the balance between bone formation and remodeling through a direct effect on osteoblast maturation.
Keywords: Fibroblast growth factor receptor 1 (FGFR1), Skeletal development, Chondrocyte, Osteoblasts
Introduction
Mutations in FGFRs account for many of the craniosynostosis and chondrodysplasia syndromes in humans (Chen and Deng, 2005; Morriss-Kay and Wilkie, 2005; Ornitz, 2005; Ornitz and Marie, 2002). Although the vast majority of mutations that cause craniosynostosis syndromes are associated with FGFR2, a single missense mutation in FGFR1 (P252R) can cause Pfeiffer syndrome and phenocopies several Pfeiffer syndrome mutations in FGFR2 (Schell et al., 1995). The same mutation has also been identified in a single patient who presented with Jackson–Weiss syndrome (Roscioli et al., 2000), a mild craniofacial syndrome. Other missense mutations in FGFR1 cause osteoglophonic dysplasia, a syndrome involving craniosynostosis, rhizomelic dwarfism and non-ossifying bone lesions (White et al., 2005). This genetic evidence establishes a role for FGFR1 in skeletal development and suggests that FGFR1, FGFR2 and FGFR3 signaling pathways may have similar or redundant functions. In addition to premature fusion of cranial sutures, several of the classic craniosynostosis syndromes have associated phenotypes that affect long bone development in the appendicular skeleton. For example, Pfeiffer syndrome patients have characteristic broad thumbs and toes, and Apert syndrome patients have soft tissue and bony syndactyly (Wilkie et al., 2002). This suggests that FGFR1 and FGFR2 are important for early limb patterning and endochondral and intramembranous skeletal development.
Fgfrs 1–3 are expressed in the developing and mature skeleton in patterns suggestive of both unique and redundant function (Ornitz and Marie, 2002). In the developing growth plate, both Fgfr1 and Fgfr2 are expressed in condensing mesenchyme that will give rise to cartilage. Fgfr2 remains expressed in reserve chondrocytes and appears to be down regulated in proliferating chondrocytes, whereas Fgfr1 is expressed in hypertrophic chondrocytes. Later in development, Fgfr1 and Fgfr2 are both expressed in the perichondrium and periosteum, tissues that give rise to osteoblasts and cortical bone. In contrast to Fgfr1 and Fgfr2, Fgfr3 is prominently expressed in proliferating chondrocytes where it regulates cell growth and differentiation (Naski et al., 1998) and in differentiated osteoblasts where it regulates bone density and cortical thickness (Valverde-Franco et al., 2004; Xiao et al., 2004).
Activating mutations in FGFR3 in humans and mice causes achondroplasia, whereas mice lacking Fgfr3 (Fgfr3−/−) showed skeletal overgrowth accompanied by increased chondrocyte proliferation and an expanded proliferating and hypertrophic chondrocyte zone (Chen et al., 1999; Colvin et al., 1996; Deng et al., 1996; Naski et al., 1998). The skeletal overgrowth is thought to result from failure of the growth plate to fully close. As adults, Fgfr3−/− mice were osteopenic, suggesting a role for FGFR3 signaling in differentiated, Fgfr3-expressing osteoblasts (Valverde-Franco et al., 2004).
Mice lacking Fgfr2 (Fgfr2−/−) die at embryonic day 10.5 (E10.5), prior to skeletal development (Xu et al., 1998). The contribution of FGFR2 signaling to skeletal development has been clarified to some extent by using splice form-specific knockouts and conditional gene deletion approaches in mice. These studies demonstrated that FGFR2 positively regulates bone growth and the anabolic function of osteoblasts. The resulting phenotype of mice lacking mesenchymal Fgfr2 included skeletal dwarfism, decreased bone density, incomplete formation of the dorsal vertebrae and tarsal joint fusion (Eswarakumar et al., 2002; Yu et al., 2003).
Embryos lacking Fgfr1 (Fgfr1−/−) die shortly after gastrulation (Yamaguchi et al., 1994), necessitating a conditional knockout approach to address function later in development. Although the contribution of Fgfr1 to specific skeletal lineages is not known, Fgfr1 has recently been shown to be important for early limb bud development and distal skeletal patterning. Fgfr1 is expressed in limb bud mesenchyme that gives rise to mesenchymal condensations and eventually to the chondrogenic and osteogenic lineage (Orr-Urtreger et al., 1991; Peters et al., 1992; Xu et al., 1999b). Hypomorphic alleles of Fgfr1 or conditional inactivation of Fgfr1 prior to limb bud initiation affects digital patterning and the formation of some skeletal elements (Li et al., 2005; Partanen et al., 1998; Verheyden et al., 2005). To address the role of FGFR1 signaling specifically in the osteo-chondrocyte lineage, we have conditionally inactivated Fgfr1 in osteo-chondro-progenitor cells and in differentiated osteoblasts. Here we provide in vivo and in vitro evidence that demonstrates multiple roles for FGFR1 signaling in endochondral bone growth and remodeling. We demonstrate that loss of signaling through FGFR1 results in a delay in terminal maturation of hypertrophic chondrocytes. In osteoblasts, we show that FGFR1 signaling differentially regulates osteoblast growth and differentiation depending on the stage of osteoblast maturation.
Materials and methods
Mice
The conditional allele for FGFR1 has been previously generated and characterized (Pirvola et al., 2002). A null allele for FGFR1 (Fgfr1Δ/+) was made by germline Cre-mediated excision of the floxed region of Fgfr1. Col2-Cre transgenic mice (Ovchinnikov et al., 2000) were used to inactivate Fgfr1 in the osteo-chondrocyte lineage. Mice heterozygous for an Fgfr1 null allele (Fgfr1Δ/+) and heterozygous for Cre (Col2-Cre/+) were mated to mice homozygous for a floxed allele of Fgfr1 (Fgfr1flox/flox). Conditional knockout mice (Fgfr1Col2-cko) with a genotype Fgfr1flox/Δ,Col2-Cre/+, were born with a normal Mendelian frequency. To selectively inactivate the floxed allele of Fgfr1 in differentiated osteoblasts, α1(I)-collagen-Cre (Col1-Cre) transgenic mice (Dacquin et al., 2002) were mated to either Fgfr1flox/flox or Fgfr1flox/Δ mice to yield Fgfr1Col1-cko conditional knockout mice. Both genotype combinations were used for different experiments and both yielded similar phenotypes.
Osteoblast and osteoclast cell culture
Osteoblasts were derived from long bones following a protocol similar to that used by Declercq et al. (2004). Briefly, femur and tibia from 1.5- to 3-month-old mice were separated from soft tissue, bone marrow was flushed out with PBS, and bones were washed several times in PBS. Bone chips were then incubated in α-minimal essential medium (α-MEM) containing 0.2 mg/ml bacterial collagenase (type 1, Sigma) for 30 min at 37°C with shaking. The medium was discarded and replaced with fresh medium containing collagenase and incubated for an additional 30 min. Released cells were collected after three collagenase digestions. Cells and digested bone pieces were collected by centrifugation and suspended in α-MEM containing 10% fetal bovine serum and antibiotics. Cells were then seeded onto 250 ml tissue culture flasks or 10 cm plates and allowed to proliferate to confluence. To measure cell proliferation, cells were trypsinized and 5000 cells were plated per well in a 96-well plate; medium was replaced with medium containing 3H-thymidine (1 μCi/well) for 4 h before lysis and harvest on glass fiber filters. Mineralization medium contained 50 μg/ml ascorbic acid and 10 mM β-glycerophosphate.
Alkaline phosphatase activity in osteoblasts
Alkaline phosphatase enzymatic activity was measured using a colorimetric assay (Sigma FAST™-pNitrophenyl Phosphate tablets, N9389, Sigma) as described (Sugiyama et al., 2003). 10,000 cells per well were plated in a 24-well plate.
Alizarin red staining of mineralized osteoblast cultures
Cell cultures were washed with PBS and then fixed in 70% ethanol for 1 h. After fixation, cells were washed with water and stained with 0.4% Alizarin red (Sigma) for 15 min. Cells were then washed with water, dehydrated with 70% ethanol and dried. Quantification of mineral content was performed as described (Murshed et al., 2005). Bound dye was dissolved in 10% glacial acetic acid and measured at 405 nm.
β-Galactosidase staining
β-Galactosidase staining was performed as previously described (Yu et al., 2003) on whole embryos or frozen sections.
In situ hybridization
Plasmids used to generate riboprobes for in situ hybridization were from the following sources: Ihh (A. McMahon); Pth1r and Opn (H. Kronenberg); Collagen X (B. Olson); Cbfa1/Runx2 (B. de Crombrugghe), Mmp9 (Z. Werb), Trap (Ek-Rylander et al., 1991), Collagen I (Metsaranta et al., 1991), Fgfr1 (Peters et al., 1992). Radioactive in situ hybridization was performed on paraffin embedded tissue as previously described (Xu et al., 1999a).
Immunohistochemistry
The following antibodies were used for immunostaining: FGFR1 (1:400, Santa Cruz), β-catenin (1:100, Santa Cruz), osteopontin (1:100, MPIIIB10, Department of Biological Sciences, University of Iowa, Iowa City). The secondary antibodies used in immunofluorescence staining were mouse anti rabbit (1:100, Chemicon), goat anti mouse (1:200, Chemicon).
Bone morphometric analysis
Bone volume was estimated by tracing diaphyseal or trabecular bone areas on photographed histological sections using Canvas X software. Cell numbers were counted within a box drawn over comparable trabecular regions of control and conditional knockout embryos.
RNA isolation, cDNA synthesis and qRT-PCR analysis
RNA was isolated from three biological samples. Each sample consisted of one 250-ml tissue culture flask of long-bone-derived cells grown in mineralization media for 1–3 weeks. Total RNAwas isolated from cell cultures or tissues using the RNAqueous™ kit (Ambion Inc.). Equal amounts of RNA were used to prepare cDNAwith random hexamers and PCR amplification using the manufacturer's protocol (Invitrogen SuperScript™). qRT-PCR amplifications used the following set of primers:
Gapdh: F-5′-TGGCAAAGTGGAGATTGTTGCC-3′;
R-5′-AAGATGGTGATGGGCTTCCCG-3′;
mFgfr1IIIc: F-5′CACATCGAGGTGAACGGGAGTAAG-3′; and
R-5′ CGCATCCTCAAAGGAGACATTCC-3′.
Fgfr2, Fgfr3 and ColI primers were as described (Chikazu et al., 2000; Mathy et al., 2003; Saghizadeh et al., 2005). QRT-PCR analysis was performed using SYBR green (BioRad) and a BioRad iCycler. Standard PCR amplification conditions were as follows: Gadph: (30 cycles, 94°C for 1 min, 55°C for 1 min, 72°C for 1.50 min). RT-PCR primers to detect the Fgfr1 delta allele (Δ) were as described (Ohkubo et al., 2004).
Bone mass measurements by dual energy x-ray absorptiometry
Bone mineral density (BMD) was measured using dual-energy X-ray absorptiometry (DEXA) on whole mice not including the head, and analyzed on a PIXImus mouse densitometer (LUNAR Corp., Madison, Wisconsin).
Results
Expression of Fgfr1 in developing bone
In developing long bones, Fgfr1 is expressed in hypertrophic chondrocytes, in the perichondrium and in the trabecular region (primary spongiosa). To identify the specific cell types that express FGFR1, histology and immunohistochemistry were used to identify both cuboidal and spindle-shaped osteoblasts and corresponding FGFR1-expressing cells. Cuboidal osteoblasts are large differentiated cells that produce an extracellular matrix, and spindle-shaped osteoblasts are thin quiescent cells that line bone trabeculae (Hoshi et al., 1999; Zhang et al., 2003). At postnatal day 0 (P0), the trabecular region contained both large cuboidal cells and spindle-shaped cells (Figs. 1A–C). Similar to what was shown previously (Calvi et al., 2003; Glass et al., 2005; Mansukhani et al., 2005; Zhang et al., 2003), both of these populations of cells express osteopontin and β-catenin (Figs. 1D–E′). Immunohistochemistry, with an antibody against the carboxy terminus of FGFR1, showed FGFR1 expression in a similar pattern to these osteoblast markers in both cuboidal and spindle-shaped osteoblasts (Figs. 1F and F′), demonstrating that FGFR1 is expressed in different stages of osteoblast maturation.
Fig. 1.
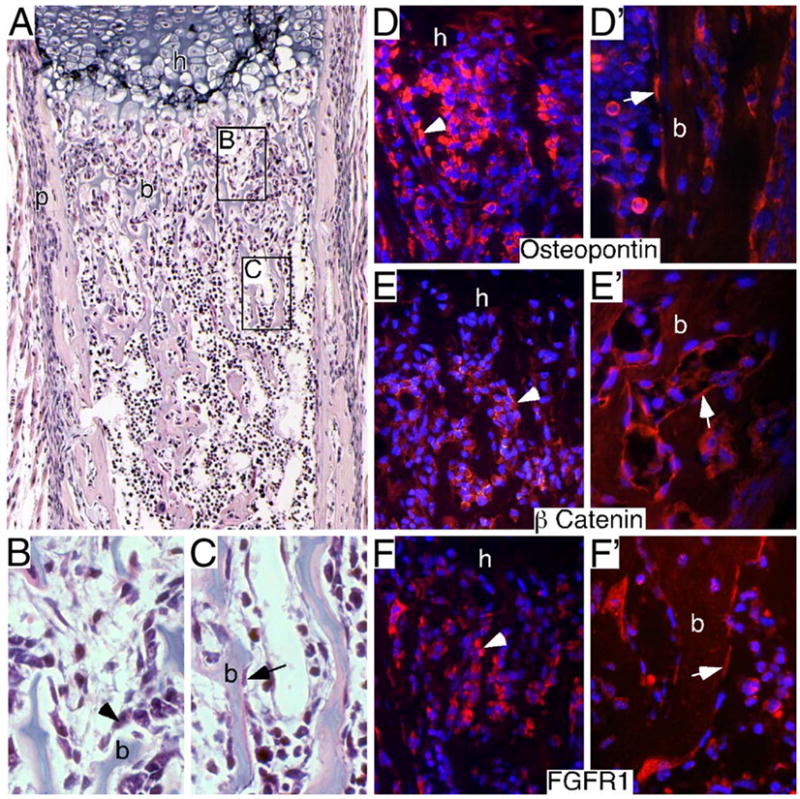
Identification of FGFR1-expressing osteoblast populations in developing endochondral bone. (A–C) Neonatal (P0) proximal femur histology stained with hematoxylin and eosin. Proximal (B) and distal (C) trabecular regions are shown at high magnification to identify cubiodal (arrowhead in panel B) and spindle-shaped (arrow in C) osteoblasts lining bone trabeculae. (D–F′) Immunohistochemical identification of osteopontin, β-catenin and FGFR1 in proximal (D–F) and distal (D′–F′) trabecular regions. Osteopontin immunohistochemistry (D and D′) and β-catenin immunohistochemistry (E and E′) identifying cuboidal osteoblasts (arrowhead) in proximal trabecular regions (D, E) and spindle-shaped osteoblasts (arrow) lining bone trabeculae in distal trabecular regions (D′, E′). FGFR1 immunohistochemistry also identifies cuboidal cells in proximal trabecular regions (F) and spindle shaped cells lining trabeculae in distal trabecular regions (F′). A, 10× objective; B–F′, 40× objective.
Conditional inactivation of Fgfr1 in osteo-chondro-progenitor cell lineages
To address the role of FGFR1 in the chondrocyte and osteoblast lineages, a floxed allele of Fgfr1 (Trokovic et al., 2003) was targeted with a proα1(II) collagen-Cre (Col2-Cre) transgene (Ovchinnikov et al., 2000). Type II collagen is an early marker of the mesenchymal condensation and, later in development, a specific marker for the chondrocyte lineage. Mating of Col2-Cre mice with ROSA26 reporter mice (R26R) (Soriano, 1999) activated β-galactosidase (β-gal) expression in both the chondrocyte and osteoblast lineages, but not in osteoclasts or bone marrow stroma (Fig. 2A, and data not shown) (Ovchinnikov et al., 2000).
Fig. 2.
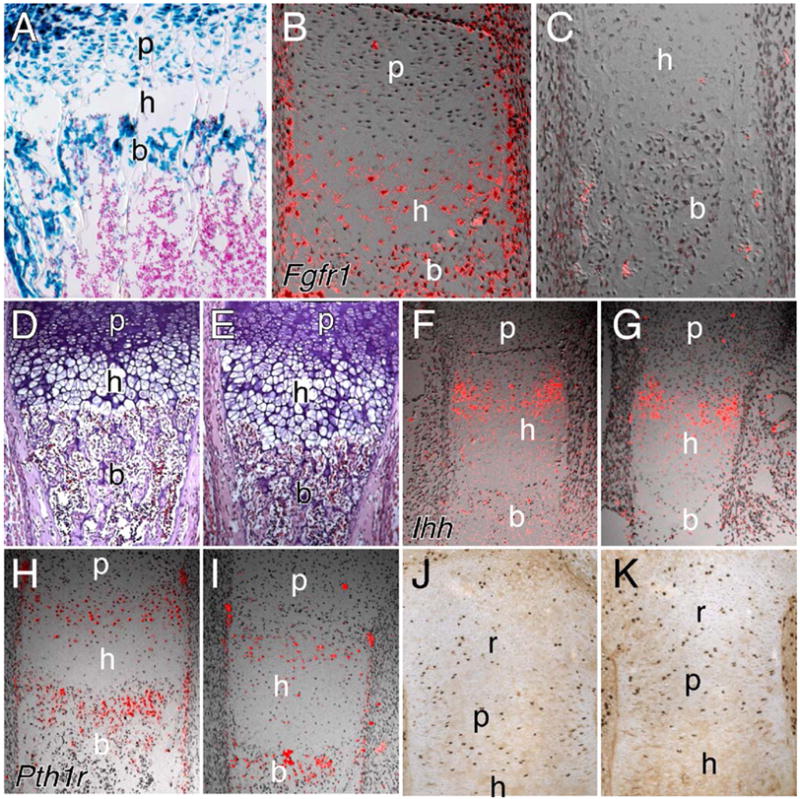
Col2-Cre-mediated inactivation of Fgfr1. (A) Histological section of the distal femur from a P10 Col2-Cre,R26R mouse showing X-gal staining in both chondrocytes and osteoblasts. (B, C) Expression of Fgfr1 at E16.5 in the femur from a control (B) and an Fgfr1Col2-cko (C) embryo showing loss of Fgfr1 expression in hypertrophic chondrocytes, and reduced expression in perichondrial and trabeculae tissue. Note that Fgfr1 expression is normally excluded from proliferating chondrocytes. (D, E) Hematoxylin and eosin-stained sections of femur from control (D) and Fgfr1Col2-cko (E) embryos at E16.5 showing an expanded HC zone in the absence of Fgfr1. (F–I) Expression analysis of Ihh (F, G) and Pth1r (H, I) in the femur at E16.5. No significant difference in expression in control (F, H) and Fgfr1Col2-cko (G, I) embryos was observed. (J, K) BrdU labeling of the proximal tibia at E18.5 showing no significant difference in proliferation in control (J) and Fgfr1Col2-cko (K) embryos. A–C, J, K, 20× objective; D–I, 10× objective. r, reserve chondrocyte zone; p, proliferating chondrocyte zone; h, hypertrophic chondrocyte zone; b, trabecular bone.
To inactivate Fgfr1 in the osteo-chondrocyte lineage, Fgfr1Δ/+,Col2-Cre/+ mice were mated to Fgfr1flox/flox mice. Conditional knockout mice (Fgfr1Col2-cko), born with a normal Mendelian frequency, were indistinguishable from wild-type littermates. Skeletal preparations of P0 mice revealed a rounded cranium but an otherwise normal appearing skeleton with no delay in the formation of ossification centers. The efficiency of Col2-Cre mediated Fgfr1 gene deletion was analyzed by RNA in situ hybridization. Histological sections of the femur of Fgfr1Col2-cko embryos showed loss of Fgfr1 expression in hypertrophic chondrocytes and trace levels of expression in trabecular bone and in the periosteum (Figs. 2B and C), suggesting nearly complete inactivation of the Fgfr1 gene in skeletal lineages.
Defects in growth plate development in Fgfr1Col2-cko mice
To examine the growth plate of Fgfr1Col2-cko mice, histological sections were prepared from E16.5-18.5 limbs. At E16.4, femur and tibia sections showed a ~60% increase (femur 58%, P = 0.0001, n = 6; tibia 68%, P = 0.0003, n = 5) in the height of the hypertrophic chondrocyte zone in Fgfr1Col2-cko mice compared to littermate controls (Figs. 2D and E). No significant change was observed in the proliferating or prehypertrophic regions. The increased size of the hypertrophic zone seen in Fgfr1Col2-cko embryos could result from increased differentiation, increased cell size or decreased loss of cells at the chondro-osseous junction. Histological sections suggested that cell size was not affected by loss of FGFR1 signaling. To examine chondrocyte differentiation, the expression of the parathyroid hormone receptor (Pth1r) and Indian hedgehog (Ihh) was examined by in situ hybridization. Both of these genes are expressed in pre-hypertrophic chondrocytes and define the pool of cells that is committed to hypertrophy. Similar levels of expression of Pth1r and Ihh were observed in Fgfr1Col2-cko and control growth plates at E16.5 and at E18.5, suggesting that the rate of chondrocyte differentiation was not significantly affected (Figs. 2F–I, and data not shown). Chondrocyte proliferation, assessed by BrdU immunohistochemistry, showed no significant difference between Fgfr1Col2-cko and control tibia (Figs. 2J and K) or femur (Supplementary Figs. 1A and B) growth plates at E18.5.
Delayed terminal differentiation of hypertrophic chondrocytes could also account for an increased hypertrophic chondrocyte zone. Type X collagen (ColX), a specific marker of the hypertrophic chondrocyte, was expressed at a similar intensity in both Fgfr1Col2-cko and control growth plates at E16.5. However, in Fgfr1Col2-cko growth plates, the area of expression was broader and ColX-positive cells were found ectopically located in the trabecular region (Figs. 3A and B). This suggested that the degradation of hypertrophic chondrocytes was delayed. Osteopontin (Opn) is expressed in distal hypertrophic zone chondrocytes and in osteoblasts (Lian et al., 1993). Opn expression at the chondro-osseous junction was significantly reduced in Fgfr1Col2-cko embryos at E18.5 (Figs. 3C and D). This suggested that maturation of hypertrophic chondrocytes and/or trabecular osteoblasts was delayed. The frequency of apoptotic cells (TUNEL labeling) in the distal hypertrophic regions was low at this stage of development but appeared similar in control and Fgfr1Col2-cko growth plates (data not shown).
Fig. 3.
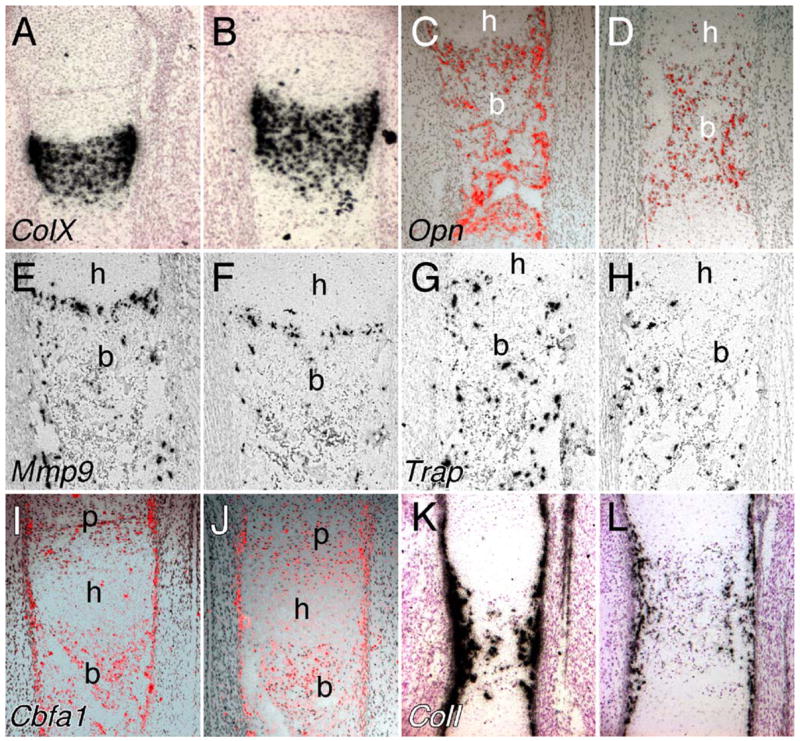
Development of the distal chondro-osseous junction of the femur of Fgfr1Col2-cko mice at E16.5. (A, B) Type X collagen expression showing an expanded hypertrophic chondrocyte zone and type X collagen-expressing cells in the trabecular region CKO tissue (B). (C, D) Osteopontin expression is reduced in Fgfr1Col2-cko femurs (D) indicating delayed osteoblast differentiation. Mmp9 (E, F) and Trap (G, H) in situ hybridization showing reduced expression of both markers at the chondro-osseous junction in Fgfr1Col2-cko femurs (F, H) compared to controls (E, G). (I–L) Expression of Cbfa1 (I, J) and type I collagen (K, L), showing similar levels of expression of Cbfa1 but reduced levels of expression of type 1 collagen in Fgfr1Col2-cko femurs (J, L) compared to controls (I, K). (A–L) 10× objective. p, proliferating chondrocyte zone; h, hypertrophic chondrocyte zone; b, trabecular bone.
Vascular invasion of the hypertrophic zone is required for the influx of both osteoblasts and osteoclasts. Osteoclasts are necessary for cartilage matrix degradation, an initial step in bone marrow cavity formation. MMP9 and MMP13 are proteases that are required for this process (Ortega et al., 2004; Stickens et al., 2004; Vu et al., 1998). Mmp9 is highly expressed in terminal hypertrophic chondrocytes, monocytes and in osteoclast/chondroclasts. Mmp13 is expressed by terminal hypertrophic chondrocytes and osteoblasts. Mice lacking either Mmp9 or Mmp13 develop an expanded hypertrophic zone and Mmp9 and Mmp13 synergize with each other in this process (Stickens et al., 2004). In Fgfr1Col2-cko embryos at E18.5, in situ hybridization analysis showed that Mmp9 expression was reduced at the chondro-osseous junction (Figs. 3E and F). This was consistent with reduced expression of Trap (tartrate-resistant acid phosphatase) mRNA (Figs. 3G and H) and a 49% decrease in the number of cells positive for TRAP enzymatic activity in the femur diaphyses of E16.5 Fgfr1Col2-cko embryos (n = 3, P = 0.006) (Supplementary Figs. 1C and D). The level of expression of Mmp13 was similar in control and Fgfr1Col2-cko embryos (data not shown). By postnatal day 14 (P14), the length of the hypertrophic chondrocyte zone returned to normal in Fgfr1Col2-cko mice (data not shown). This transient expansion of the hypertrophic zone is similar to what has been seen in Mmp9−/−and Mmp13−/− mice (Stickens et al., 2004).
Fgfr1 regulates osteoblast function but not cell fate specification
Cbfa1/Runx2 is a transcription factor required for chondrogenesis and osteogenesis. Cbfa1 is expressed in osteopro-genitor cells, prior to the earliest stages of ossification, in proliferating chondrocytes and in osteoblasts (Otto et al., 1997; Yoshida et al., 2002). Histological analysis of E16.5 Fgfr1Col2-cko femurs showed normal levels of Cbfa1 expression in chondrocytes and in periosteal and trabecular regions, indicating normal specification of the chondrocyte and osteoblast lineage (Figs. 3I and J). Interestingly, expression of Type I collagen, a marker of differentiating osteoblasts, was reduced in Fgfr1Col2-cko embryos (Figs. 3K and L), suggesting a delay in osteoblast differentiation or an anabolic deficit in the osteoblast. Osteocalcin (Oc), a marker of differentiated osteoblasts, was expressed at very low levels in both control and Fgfr1Col2-cko embryos at this stage, and no difference in expression could be detected (data not shown). However, by postnatal day 14, Oc expression was reduced in the secondary ossification center of Fgfr1Col2-cko mouse femurs (Supplementary Figs. 1E and F).
Conditional inactivation of Fgfr1 in the osteoblast compartment
To assess the function of Fgfr1 in differentiated osteoblasts that express type I collagen, an α1(I)-collagen-Cre (Col1-Cre) transgenic line (Dacquin et al., 2002) was used to selectively inactivate the floxed allele of Fgfr1. ColI-Cre transgenic mice, when crossed with a β-galactosidase reporter line (R26R), showed β-gal activity in the periosteum and trabecular region, indicating that Cre activity was restricted to differentiated osteoblasts (Fig. 4A). Specificity of the Fgfr1 gene deletion was demonstrated by genomic PCR, which showed the presence of a deleted Fgfr1 allele (Fgfr1Δ) in bone but not kidney tissue from Fgfr1flox/flox,ColI-Cre/+ mice (Fig. 4B). Immunohistochemical analysis of FGFR1 expression in trabecular bone also showed a reduction in FGFR1 staining intensity in Fgfr1flox/Δ,ColI-Cre/+ conditional knockout (Fgfr1ColI-cko) tissue compared to control tissue (Figs. 4C and D). To further examine Fgfr1 gene inactivation in osteoblasts, cultured long-bone-derived osteoblasts were allowed to differentiate in vitro and were then analyzed for the Fgfr1 gene deletion. The Fgfr1Δ allele was detected in osteoblast cultures derived from Fgfr1ColI-cko mice but not in osteoclast cultures derived from the same mice (Fig. 4E). RT-PCR analysis showed that mRNA containing the Cre-induced deletion was expressed in osteoblast cultures derived from Fgfr1ColI-cko mice but not from control Fgfr1flox/flox mice (Fig. 4F). Interestingly, higher levels of Fgfr1Δ were detected in osteoblasts that were induced to differentiate with dexamethasone, consistent with expression of Col1 in more differentiated osteoblasts.
Fig. 4.
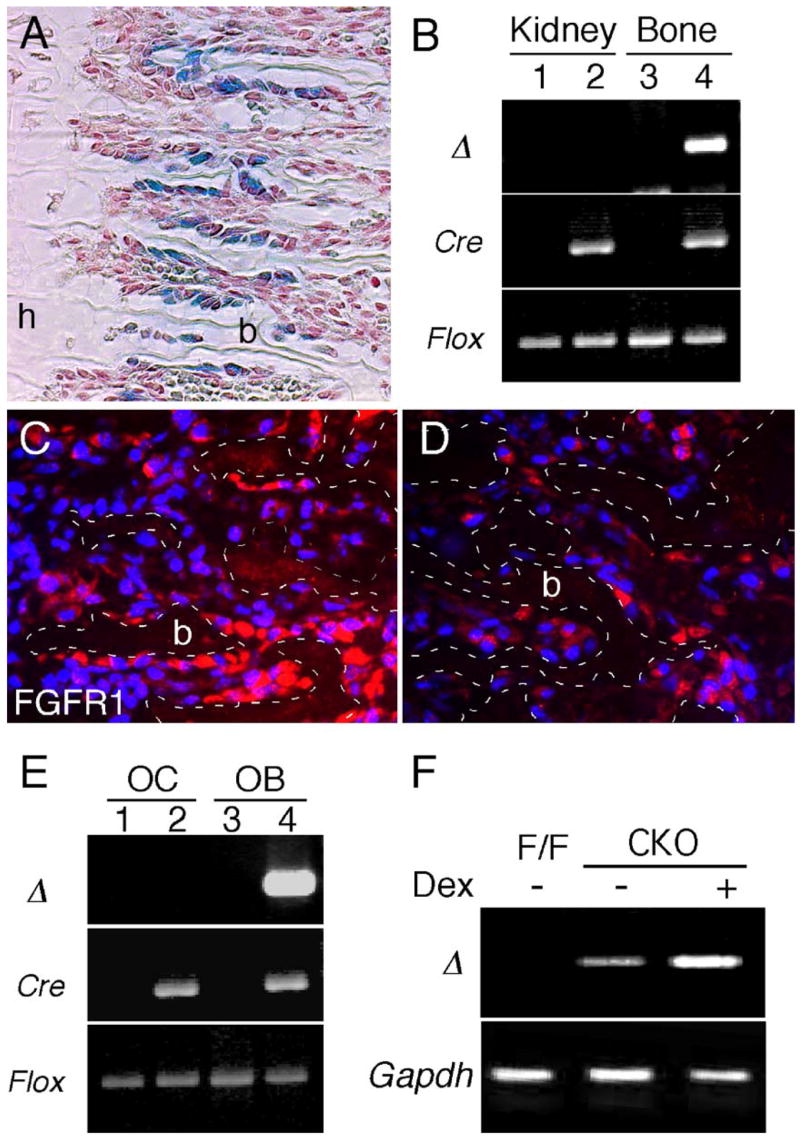
Col1-Cre-mediated inactivation of Fgfr1. (A) Histological section of the distal femur from a P7 ColI-Cre, R26R animal showing X-gal staining in osteoblasts lining trabecular bone. (B) Genomic PCR analysis showing Cre-mediated deletion of the Fgfr1 floxed allele in two month-old Fgfr1ColI-cko and control mice. PCR amplification of the deleted allele of Fgfr1 (Δ) was detected in bone but not kidney from Fgfr1ColI-cko mice (lanes 2 and 4). The Fgfr1 (Δ) allele was not detected in the absence of Cre (lanes 1 and 3). (C, D) Immunohistochemical detection of FGFR1 in P0 Fgfr1flox/flox (C) and Fgfr1ColI-cko (D) femurs showing decreased expression in Fgfr1ColI-cko tissue. Dotted lines outline trabecular bone (b). (E) Genomic PCR analysis of primary osteoclasts (OC, lanes 1 and 2) and osteoblasts (OB, lanes 3 and 4) showing Cre-mediated deletion of the Fgfr1 floxed allele only in osteoblast cultures from Fgfr1ColI-cko mice (lane 4). (F) RT-PCR analysis showing expression of the deleted Fgfr1 allele (Δ) in Fgfr1ColI-cko-derived osteoblasts (cko) but not in control (F/F) osteoblasts. Dexamethasone (Dex)-treated Fgfr1ColI-cko osteoblasts increased the level of Fgfr1Δ expression. Gapdh expression was used as a control for cDNA synthesis and PCR amplification. A, C and D, 10× objective.
Increased trabecular bone volume in Fgfr1ColI-cko neonatal mice
Histological sections, prepared from the femur and tibia of neonatal mice, showed increased trabecular bone volume and a decreased marrow space in Fgfr1ColI-cko embryos (Figs. 5A and B). Morphometric analysis of histological sections showed a 54% increase in the trabecular bone volume/total volume ratio in neonatal Fgfr1ColI-cko mice (Fig. 5C). Examination of trabecular architecture showed decreased cartilage matrix and increased osteoid in Fgfr1ColI-cko embryos. This suggested an enhanced mineralization of cartilage matrix and increased deposition of bone matrix in mice with Fgfr1-deficient osteoblasts. Consistent with this, von Kossa staining showed increased mineralization in the trabecular region of Fgfr1ColI-cko mice compared to control mice (Figs. 5E and F). Morphometric analysis of Fgfr1ColI-cko neonatal limbs showed a 52% reduction in the number of cuboidal-shaped osteoblasts in the trabecular region (Fig. 5D). At high magnification, these osteoblasts appeared to have an increased cytoplasmic area compared to wild-type osteoblasts (data not shown).
Fig. 5.

Increased trabecular bone volume and enhanced osteoblast ossification in Fgfr1ColI-cko mice. (A, B) Hematoxylin and eosin-stained sections of the distal femur at P0 mice showing increased trabecular bone in Fgfr1ColI-cko (B) compared to control (A) mice. (C, D) Morphometric analysis of P0 mouse femur trabecular bone showing increased bone volume (BV) to total volume (TV) ratio and decreased number of cuboidal osteoblasts in Fgfr1ColI-cko (CKO) compared to control (F/F) mice. *P < 0.05, **P = 0.004. (E, F) Mineralization of the distal femur at P0 detected by von Kossa staining, showing increased mineralization in the trabecular region in Fgfr1ColI-cko (F) compared to control (E) mice. (G, H) TRAP stain of distal femur sections from control mice showing osteoclasts spread over bone trabeculae in control bone (G, G′) but appearing more rounded and less adherent in Fgfr1ColI-cko bone (H, H′). (I–L) Detection of maturing osteoblasts with X-gal staining of P0 control (Col1-Cre,R26R) and Fgfr1Col1-cko,R26R femur. (I, J) Whole-mount X-gal staining analysis showing intense staining in the trabeculae and cortical region in Fgfr1ColI-cko femur. (K, L) Histological sections showing increased numbers of X-gal-positive osteoblasts in Fgfr1ColI-cko (L) compared to control (K) femurs. A, B, E–H, 10× objective; K, L, G′, H′, 20× objective. h, hypertrophic chondrocyte zone; b, trabecular bone; p, periosteum.
The observed increased trabecular bone volume could result from a combination of either increased deposition of bone matrix or decreased degradation and remodeling of bone. Because osteoclasts are essential for the bone remodeling process and are required to maintain the bone marrow cavity, we examined osteoclast number and morphology in bones from neonatal Fgfr1ColI-cko embryos by staining for tartrate-resistant acid phosphatase (TRAP) activity. The number of TRAP-positive osteoclasts in Fgfr1ColI-cko embryos was similar to that in wild-type embryos, suggesting normal numbers of osteoclast progenitor cells (Figs. 5G and H). However, in Fgfr1ColI-cko embryos, TRAP-positive cells were smaller and more rounded, whereas in control embryos, these cells were larger, flattened and spread over bone spicules (Figs. 5G′ and H′). Given that the Col1-Cre transgene showed no recombinase activity in cultured osteoclasts (Fig. 4E), the observed morphologic defect in osteoclast differentiation is likely to be secondary to FGFR1-dependent activities in osteoblasts.
To assess the extent of osteoblast maturation in control and Fgfr1ColI-cko embryos, we introduced the R26R reporter allele into this genetic background. β-Gal staining acts as a marker to identify differentiated osteoblasts that have expressed Col1-Cre. In Col1-Cre,R26R mice, β-gal staining can be detected by postnatal day 5 (P5) in trabecular zone osteoblasts (Dacquin et al., 2002). Interestingly, whole-mount and histological sections of P0 femurs from Fgfr1ColI-cko, R26R embryos showed an increase in the intensity of β-gal staining compared to that of femurs from Col1-Cre,Fgfr1flox/+,R26R control embryos (Figs. 5I–L). Because Col1-Cre should be inactivating, Fgfr1and activating R26R simultaneously only in differentiated osteoblasts, this observation suggests that differentiated osteoblasts lacking FGFR1 may enhance maturation of neighboring immature osteoblasts, thus increasing the number of cells expressing Cre recombinase. To further examine the affects of loss of FGFR1 on osteoblast maturation, we examined osteoblast maturation in vitro.
Osteoblast differentiation, in vitro
To examine the effect of FGFR1 on the ability of osteoblasts to differentiate in vitro, osteoblast cultures were prepared from the femur and tibia of 1.5- to 2-month-old wild-type and Fgfr1 conditional knockout mice. Osteoblast cultures prepared from Fgfr1Col2-cko mice showed a 39% increase in the rate of proliferation compared to cultures prepared from control mice (Fig. 6A). Furthermore, alkaline phosphatase activity was significantly lower in Fgfr1Col2-cko-derived cultures (Fig. 6B). These data suggest that FGFR1 signaling normally acts to inhibit proliferation and enhance commitment to the osteoblast lineage in osteo-chondro-progenitor cells. To assess the ability of FGFR1-deficient osteo-progenitor cells to differentiate and mineralize, cultures were treated with medium containing β-glycerophosphate and ascorbic acid. After three weeks in mineralization medium, mineralized bone nodules were identified by alizarin red staining. Consistent with delayed osteoblast maturation, Alizarin red binding was decreased by 3.3-fold in Fgfr1Col2-cko cultures compared to control cultures (Figs. 6C–E).
Fig. 6.
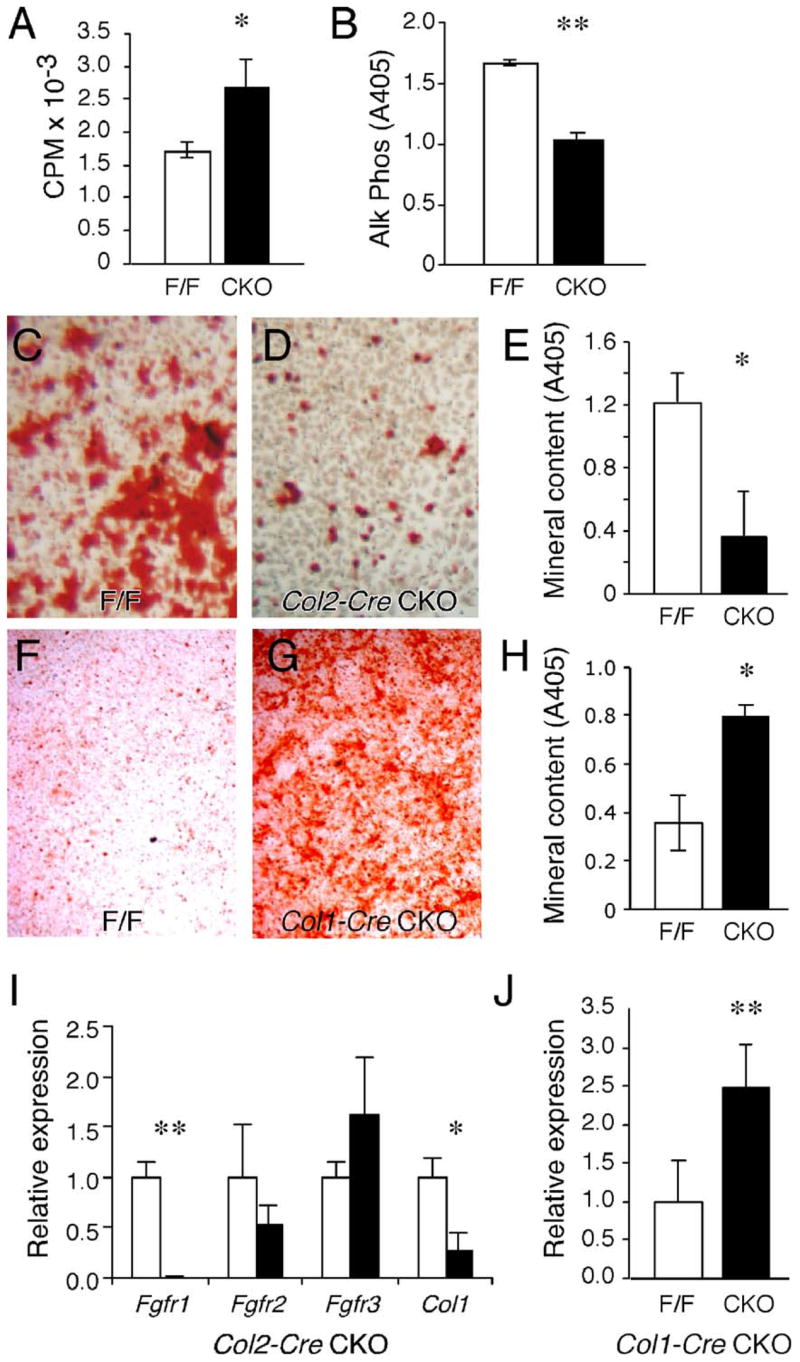
Growth and differentiation of osteoprogenitor cells in vitro. (A–E) Osteoprogenitor cell cultures derived from control and Fgfr1Col2-cko long bones. (A) Cell proliferation, assessed by 3H-thymidine incorporation, is increased in Fgfr1Col2-cko cells compared to control cells (n = 3, * P = 0.02). (B) Alkaline phosphatase activity is decreased in Fgfr1Col2-cko cells compared to control cells (n = 3, ** P < 0.001). (C–E) Alizarin red staining of osteoprogenitor cells maintained in mineralization medium for 3 weeks showing fewer mineralized nodules in Fgfr1Col2-cko cultures (D) compared to control cultures (C). (E) Quantification of the mineral deposition in Fgfr1Col2-cko and control cultures (n = 3, * P = 0.01). (F–H) Alizarin red staining of osteoprogenitor cells derived from control and Fgfr1Col1-cko long bones maintained in mineralization medium for 3 weeks showing increased numbers of mineralized nodules in Fgfr1Col1-cko cultures (F) compared to control cultures (G). (H) Quantification of the mineral deposition in Fgfr1Col1-cko and control cultures (n = 3, * P = 0.01). (I) Quantitative RT-PCR analysis of osteoblast cultures maintained in mineralization medium for 1 week showing decreased expression of Fgfr1 (n = 3, ** P < 0.0001) and Col1 (n = 3, * P < 0.01) in Fgfr1Col2-cko cultures (solid bars) compared to control cultures (open bars). (J) Quantitative RT-PCR analysis of osteoblast cultures maintained in mineralization medium for 2 weeks showing increased expression of Fgfr3 in Fgfr1Col1-cko cultures (solid bar) compared to control cultures (open bar) (n = 4, ** P < 0.008).
In contrast to cultures prepared from Fgfr1Col2-cko mice, cultures prepared from the femur and tibia of 1.5- to 2-month-old Fgfr1Col1-cko mice showed similar levels of alkaline phosphatase activity and no change in the rates of proliferation after one week in culture. These data suggest that differentiation of osteo-chondro-progenitor cells were not affected by Col1-Cre, consistent with Cre expression only in differentiated osteoblasts. However, when Fgfr1Col1-cko cultures were placed in mineralization medium for three weeks, alizarin red staining showed significantly more intense staining than control cells (2.3-fold increase in alizarin red binding) (Figs. 6F–H). These data indicate that once osteoblasts are induced to mature, inactivation of Fgfr1 can further enhance their differentiation.
Osteoblast cultures lacking Fgfr2 or Fgfr3 show decreased mineralization (Valverde-Franco et al., 2004 and data not shown). Fgfr3−/− cultures also showed increased expression of Fgfr1 (Valverde-Franco et al., 2004). Another mechanism by which FGFR1 signaling could regulate osteoblast maturation could be by regulating the expression of Fgfr2 and Fgfr3. We therefore examined the expression of Fgfr2 and Fgfr3 in Fgfr1ColI-cko osteoblasts cultured under ossifying conditions. In cultures prepared from Fgfr1Col2-cko mice, no significant change in the levels of expression of Fgfr2 and Fgfr3, but a significant decrease in the level of Col1 was observed (Fig. 6I). In cultures prepared from Fgfr1Col1-cko mice, an increased level of expression of Fgfr3 was observed (Fig. 6J). This is consistent with FGFR3 acting in differentiated osteoblasts to stimulate mineralization (Valverde-Franco et al., 2004).
Bone pathology during skeletal maturation
Histological analysis of the growth plate of P10 to P15 Fgfr1Col1-cko animals showed a partial resolution of the trabecular bone overgrowth phenotype seen at earlier ages. However, the trabecular region remained disorganized with persistent islands of hypertrophic chondrocytes trapped within trabecular bone and vascular sinuses encroaching on the chondro-osseous junction (Figs. 7A and B). By P10, both control and Fgfr1Col1-cko bones showed normalized osteoclast morphology, with multinucleated osteoclasts attached and spread out over trabeculae (data not shown). Examination of histological sections of femur and tibia of eight-month-old Fgfr1Col1-cko mice showed increased trabecular bone compared to age matched control mice (Figs. 7C and D). Morphometric analysis of the femurs revealed a 3 ± 0.5-fold increase in the trabeculae bone volume compared to control mice (Fig. 7E).
Fig. 7.
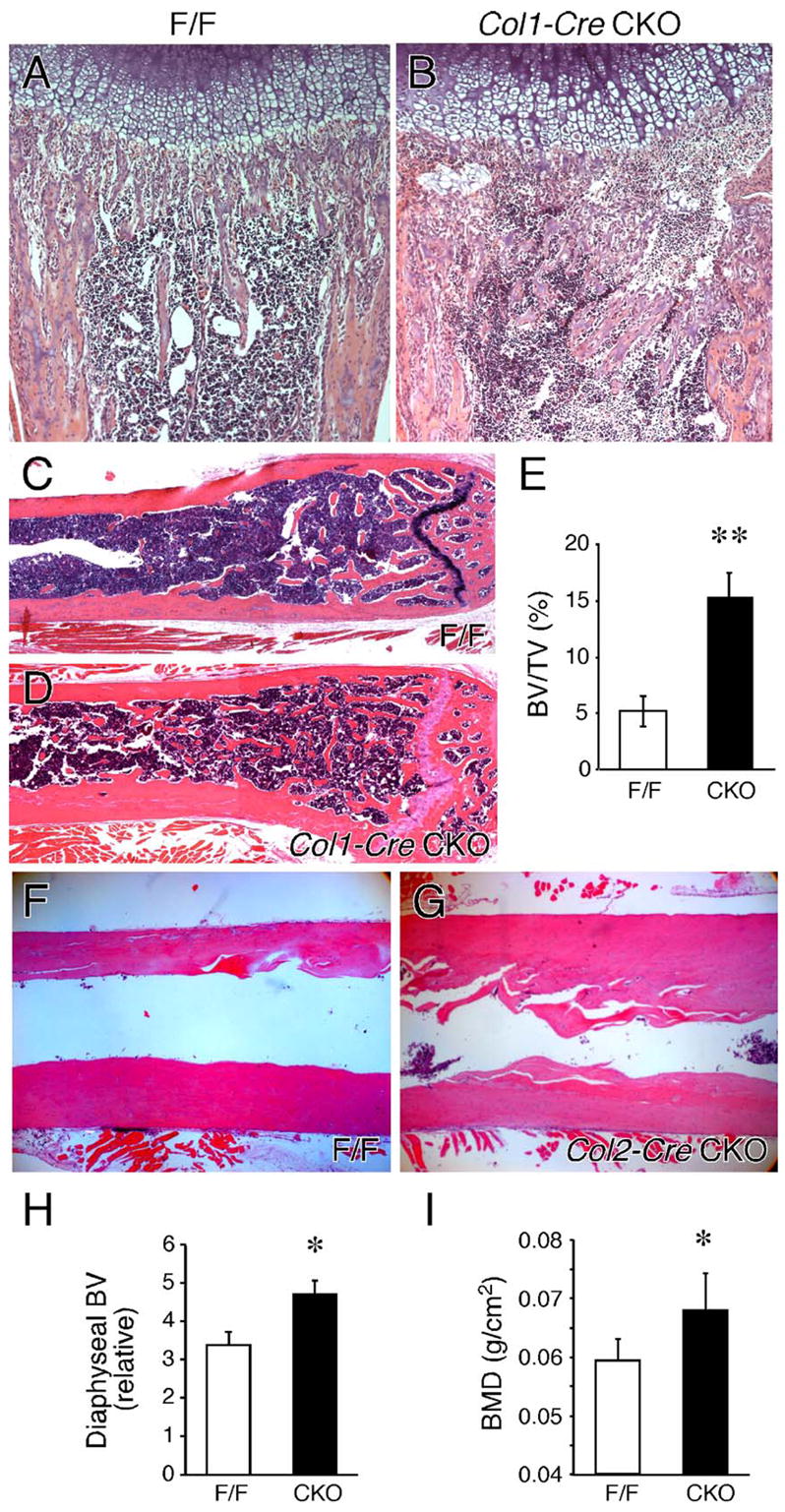
Skeletal maturation. (A, B) Hematoxylin and eosin-stained sections from control (A) and Fgfr1ColI-cko (B) distal femurs at postnatal day 10 (P10) showing irregular formation of the trabecular region, ectopic islands of hypertrophic chondrocytes and the presence of vascular sinuses adjacent to the chondro-osseous junction in Fgfr1ColI-cko tissue. (C–I) Increased bone formation in adult mice lacking Fgfr1. Histological sections of the distal femur of an eight-month-old control (C) and Fgfr1ColI-cko (D) mouse. Increased trabecular bone formation and trabeculae-like structures lining the cortical bone are evident in Fgfr1ColI-cko tissue. (E) Morphometric analysis of femur trabecular bone showing a 3 ± 0.5-fold increase in the bone volume (BV) to total volume (TV) ratio in Fgfr1ColI-cko (CKO) compared to control (F/F) mice (n = 4, **P < 0.001). Histological sections of the distal femur of a five-month-old control (F) and Fgfr1Col2-cko (G) mouse showing increased cortical bone formation. (H) Morphometric analysis of the femur diaphysis showing a 40% increase in the diaphyseal bone volume (BV) in Fgfr1Col2-cko (CKO) compared to control (F/F) mice (n = 3, *P = 0.01). (I) DEXA analysis showing a 14% increase in bone mineral density of Fgfr1Col2-cko compared to control mice (n = 5, WT; n = 6, CKO, *P = 0.02). A and B, 10×; C and D, 2.5×; F and G, 4×.
Histological analysis of femur and tibia sections of six month and older Fgfr1Col2-cko mice showed increased cortical bone thickness compared to age-matched controls (Figs. 7F–H); however, the trabecular regions appeared normal. Radiological examination of Fgfr1Col2-cko mice also showed increased cortical thickness of the femur and tibia (not shown). Consistent with increased cortical thickness, whole body bone mineral density (DEXA) was increased by 14% in female Fgfr1Col2-cko mice compared to age matched controls (Fig. 7I).
Discussion
Mutations in FGFR1 that cause Pfeiffer syndrome, Jackson–Weiss syndrome and osteoglophonic dysplasia in humans (Roscioli et al., 2000; Schell et al., 1995; White et al., 2005) provide genetic evidence for a role for FGFR1 in skeletal development. To define the function of FGFR1 in chondrocytes and in early and late stages of osteoblast development, we have conditionally inactivated a floxed allele of Fgfr1 in osteo-chondro-progenitor cells using Col2-Cre and in differentiated osteoblasts using Col1-Cre. Fgfr1Col2-cko mice developed defects in both chondrocytes and osteoblasts, whereas the phenotype of Fgfr1Col1-cko mice was restricted to osteoblasts.
Although Fgfr1Col2-cko mice developed an expanded hypertrophic chondrocyte zone, the precise role for FGFR1 signaling in the hypertrophic chondrocyte cannot be determined from these experiments because Fgfr1 is expressed and inactivated in both chondrocytes and cells within the perichondrium and chondro-osseous junction. Nevertheless, the experiments shown here suggest that loss of FGFR1 signaling primarily affects hypertrophic chondrocytes by acting on the chondro-osseous junction to limit mechanisms that degrade hypertrophic chondrocytes. For example, Fgfr1Col2-ckomice had decreased expression of MMP9 and decreased numbers of osteoclasts at the chondro-osseous junction, which could delay the degradation of hypertrophic chondrocytes (Ortega et al., 2004; Vu et al., 1998). Additionally, maturation of hypertrophic chondrocytes may also be delayed, as indicated by reduced expression of osteopontin. Interestingly, in mice in which Fgfr2 was inactivated in a similar pattern with Dermo1-Cre, the hypertrophic chondrocyte zone was decreased at later stages of development (P7 versus E16.5), and osteoclast numbers at the chondro-osseous junction were increased (Eswarakumar et al., 2002; Yu et al., 2003). Thus, although acting most potently at different stages of skeletal development, FGFR1 and FGFR2 appear to have opposite affects on distal growth plate development. This could be due to differential activation of FGFR1 and FGFR2 by FGF ligands, differences in the strength or downstream components of the FGFR signals, or expression of FGFR1 and FGFR2 at different stages of osteoblast and chondrocyte development.
Fgfr regulation of osteoblast proliferation and differentiation
Inactivation of FGFR1 in immature osteoblasts (Fgfr1Col2-cko) did not affect formation of osteoprogenitor cells, as indicated by normal levels of expression of Cbfa1, but did cause a delay in the maturation of osteoblasts, as shown by reduced expression of type 1 collagen and osteopontin. Consistent with this, osteoprogenitor cell cultures derived from cortical bone of Fgfr1Col2-cko mice showed increased rates of proliferation and decreased differentiation (decreased alkaline phosphatase expression and delayed mineralization). These data suggest that FGFR1 signaling in the osteoprogenitor cell normally acts to suppress proliferation and stimulate differentiation (Fig. 8A). In contrast, when Fgfr1ColI-cko osteoblasts were cultured under ossifying conditions, they showed accelerated differentiation, demonstrating that in differentiated osteoblasts, FGFR1 functions to suppress differentiation (Fig. 8B). Thus, FGFR1 signaling has stage-specific effects on osteoblast maturation.
Fig. 8.
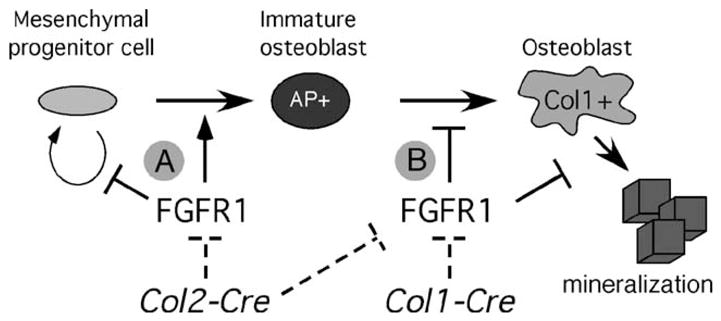
Model for FGFR1 regulation of osteoblast maturation. Inactivation of Fgfr1 at different stages of osteoblast development was achieved by using two different Cre-expressing transgenic lines. Col2-Cre inactivates Fgfr1 before commitment to the osteoblast lineage whereas Col1-Cre inactivates Fgfr1 after commitment to the osteoblast lineage (dashed lines). (A) FGFR1 functions at early stages of osteoprogenitor development to inhibit cell proliferation and promote differentiation. (B) FGFR1 also functions in committed osteoblasts to prevent further differentiation and mineralization. AP, alkaline phosphatase; Col1, type I collagen.
Regulation of osteoclast activation by osteoblast-FGFR1 signaling
Osteoclasts must spread out and attach to bone matrix to carry out their normal catabolic function (Teitelbaum, 2000). The observation that osteoclasts are smaller and not spread out over bone trabeculae in neonatal Fgfr1ColI-cko mice correlated with the observed increase in trabecular bone volume. This phenotype supports the hypothesis that Fgfr1-deficient osteo-blasts cannot maintain normal osteoclast activity during this developmental period. We therefore hypothesized that FGFR1 signaling through the osteoblast must be involved in the regulation of osteoclastogenic cytokine production. Consistent with this, preliminary data shows that Fgfr1ColI-cko osteoblast cultures express less RANKL (receptor activator of NFκβ ligand) and are not able to support osteoclastogenesis in in vitro co-culture. Further experiments will explore the stage-specific role of FGFR signaling in undifferentiated and differentiated osteoblasts’ ability to produce RANKL and other cytokines that regulate osteoclast activity.
FGF signaling regulates bone growth
Increased anabolic activity in Fgfr1ColI-cko osteoblasts was supported by the observation of increased numbers of β-gal-positive osteoblasts in Fgfr1Col1-cko;R26R embryos compared to ColI-Cre;R26R embryos. In contrast to mice lacking Fgfr1, mice lacking Fgfr2 or Fgfr3 became osteopenic as adults (Eswarakumar et al., 2002; Valverde-Franco et al., 2004; Yu et al., 2003). Thus, signaling through FGFR2 and FGFR3 acts to enhance bone formation. Consistent with this activity, osteoblast cultures derived from Fgfr2Col1-cko mice or Fgfr3−/− mice showed decreased mineralization when compared to control cultures (Valverde-Franco et al., 2004 and data not shown). The accelerated mineralization of Fgfr1ColI-cko osteoblasts in culture may be further enhanced by increased expression and activity of Fgfr3.
The FGF ligands that signal to FGFR1 in osteoblasts are not known; however, three FGFs (FGFs 2, 9 and 18) are likely candidates for this role. FGF9 and FGF18 are expressed in the perichondrium/periosteum, and mice lacking these FGFs show delayed ossification during mid-gestation skeletogenesis (Colvin et al., 1999; Liu et al., 2002; Ohbayashi et al., 2002) (I. Hung, DMO, unpublished data). FGF2 is expressed in periosteal cells and osteoblasts (Hurley et al., 1999; Sabbieti et al., 1999) and adult Fgf2−/− mice showed a loss of trabecular bone volume; however, no skeletal dysmorphology was reported in neonatal Fgf2−/− mice (Montero et al., 2000). These observations suggest that FGFs 2, 9 and 18 may act alone or redundantly to regulate osteoblast activity and physiology and that FGFs 9 and 18 may constitute the predominant signals during embryonic development, whereas FGF2 may be more important during postnatal stages. Consistent with a role for FGF2 in more differentiated osteoblasts, bone marrow stromal cultures from Fgf2−/− mice showed a significantly decreased ability to mineralize in vitro (Montero et al., 2000). The phenotypic similarities in Fgf2−/− mice and Fgfr3−/− mice suggest that FGF2 may signal through FGFR3 in osteoblasts to enhance mineralization.
The increased formation of bone at early ages and the increased number and rate of mineralization of differentiated osteoblasts could lead to the observed increase in bone volume observed in older mice. Furthermore, because in older mice chondrogenesis is not an active process and osteoblasts have progressed towards maturity, similarities between adult Fgfr1ColI-cko and Fgfr1Col2-cko mice suggest that the predominant role for FGFR1 signaling in mature bone is to decrease anabolic activity and slow terminal differentiation (mineralization) of osteoblasts (Fig. 8B). A component of this mechanism may be through suppression of FGFR3 expression.
Supplementary Material
Acknowledgments
We thank S. Teitelbaum and P. Ross for helpful discussions. This work was supported by funds from Washington University School of Medicine, NIH grant HD39952, HD049808 and the Alice and Julius Kantor Charitable Trust.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.ydbio.2006.05.031.
References
- Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR, Milner LA, Kronenberg HM, Scadden DT. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- Chen L, Deng CX. Roles of FGF signaling in skeletal development and human genetic diseases. Front Biosci. 2005;10:1961–1976. doi: 10.2741/1671. [DOI] [PubMed] [Google Scholar]
- Chen L, Adar R, Yang X, Monsonego EO, Li C, Hauschka PV, Yayon A, Deng CX. Gly369Cys mutation in mouse FGFR3 causes achondroplasia by affecting both chondrogenesis and osteogenesis. J Clin Invest. 1999;104:1517–1525. doi: 10.1172/JCI6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikazu D, Hakeda Y, Ogata N, Nemoto K, Itabashi A, Takato T, Kumegawa M, Nakamura K, Kawaguchi H. Fibroblast growth factor (FGF)-2 directly stimulates mature osteoclast function through activation of FGF receptor 1 and p42/p44 MAP kinase. J Biol Chem. 2000;275:31444–31450. doi: 10.1074/jbc.M910132199. [DOI] [PubMed] [Google Scholar]
- Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet. 1996;12:390–397. doi: 10.1038/ng0496-390. [DOI] [PubMed] [Google Scholar]
- Colvin JS, Feldman B, Nadeau JH, Goldfarb M, Ornitz DM. Genomic organization and embryonic expression of the mouse fibroblast growth factor 9 gene. Dev Dyn. 1999;216:72–88. doi: 10.1002/(SICI)1097-0177(199909)216:1<72::AID-DVDY9>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Dacquin R, Starbuck M, Schinke T, Karsenty G. Mouse alpha1(I)-collagen promoter is the best known promoter to drive efficient Cre recombinase expression in osteoblast. Dev Dyn. 2002;224:245–251. doi: 10.1002/dvdy.10100. [DOI] [PubMed] [Google Scholar]
- Declercq H, Van den Vreken N, De Maeyer E, Verbeeck R, Schacht E, De Ridder L, Cornelissen M. Isolation, proliferation and differentiation of osteoblastic cells to study cell/biomaterial interactions: comparison of different isolation techniques and source. Biomaterials. 2004;25:757–768. doi: 10.1016/s0142-9612(03)00580-5. [DOI] [PubMed] [Google Scholar]
- Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Ek-Rylander B, Bill P, Norgard M, Nilsson S, Andersson G. Cloning, sequence, and developmental expression of a type 5, tartrate-resistant, acid phosphatase of rat bone. J Biol Chem. 1991;266:24684–24689. [PubMed] [Google Scholar]
- Eswarakumar VP, Monsonego-Ornan E, Pines M, Antonopoulou I, Morriss-Kay GM, Lonai P. The IIIc alternative of Fgfr2 is a positive regulator of bone formation. Development. 2002;129:3783–3793. doi: 10.1242/dev.129.16.3783. [DOI] [PubMed] [Google Scholar]
- Glass DA, II, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, Karsenty G. Canonical wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8:751–764. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- Hoshi K, Komori T, Ozawa H. Morphological characterization of skeletal cells in Cbfa1-deficient mice. Bone. 1999;25:639–651. doi: 10.1016/s8756-3282(99)00223-9. [DOI] [PubMed] [Google Scholar]
- Hurley MM, Tetradis S, Huang YF, Hock J, Kream BE, Raisz LG, Sabbieti MG. Parathyroid hormone regulates the expression of fibroblast growth factor-2 mRNA and fibroblast growth factor receptor mRNA in osteoblastic cells. J Bone Miner Res. 1999;14:776–783. doi: 10.1359/jbmr.1999.14.5.776. [DOI] [PubMed] [Google Scholar]
- Li C, Xu X, Nelson DK, Williams T, Kuehn MR, Deng CX. FGFR1 function at the earliest stages of mouse limb development plays an indispensable role in subsequent autopod morphogenesis. Development. 2005;132:4755–4764. doi: 10.1242/dev.02065. [DOI] [PubMed] [Google Scholar]
- Lian JB, McKee MD, Todd AM, Gerstenfeld LC. Induction of bone-related proteins, osteocalcin and osteopontin, and their matrix ultrastructural localization with development of chondrocyte hypertrophy in vitro. J Cell Biochem. 1993;52:206–219. doi: 10.1002/jcb.240520212. [DOI] [PubMed] [Google Scholar]
- Liu Z, Xu J, Colvin JS, Ornitz DM. Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev. 2002;16:859–869. doi: 10.1101/gad.965602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansukhani A, Ambrosetti D, Holmes G, Cornivelli L, Basilico C. Sox2 induction by FGF and FGFR2 activating mutations inhibits Wnt signaling and osteoblast differentiation. J Cell Biol. 2005;168:1065–1076. doi: 10.1083/jcb.200409182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathy JA, Lenton K, Nacamuli RP, Fong KD, Song HM, Fang TD, Yang GP, Longaker MT. FGF-2 stimulation affects calvarial osteoblast biology: quantitative analysis of nine genes important for cranial suture biology by real-time reverse transcription polymerase chain reaction. Plast Reconstr Surg. 2003;112:528–539. doi: 10.1097/01.PRS.0000070729.05978.BB. [DOI] [PubMed] [Google Scholar]
- Metsaranta M, Toman D, De Crombrugghe B, Vuorio E. Specific hybridization probes for mouse type I, II, III and IX collagen mRNAs. Biochim Biophys Acta. 1991;1089:241–243. doi: 10.1016/0167-4781(91)90014-d. [DOI] [PubMed] [Google Scholar]
- Montero A, Okada Y, Tomita M, Ito M, Tsurukami H, Nakamura T, Doetschman T, Coffin JD, Hurley MM. Disruption of the fibroblast growth factor-2 gene results in decreased bone mass and bone formation. J Clin Invest. 2000;105:1085–1093. doi: 10.1172/JCI8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morriss-Kay GM, Wilkie AO. Growth of the normal skull vault and its alteration in craniosynostosis: insights from human genetics and experimental studies. J Anat. 2005;207:637–653. doi: 10.1111/j.1469-7580.2005.00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshed M, Harmey D, Millan JL, McKee MD, Karsenty G. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 2005;19:1093–1104. doi: 10.1101/gad.1276205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naski MC, Colvin JS, Coffin JD, Ornitz DM. Repression of hedgehog signaling and BMP4 expression in growth plate cartilage by fibroblast growth factor receptor 3. Development. 1998;125:4977–4988. doi: 10.1242/dev.125.24.4977. [DOI] [PubMed] [Google Scholar]
- Ohbayashi N, Shibayama M, Kurotaki Y, Imanishi M, Fujimori T, Itoh N, Takada S. FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes Dev. 2002;16:870–879. doi: 10.1101/gad.965702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkubo Y, Uchida AO, Shin D, Partanen J, Vaccarino FM. Fibroblast growth factor receptor 1 is required for the proliferation of hippocampal progenitor cells and for hippocampal growth in mouse. J Neurosci. 2004;24:6057–6069. doi: 10.1523/JNEUROSCI.1140-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM. FGF signaling in the developing endochondral skeleton. Cytokine Growth Factor Rev. 2005;16:205–213. doi: 10.1016/j.cytogfr.2005.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16:1446–1465. doi: 10.1101/gad.990702. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Givol D, Yayon A, Yarden Y, Lonai P. Developmental expression of two murine fibroblast growth factor receptors, flg and bek. Development. 1991;113:1419–1434. doi: 10.1242/dev.113.4.1419. [DOI] [PubMed] [Google Scholar]
- Ortega N, Behonick DJ, Werb Z. Matrix remodeling during endochondral ossification. Trends Cell Biol. 2004;14:86–93. doi: 10.1016/j.tcb.2003.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB, Owen MJ. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- Ovchinnikov DA, Deng JM, Ogunrinu G, Behringer RR. Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis. 2000;26:145–146. [PubMed] [Google Scholar]
- Partanen J, Schwartz L, Rossant J. Opposite phenotypes of hypomorphic and Y766 phosphorylation site mutations reveal a function for Fgfr1 in anteroposterior patterning of mouse embryos. Genes Dev. 1998;12:2332–2344. doi: 10.1101/gad.12.15.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters KG, Werner S, Chen G, Williams LT. Two FGF receptor genes are differentially expressed in epithelial and mesenchymal tissues during limb formation and organogenesis in the mouse. Development. 1992;114:233–243. doi: 10.1242/dev.114.1.233. [DOI] [PubMed] [Google Scholar]
- Pirvola U, Ylikoski J, Trokovic R, Hebert JM, McConnell SK, Partanen J. FGFR1 is required for the development of the auditory sensory epithelium. Neuron. 2002;35:671–680. doi: 10.1016/s0896-6273(02)00824-3. [DOI] [PubMed] [Google Scholar]
- Roscioli T, Flanagan S, Kumar P, Masel J, Gattas M, Hyland VJ, Glass IA. Clinical findings in a patient with FGFR1 P252R mutation and comparison with the literature. Am J Med Genet. 2000;93:22–28. doi: 10.1002/1096-8628(20000703)93:1<22::aid-ajmg5>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Sabbieti MG, Marchetti L, Abreu C, Montero A, Hand AR, Raisz LG, Hurley MM. Prostaglandins regulate the expression of fibroblast growth factor-2 in bone. Endocrinology. 1999;140:434–444. doi: 10.1210/endo.140.1.6442. [DOI] [PubMed] [Google Scholar]
- Saghizadeh M, Kramerov AA, Tajbakhsh J, Aoki AM, Wang C, Chai NN, Ljubimova JY, Sasaki T, Sosne G, Carlson MR, Nelson SF, Ljubimov AV. Proteinase and growth factor alterations revealed by gene microarray analysis of human diabetic corneas. Invest Ophthalmol Visual Sci. 2005;46:3604–3615. doi: 10.1167/iovs.04-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell U, Hehr A, Feldman GJ, Robin NH, Zackai EH, de Die-Smulders C, Viskochil DH, Stewart JM, Wolff G, Ohashi H, et al. Mutations in FGFR1 and FGFR2 cause familial and sporadic Pfeiffer syndrome. Hum Mol Genet. 1995;4:323–328. doi: 10.1093/hmg/4.3.323. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Stickens D, Behonick DJ, Ortega N, Heyer B, Hartenstein B, Yu Y, Fosang AJ, Schorpp-Kistner M, Angel P, Werb Z. Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development. 2004;131:5883–5895. doi: 10.1242/dev.01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama O, Orimo H, Suzuki S, Yamashita K, Ito H, Shimada T. Bone formation following transplantation of genetically modified primary bone marrow stromal cells. J Orthop Res. 2003;21:630–637. doi: 10.1016/S0736-0266(02)00260-7. [DOI] [PubMed] [Google Scholar]
- Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289:1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- Trokovic R, Trokovic N, Hernesniemi S, Pirvola U, Vogt Weisenhorn DM, Rossant J, McMahon AP, Wurst W, Partanen J. FGFR1 is independently required in both developing mid- and hindbrain for sustained response to isthmic signals. EMBO J. 2003;22:1811–1823. doi: 10.1093/emboj/cdg169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde-Franco G, Liu H, Davidson D, Chai S, Valderrama-Carvajal H, Goltzman D, Ornitz DM, Henderson JE. Defective bone mineralization and osteopenia in young adult FGFR3−/− mice. Hum Mol Genet. 2004;13:271–284. doi: 10.1093/hmg/ddh034. [DOI] [PubMed] [Google Scholar]
- Verheyden JM, Lewandoski M, Deng C, Harfe BD, Sun X. Conditional inactivation of Fgfr1 in mouse defines its role in limb bud establishment, outgrowth and digit patterning. Development. 2005;132:4235–4245. doi: 10.1242/dev.02001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White KE, Cabral JM, Davis SI, Fishburn T, Evans WE, Ichikawa S, Fields J, Yu X, Shaw NJ, McLellan NJ, McKeown C, Fitzpatrick D, Yu K, Ornitz DM, Econs MJ. Mutations that cause osteoglo-phonic dysplasia define novel roles for FGFR1 in bone elongation. Am J Hum Genet. 2005;76:361–367. doi: 10.1086/427956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie AO, Patey SJ, Kan SH, van den Ouweland AM, Hamel BC. FGFs, their receptors, and human limb malformations: clinical and molecular correlations. Am J Med Genet. 2002;112:266–2678. doi: 10.1002/ajmg.10775. [DOI] [PubMed] [Google Scholar]
- Xiao L, Naganawa T, Obugunde E, Gronowicz G, Ornitz DM, Coffin JD, Hurley MM. Stat1 controls postnatal bone formation by regulating fibroblast growth factor signaling in osteoblasts. J Biol Chem. 2004;279:27743–27752. doi: 10.1074/jbc.M314323200. [DOI] [PubMed] [Google Scholar]
- Xu X, Weinstein M, Li C, Naski M, Cohen RI, Ornitz DM, Leder P, Deng C. Fibroblast growth factor receptor 2 (FGFR2)-mediated regulation loop between FGF8 and FGF10 is essential for limb induction. Development. 1998;125:753–765. doi: 10.1242/dev.125.4.753. [DOI] [PubMed] [Google Scholar]
- Xu J, Lawshe A, MacArthur CA, Ornitz DM. Genomic structure, mapping, activity and expression of fibroblast growth factor 17. Mech Dev. 1999a;83:165–178. doi: 10.1016/s0925-4773(99)00034-9. [DOI] [PubMed] [Google Scholar]
- Xu XL, Weinstein M, Li CL, Deng CX. Fibroblast growth factor receptors (FGFRs) and their roles in limb development [Review] Cell Tissue Res. 1999b;296:33–43. doi: 10.1007/s004410051264. [DOI] [PubMed] [Google Scholar]
- Yamaguchi TP, Harpal K, Henkemeyer M, Rossant J. FGFR-1 is required for embryonic growth and mesodermal patterning during mouse gastrulation. Genes Dev. 1994;8:3032–3044. doi: 10.1101/gad.8.24.3032. [DOI] [PubMed] [Google Scholar]
- Yoshida CA, Furuichi T, Fujita T, Fukuyama R, Kanatani N, Kobayashi S, Satake M, Takada K, Komori T. Core-binding factor beta interacts with Runx2 and is required for skeletal development. Nat Genet. 2002;32:633–638. doi: 10.1038/ng1015. [DOI] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130:3063–3074. doi: 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]
- Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, Ross J, Haug J, Johnson T, Feng JQ, Harris S, Wiedemann LM, Mishina Y, Li L. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–841. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.