

Abstract
Dmp1 (cyclin D binding myb-like protein 1; also called Dmtf1) is a transcription factor that was isolated in a yeast two-hybrid screen through its binding property to cyclin D2. Although it was initially predicted to be involved in the cyclin D-Rb pathway, overexpression of Dmp1 in primary cells induces cell cycle arrest in an Arf, p53-dependent fashion. Dmp1 is a unique Arf regulator, the promoter of which is activated by oncogenic Ras-Raf signaling. Dmp1 expression is repressed by physiological mitogenic stimuli as well as by overexpressed E2F proteins; thus, it is a novel marker of cells that have exited from the cell cycle. Spontaneous and oncogene-induced tumor formation is accelerated in both Dmp1+/− and Dmp1−/− mice; the Dmp1+/− tumors often retain and express the wild-type allele; thus, Dmp1 is haplo-insufficient for tumor suppression. Tumors from Dmp1+/− and Dmp1−/− mice often retain wild-type Arf and p53, suggesting that Dmp1 is a physiological regulator of the Arf-p53 pathway. The human DMP1 (hDMP1) gene is located on chromosome 7q21, the locus of which is often deleted in myeloid leukemia and also in some types of solid tumors. Post-translational modification of Dmp1 and its role in human malignancy remain to be investigated.
Keywords: Dmp1, cyclin D, Arf, p53, Ras, haplo-insufficiency
Discovery of Dmp1
D-type cyclins (D1, D2 and D3) are induced in the context of a delayed early response to growth factor stimulation, and their synthesis and assembly with their catalytic partners, cyclin-dependent kinase 4 (Cdk4) and Cdk6, depend upon the presence of mitogens (Sherr, 2000; Sherr and Robers, 2004; Giacinti and Giordano, 2006). Cyclin D-Cdk holoenzymes play two established roles in facilitating progression through the G1 phase of the cell division cycle:(1) they catalyse the phosphorylation of the retinoblastoma protein (pRb), and (2) during G1 progression, accumulating cyclin DCdk holoenzymes recruit Cdk inhibitors, such as p27Kip1 and p21Cip1, into higher order complexes, thereby neutralizing their effects on other Cdks and facilitating the activation of cyclin E-Cdk2 later in the G1 phase (Sherr, 2000; Giacinti and Giordano, 2006). However, this established concept did not exclude the possibility of D-type cyclins of interacting with other proteins. Using a yeast two-hybrid interactive screen, Hirai and Sherr isolated a novel cyclin D binding myb-like protein 1 (designated as Dmp1; also called Dmtf1, cyclin D binding myb-like transcription factor 1), which binds specifically to the nonamer DNA consensus sequences CCCG(G/T)ATGT to activate transcription. Although Dmp1 is structurally related to the myb family proteins (c-myb, A-myb and B-myb), a subset of these Dmp1 recognition sequences contains a GGA trinucleotide core, which additionally functions as Ets-responsive elements. Dmp1 binds to all of the D-type cyclins directly in vitro and when co-expressed in insect Sf9 cells. Dmp1 can be phosphorylated by cyclin D-dependent kinases in Sf9 cells, suggesting that its transcriptional activity might be regulated through cyclin D/Cdks holoenzymes (Hirai and Sherr, 1996). Hirai and Sherr's very first report raised the possibility that cyclin D/Cdks might regulate gene expression in an Rb-independent manner, suggesting that D-cyclins might link other genetic programs to the cell cycle progression (Hirai and Sherr, 1996).
Dmp1 arrests cell cycle progression in rodent fibroblasts, the activity of which is antagonized by D-type cyclins in Cdk-independent fashion
Dmp1 contains a central DNA binding domain that includes three imperfect Myb-like repeats flanked by two acidic transactivation domains at the amino- and carboxyl-termini (Figure 1; Inoue and Sherr, 1998). Although Dmp1 does not belong to any gene family, it is structurally related to Myb proteins in the DNA-binding domain and transcription termination factor-1 in the transactivation domain (Evers and Grummt, 1995). So far, the negative regulatory domain in the c-Myb protein has not been identified in Dmp1 (Oh and Reddy, 1999). The Dmp1 protein migrates at 120–130 kDa, although the expected molecular weight is 85 kDa (Inoue and Sherr, 1998). This is considered to be due to extensive post-translational modification of the protein, possibly due to phosphorylation, at least in Sf9 cells (Hirai and Sherr, 1996). One preliminary report suggests the possible involvement of ERK in this process (Cheng M and Sherr CJ, personal communication). D-type cyclins associate with a region of the Dmp1 DNA-binding domain immediately adjacent to the Myb-like repeats to form heteromeric complexes that do not detectably interact with Cdk4 or with DNA (Figure 1). The segment of D-type cyclins required for its interaction with Dmp1 was mapped outside the ‘cyclin box’, which contains the residues predicted to contact Cdk4. Interestingly, the estrogen receptor and the steroid receptor co-activator bind with cyclin D1 outside the cyclin box, suggesting that the carboxyl-terminal half of cyclin D1 is important for its interaction with DNA-binding proteins (Bernards, 1999; Zwijsen et al., 1997). Coexpression of any of three D-type cyclins (D1, D2 or D3) with Dmp1 in mammalian cells canceled its ability to activate gene expression, which was independent of Cdks (Inoue and Sherr, 1998). Over-expression of Dmp1 into mouse fibroblasts inhibits their entry into S phase. Cell cycle arrest depended upon the ability of Dmp1 to bind to DNA and to transactivate gene expression and was specifically antagonized by co-expression of D-type cyclins, including a D1 point mutant (D1KE) that does not bind to Cdk4 (Inoue and Sherr, 1998). From these early studies, it was predicted that Dmp1 induces genes that interfere with S phase entry and that D-type cyclins can override Dmp1-mediated growth arrest in a Cdk-independent manner. A study from another group showed that cyclin D1 and D2 specifically inhibited transcription through the v-Myb DNA-binding domain, but not the c-Myb DNA-binding domain (Ganter et al., 1998). Analysis of a cyclin D1 mutant and a dominant-negative Cdk4 mutant suggested that this repression was independent of Cdks. Interestingly, the cyclin D-interacting domain was mapped within the v-Myb DNA-binding domain (Ganter et al., 1998). Cyclin D1 also inhibits the activity of B-Myb, the promoter of which is a direct target for E2Fs (Horstmann et al., 2000).
Figure 1.

The structure of the Dmp1 (Dmtf1) transcription factor. Both murine and human Dmp1 has three tandem myb-like repeats with two transactivation domains. Mutation of the lysine residue into glutamic acid abolishes its DNA binding. The cyclin Dinteraction domain has been mapped to the amino-terminal segment of the DNA-binding domain. The negative regulatory domain (NR) that is found in c-Myb has not been identified in Dmp1.
Although the physiological roles for cyclin D as a Cdk-independent repressor of transcriptional activation by Myb proteins remain to be determined, it was recently reported that the cyclin D1-associated kinase activity is largely dispensable for the development of retina and breast, but is essential for HER2/neu-induced breast tumorigenesis (Landis et al., 2006). Thus, cyclin D1 has some physiological activity in normal tissue development that is independent of Cdk activation.
Direct binding and activation of the Arf promoter by Dmp1
Through the extensive search for Dmp1-consensus sequences on naturally occurring promoters, it was found that the human CD13/Aminopeptidase N and the murine and human Arf promoters have high-affinity Dmp1-binding sequences (Inoue et al., 1998a, 1999; for INK4a/ARF reviews, see Kim and Sharpless, 2006; Sherr 2000, 2001, 2006). Dmp1 directly binds to a unique consensus site (5′-CCCGGATGC-3′) on the murine Arf promoter to activate its gene expression (Inoue et al., 1999). Dmp1-mediated Arf promoter activation was dependent on the consensus sequence, since the mutant reporter was not activated by Dmp1. Other Ets family proteins (Ets1, Ets2, Elf1 and Fli1) did not activate the Arf promoter by themselves, although the Dmp1/Ets site showed high affinity binding to the recombinant Ets protein. When Dmp1:ER virus-infected cells were stimulated with 4-HT, they increased both Arf mRNA and protein, and thereby induced Arf-, p53-dependent cell cycle arrest within 48 h (Inoue et al., 1999). Although Dmp1 and E2F-1 bind to different sites on the Arf promoter and act additively in a transactivation assays, Dmp1 induces cell cycle arrest but does not provoke programmed cell death. The data suggest that Dmp1 activates Arf transcription but does not stimulate other collateral pathways, such as Apaf-1 or caspases that are essential for E2F1-mediated apoptosis. Therefore, apart from its established role in protecting cells from potentially oncogenic signals, p19Arf can be induced in response to anti-proliferative stimuli that do not obligatorily lead to cell death (Inoue et al., 1999).
p19Arf activity is compromised but not eliminated in Dmp1-null cells
To study the role of Dmp1 in vivo, mice that lack Dmp1 by disrupting exons that encode the Myb-like repeats were created (Inoue et al., 2000). Dmp1-null animals are 20–30% smaller than their wild-type littermates at birth. Male Dmp1−/− mice remained smaller even in adults, however, female knockout mice eventually became indistinguishable from their Dmp1+/+ or Dmp1+/− littermates. Dmp1-null mice have other miscellaneous phenotypes, such as generalized seizures, seminal vesicle dilatation caused by urologic syndromes, and poor mammary gland development in females (Inoue et al., 2000). The growth of Dmp1-null MEFs (murine embryonic fibroblasts) is progressively retarded as cells are passaged in culture, however, unlike normal cells, p19Arf and p53 levels remain relatively low and the MEFs continued to grow slowly without reaching replicative senescence. The rate of p16Ink4a induction in Dmp1-null cells remained largely identical with those in Dmp1+/+ and Dmp1+/− cells. The levels of Dmp1 dramatically increased from passage 2 to passage 3 in both Dmp1+/+ and Dmp1+/− cells, and the accumulation of Dmp1 preceded that of p19Arf (Inoue et al., 2000). The data suggested that both Dmp1 and p19Arf are induced in response to stress signaling caused by non-physiological cell culture conditions. When wild-type MEFs were cultured beyond the period of replicative senescence, immortalized cell lines that had either a mutant p53 (∼80%) or deleted Arf locus (∼20%) were obtained (Inoue et al., 2000). However, Dmp1−/− cells readily gave rise to established cell lines that retained wild-type Arf and functional p53 without overexpression of Mdm2, suggesting that the activity of the Arf-Mdm2-p53 pathway is strikingly impaired in Dmp1−/− cells (Inoue et al., 2000).
Ras-mediated signaling pathways are critical for the mitogen-dependent induction of cyclin D1 and its assembly with Cdk4 (Cheng et al., 1998). Overexpression of activated Ras initiates DNA synthesis independent of growth factor stimulation. Paradoxically, continued overexpression of oncogenic Ras and its various effectors elicit irreversible cell cycle arrest by upregulating the levels of p16Ink4a, p19Arf and p53 (Lin et al., 1998; Palmero et al., 1998; Serrano et al., 1997; for review, McMahon and Woods, 2001). Early passage Dmp1-null cells, like MEFs from either Arf-null or p53-null mice were transformed by oncogenic Ha-Ras alone, thus bypassing the effects of immortalizing oncogenes. These data suggest that loss of Dmp1 compromises the Arf-, p53-dependent senescence response that suppresses oncogenic transformation. These activities are consistent with the role of Dmp1 as a tumor suppressor.
MEFs were not only the tissue that caused hyper-proliferation in culture. Splenic pre-B lymphocytes or T-lymphocytes isolated from Dmp1-null mice showed much higher proliferative capacity than Dmp1+/+ cells when stimulated with appropriate mitogens (Inoue et al., 2000; Inoue K and Sherr CJ, unpublished data). Interestingly, the T-cell phenotype was even stronger in Dmp1-null T-cells than in Arf-null cells, suggesting that Dmp1 must have different target genes than p19Arf for T-cell proliferation (Inoue K et al., unpublished data).
Both Dmp1-null and heterozygous mice are prone to tumor development; haploid-insufficiency of Dmp1 in tumor suppression
Consistent with the data obtained in MEF studies, Dmp1-null mice spontaneously developed lethal tumors in their second year of life with a mean latency of 83 weeks (Inoue et al., 2001). The most frequently encountered tumors were pulmonary adenomas/adenocarcinomas (42%). Vascular tumors, including hemangiomas and hemangiosarcomas (24%), hepatocellular adenomas/adenocarcinomas (18%) and B-cell lymphomas (15%) were also relatively common (Inoue et al., 2001). The time of appearance and variety of tumors observed in Dmp1-null mice bore no obvious relationship to those in Arf-null or p53-null mice, which exhibit a different spectrum (Donehower et al., 1992; Kamijo et al., 1999; Inoue et al., 2001). Treating neonatal Dmp1-null mice with dimethylbenzanthracene (DMBA) or ionizing radiation accelerated tumorigenesis, and many such animals developed multiple tumors of more than one histological type (Inoue et al., 2000, 2001). In addition to lung and skin carcinomas, DMBA-treated Dmp1−/− and Dmp1+/− mice often developed ovarian tumors, T-cell leukemia/lymphomas, hepatocellular carcinomas and melanomas (Inoue et al., 2001). Since these tumors were not found in the control Dmp1+/+ mice that received the same treatment, Dmp1-inactivation apparently contributed to the change of tumor spectra. In humans, epithelial tumors such as epidermoid carcinomas and adenocarcinomas are much more common than sarcomas or lymphomas, and carcinomas are almost exclusively found after 40 years of age. Given that Dmp1-null mice developed epithelial tumors in their second year of life, they might be useful as an animal model of human carcinogenesis.
Eμ-Myc transgenic mice develop Burkitt-type B-cell tumors with a mean latency of about 6 months. When crossed onto a Dmp1+/− or Dmp1−/− background, lymphomas induced by the Eμ-Myc transgene were greatly accelerated (mean latency, 12 weeks) with no differences between cohorts lacking one or two Dmp1 alleles (Inoue et al., 2001). Intriguingly, the latency in the Dmp1+/− or Dmp1−/− strains mimicked that of Arf+/−, Eμ-Myc transgenic mice (Eischen et al., 1999), consistent with the idea that Dmp1 loss lowers p19Arf expression (Inoue et al., 1999, 2000). Tumors from Dmp1 heterozygotes retained and expressed the wild-type Dmp1 allele, and most contained detectable Dmp1 protein (Inoue et al., 2001). Direct nucleotide sequencing of Dmp1 reverse transcription–polymerase chain reaction (RT–PCR) products from five such tumors identified no mutations in the DNA-binding domain. The results provide strong evidence that Dmp1 is haplo-insufficient for tumor suppression (Inoue et al., 2001; reviewed by Brooksbank, 2001; Quon and Berns, 2001). The combined frequencies of p53 mutation and Arf deletion in the Dmp1−/− and Dmp1+/− cohorts were ∼10 versus ∼50% in Dmp1+/+ littermates (Figure 2). These results provided strong genetic evidence that loss of even a single Dmp1 allele alleviates the selection for p53 mutation and Arf loss that otherwise occurs during Eμ-Myc-induced lymphoma generation, indicating that Dmp1 is a physiological regulator of the Arf-p53 pathway in vivo.
Figure 2.

Disruption of the Arf-Mdm2-p53 pathway in Eμ-Myc lymphomas. In Dmp1+/− and Dmp1−/− Eμ-Myc lymphomas, there is a striking reduction in the frequencies of p53 mutations and Arf deletions. This suggests that Dmp1 is a regulator of the Arf-p53 pathway in vivo.
Regulation of the Dmp1 promoter by oncogenic Ras
As transfected Dmp1 protein has a relatively long half-life (∼12 h), it was speculated that transcriptional control played important roles in its regulation. In cultured primary cells, the Dmp1 promoter was efficiently activated by oncogenic Ha-RasV12, but not by overexpressed c-Myc or E2F-1 (Sreeramaneni et al., 2005). The Dmp1 promoter activation by RasV12 depended on Raf-MEK-ERK signaling because the double mutant RasV12S35 activated the promoter and because U0126 completely blocked the promoter activation (Sreeramaneni et al., 2005). Induction of p19Arf and p21Cip1 by oncogenic Raf was compromised in Dmp1-null cells, which were resistant to Raf-mediated premature senescence (Sreeramaneni et al., 2005). This indicated that Dmp1 is a critical target for oncogenic Raf-induced premature senescence. A RasV12-responsive element was mapped to the 5′ leader sequence of the murine Dmp1 promoter, where endogenous fos and jun family proteins bind. The Dmp1 promoter activation by RasV12 was strikingly impaired in c-jun as well as in junB knock-down cells, suggesting the critical role of jun proteins in the Dmp1 promoter activation.
Importantly, a RasV12-responsive element was mapped to the unique Dmp1/Ets site on the Arf promoter, where endogenous Dmp1 proteins bind upon oncogenic Raf activation (Sreeramaneni et al., 2005). Therefore, activation of the Arf promoter by Ras/Raf signaling is mediated by Dmp1, and this is why Dmp1-null primary cells are highly susceptible to Ras-induced transformation. Although oncogenic Ras activates the E2F transcription factors, E2Fs do not play important roles in Arf induction by Ras (Palmero et al., 2002; Rowland et al., 2002). Collectively, these three reports indicate the presence of a novel jun-Dmp1 pathway that directly links oncogenic Ras-Raf signaling and p19Arf, independent of the classical cyclin D1/Cdk4-Rb-E2F pathway (Figure 3).
Figure 3.
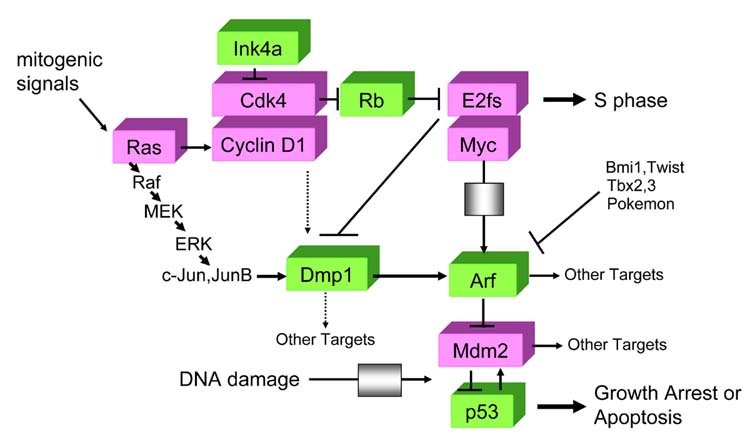
Dmp1 links the Rb and p53 pathways. The novel ‘Jun-Dmp1’ pathway links oncogenic Ras-Raf signaling and the Arf-p53 pathway. This pathway is independent of the classical cyclin D1/Cdk4-Rb-E2F signaling. E2Fs directly bind to the Dmp1 promoter and causes repression; thus they have differential effects on the Dmp1 and on the Arf promoter. Overexpression of D-cyclins inhibits the activity of Dmp1 in a Cdk-independent fashion; however, on the Arf promoter, Dmp1 and cyclin D1 act synergistically, and this activity is dependent on Cdks. Dmp1 might have other targets than Arf, especially in T lymphocytes.
One interesting piece of data is the response of the Arf promoter to D-cyclins. Overexpressed D-type cyclins antagonize Dmp1 transcriptional activity in a Cdk-independent fashion when tested with artificial promoter-reporter plasmids containing concatamerized Dmp1 consensus binding sequences or with some natural promoters, such as those derived from the CD13/Aminopeptidase N gene (Inoue and Sherr, 1998; Inoue et al., 1998a). However, the results were reversed for the Arf promoter, where D-type cyclins cooperated to enhance the activity of Dmp1 in a Cdk4-dependent manner. The Arf promoter contains both Dmp1- and E2F-binding sites, enabling RasV12-induced cyclin D1 to assemble with Cdk4, promote the release of E2Fs from Rb, and thereby collaborate with Dmp1 in activating Arf gene expression (Inoue et al., 1999). On the other hand, the CD13/Aminopeptidase N promoter, which lacks E2F-consensus sequences, can be experimentally suppressed by D-type cyclins, which, when overexpressed, can interfere with Dmp1 binding to DNA. The Dmp1/Ets-consensus sequences found within these two promoters is completely identical (CCCGGATGC) (Inoue et al., 1998a,1999), consistent with the hypothesis that sequences flanking the Dmp1-binding site determine the responsiveness of the promoter to D-type cyclins. It is important to emphasize that interference of Dmp1 activity by D-type cyclins has not been demonstrated in situations where D-type cyclins accumulate to physiological levels. Indeed, the level of cyclin D1 achieved after Ha-RasV12 expression was ten times lower than that generated by the cyclin D1 expression vector itself (Sreeramaneni R and Inoue K, unpublished data).
Regulation of Dmp1 by E2Fs and its expression in specific differentiated, non-proliferating cells
Although the Dmp1 promoter is activated by oncogenic Ras signaling, it was repressed when the cells entered the S to G2/M phase of the cell cycle when both Dmp1 and Arf expressions were downregulated (Mallakin et al., 2006). Subsets of E2Fs were specifically bound to the Dmp1 promoter upon mitogenic signaling, and E2Fs 1–4 inhibited the Dmp1 promoter in a reporter assay. It has been reported that E2F-DB mutant binds to endogenous E2F sequences and blocks their repressor as well as activator activity (Rowland et al., 2002). The Dmp1 mRNA was not downregulated by serum in E2F-DB( + ) cells, suggesting that the Dmp1 promoter repression by serum is E2F-dependent. It was reported that endogenous activating E2Fs, E2F1 and E2F3a were recruited to the Arf promoter in response to hyperproliferative oncogenic signaling, indicating that distinct subsets of E2F proteins contribute to the normal repression and oncogenic activation of Arf (Aslanian et al., 2004). Surprisingly, all of the E2F1, E2F2 and E2F3a proteins were repressors on the Dmp1 promoter, especially the former two. Thus, E2F1 has differential effects on the Dmp1 promoter (repression) and the Arf promoter (activation) when overexpressed in rodent fibroblasts (Inoue et al., 1999; Mallakin et al., 2006). The E2F-mediated regulation of the Dmp1 promoter is conserved in humans since the hDMP1 promoter has a typical E2F site (5′-TTTCGCGC) and is efficiently repressed by E2Fs (Inoue K et al., unpublished data). It is important to note that the Dmp1 promoter is not the only promoter repressed by ‘activating’ E2Fs; repression of the human telomerase promoter as well as the tumor suppressor ARHI promoter by E2F1 have also been reported (Crowe et al., 2001; Lu et al., 2006). Although the detailed mechanism of E2F-mediated repression of the Dmp1 promoter remains to be determined, one preliminary experiment suggests that the repression is Rb-independent since the E2F1 Y411C mutant that does not interact with Rb (Rowland et al., 2002) inhibited the Dmp1 promoter as well (Mallakin et al., 2006).
Immunohistochemical staining was conducted to identify the pattern of Dmp1 expression in normal murine tissues compared with the proliferation marker Ki67, to search for Dmp1-expressing cells in vivo (Mallakin et al., 2006). In thymus, the nuclei of mature T lymphocytes in the medulla were strongly positive for Dmp1, whereas Ki67 was detected only in the cortex. In intestine, Dmp1 was detected in the nuclei of superficial layers of the villi, whereas Ki67-positive cells were confined to the lower one-third of the crypt. Double staining for Dmp1 and Ki67 revealed that these two proteins were expressed in a mutually exclusive fashion in nearly all the tissues examined. This pattern of expression of Dmp1 is in contrast to that of c-myb protein in normal murine tissues, which is expressed specifically in actively proliferating cells in the testis and the thymus (Oh and Reddy, 1999). Interestingly, c-Myb and Dmp1 collaborate to activate the CD13/Aminopep-tidase N promoter (Inoue et al., 1998a). Thus, c-myb and Dmp1 seem to play complementary roles in regulating expression of genes involved in cell differentiation.
The human DMP1 gene and cancer
In striking contrast to the accumulating information on murine Dmp1, very little is known about the involvement of human DMP1 (hDMP1) in human cancer. hDMP1 has very high structural homology with its murine counterpart (760 amino acids, 96% similarity and 95% identity with murine Dmp1 at protein level). The hDMP1 gene is located on human chromosome 7q21, a locus often deleted in human malignancies (Bieche et al., 1992; Kerr et al., 1996; Bodner et al., 1999; Trovato et al., 2004). One copy of the genes at the hDMP1 locus was reportedly deleted in all the tumor cells with chromosome 7q abnormalities regardless of the detailed karyotype, suggesting that one allele loss of hDMP1 could contribute to 7q- malignancies that are refractory to conventional chemotherapy (Bodner et al., 1999). It has been reported that the hDMP1 locus encodes at least three splicing variants, that is hDMP1α, β and γ (Tschan et al., 2003). The full-length hDMP1α corresponds to murine Dmp1, which positively regulates the p19Arf-p53 pathway (Tschan et al., 2003). Therefore, hDMP1α is considered to have tumor-suppressor activity. The β- and γ-splicing variants do not bind to DNA, but they can make heterodimers with hDMP1α. Therefore, hDMP1β and γ proteins are dominant negative forms for hDMP1α when they are over-expressed (Tschan et al., 2003). They are specifically expressed in immature hematopoietic cells. U937 cells that constitutively express hDMP1β isoform showed reduced cell surface expression of CD13/Aminopeptidase N and continued to proliferate even after phorbol 12-myristate 13-acetate treatment (Tschan et al., 2003). Therefore, it is highly possible that splicing abnormalities that result in the overexpression of β/γ isoforms of hDMP1 contribute to human leukemogenesis. Although the endogenous products of hDMP1β and γ have not been reported, these proteins may bind to D-type cyclins, since they retain the amino-terminal DNA-binding domain required for cyclin D interaction (Inoue and Sherr, 1998; Tschan et al., 2003).
Recently, it was reported that hDMP1 is a potential target for Wilms tumor gene WT1 as well as for miR-15a (Kiriakidou et al., 2004; Elmaagacli et al., 2005). Although the WT1 gene has been isolated as a tumor suppressor of Wilms tumor, it is aberrantly over-expressed in human leukemic cells, especially in acute myelocytic leukemia and in blastic crisis of chronic myelocytic leukemia (Inoue et al., 1994, 1997). Antisense oligonucleotides to WT1 inhibit the growth of human leukemic cells and overexpression of WT1 interferes with myeloid differentiation programs (Inoue et al., 1998b; Yamagami et al., 1996). It was reported that siRNA treatment of the WT1 gene in a K562 cell line upregulated hDMP1 (∼3-fold) when the cells stopped proliferating and underwent apoptosis (Elmaagacli et al., 2005). The miR-15a gene is located on human chromosome 13q14.3, which is often deleted in patients with B-cell chronic lymphocytic leukemia and is involved in apoptosis (Calin and Croce, 2006; Cimmino et al., 2005). A combined computational-experimental approach predicted that hDMP1 could be a potential target for miR-15a (Kiriakidou et al., 2004). Although these experimental results were correlative, it is intriguing to speculate about a role for hDMP1 as a tumor suppressor in human leukemic cells.
Dmp1, on the cusp between oncogene and tumor suppressor signaling – future prospects
Accumulating evidence suggests that Dmp1 is a regulator of the Arf-p53 pathway, although the gene had been isolated through its binding to cyclin D2. The role of Dmp1 in Ink4-cyclin D/Cdk-Rb signaling remains to be determined. Dmp1 is a transcription factor located at the junction of oncogene-tumor suppressor gene signaling; thus, it is highly possible that the gene is inactivated in a significant percentage of human cancers. The molecular mechanisms of haploid insufficiency may be determined by the identification of novel signaling pathways regulated by Dmp1. The mechanism of post-translational modification and identification of Dmp1-binding partners is an undiscovered area, which might yield further information about how this mysterious transcription factor is regulated at protein levels.
Acknowledgments
We thank Ms Karen Klein for critical reading of this review and Drs Bruce Torbett, Mario Tschan and Hiroshi Hirai for sharing unpublished data. We are very grateful to Drs Charles Sherr, Martine Roussel and John Cleveland for collaborative work and continuous encouragement on Dmp1 projects. We also thank Pankaj Taneja, Lauren Matise, Mark Willingham, Mayur Choudhary, Samantha Allen, Scott Barton, Asif Chaudhry and Ramesh Sreeramaneni for collaboration. K Inoue is supported by NIH/NCI 5R01CA106314.
References
- Aslanian A, Iaquinta PJ, Verona R, Lees JA. Repression of the Arf tumor suppressor by E2F3 is required for normal cell cycle kinetics. Genes Dev. 2004;18:1413–1422. doi: 10.1101/gad.1196704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernards R. CDK-independent activities of D type cyclins. Biochem Biophys Acta. 1999;1424(2–3):M17–M22. doi: 10.1016/s0304-419x(99)00024-4. [DOI] [PubMed] [Google Scholar]
- Bieche I, Champeme MH, Matifas F, Hacene K, Callahan R, Lidereau R. Loss of heterozygosity on chromosome 7q and aggressive primary breast cancer. Lancet. 1992;339:139–143. doi: 10.1016/0140-6736(92)90208-k. [DOI] [PubMed] [Google Scholar]
- Bodner SM, Naeve CW, Rakestraw KM, Jones BG, Valentine VA, Valentine MB, et al. Cloning and chromosomal localization of the gene encoding human cyclin D-binding Myb-like protein (hDMP1) Gene. 1999;229:223–228. doi: 10.1016/s0378-1119(98)00591-5. [DOI] [PubMed] [Google Scholar]
- Brooksbank C. Tumor suppressors. One-hit wonders? Nature Rev Cancer. 2001;1:174. [Google Scholar]
- Calin GA, Croce CM. Genomics of chronic lymphocytic leukemia microRNAs as new players with clinical significance. Semin Oncol. 2006;33:167–173. doi: 10.1053/j.seminoncol.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Cheng M, Sexl V, Sherr CJ, Roussel MF. Assembly of cyclin D-dependent kinase and titration of p27kip1 regulated by mitogen-activated protein kinase kinase (MEK1) Proc Natl Acad Sci USA. 1998;95:1091–1096. doi: 10.1073/pnas.95.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe DL, Nguyen DC, Tsang KJ, Kyo S. E2F-1 represses transcription of the human telomerase transcriptase gene. Nucl Acid Res. 2001;29:2789–2794. doi: 10.1093/nar/29.13.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13:2658–2669. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmaagacli AH, Koldehoff M, Peceny R, Klein-Hitpass L, Ottinger H, Beelen DW, et al. WT1 and BCR-ABL specific small interfering RNA have additive effects in the induction of apoptosis in leukemic cells. Leukemogenesis. 2005;90:326–334. [PubMed] [Google Scholar]
- Evers R, Grummt I. Molecular coevolution of mammalian ribosomal gene terminator sequences and the transcription termination factor TTF-1. Proc Natl Acad Sci USA. 1995;92:5827–5831. doi: 10.1073/pnas.92.13.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganter B, Fu S, Lipsick JS. D-type cyclins repress transcriptional activation by the v-Myb but not the c-Myb DNA-binding domain. EMBO J. 1998;17:255–268. doi: 10.1093/emboj/17.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220–5227. doi: 10.1038/sj.onc.1209615. [DOI] [PubMed] [Google Scholar]
- Hirai H, Sherr CJ. Interaction of D-type cyclins with a novel myb-like transcription factor, DMP1. Mol Cell Biol. 1996;16:6457–6467. doi: 10.1128/mcb.16.11.6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horstmann S, Ferrari S, Klempnauer KH. Regulation of B-Myb activity by cyclin D1. Oncogene. 2000;19:298–306. doi: 10.1038/sj.onc.1203302. [DOI] [PubMed] [Google Scholar]
- Inoue K, Ogawa H, Sonoda Y, Kimura T, Sakabe H, Oka Y, et al. Aberrant overexpression of the Wilms tumor gene (WT1) in human leukemia. Blood. 1997;89:1405–1412. [PubMed] [Google Scholar]
- Inoue K, Roussel MF, Sherr CJ. Induction of ARF tumor suppressor gene expression and cell cycle arrest by transcription factor DMP1. Proc Natl Acad Sci USA. 1999;96:3993–3998. doi: 10.1073/pnas.96.7.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Sherr CJ. Gene expression and cell cycle arrest mediated by transcription factor DMP1 is antagonized by D-type cyclins through a cyclin-dependent-kinase-independent mechanism. Mol Cell Biol. 1998;18:1590–1600. doi: 10.1128/mcb.18.3.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Sherr CJ, Shapiro LH. Regulation of the CD13/aminopeptidase N gene by DMP1, a transcription factor antagonized by D-type cyclins. J Biol Chem. 1998a;273:29188–29194. doi: 10.1074/jbc.273.44.29188. [DOI] [PubMed] [Google Scholar]
- Inoue K, Sugiyama H, Ogawa H, Nakagawa M, Yamagami T, Miwa H, et al. WT1 as a new prognostic factor and a new marker for the detection of minimal residual disease in acute leukemia. Blood. 1994;84:3071–3079. [PubMed] [Google Scholar]
- Inoue K, Tamaki H, Ogawa H, Oka Y, Soma T, Tatekawa T, et al. Wilms' tumor gene (WT1) competes with differentiation-inducing signal in hematopoietic progenitor cells. Blood. 1998b;91:2969–2976. [PubMed] [Google Scholar]
- Inoue K, Wen R, Rehg JE, Adachi M, Cleveland JL, Roussel MF, et al. Disruption of the ARF transcriptional activator DMP1 facilitates cell immortalization, Ras transformation, and tumorigenesis. Genes Dev. 2000;14:1797–1809. [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Zindy F, Randle DH, Rehg JE, Sherr CJ. Dmp1 is haplo-insufficient for tumor suppression and modifies the frequencies of Arf and p53 mutations in Myc-induced lymphomas. Genes Dev. 2001;15:2934–2939. doi: 10.1101/gad.929901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Tumor spectrum in ARF-deficient mice. Cancer Res. 1999;59:2217–2222. [PubMed] [Google Scholar]
- Kerr J, Leary JA, Hurst T, Shih YC, Antalis TM, Friedlander M, et al. Allelic loss on chromosome 7q in ovarian adenocarcinomas: two critical regions and a rearrangement of the PLANH1 locus. Oncogene. 1996;13:1815–1818. [PubMed] [Google Scholar]
- Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–275. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Kiriakidou M, Nelson PT, Kouranov A, Fitziev P, Bouyioukos C, Mourelatos Z, et al. A combined computational-experimental approach predicts human microRNA targets. Genes Dev. 2004;18:1165–1178. doi: 10.1101/gad.1184704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell. 2006;9:13–22. doi: 10.1016/j.ccr.2005.12.019. [DOI] [PubMed] [Google Scholar]
- Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998;12:3008–3019. doi: 10.1101/gad.12.19.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Luo RZ, Peng H, Huang M, Nishimoto A, Hunt KK, et al. E2F-HDAC complexes negatively regulate the tumor suppressor gene ARHI in breast cancer. Oncogene. 2006;25:230–239. doi: 10.1038/sj.onc.1209025. [DOI] [PubMed] [Google Scholar]
- Mallakin A, Taneja P, Matise LA, Willingham MC, Inoue K. Expression of Dmp1 in specific differentiated, nonproliferating cells and its repression by E2Fs. Oncogene. 2006;25:7703–7713. doi: 10.1038/sj.onc.1209750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon M, Woods D. Regulation of the p53 pathway by Ras, the plot thickens. Biochem Biophys Acta. 2001;1471:M63–M71. doi: 10.1016/s0304-419x(00)00027-5. [DOI] [PubMed] [Google Scholar]
- Oh IH, Reddy EP. The myb gene family in cell growth, differentiation, and apoptosis. Oncogene. 1999;18:3017–3033. doi: 10.1038/sj.onc.1202839. [DOI] [PubMed] [Google Scholar]
- Palmero I, Pantoja C, Serrano M. p19ARF links the tumour suppressor p53 to ras. Nature. 1998;395:125–126. doi: 10.1038/25870. [DOI] [PubMed] [Google Scholar]
- Palmero I, Murga M, Zubiaga A, Serrano M. Activation of ARF by oncogenic stress in mouse fibroblasts is independent of E2F1 and E2F2. Oncogene. 2002;21:2939–2947. doi: 10.1038/sj.onc.1205371. [DOI] [PubMed] [Google Scholar]
- Quon KC, Berns A. Haplo-insufficiency? Let me count the ways. Genes Dev. 2001;15:2917–2921. doi: 10.1101/gad.949001. [DOI] [PubMed] [Google Scholar]
- Rowland BD, Denissov SG, Douma S, Stunnenberg HG, Bernards R, Peeper DS. E2F transcriptional repressor complexes are critical downstream targets of p19(ARF)/p53-induced proliferative arrest. Cancer Cell. 2002;2:55–65. doi: 10.1016/s1535-6108(02)00085-5. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. The Pezcoller lecture: cancer cell cycle revisited. Cancer Res. 2000;60:3689–3695. [PubMed] [Google Scholar]
- Sherr CJ. The INK4a/ARF network in tumor suppression. Nat Rev Mol Cell Biol. 2001;2:731–737. doi: 10.1038/35096061. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6:663–673. doi: 10.1038/nrc1954. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Robers JM. Living with or without cyclin and cyclin-dependent kinases. Genes Dev. 2004;18:2699–2711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- Sreeramaneni R, Chaudhry A, McMahon M, Sherr CJ, Inoue K. Ras-Raf-Arf signaling critically depends on Dmp1 transcription factor. Mol Cell Biol. 2005;25:220–232. doi: 10.1128/MCB.25.1.220-232.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trovato M, Ulivieri A, Dominici R, Ruggeri RM, Vitarelli E, Benvenga S, et al. Clinico-pathological significance of cell-type-specific loss of heterozygosity on chromosome 7q21: analysis of 318 microdissected thyroid lesions. Endocr Relat Cancer. 2004;11:365–376. doi: 10.1677/erc.0.0110365. [DOI] [PubMed] [Google Scholar]
- Tschan MP, Fischer KM, Fung VS, Pirnia F, Borner MM, Fey MF, et al. Alternative splicing of the human cyclin D-binding Myb-like protein (hDMP1) yields a truncated protein isoform that alters macrophage differentiation patterns. J Biol Chem. 2003;278:42750–42760. doi: 10.1074/jbc.M307067200. [DOI] [PubMed] [Google Scholar]
- Yamagami T, Sugiyama H, Inoue K, Ogawa H, Tatekawa T, Hirata M, et al. Growth inhibition of human leukemic cells by WT1 antisense oligonucleotides. Blood. 1996;87:2878–2884. [PubMed] [Google Scholar]
- Zwijsen R, Wientjens E, Klompmaker R, van der Sman J, Bernards R, Michalides R. CDK-independent activation of estrogen receptor by cyclin D1. Cell. 1997;88:405–415. doi: 10.1016/s0092-8674(00)81879-6. [DOI] [PubMed] [Google Scholar]