

Abstract
Dmp1 is a Myb-like transcription factor that transmits oncogenic Ras-Raf signaling to the Arf-p53 pathway and induces cell cycle arrest. Immunohistochemical staining was performed to identify the pattern of Dmp1 expression in normal murine tissues compared with the proliferation marker, Ki67. In thymus, the nuclei of mature T lymphocytes in the medulla were strongly positive for Dmp1, whereas Ki67 was detected only in the cortex. In intestine, Dmp1 was detected in the nuclei of superficial layers of the villi, whereas Ki67-positive cells were confined to the lower one-third of the crypt. Double staining for Dmp1 and Ki67 revealed that these two proteins were expressed in mutually exclusive fashion in nearly all the tissues examined. Subsets of E2Fs were specifically bound to the Dmp1 promoter upon mitogenic signaling and E2Fs 1–4 inhibited the Dmp1 promoter in a reporter assay. The Dmp1 promoter was repressed when the cells entered the S to G2/M phase of the cell cycle when both Dmp1 and Arf expressions were downregulated. The Dmp1 mRNA was not downregulated by serum in E2F-DB(+) cells, suggesting that the Dmp1 promoter repression is E2F-dependent. This explains why the Dmp1 and Ki67-positive cells are stained in mutually exclusive fashion in normal tissues.
Keywords: Dmp1, Ki67, Arf, E2F, cell cycle, immunohistochemistry
Introduction
The INK4a/ARF locus on human chromosome 9p21 encodes two tumor-suppressor genes known as p16INK4a and ARF (p14ARF in humans and p19Arf in mice). As one locus encodes two independent tumor suppressors, the locus is very frequently disrupted in human cancers (Ruas and Peters, 1998). p16Ink4a responds to conditions of cellular stress to turn off Cdk4/6 activity and, thus, keeps pRb in its active, antiproliferative state (Sherr and Roberts, 1999). p19Arf binds to the p53 negative regulator, Mdm2, thereby stabilizing and activating p53 (Sherr, 2001; Lowe and Sherr, 2003). p19Arf is induced by potentially harmful growth-promoting signals resulting from over-expression of a variety of oncoproteins, including c-myc, E2F1, mutated Ras, v-Abl and β-catenin. These stimulate the cancer cells to undergo p53-dependent and p53-independent cell cycle arrest or apoptosis, providing a powerful mode of tumor suppression (Sherr, 2001; Lowe and Sherr, 2003). In turn, Arf-null mice are highly prone to spontaneous tumor development and most of them die of various forms of cancer within 12 months of age (Kamijo et al., 1997, 1999). Recently created Arf-GFP knock-in mice provided direct experimental evidence that the Arf promoter monitors dormant oncogenic signals in vivo (Zindy et al., 2003).
One of the Arf-activating transcription factor Dmp1 (cyclin D binding myb-like protein 1; also called Dmtf1, cyclin D binding myb-like transcription factor 1) was originally isolated in a yeast two-hybrid screen of a murine T-lymphocyte library with cyclin D2 as bait (Hirai and Sherr, 1996). Dmp1 binds to the DNA consensus sequences CCCG (G/T) ATG (T/C), a subset of which is also recognized by proteins of the Ets family (Hirai and Sherr, 1996). Dmp1 can physically interact with any of the three D-type cyclins, each of which can interfere with Dmp1’s ability to bind to DNA independent of Cdks when overexpressed (Hirai and Sherr, 1996; Inoue and Sherr, 1998; Inoue et al., 1998). Dmp1 protein levels are high in the testis, thymus, brain and lung of wild-type mice when studied with antibodies to the carboxyl-terminal end of the protein (Inoue et al., 2000). Dmp1 directly binds to the Arf promoter to activate its gene expression, upregulates Arf and thereby, induces Arf-, p53-dependent cell cycle arrest (Inoue et al., 1999). The Dmp1−/− MEFs have biological features that mimic those of Arf−/− cells (Inoue et al., 2000). More than 80% of Dmp1-null MEFs gave rise to immortalized cell lines that retained wild-type Arf and p53 and, therefore, the cells were transformed by oncogenic Ras alone (Inoue et al., 2000). Both Dmp1−/− and Dmp1+/− mice often develop tumors when left untreated or when the mice were neonatally treated with dimethylbenzanthracene or by ionizing radiation (Inoue et al., 2000, 2001). Tumors from Dmp1+/− mice often retain the wild-type Dmp1 locus and express the protein. Therefore, Dmp1 is a haplo-insufficient tumor suppressor in mice (Quon and Berns, 2001). Dmp1 has been considered to be a physiological regulator of the Arf-p53 pathway in vivo since the combined frequency of p53 mutation and Arf deletion in the Dmp1−/− and Dmp1+/− Eμ-Myc lymphomas were significantly lower than that in Dmp1+/+tumors (Inoue et al., 2001). Recently, we found that the murine Dmp1 promoter was efficiently activated by oncogenic RasV12 as well as by constitutively active c-Raf in primary culture cells (Sreeramaneni et al., 2005). p19Arf and p53 did not accumulate in response to activated Raf signaling in Dmp1-null cells and the cells were refractory to Raf-mediated cell cycle arrest. Our data suggested the critical role of Dmp1 between Ras-Raf-MEK-ERK oncogenic signaling and the Arf-p53 tumor-suppressor pathway (Sreeramaneni et al., 2005). Although Dmp1 does not belong to any gene family, it is structurally related to Myb-family proteins (Hirai and Sherr, 1996). Interestingly, two of the Myb proteins, c-Myb and B-Myb, are the direct target of E2Fs (Campanero et al., 1999; Catchpole et al., 2002), and their roles in cell cycle regulation have been actively studied (Oh and Reddy, 1999; Sala and Watson, 1999; Joaquin and Watson, 2003).
The E2F family of transcription factors plays essential roles in cell cycle progression and DNA replication (DeGregori, 2002; Trimarch and Lees, 2002). They can be divided into two major subgroups based on their function and the mechanism of action. E2Fs 1–3, the ‘activating’ E2Fs, are required for the transactivation of target genes involved in the G1 to S phase transition and, hence, for accurate progression through the cell cycle (DeGregori, 2002; Trimarch and Lees, 2002). Indeed, acute loss of all three activating E2F proteins results in cell cycle arrest (Wu et al., 2001). Furthermore, over-expression of E2F1, 2, or 3 in quiescent fibroblasts is sufficient to drive them into S phase (Johnson et al., 1993). The E2F3 locus can encode for two proteins, E2F3a and E2F3b, by alternative splicing (Leone et al., 2000). E2F3a is a transcriptional activator mainly expressed during S phase, while E2F3b acts as a transcriptional repressor and is continuously expressed during the cell cycle (Leone et al., 2000). In contrast, E2F4 and E2F5 possess repressive activity and their major roles have been considered to be the induction of cell cycle exit and differentiation rather than cell cycle progression (DeGregori, 2002; Trimarch and Lees, 2002).
In this study, we prepared specific antibodies to Dmp1 that can detect the endogenous protein in tissues. Here, we show that Dmp1 and a proliferation marker Ki67 expression are essentially mutually exclusive in normal murine tissues. We also show that the Dmp1 promoter is a target for E2Fs and is negatively regulated when cells enter the S to G2/M phase of the cycle.
Results
Detection of the Dmp1 protein in normal murine tissues by Western blotting
Western blotting analysis was performed to verify the presence of the Dmp1 protein in normal murine tissues with newly generated polyclonal antibodies to Dmp1 (RAX) that recognize the DNA-binding domain of the protein (amino acid (a.a.) 136 to a.a. 150). The assay with RAX antibodies showed the presence of Dmp1 as multiple 120–130 kDa bands due to post-translational modification (Hirai and Sherr, 1996; Inoue and Sherr, 1998; Inoue et al., 2000) in most normal murine tissues examined (Figure 1a, top panel). Competition of RAX with the antigenic peptide dramatically reduced the intensity of the bands, suggesting that these multiple bands were specific to Dmp1 (Figure 1a, middle panel). Densitometric analysis was performed and the values normalized to actin levels are presented in the graph (Figure 1b). The highest expression levels of Dmp1 were observed in the testis followed by thymus and lung; moderate expression level of Dmp1 was found in the brain, spleen, intestines and kidney of wild-type mice. The Dmp1 protein was barely detectable in the muscle (heart and tongue), the liver, the pancreas and the bone marrow. The results were consistent with tissue distribution of Dmp1 with different Dmp1 antibodies, RAF (Inoue et al., 2000; Supplementary Material 1). The specificity of the RAX antibodies was also confirmed by Western blotting of tissue lysates from a Dmp1−/− mouse (Figure 1c).
Figure 1.
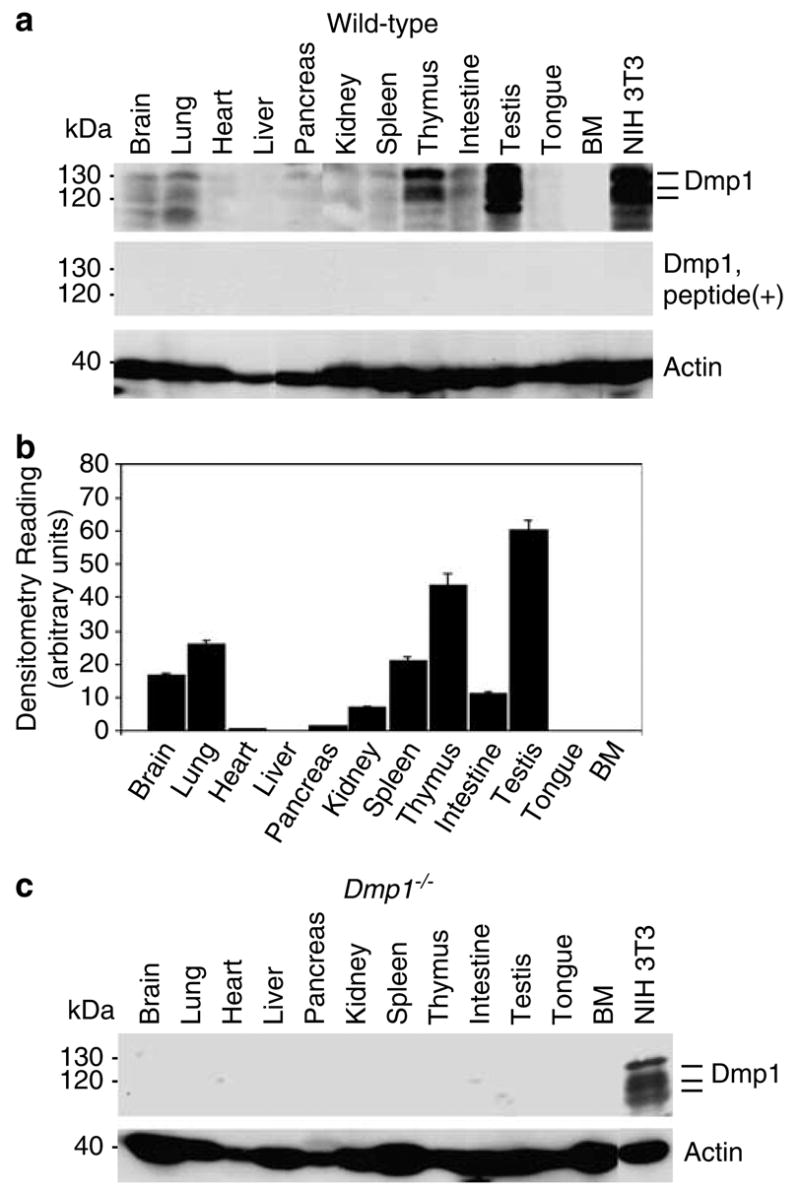
Distribution of the Dmp1 protein in normal murine tissues and the specificity of the anti-Dmp1 antibodies for immunohistochemistry. (a) Detection of the Dmp1 protein in an 8-week-old mouse using tissue samples with RAX antibodies. Indicator bars at right show the positions of multiple Dmp1 isoforms. Lysate from NIH 3T3 cells was used as a positive control. Equal protein loading was verified by blotting the filter with antibodies to actin. Top panel, Western blotting with RAX without antigenic peptide; middle panel, Western blotting with RAX preincubated with the antigenic peptide. (b) Histogram of densitometry of the immunoblots shown in A. The highest level of expression can be found in the testis, thymus, and lung. The Dmp1 protein was barely detectable in the heart, liver, pancreas, tongue, and bone marrow. The intensity of signals was calculated by using densitometric readings, and bars show mean optical density normalized to the control samples for each group ± s.e.m., n = 2, *P<0.05. (c) The Dmp1 protein is not detectable with RAX in tissues from an 8-week-old Dmp1−/− mouse.
Detection of Dmp1 by immunohistochemistry in different mice tissues
The normal murine tissues from an 8-week-old male mouse were stained with RAX antibodies to study the distribution of the Dmp1 protein by using formalin-fixed, paraffin-embedded tissues (the testis, thymus, lung, kidney and pancreas). Brown labeling represented the presence of Dmp1 (DAB substrate). The Dmp1 protein was detected in a wide range of normal mouse tissues, the intensity of which was proportional to the signals of the Western blots (Figures 1 and 2). No detectable labeling was observed in the tissues in the absence of primary antibody or with primary antibodies that had been pre-incubated with the peptide (data not shown). In testis, the nuclei of terminally differentiated sperm and spermatids as well as those of interstitial cells (arrow) were positively stained for Dmp1 (Figure 2a). These signals were not found in the testis from the Dmp1−/− mouse (Figure 2b), showing the specificity of the RAX antibodies.
Figure 2.
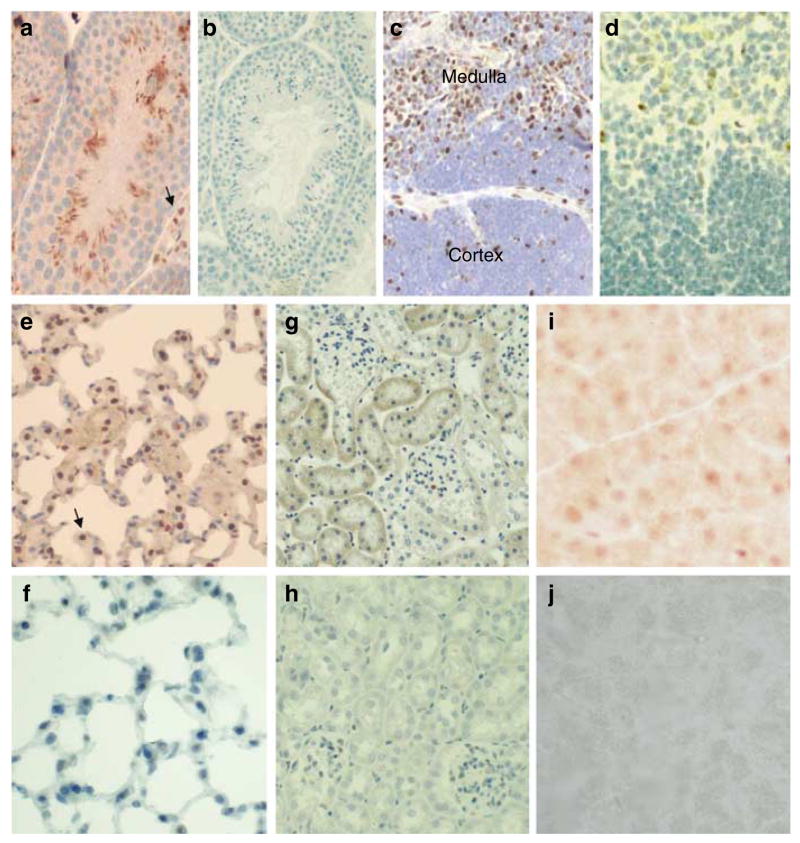
Immunohistochemical detection of Dmp1 in tissues. (a) Immunohistochemical analysis of Dmp1 localization visualized with DAB (brown staining) in the testis and counterstained with hematoxylin. (b) Testis from a Dmp1−/− mouse stained with RAX antibody followed by hematoxylin counterstain. (c) Thymus with positive Dmp1 staining in the medulla. (d) Thymus from a Dmp1−/− mouse stained with RAX, then with hematoxylin counterstain. (e) Dmp1 staining of the lung. Dmp1 localization is visualized with DAB in alveolar macrophages and epithelial cells. (f) Lung from a Dmp1−/− mouse stained with RAX, then with hematoxylin counterstain. (g) Dmp1 staining in the kidney. (h) Kidney from a Dmp1−/− mouse. (i) Weak Dmp1 staining in the pancreas. (j) Pancreas from a Dmp1−/− mouse.
In thymus, Dmp1 appeared mainly in the nuclei of the vast majority of lymphoid cells in the medullary compartment (Figure 2c); some peripheral epithelial-like cells in the Hassall’s bodies appeared also stained (data not shown). Moreover, weak but specific Dmp1 immunoreactivity was also noted in a few subcapsular and cortical epithelial-like cells (data not shown). No detectable labeling was observed in the thymus from Dmp1−/− mice (Figure 2d). A representative image of the normal mouse lung showed prominent Dmp1 signals in alveolar macrophages (arrow), although a significant percentage of type I and type II alveolar cells were weakly positive for Dmp1 as well (Figure 2e). Bronchial epithelial cells also showed Dmp1 immunoreactivity (data not shown). In kidney, there was some staining in the cytoplasm of renal tubules with minimal staining in glomeruli (Figure 2g). The Dmp1 protein was barely detectable in the pancreas which showed very low signals on Western blotting (Figures 1 and 2i). No significant staining was observed in the lung, kidney and pancreas from a Dmp1−/− mouse (Figure 2f, h, and j). Thus, the RAX antibodies specifically detect the Dmp1 protein in formalin-fixed, paraffin-embedded murine tissues.
Detection of the Dmp1 protein in differentiated spermatids, sperm and interstitial cells in testis suggests that the Dmp1 expression may be dependent on cell differentiation or cell cycle. In order to clarify this point, the thymus and the spleen were double stained for Dmp1 (brown) and the proliferation marker Ki67 (blue; Lopez et al., 1991; Kindblom et al., 1995) (Figure 3). In thymus, nearly all the cells were positive for Dmp1 in the medulla, where T-lymphocytes that exited from the cell cycle migrate and accumulate. On the other hand, very few cells were positive for Dmp1 in the cortex, and those cells were negative for Ki67 (Figure 3a and b). In short, the Dmp1 staining and Ki67 staining was mutually exclusive in the thymus. In spleen, Dmp1-positive cells were found in the nuclei of small lymphocytic cells in the medulla (Figure 3c and d), while proliferating Ki67 positive cells were clustered in large lymphoid cells located at the cortex (Figure 3c and d). Dmp1-positive cells and Ki67-positive cells were not overlapping in most cases, although a minor population (20–30%) of the Ki67-positive cells were also positive for Dmp1.
Figure 3.
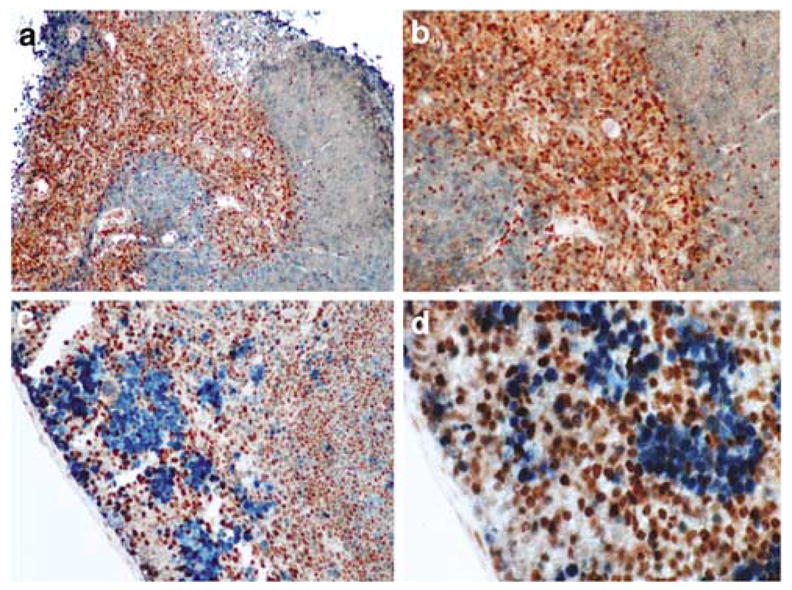
Immunohistochemical detection of Dmp1 in normal thymus and spleen. (a, b) Thymic tissue sections were stained with the Dmp1 and Ki67 antibodies, using peroxidase-conjugate and DAB for Dmp1 (brown) and alkaline phosphatase-conjugate for Ki67 (blue). Note that the staining for Dmp1 and Ki67 are very exclusive. (a) × 10; (b) × 20. (c, d) splenic sections from formalin-fixed and paraffin-embedded mouse tissue were stained with the Dmp1 and Ki67 antibodies, using peroxidase-conjugate and DAB for Dmp1 (brown) and alkaline phosphatase-conjugate for Ki67 (blue). Dmp1 nuclear expression is found mainly in the germinal centers of the organ, while Ki67 was expressed in mononuclear cells in the subcortical area. Magnification, (c) × 10; (d) × 20.
Generally, Dmp1 was detected in the epithelial, interstitial (mostly hematopoietic and vascular endothelial cells), and smooth muscle cells in small and large intestines. Representative photomicrographs are presented in Figure 4. Dmp1 was mostly localized in the nuclei of epithelial cells and interstitial section of the villi with a progressive decrease in staining intensity towards the lower portions (Figure 4a and b). The Dmp1 immunoreactivity was also observed in the nuclei of Goblet cells (Figure 4e, brown signal), Paneth cells, and smooth muscle cells (data not shown). When the neighboring section was stained with the Ki67 proliferation marker, a strong nuclear staining of nearly all cells in the lower half to 1/3 of the crypts was observed (Figure 4c and d). The Ki67-positive cells were rarely found along the villous surface. Double staining of the small intestine revealed that there was very little overlap between Dmp1-positive cells and Ki67-positive cells (Figure 4e). Staining for Dmp1 and another proliferation marker PCNA was also exclusive (data not shown). In colon, the nuclei of epithelial cells, Goblet cells, and submucosal area show strong Dmp1 immunoreactivity (Figure 4f). On the other hand, Ki67 staining was confined to the lower proliferative portion of the crypt. Again, there was very little overlap between Dmp1-positive cells and Ki67-positive cells (Figure 4f).
Figure 4.
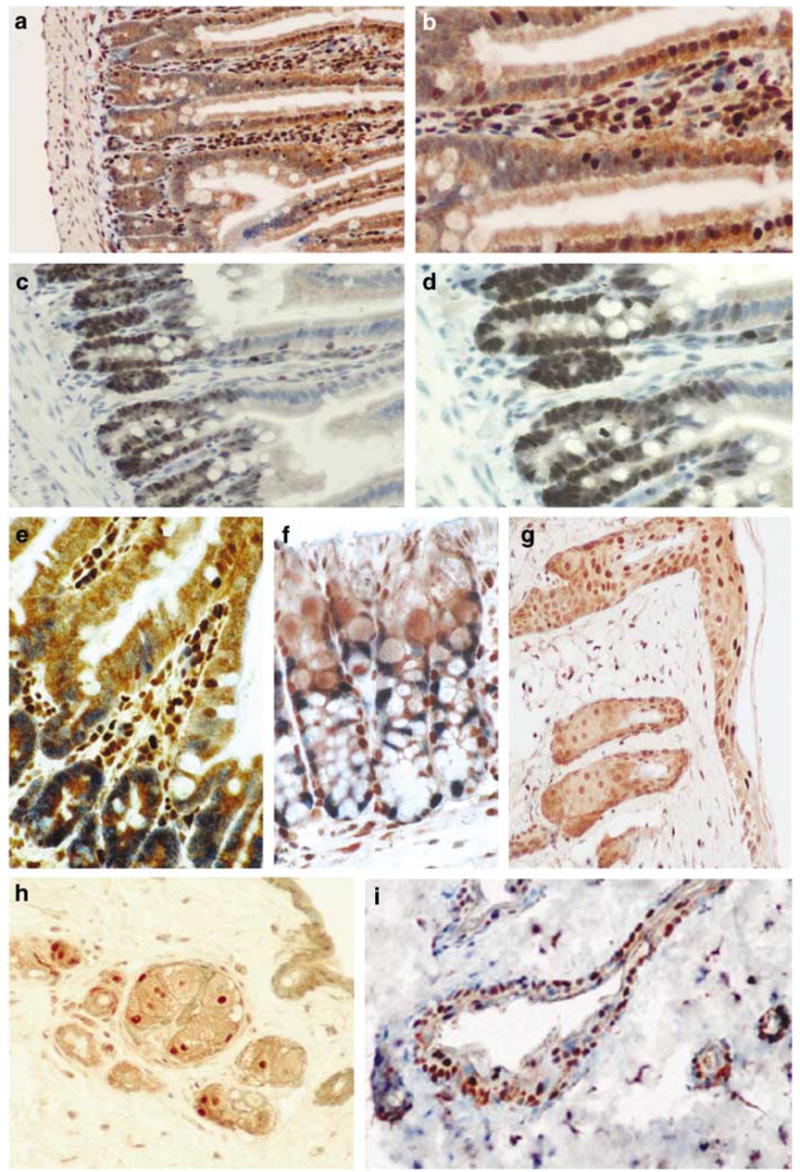
Mutually exclusive staining of Dmp1 and Ki67 in normal intestines (a–f) and skin (g–i). (a, b) Staining with the Dmp1 antibody in the small intestine of a wild-type mouse using peroxidase-conjugate and DAB substrate (brown). Magnification: (a) ×10; (b) × 20. (c, d) Showing the positive expression of Ki67 in the small intestine (dark blue) (c, ×10; d, ×20). (e) Showing double staining with Dmp1 and Ki67 in small intestine (× 40). (f) Showing Dmp1 (brown) and Ki67 (blue) double staining in mouse colon (× 40). (g) Showing Dmp1 staining in the skin (brown, × 20). (h) Showing Ki67 staining of the skin in a cross section of a hair root (brown, ×20). (i) Showing the Dmp1 (brown) and Ki67 (blue) double staining of the skin (× 20). The Dmp1 expression is more localized at the epidermis.
The localization of Dmp1 and Ki67 within the normal mouse skin was also examined (Figure 4g–i). Although the immunoreactivity for Dmp1 was detected in the nuclei of the keratinocytes in the whole epidermis, the staining was most intense in the most superficial layers of the epidermis, where the cells completely exited from the cell cycle (Figure 4g). In contrast, the staining for Ki67 was localized to the nuclei of basal cells at the inner root sheath of the hair shaft (brown, Figure 4h), and it was not detected at the epidermis (Figure 4h). The Dmp1 staining and Ki67 staining appeared not to overlap (Figure 4i; Dmp1 was stained brown, Ki67 was stained blue).
Responsiveness of the murine Dmp1 promoter to E2Fs and mitogenic stimuli
As we detected very specific expression of Dmp1 in the nuclei of well differentiated, nonproliferating cells in normal tissues, we were interested to know whether the Dmp1 promoter responds to E2Fs. The murine Dmp1 promoter contains three possible E2F-binding sequences around the transcriptional initiation site (Figure 5a). The Dmp1 promoter was efficiently repressed by all of the E2Fs studied (E2F1, E2F2, E2F3a, E2F3b and E2F4) (Figure 5b, left panel). The repression of the Dmp1 promoter by E2Fs was dependent on the histone deacetylases since trichostatin A reversed the repression (data not shown). The result of the reporter assay was confirmed by quantitation of the Dmp1 mRNA in 3T3 cells transfected with E2F expression vectors (Figure 5b, right panel). Then we mapped the E2F-responsive element on the Dmp1 promoter. Cloning of the other two E2F sites (#2 and #3) into the –374 NsiI reporter construct did not influence the responsiveness of the Dmp1 promoter to E2F1 (Figure 5c). On the other hand, the responsiveness of the Dmp1 promoter to E2F1 was completely lost in the construct where the E2F site #1 was mutated (–374 NsiI E2F#1M, Figure 5c). Thus, the E2F site #1 plays the most important role in E2F-mediated repression of the Dmp1 promoter. Next, we investigated whether endogenous E2Fs bind to the Dmp1 promoter upon physiological mitogenic stimulation. Proteins that specifically react with anti-E2F1, E2F3a + b and E2F4 antibodies were specifically found on the Dmp1 promoter upon serum stimulation of the cells (Figure 5d). In E2F3, E2F3b was considered to be on the Dmp1 promoter since antibodies that specifically bind to E2F3a did not give any specific signals (Figure 5d).
Figure 5.

Binding of E2F proteins to the Dmp1 promoter. (a) Nucleotide sequences of the mouse Dmp1 promoter proximal region (−115 to +184). The major transcription intiation site determined by 5′-RACE is shown in a bold letter G. Consensus sequences for possible transcription factor binding including Sp1 and E2F sites are also shown. Major restriction enzyme sites and the positions of PCR primers used for ChIP are shown in bold italic characters. (b) Left panel: responsiveness of the Dmp1 promoter to E2Fs. Mouse 3T3 cells were transfected with the Dmp1 luciferase reporter with expression vectors for E2F1, E2F2, E2F3a, E2F3b and NLS(+) E2F4 driven by the CMV promoter. The numbers show the relative luciferase activity corrected by the internal control secreted endocrine alkaline phosphatase levels. (b) Right panel: quantification of the Dmp1 mRNA by real-time PCR in 3T3 cells transfected with E2F expression vectors. (c) Mapping of the E2F-responsive element on the Dmp1 promoter. Cloning of the two additional E2F sites #2 and #3 (–374 NsiI (+)) did not change the responsiveness of the Dmp1 promoter to E2F1 while the responsiveness was totally lost in the construct where E2F#1 site was mutated (–374 NsiI E2F#1M). (d) ChIP assay on the Dmp1 promoter. Endogenous proteins that bind to E2F1, E2F3a +b, and E2F4 antibodies were detected on the Dmp1 promoter with primers that cover the possible E2F sites in response to serum stimulation of MEFs (upper panel, Dmp1). No significant signals were obtained with control primers that amplify 2 kb upstream Dmp1 promoter sequence from the region of interest (lower panel, Control). The last two lanes present signals from total chromatin samples.
In order to test the responsiveness of the Dmp1 promoter to serum stimulation, rodent fibroblasts were synchronized and were stimulated with serum for various length of time to perform the Dmp1 promoter reporter assay (Figure 6a). The Dmp1 promoter activity was repressed and reached a minimum at 16 h after serum stimulation when most of the cells had entered the S phase (~60%). The levels remained low when the cells cycled to the G2/M phase of the cell cycle (Figure 6a upper panel). The serum-responsiveness of the Dmp1 promoter was lost in the –374 NsiI E2F#1M construct, suggesting the importance of this site for cell cycle-dependent regulation as well (Figure 6a, lower panel). The inhibition of the Dmp1 promoter activity resulted in the decline of the Dmp1 mRNA in primary MEFs (70–90% repression by semi-quantitative PCR and quantitative real-time PCR at S to G2/M phase; Figure 6b and c). Significant decrease of the Dmp1 target Arf mRNA was also observed when the cells entered the S phase of the cell cycle (~55%), while Twist (data not shown) or β-actin levels did not change (Figure 6b and c). The decrease of the Dmp1 mRNA resulted in the decline of the Dmp1 protein levels several hours following the decrease of the Dmp1 mRNA (at 16–28 h after addition of the serum) (Figure 6d, top panel). The p19Arf proteins also decreased when the Dmp1 levels were downregulated (Figure 6d, the second panel). The PCNA protein levels increased several hours earlier than the decrease of the Dmp1 protein, suggesting the activity of E2F family proteins (Figure 6d, the third panel) (DeGregori et al., 1995). In order to confirm the E2F-dependence of the Dmp1 promoter repression in S to G2/M phase of the cell cycle, we infected wild-type MEFs with E2F-DB retroviruses (Figure 6e). E2F-DB is a deletion mutant of E2F1 that lacks both transactivation domain and Rb-binding domain, thus, it inhibits the activity of all the repressor functions of E2F proteins (Rowland et al., 2002). In E2F-DB(+) cells, the Dmp1 promoter was not inhibited when the cells entered S to G2/M phase of the cell cycle (Figure 6f). Real-time PCR indicated that there was no change of the Dmp1 levels throughout the cell cycle (Figure 6g). Our data suggest that upon serum stimulation, subsets of E2F proteins bind to the Dmp1 promoter and cause its repression, and this, in turn, downregulates the Dmp1 mRNA and protein levels in the S to G2/M phase of the cell cycle.
Figure 6.

Both Dmp1 mRNA and protein are downregulated as the cells enter the S to G2/M phase of the cell cycle. (a) Upper panel: the Dmp1 reporter assay in synchronized 3T3 cells. Cells were transfected with the Dmp1-luciferase reporter (–374 NsiI) and the internal control actin promoter-SEAP vector. The cells were starved for serum for 24 h and were harvested at different time points after addition of 10% serum. (a) Lower panel: mapping of serum-responsive element on the Dmp1 promoter. The Dmp1 promoter is repressed by serum both in the wild-type construct (–374 NsiI) and also in the – NsiI (+) construct, but not in that with disrupted E2F sequence #1 (–374 NsiI E2F#1M). (b) Semi-quantitative RT–PCR assay for Dmp1, Arf and β-actin mRNA in synchronized MEFs. (c) Real-time PCR analysis of the Dmp1 and Arf gene expression in synchronized MEFs. The Dmp1 mRNA was dramatically downregulated at S to G2/M phase in primary MEFs. Significant decrease of the Dmp1 target, Arf mRNA was also observed when the cells entered the S phase of the cell cycle (~55%). (d) Western blotting analysis of synchronized MEFs for Dmp1, p19Arf, PCNA and actin. The arrows indicate the peaks of the S phase of the cell cycle. (e) Detection of the E2F-DB protein in retrovirus-infected MEFs. Lane 1, pBabe-puro empty vector virus; lane 2, pBabepuro-E2F-DB virus. (f) The Dmp1 promoter is not repressed by serum in E2F-DB(+) cells. Wild-type MEFs were infected with pBabe puro-vector virus or pBabepuro-E2F-DB virus were synchronized and stimulated with serum for indicated hours. (g) Real-time PCR analysis of the Dmp1 mRNA in E2F-DB(+) cells. There was no significant change of the Dmp1 mRNA in E2F-DB cells when cells entered the S to G2/M phase of the cell cycle.
Discussion
Our previous studies have demonstrated that overexpression of Dmp1 causes p19Arf-, p53-dependent cell cycle arrest in rodent fibroblasts (Inoue et al., 1999). Disruption of the Arf-p53 pathway is less frequent in tumors from Dmp1-knockout or heterozygous mice and, therefore, Dmp1 has been considered to be a physiological regulator of the Arf-p53 pathway (Inoue et al., 2001). In this study, we have raised specific antibodies to Dmp1 for the immunohistochemical detection of this protein in tissues. We have found that significant levels of the Dmp1 protein were expressed in differentiated cells in the testis, thymus, spleen, intestine, skin and lung tissues of mice. Double staining of tissues with Dmp1 and the proliferation marker Ki67 (or PCNA) antibodies indicated that Dmp1 and proliferation markers are expressed in mutually exclusive fashion, suggesting that Dmp1 is a novel marker of cells that have exited from the cell cycle in some cell types. Molecular biology studies demonstrated that both Dmp1 promoter activity and the mRNA/protein levels were downregulated when the cells entered the S to G2/M phase. In an attempt to detect the regulation of the Dmp1 promoter by different transcription factors, several experiments have been conducted. Chromatin immunoprecipitation (ChIP) assay with specific antibodies to E2F proteins showed unambiguous binding of E2F1, E2F3b and E2F4 to the Dmp1 promoter when the cells entered the S to G2/M phase of the cell cycle. Importantly, the Dmp1 mRNA levels did not change upon mitogenic stimulation in E2F-DB(+) cells where all the E2F repressor complexes were inactivated (Rowland et al., 2002), indicating that the repression of the Dmp1 promoter was dependent on E2Fs. These assays explain how Dmp1 and Arf are downregulated as the cells enter the S to G2/M phase of the cell cycle and why Dmp1 and Ki67 are stained in different subsets of cells in normal tissues.
On the Dmp1 promoter, subsets of E2Fs were specifically bound when the synchronized MEFs entered the S phase of the cell cycle. The Arf promoter was reported to be occupied by E2F3b, and not by other E2F family members, E2F1, E2F4 (Aslanian et al., 2004) in quiescent wild-type MEFs. It was also reported that endogenous activating E2Fs, E2F1 and E2F3a were recruited to the Arf promoter in response to hyperproliferative oncogenic signaling, indicating that distinct subsets of E2F proteins contribute to the normal repression and oncogenic activation of Arf (Aslanian et al., 2004). Surprisingly, all of the E2F1, E2F2 and E2F3a proteins were repressors on the Dmp1 promoter, especially the former two. Thus, E2F1 has differential effects on the Dmp1 promoter (repression) and the Arf promoter (activation) when overexpressed in rodent fibroblasts (Figure 5b, Inoue et al., 1999). The E2F-mediated regulation of the Dmp1 promoter is conserved in humans since the hDMP1 promoter has a typical E2F site (5′-TTTCGCGC) and is efficiently repressed by E2Fs (Inoue et al., unpublished data). It is important to note that the Dmp1 promoter is not the only promoter repressed by ‘activating’ E2Fs; repression of the human telomerase promoter as well as the tumor suppressor ARHI promoter by E2F1 have also been reported (Crowe et al., 2001; Lu et al., 2006). Although the detailed mechanism of E2F-mediated repression of the Dmp1 promoter in mitogen-stimulated cells remains to be determined, our preliminary data suggest that the repression is Rb-independent since the E2F1 Y411C mutant that does not bind to Rb (Rowland et al., 2002) inhibited the Dmp1 promoter as well. However, the repression is still histone deacetylase-dependent since trichostatin A reversed the repression. Further studies will be required to understand the requirement of pocket-proteins for the Dmp1 promoter repression by E2Fs.
In normal tissues, the Arf mRNA is detectable in the testis, thymus, spleen and lung only by very sensitive reverse transcriptase–polymerase chain reaction (RT–PCR) assays (Zindy et al., 1997). However, a recent study with rat monoclonal antibodies to mouse p19Arf (5-C3-1) demonstrated that the protein was detected in the nucleoli of spermatogonia, suggesting that p19Arf might be involved in facilitating the commitment of mitotic progenitor cells to meiosis (Bertwistle et al., 2004). However, the monoclonal antibodies did not detect significant signals in the differentiated sperm or interstitial cells in the testis nor in any other tissues where Dmp1 protein is detected (Bertwistle et al., 2004). Our interpretation is that the p19Arf expression in normal tissues might be too low to be detected even with these Arf-specific monoclonal antibodies and different transcription factors must be regulating the Arf expression in the nucleoli of spermatogonia. This could be because p19Arf is an extremely potent inhibitor of cell cycle progression, and even undetectable levels of p19Arf levels may play certain roles in normal cell cycle progression.
Although the Dmp1 protein binds to some subsets of DNA sequences recognized by the Ets family (XXCGGATGT/C), it is structurally more related to the Myb-family proteins. Indeed, c-Myb and Dmp1 collaborate to activate the myeloid/fibroblast-specific CD13/aminopeptidase N promoter (Inoue et al., 1998). The Dmp1 expression is high in the thymus and spleen, while it is almost undetectable in bone marrow cells. The Dmp1 protein is specifically expressed in the medulla of the thymus, where the proliferation marker Ki67 was completely negative. It was previously found that Dmp1−/− lymphocytes are hyperproliferative in culture, and also T-cell lymphomas/leukemias are frequently observed in Dmp1-knockout mice in response to dimethylbenzanthracene and γ-irradiation (Inoue et al., 2000, 2001). Therefore, Dmp1 may be playing critical roles in the maintenance of the resting state of differentiated T lymphocytes. Among the Myb family proteins (A-myb, B-myb, c-myb), c-myb expression has been considered to be an indicator of hematopoietic cell proliferation since the level of expression is high in the thymus and in the bone marrow, and its downregulation occurs as an early event in cellular differentiation (Oh and Reddy, 1999). In thymus, c-myb is selectively expressed in the cortex, and the expression is much lower in the medulla which behaves like immune competent peripheral T cells (Sheiness and Gardinier, 1984). Taken together, Dmp1 and c-myb may play complementary roles in early (c-myb) and late (Dmp1) thymic T-lymphocyte development and proliferation.
In intestines, Dmp1 protein was specifically expressed in the nuclei of differentiated epithelial cells, Goblet cells and in Paneth cells. On the other hand, Ki67 staining was strictly localized to the basal layers of the intestinal crypts, and the double staining for Dmp1 and Ki67 showed that there is very little overlap in cells that express Dmp1 and Ki67, indicating that Dmp1 is a marker for post-mitotic cells in the intestines, as well. However, in small intestine, the strongest signals were obtained in the interstitial cells of villi. These Dmp1-positive interstitial cells are mostly peripheral leukocytes (eosinophils and mature lymphocytes). It is interesting to note that most of these Dmp1-positive cells in the interstitium were negative for Ki67 and, again, were post-mitotic. As epithelial cells migrate up the crypts, the Ki67-expressing proliferating compartment near the crypt base ends abruptly, with the coincident appearance of a non-proliferating compartment expressing p21Cip1 (el-Deiry et al., 1995). Thus, the expression pattern of p21Cip1 in intestines is very close to that of Dmp1.
In conclusion, our study showed differential expression of Dmp1 and Ki67 in all the tissues examined and, thus, Dmp1 is a novel biomarker of some cells that have exited from the cell cycle. The mutually exclusive staining pattern for Dmp1 and Ki67 was very prominent in the thymus, intestines and skin. Although we cannot explain why there are non-proliferating tissues that do not express Dmp1 (such as kidney, liver, pancreas and muscle), our data suggest new fundamental roles of Dmp1 in cell cycle regulation in normal tissues than previously reported. Dmp1 could, thus, be subject of precise cellular control, and escape from this regulation may be a critical feature of neoplastic transformation.
Materials and methods
Cell culture and reporter assays
Wild-type MEFs were established from 13.5-day-old embryos and maintained as described previously (Inoue et al., 2000). For reporter assays, 3 × 105 mouse 3T3 cells were seeded onto 60 mm diameter culture dishes 24 h before transfection. In order to study the responsiveness of the Dmp1 promoter to the serum, 1.5 μg of luciferase reporter DNA and 1.5 μg of the internal control β-actin promoter-secreted endocrine alkaline phosphatase vector were used (a gift from Dr Michael Ostrowski). At 24 h after transfection, cells were serum starved for another 24 h and then 10% of FBS was added to the dishes for the indicated time period. In order to test the responsiveness of the Dmp1 promoter to E2Fs, 1.5 μg of the Dmp1 reporter and 0.25 μg and 0.75 μg of E2F expression vectors were used per 60 mm dish. Genejuice (EMD Biosciences, San Diego, CA, USA) was used in all transfections.
Plasmids
The E2F expression vectors (E2F1, 2, 3a, and 3b) driven by the CMV promoter were gifts from Dr Joseph Nevins (Johnson et al., 1993; Leone et al., 2000). The NLS(+)E2F4 expression vector was from Dr Geoffrey Lindeman (Lindeman et al., 1997). The E2F-DB and E2F1Y411C expression vectors were obtained from Dr Rene Bernards (Rowland et al., 2002).
In vitro mutagenesis
In order to create the Dmp1 promoter that contains all the possible E2F-binding consensus sequences (–374 NsiI (+)), the 5′RACE product that contains the 5′leader sequence of the Dmp1 gene was digested with PstI and XbaI, and the 216 bp fragment was recloned into the PstI–NheI site of the –374 NsiI wt construct. In order to mutate the E2F site #1 on the Dmp1 promoter (–374 NsiI E2F#1M), PCR was performed by using oligonucleotides 5′-GCCTCGCGGCTC CGTCGTAGGTGGCTGGTTGCGC-3′ and its reverse complementary sequence. The mutated E2F site is underlined.
Generation of anti-Dmp1 antibodies
Polyclonal antibodies to the Dmp1 DNA-binding domain were generated by injecting rabbits with KLH-conjugated peptides NH2-(GC)TTKEDKDSLTNKGHK-COOH (Bio-synthesis, Lewisville, TX, USA). This sequence corresponds to a.a. 136–150 of the murine and human DMP1 protein. The anti-Dmp1 antiserum (RAX) was affinity-purified and was used for Western blotting and for immunohistochemistry.
Immunohistochemical staining
Organs were isolated from 8-week-old male mice, and were fixed immediately in 10% formalin (3.7% formaldehyde in PBS) for 24 h, and then embedded in paraffin and cut into 4-μm thick sections. Tissue sections were deparaffinized with xylene, and re-hydrated in a descending ethanol series. Antigen retrieval was carried out by using Dakocytomation Target Retrieval Solution (Dakocytomation, Carpinteria, CA, USA) (Barrett et al., 2001). The sections were then incubated with primary antibodies (Dmp1, PCNA (sc-56, Santa Cruz), or Ki67 (Ab-3, NeoMarkers)) at optimum dilution (1:100–1:250) for 30 min, rinsed three times with washing buffer PBS, followed by 30 min incubation with alkaline-phosphatase conjugated affinity purified goat anti-mouse IgG for PCNA and horseradish peroxidase (HRP) conjugated affinity purified goat anti-rabbit IgG secondary antibody for Dmp1 and Ki67 staining (Jakson Immunoresearch Laboratories, West Grove, PA, USA). After substrate reactions using Vector™ Elite ABC and AP kits (Vector Laboratories, Burlingame, CA, USA) according to the manufacturer’s instructions, sections were counterstained with Mayer’s hematoxylin and mounted with coverslips using an aqueous mounting media. Sections for negative controls were treated as above except for omission of the primary antibodies.
Double staining immunohistochemistry
After the first color reaction (DAB) was developed by using a DAKO LSAB System (DAKO), the secondary antibodies for the second antigen were applied in the same manner as that used for the first antigen, and the second color reaction was developed with an Alkaline Phosphatase Substrate Kit III (blue) (Vector Laboratories).
Semi-quantitative RT–PCR
Semi-quantitative RT–PCR was employed with specific primers for Dmp1 (32 cycles), murine Arf exon 1β (30 cycles), and for β-actin (21 cycles) (Inoue et al., 1994, 1999). The sequences of the primers were sense 5′-CTGCAACACAGG GAAATGGA-3′and anti-sense 5′-ACGGACACTGCTC CATCCTT-3′for Dmp1; sense 5′-ATGGGTCGCAGGTTCT TGGTC-3′and anti-sense 5′-CTGGTCCAGGATTCCGGT GC-3′for Arf; sense 5′-GTGGGCCGCCCTAGGCACCAG-3′and antisense 5′-CTCTTTGATGTCACGCACGATTTC-3′for β-actin.
Quantitative real-time PCR assay
The ABI PRISM 7000 instrument (Applied Biosystems, Foster City, CA, USA) was used for quantitative real-time PCR amplification and detection. Quantitative PCR was prepared in triplicates of 50 μl reaction mixture in MicroAmp optical 96-well reaction plates and sealed with optical adhesive covers (Applied Biosystems). Master mix components and PCR cycling conditions were similar to the one explained previously (Bentley et al., 2005). If a sample amplification curve crosses the threshold line to give a threshold value (Ct), the sample would be interpreted as valid regardless of the PCR result of the respective Internal Positive Control. Serial dilutions of cDNA template were conducted in triplicates to establish the standard curves. The standard curve is a plot of the threshold cycle (Ct) versus log concentration (Co). For any unknown total cDNA sample, by interpolating its Ct value against the standard curve, the absolute quantity of DNA template was obtained. Also relative quantitative analysis was conducted on the cDNA samples. The expression assay for Dmp1 was Applied Biosystems’ Custom TaqMan that was initially designed by our laboratory (forward primer: 5′-TGACCA CAAATCCCACAGTAACC-3′and reverse primer: 5′-GTG ACATTATCGCTTGTGTTCAACA-3′). The ABI assay ID for p19Arf was Mm00494449_ml and that for β-actin was Mm00607939_s1.
Western blotting
For analysis of Dmp1, p19Arf, PCNA, E2F-DB, and actin, frozen cell pellets were dissolved in ice-cold EBC buffer (Hirai and Sherr, 1996) and left on ice for 1 h. After sonication at 4°C (7 s × 2), nuclei and cellular debris were removed by centrifugation in a microcentrifuge at 15 000 r.p.m. for 15 min at 4°C. Proteins (80 μg/lane) were separated electrophoretically on denaturing polyacrylamide gels containing SDS and transferred onto nitrocellulose (MSI, Westboro, MA, USA). Proteins were visualized by direct immunoblotting with affinity-purified polyclonal antibodies to the mouse Dmp1 (RAX), p19Arf (sc-32748, Santa Cruz), PCNA (sc-56), E2F1 (05-379, Upstate, Charlottesville, VA, USA), or actin (sc-1615), followed by incubation of the filters with HRP-conjugated second antibodies, reaction with the enhanced ECL detection kit (Perkin Elmer, Boston, MA, USA).
Densitometry
The bands on the developed immunoblot films were scanned using a Hewlett Packard laser scanner. Densitometry was carried out using automated digitizing software UN-SCA-NIT™ Gel Version 4.3 (Silk Scientific Inc., Orem, UT, USA). The generated total relative densitometric units for each sample were analysed. Any background noise was subtracted from each lane. The molecular weights of the bands in the standards lane were defined. Finally the relative densitometric units for the protein of interest were normalized to relative densitometric units for actin from the same sample. An output of peak intensity and estimated molecular weight for each band was produced, and the average levels of Dmp1 protein expression from two different 8-week-old C57BL/6 male mice were shown.
Chromatin immunoprecipitation
ChIP was performed as described previously (Chadee et al., 1999; Sreeramaneni et al., 2005). Briefly, confluent wild-type MEFs were starved for serum for 24 h, and then were split 1:3 with complete medium for 18 h. The lysates were precipitated with specific antibodies to E2F proteins (E2F-1: sc-193x; E2F2, sc-632 and sc-633; E2F3a, sc-879x; E2F3a + b, sc-878; and E2F4, sc-1082x, all from Santa Cruz Biotech) and were incubated at 4°C overnight. The antibodies were recovered by using Protein A-sepharose, and washed extensively. The immunoprecipitated DNA was recovered by digestion of the samples with proteinase K and RNase. The purified DNA was detected by PCR, including 1 μCi of [α-32P] dATP (GE Healthcare), separated on a 10% nondenaturing polyacrylamide gel. For detection of the endogenous E2F family transcription factors on the murine Dmp1 promoter, sense primer 5′-CTCGCGGCTCCGTTTCCG-3′and antisense primer 5′-CGGACCTGAAGGTTCCATCG-3′were used. The primer sequences for the control amplification of 2-kb upstream sequence on the Dmp1 promoter were described (Sreeramaneni et al., 2005).
Supplementary Material
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
Acknowledgments
We are grateful to Dr Tim Kute for helpful discussions. We thank Asif Chaudhry and Oktay Kaplan for technical assistance, Drs Charles Sherr, Martine Roussel, Joseph Nevins, Geoffrey Lindeman, Rene Bernards, and Michael Ostrowski for plasmid DNAs. We also thank Dr Michael Robbins for editing and critical reading of the manuscript. This work was supported by National Institute of Health Grant 5R01CA106314-02 (K Inoue) and WFUCCC push grant CA12197-31 (K Inoue). Supplementary material 1 is available at Oncogene’s website.
References
- Aslanian A, Iaquinta PJ, Verona R, Lees JA. Genes Dev. 2004;18:1413–1422. doi: 10.1101/gad.1196704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett K, Willingham JM, Garvin AJ, Willingham MC. J Histochem Cytochem. 2001;49:821–832. doi: 10.1177/002215540104900703. [DOI] [PubMed] [Google Scholar]
- Bentley HA, Belloni DR, Tsongalis GJ. Clin Biochem. 2005;38:183–186. doi: 10.1016/j.clinbiochem.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Bertwistle D, Zindy F, Sherr CJ, Roussel MF. Hybrid Hybridomics. 2004;23:293–300. doi: 10.1089/hyb.2004.23.293. [DOI] [PubMed] [Google Scholar]
- Campanero M, Armstrong M, Flemington E. Mol Cell Biol. 1999;19:8442–8450. doi: 10.1128/mcb.19.12.8442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchpole S, Tavner F, Cam LE, Sardet C, Watson RJ. J Biol Chem. 2002;277:39015–39024. doi: 10.1074/jbc.M202960200. [DOI] [PubMed] [Google Scholar]
- Chadee DN, Hendzel MJ, Tylipski CP, Allis CD, Bazett-Jones DP, Wright JA, et al. J Biol Chem. 1999;274:24914–24920. doi: 10.1074/jbc.274.35.24914. [DOI] [PubMed] [Google Scholar]
- Crowe DL, Nguyen DC, Tsang KJ, Kyo S. Nucleic Acid Res. 2001;29:2789–2794. doi: 10.1093/nar/29.13.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGregori J, Kowalik T, Nevins JR. Mol Cell Biol. 1995;15:4215–4224. doi: 10.1128/mcb.15.8.4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGregori J. Biochem Biophys Acta. 2002;1602:131–150. doi: 10.1016/s0304-419x(02)00051-3. [DOI] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Waldman T, Oliner JD, Velculesu VE, Burrell M, et al. Cancer Res. 1995;55:2910–2919. [PubMed] [Google Scholar]
- Hirai H, Sherr CJ. Mol Cell Biol. 1996;16:6457–6467. doi: 10.1128/mcb.16.11.6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Roussel MF, Sherr CJ. Proc Natl Acad Sci USA. 1999;96:3993–3998. doi: 10.1073/pnas.96.7.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Sherr CJ. Mol Cell Biol. 1998;18:1590–1600. doi: 10.1128/mcb.18.3.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Sherr CJ, Shapiro LH. J Biol Chem. 1998;273:29188–29194. doi: 10.1074/jbc.273.44.29188. [DOI] [PubMed] [Google Scholar]
- Inoue K, Sugiyama H, Ogawa H, Yamagami T, Azuma T, Oka Y, et al. Blood. 1994;84:2672–2680. [PubMed] [Google Scholar]
- Inoue K, Wen R, Rehg JE, Adachi M, Cleveland JL, Roussel MF, et al. Genes Dev. 2000;14:1797–1809. [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Zindy F, Randle DH, Rehg JE, Sherr CJ. Genes Dev. 2001;15:2934–2939. doi: 10.1101/gad.929901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joaquin M, Watson RJ. Cell Mol Life Sci. 2003;60:2389–2401. doi: 10.1007/s00018-003-3037-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DG, Schwartz JK, Cress WD, Nevins JR. Nature. 1993;365:349–352. doi: 10.1038/365349a0. [DOI] [PubMed] [Google Scholar]
- Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Cancer Res. 1999;59:2217–2222. [PubMed] [Google Scholar]
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, et al. Cell. 1997;91:649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- Kindblom LG, Ahlden M, Meis-Kindblom JM, Stenman G. Virchows Arch. 1995;427:19–26. doi: 10.1007/BF00203733. [DOI] [PubMed] [Google Scholar]
- Leone G, Nuckolls F, Ishida S, Adams M, Sears R, Jakoi L, et al. Mol Cell Biol. 2000;20:3626–3632. doi: 10.1128/mcb.20.10.3626-3632.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeman GJ, Gaubatz S, Livingston DM, Ginsberg D. Proc Natl Acad Sci USA. 1997;94:5095–5100. doi: 10.1073/pnas.94.10.5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez F, Belloc F, Lacombe F, Dumain P, Reiffers J, Bernard P, et al. Cytometry. 1991;12:42–49. doi: 10.1002/cyto.990120107. [DOI] [PubMed] [Google Scholar]
- Lowe S, Sherr CJ. Curr Opin Genet Dev. 2003;13:77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- Lu Z, Luo RZ, Peng H, Huang M, Nishimoto A, Hunt KK, et al. Oncogene. 2006;25:230–239. doi: 10.1038/sj.onc.1209025. [DOI] [PubMed] [Google Scholar]
- Oh I-H, Reddy EP. Oncogene. 1999;18:3017–3033. doi: 10.1038/sj.onc.1202839. [DOI] [PubMed] [Google Scholar]
- Quon K, Berns A. Genes Dev. 2001;15:2917–2921. doi: 10.1101/gad.949001. [DOI] [PubMed] [Google Scholar]
- Rowland B, Denissov SG, Douma S, Stunnenburg HG, Bernards R, Peeper DS. Cancer Cell. 2002;2:55–65. doi: 10.1016/s1535-6108(02)00085-5. [DOI] [PubMed] [Google Scholar]
- Ruas M, Peters G. Biochem Biophys Acta Rev Cancer. 1998;1378:F115–F177. doi: 10.1016/s0304-419x(98)00017-1. [DOI] [PubMed] [Google Scholar]
- Sala A, Watson R. J Cell Physiol. 1999;179:245–250. doi: 10.1002/(SICI)1097-4652(199906)179:3<245::AID-JCP1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Sheiness D, Gardinier M. Mol Cell Biol. 1984;4:1206–1212. doi: 10.1128/mcb.4.7.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ. Nat Rev Mol Cell Biol. 2001;2:731–737. doi: 10.1038/35096061. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Sreeramaneni R, Chaudhry A, McMahon M, Sherr CJ, Inoue K. Mol Cell Biol. 2005;25:220–232. doi: 10.1128/MCB.25.1.220-232.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimarch JM, Lees JA. Nat Rev Mol Cell Biol. 2002;3:11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- Wu L, Timmers C, Maiti B, Saavedra HI, Sang L, Chong GT, et al. Nature. 2001;414:457–462. doi: 10.1038/35106593. [DOI] [PubMed] [Google Scholar]
- Zindy F, Quelle DE, Roussel MF, Sherr CJ. Oncogene. 1997;15:203–211. doi: 10.1038/sj.onc.1201178. [DOI] [PubMed] [Google Scholar]
- Zindy F, Williams RT, Baudino TA, Rehg JE, Skapek SX, Cleveland JL, et al. Proc Natl Acad Sci USA. 2003;100:15930–15935. doi: 10.1073/pnas.2536808100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)