

Abstract
Protein kinase D2 (PKD2), a member of the PKD family of serine/threonine kinases, is localized in various subcellular compartments including the nucleus where the kinase accumulates upon activation of G-protein-coupled receptors. We define three critical post-translational modifications required for nuclear accumulation of PKD2 in response to activation of the CCK2 receptor (CCK2R): phosphorylation at Ser706 and Ser710 within the activation loop by PKCη leading to catalytic activity and phosphorylation at Ser244 within the zinc-finger domain, which is crucial for blocking nuclear export of active PKD2 by preventing its interaction with the Crm-1 export machinery. We identify CK1δ and ɛ as upstream activated kinases by CCK2R that phosphorylate PKD2 at Ser244. Moreover, nuclear accumulation of active PKD2 is a prerequisite for efficient phosphorylation of its nuclear substrate, HDAC7. Only nuclear, active PKD2 mediates CCK2R-induced HDAC7 phosphorylation and Nur77 expression. Thus, we define a novel, compartment-specific signal transduction pathway downstream of CCK2R that phosphorylates PKD2 at three specific sites, results in nuclear accumulation of the active kinase and culminates in efficient phosphorylation of nuclear PKD2 substrates in human gastric cancer cells.
Keywords: CK1, compartment-specific signalling, gastric cancer, HDACs, PKD2
Introduction
Protein kinase D2 (PKD2) is a member of the PKD family of serine/threonine kinases, which belongs to the CAMK superfamily (Manning et al, 2002) and comprises PKD1/PKCμ, PKD2 (Sturany et al, 2001) and PKD3/PKCν (Hayashi et al, 1999). PKDs exhibit a homologous catalytic domain, but vary with respect to their subcellular localization and expression (Rykx et al, 2003; Rozengurt et al, 2005). They are major targets for tumor-promoting phorbol esters and activated by G-protein-coupled receptors (GPCRs) via PKCs (Rozengurt et al, 2005). The best-characterized isoform is PKD1, which has been implicated in the regulation of Golgi function, cell proliferation, apoptosis and migration (Van Lint et al, 2002). Recently, we demonstrated that gastrin, an important acid secretagogue and a trophic factor for the gastrointestinal mucosa, activates PKD2 in human gastric cancer cells expressing the CCK2 receptor (CCK2R) (Sturany et al, 2001).
The subcellular localization plays an important role in defining the biological effect of a given kinase signal. The biological function of PKD1 is regulated, for example, by its localization at the plasma membrane (Oancea et al, 2003). PKD1 has also been demonstrated to mediate nuclear export of class IIa histone deacetylases in thymocytes and lymphocytes (Vega et al, 2004; Dequiedt et al, 2005; Matthews et al, 2006), but the compartment(s) in which this process occurs are not defined. In resting human epithelial cells, PKD2 is largely detectable in the cytoplasm. It is not actively retained there, but shuttles continuously between cytoplasm and nucleus most likely due to the combined activity of a nuclear localization signal in the linker region between the C1a and C1b domains and a nuclear export signal in the C1a domain. The activity of the nuclear export signal is dependent on the nuclear transport receptor, Crm-1. Upon activation of the CCK2R, PKD2 accumulates in the nucleus by an as yet unknown mechanism (Auer et al, 2005). The potential role of PKD2 nuclear accumulation for efficient substrate phosphorylation is currently elusive.
Here we demonstrate that nuclear accumulation of PKD2 mediated by CCK2R requires three phosphorylation events, phosphorylation at Ser706 and Ser710, that is catalytic activity, and phosphorylation at a novel site in the zinc-finger linker domain of PKD2, Ser244. We identify casein kinase 1 (CK1) δ and ɛ as kinases downstream of the CCK2R that phosphorylate PKD2 at Ser244. Our data show that PKD2 interacts with and phosphorylates HDAC7, leading to its nuclear exclusion and concomitant derepression of HDAC7-regulated genes such as nur77. Only active PKD2 that also accumulates in the nucleus induces efficient translocation of HDAC7 from the cytoplasm to the nucleus and maximum Nur77 reporter activity. Gastrin-induced nuclear exclusion of HDAC7 and Nur77 reporter activity is critically dependent on PKD2. In conclusion, we establish a novel pathway that includes the coordinate regulation of PKD2 by CK1δ/ɛ and PKCη downstream of the CCK2R, leading to nuclear accumulation of active PKD2, which results in nuclear exclusion of HDAC7 and transcriptional activation of nur77. Thus, compartment-specific signaling accomplished by phosphorylation at three critical sites determines the precise function of PKD2 as a mediator of CCK2R-induced transcriptional activation in gastric cancer cells.
Results
Role of the catalytic activity for nuclear accumulation of PKD2
Work from our laboratory established that PKD2 shuttles continuously between the cytoplasm and the nucleus in human epithelial tumor cells (Auer et al, 2005). In AGS-B cells, endogenous PKD2 as well as ectopically expressed enhanced green fluorescence (eGFP)-PKD2 is localized predominantly in the cytoplasm and relocalizes to the nucleus upon treatment of cells with gastrin (Figure 1A and B). Nuclear accumulation of PKD2 in response to gastrin was time dependent, reaching a maximum between 20 and 30 min of incubation (Supplementary Figure S1A). Treatment with leptomycin B (LMB) that blocks Crm-1-dependent nuclear export (Kudo et al, 1999) led to nuclear accumulation of both endogenous and ectopically expressed PKD2 (Figure 1A and B).
Figure 1.

Role of catalytic activity for nuclear accumulation of PKD2. (A) Subcellular localization of endogenous PKD2. AGS-B cells incubated with solvent (−) or 100 nM gastrin (G) for 40 min or 10 ng/ml LMB for 1 h were fixed followed by anti-PKD2/Alexa 488 immunostaining and confocal laser scanning microscopy (LSCM). Fin was calculated. Size bar indicates 5 μm. (B) Subcellular localization of eGFP-PKD2 and its mutants in the presence and absence of gastrin. AGS-B cells were transfected with eGFP-PKD2 (wt-PKD2), eGFP-PKD2-S706/710E or eGFP-PKD2-D695A and treated as described in panel A. Images were captured by in vivo LSCM and Fin was calculated. (C) Lysates of AGS-B cells transfected with eGFP-PKD2 together with empty vector (−) or constitutively active PKCη for 48 h were analyzed by anti-pPKD2-Ser706/710 (pPKD2), anti-PKD2 or anti-PKCη western analysis (WB). (D) Effect of active PKCη on PKD2 nuclear accumulation. AGS-B cells transfected with constitutively active PKCη, eGFP-PKD2, eGFP-PKD2-S706/710E or eGFP-PKD2-D695A for 48 h were incubated with 100 nM gastrin (G) or solvent (−) for 40 min. Images were captured by LSCM in live cells and Fin was calculated. (E) AGS-B cells transfected with eGFP-PKD2 for 48 h were incubated with 400 nM TPA (TPA, +) or 100 nM gastrin (G, +) for 40 min or solvent (−) followed by anti-GFP immunoprecipitation and anti-pPKD2-Ser706/710 (pPKD2) or anti-GFP western analysis. (F) AGS-B cells were transfected with eGFP-PKD2-WT, eGFP-PKD2-S706/710E, eGFP-PKD2-D695A and incubated with solvent (−) or 400 nM TPA and further analyzed as described in (D).
Next, we determined whether gastrin-induced catalytic activity of PKD2 (Sturany et al, 2002) was required and sufficient for its nuclear accumulation. Similar to wild-type (wt) PKD2, constitutively active eGFP-PKD2-S706/710E was mainly localized in the cytoplasm in the absence of gastrin (Figure 1B). Thus, catalytic activity alone is not sufficient to trigger nuclear accumulation of PKD2. Kinase-dead eGFP-PKD2-D695A was also largely localized in the cytoplasm of untreated AGS-B cells. Upon incubation with gastrin, PKD2-S706/710E accumulated in the nucleus, whereas PKD2-D695A remained in the cytoplasm (Figure 1B). The lack of nuclear accumulation of kinase-inactive PKD2 was largely due to increased nuclear export since LMB treatment led to a marked nuclear accumulation of PKD2-D695A. This indicates that catalytically inactive PKD2 also shuttles continuously between cytoplasm and nucleus. Thus, nuclear accumulation of PKD2 is regulated by nuclear export rather than import and catalytic activity of PKD2 is required but not sufficient to trigger its nuclear accumulation in response to gastrin.
PKCη and phorbol esters activate PKD2 but fail to induce nuclear accumulation of the kinase
Next, we examined which signaling pathways mediate nuclear accumulation of PKD2 in response to gastrin. PKCη is a known downstream target of the CCK2R. Constitutively active PKCη induces phosphorylation of PKD2 at Ser706/710 (Sturany et al, 2002), the critical sites for catalytic activity of PKD2 in the activation loop (Figure 1C). Active PKCη induced phosphorylation of PKD2 at Ser706/710 (Figure 1C), but failed to induce nuclear accumulation of wt, constitutively active or catalytically inactive PKD2 (Figure 1D). Only upon treatment of PKCη-transfected cells with gastrin, wt and constitutively active PKD2, but not PKD2-D695A, accumulated in the nucleus (Figure 1D).
TPA or PdBu induces phosphorylation of PKD2 at Ser706/710 to a similar degree as gastrin (Figure 1E and Supplementary Figure S1B). However, TPA failed to induce nuclear accumulation of wt-PKD2, PKD2-S706/710E or PKD2-D695A in AGS-B cells (Figure 1F). Similarly, PdBu did not induce nuclear accumulation of endogenous or transfected PKD2 (Supplementary Figure S1B). In contrast, both PdBu and TPA induced membrane association of PKD2 (Figure 1F, Supplementary Figure S1B and data not shown). These data indicate that gastrin confers an additional modification to PKD2 that enables nuclear accumulation of the kinase and that is not inducible by PKCη or other phorbol ester-sensitive kinases that activate PKD2.
Gastrin induces serine phosphorylation in the cysteine-rich domain of PKD2
To identify additional sites phosphorylated in PKD2 upon gastrin treatment, we performed an extensive phosphorylation analysis of PKD2 from HEK293 cells expressing the CCK2R. The phosphorylation pattern of PKD2 from cells treated with 100 nM gastrin or solvent was examined by nanoLC/ES-MS/MS (Figure 2). Only when PKD2 was extracted from gastrin-treated cells, Ser706 and Ser710 were found to be phosphorylated (data not shown) and a tryptic peptide corresponding to residues 233–255 in PKD2 was identified that contained a single phosphorylated residue. For target MS/MS analysis of the unphosphorylated and the mono-phosphorylated tryptic peptide RPPSSSSSSSASSYTGRPIELDK, the triply charged precursor ions at m/z 799.7 and 826.4 were chosen as preferred masses (Figure 2A). In the tandem mass spectrum of the m/z 826.4 precursor ion, an intense peak at m/z 793.7 with a 32.7 Da mass difference (98/3 Da) could be observed, indicating the neutral loss of phosphoric acid from the precursor ion (Figure 2B). Based on the MS/MS data, the phosphorylation site could be determined at Ser244 that is exclusively phosphorylated in PKD2 extracted from gastrin-treated cells (Figure 2C). A sequence comparison between PKD1 and PKD2 reveals that the peptide around Ser244 in PKD2 corresponds to some degree to a peptide in the cysteine-rich region of PKD1 in which Ser255 is trans-phosphorylated (Vertommen et al, 2000). However, this motif is not found in human PKD3, Drosophila PKD or DKF-1, the PKD2 ortholog in Caenorhabditis elegans (Figure 2D).
Figure 2.

NanoLC/ES-MS/MS analysis of digested GST-PKD2 fusion proteins. HEK293 cells co-transfected with pGMEX-PKD2 and the CCK2R and treated with solvent or 100 nM gastrin as indicated were lysed and GST-PKD2 was purified and digested with trypsin. (A) Top panel: base peak chromatogram. Bottom panels: ES mass spectra acquired during the elution of the unphosphorylated (left figure) and the mono-phosphorylated (right figure) tryptic peptide RPPSSSSSSSASSYTGRPIELDK (corresponding to residues 233–255 in full-length PKD2). (B) Positive-ion tandem mass spectrum of the [M+3H]3+ precursor ion at m/z 826.4 from the mono-phosphorylated tryptic peptide RPPSSSSSSSASSYTGRPIELDK (corresponding to residues 233–255 in full-length PKD 2). As a result of gas-phase elimination of H3PO4 from the precursor ion (loss of 98/3 Da), an intense [M+3H-H3PO4]3+ neutral loss fragment ion at m/z 793.7 was formed (Th=Thompson). (C) Interpretation of the tandem mass spectrum of the triply charged [M+3H]3+ precursor ion at m/z 826.4 from the mono-phosphorylated tryptic peptide shown in panel B. Based on the acquired fragment ions, the phosphorylation site could be determined at Ser244. (D) The sequence around Ser244 is unique in PKD2. Sequence comparison of the regions around S244 in human PKD1, PKD2, PKD3, Drosophila PKD and C. elegans DKF-1. Conserved serines (S249 in PKD1, S244 in PKD2) are shown in red. Sequence alignments were performed with the CLC Free Workbench alignment software.
Role of Ser244 in nucleocytoplasmic shuttling of PKD2
To examine whether phosphorylation of PKD2 at Ser244 plays a role in nucleocytoplasmic shuttling of the kinase, a phosphomimetic PKD2 mutant (PKD2-S244E) was employed. Similar to wt-PKD2, PKD2-S244E was predominantly localized in the cytoplasm of unstimulated cells. Upon incubation of cells with gastrin, PKD2-S244E accumulated in the nucleus (Figure 3A). Thus, phosphorylation of PKD2 at Ser244 alone is not sufficient to trigger its nuclear accumulation. However, a PKD2-S244/706/710E triple mutant exhibited maximum nuclear accumulation that was not increased in the presence of gastrin (Fin 2.390 versus 2.554; Figure 3A), whereas constitutively active PKD2 lacking a phosphorylatable serine residue at position 244 (PKD2-S244A-S706/710E) was mainly localized in the cytoplasm and substantially impaired in its ability to accumulate in the nucleus in response to gastrin. A kinase-inactive PKD2-S244E-S706/710A mutant failed to accumulate in the nucleus even in the presence of gastrin (Fin 0.307 versus 0.346). There was no difference in basal and gastrin-induced catalytic activity of PKD2-S244A, PKD2-S244E and wt-PKD2. The catalytic activity of the PKD2-S244/706/710E triple mutant was comparable to PKD2-S706/710E and to PKD2-S244A/S706/710E. Gastrin treatment could not further enhance the catalytic activity of these mutants (Figure 3B and Supplementary Figure S2A). This indicates that pSer244 does not modify the catalytic activity of PKD2. Thus, optimum nuclear accumulation of PKD2 in response to gastrin requires phosphorylation of Ser244 and catalytic activity, which is accomplished by phosphorylation of Ser706/710.
Figure 3.

(A) Phosphorylation of Ser244 in PKD2 is required for nuclear accumulation of PKD2. AGS-B cells transfected with eGFP-PKD2 (wt-PKD2), eGFP-PKD2-S244E (D2-S244E), eGFP-PKD2-S244/706/710E (D2-S244/706/710E), eGFP-PKD2-S244A/S706/710E (D2-S244A/S706/710E) and eGFP-PKD2-S244E/S706/710A (D2-S244E/S706/710A) for 48 h were incubated with solvent (−) or 100 nM gastrin (G) followed by in vivo LSCM. Fin was calculated. (B) Phosphorylation at Ser244 does not affect PKD2 catalytic activity. AGS-B cells transfected as indicated were incubated for 10 min with 100 nM gastrin (G+) or solvent (−). Lysates were subjected to IP with an anti-GFP mAb followed by in vitro kinase assays (IVKs) using histone (His) as substrate or to anti-GFP western analysis (WB, PKD2). (C) Role of Ser244 phosphorylation in nucleocytoplasmic shuttling of PKD2. Left: AGS-B cells transfected with eGFP-PKD2 for 48 h were treated with 100 nM gastrin (G) or 10 ng/ml LMB for 20 min followed by photobleaching and FLIP measurements. An area of the cytoplasm (dotted line) was repeatedly bleached and the loss of fluorescence in the nuclear region was determined by LSCM. Bottom panel: FLIP was performed in AGS-B cells transfected with eGFP-PKD2-S244/706/710E. Right: Quantitative analysis of the changes in nFi of eGFP-PKD2 (wt-PKD2) or eGFP-PKD2-S244/706/710E (D2-S244/706/710E) during photobleaching of the cytoplasm. (D) Effect of Ser244 phosphorylation on the binding of PKD2 to Crm-1 and GST-14-3-3η. AGS-B cells expressing eGFP-PKD2 (wt-PKD2) or eGFP-PKD2-S244/706/710E (D2-S244/706/710E) were lysed and subjected to IP with an anti-GFP mAb followed by anti-Crm-1 or anti-PKD2 western analysis. (E) GST-14-3-3η on glutathione sepharose beads was incubated with lysates from AGS-B cells expressing eGFP-PKD2 (wt-PKD2) and treated with gastrin (G+) or solvent (−), eGFP-PKD2-S706/710E (D2-S706/710E) and eGFP-PKD2-S244/706/710E (D2-S244/706/710E). Pull-down reactions were analyzed by anti-PKD2 and anti-GST western blotting. Expression of the various eGFP-tagged PKD2 plasmids was determined by anti-PKD2 western blotting in aliquots of cell lysates obtained prior to incubation with the GST-14-3-3η beads (PKD2 input).
To determine how phosphorylation at Ser244 affects nucleocytoplasmic shuttling of PKD2, we first examined the effect of Ser244 phosphorylation on the nuclear import of the kinase. In the presence of LMB, the kinetics of nuclear accumulation of wt-PKD2, PKD2-S244A and PKD2-S244E were virtually identical (Supplementary Figure S2B). Fluorescence loss in photobleaching (FLIP) is a powerful approach to determine whether distinct cellular compartments are in equilibrium (White and Stelzer, 1999; Lippincott-Schwartz et al, 2001). AGS-B cells were transfected with the respective eGFP-PKD2 expression plasmids and the cytoplasm of eGFP-positive cells was repetitively and selectively bleached. A decrease in nuclear fluorescence is the consequence of nuclear export of the protein. These experiments demonstrated that eGFP-PKD2 nuclear fluorescence intensity (nFi) decreases by 50% in the presence of gastrin, reaching a plateau 600 s after bleaching of the cytoplasm (Figure 3C). Thus, PKD2 leaves the nucleus also in the presence of gastrin, which induces maximal nuclear steady-state localization of PKD2. There was only a 10% decrease in nFi of eGFP-PKD2 in the presence of LMB and a comparable, minor decrease in nFi of cells transfected with eGFP-PKD2-S244/706/710E (Figure 3C), indicating that phosphorylation of active PKD2 at Ser244 prevents the nuclear export of the kinase.
PKD2 interacts with Crm-1 and is exported from the nucleus by a Crm-1-dependent mechanism (Auer et al, 2005). 14-3-3 proteins act in cooperation with Crm-1 as a ligand-dependent nuclear export machine (Kudo et al, 1999; Rittinger et al, 1999). To determine whether phosphorylation of PKD2 changes its interaction with the Crm-1 machinery, we examined the interaction of wt-PKD2 and PKD2-S244/706/710E with Crm-1. Compared to wt- PKD2, the ability of PKD2-S244/706/710E to interact with Crm-1 was substantially decreased (Figure 3D). Pull-down assays using a GST-14-3-3η fusion protein revealed that wt-PKD2 interacts with GST-14-3-3η. Treatment of cells with gastrin prevents the interaction of PKD2 with GST-14-3-3η. Similar to wt-PKD2, catalytically active PKD2-S706/710E strongly interacts in the absence and does not interact with GST-14-3-3η in the presence of gastrin (Figure 3E and Supplementary Figure S2B). PKD2-S244/706/710E does not interact with GST-14-3-3η even in the absence of gastrin (Figure 3E). Conversely, PKD2-S244A strongly interacts with 14-3-3η even in the presence of gastrin (Supplementary Figure S2C), indicating that phosphorylation at Ser244 is critical for disrupting the interaction of 14-3-3 with PKD2 and thereby affects its nuclear export.
CK1 phosphorylates PKD2 at Ser244
To identify the upstream kinase(s) that phosphorylate PKD2 at Ser244, we first examined the effect of various kinase inhibitors on gastrin-induced nuclear accumulation of PKD2. Selective inhibition of MEK, PI-3 kinase or p38 MAPK had no effect on gastrin-induced nuclear accumulation of PKD2 (Supplementary Figure S3A). As expected, treatment of cells with a PKC inhibitor, GF109203X, prevented nuclear accumulation of PKD2. Furthermore, the selective CK1δ/ɛ inhibitor IC261 (Mashhoon et al, 2000) also blocked gastrin-induced accumulation of PKD2 in the nucleus (Figure 4A). These data were confirmed using another two selective CK1 inhibitors, CKI-7 and D4476. Next we employed CK1δ/ɛ-specific siRNA constructs that also blocked nuclear accumulation of PKD2 in response to gastrin. All CK1 inhibitors as well as the CK1-specific siRNA constructs had no effect on PKC-dependent phosphorylation of PKD2 at Ser706/710, which was prevented by GF109203X (Supplementary Figure S3A).
Figure 4a.

(A) Various CK1 inhibitors and CK1 RNAi inhibit nuclear accumulation of PKD2 in response to gastrin. Upper panels: AGS-B cells transfected with eGFP-PKD2 for 48 h were incubated with solvent (−) or 3.5 μM GF109203x, 1.6 μM IC261, 10 μM CKI-7 or 100 μM D4476 for 1 h and subsequently incubated with 100 nM gastrin (G) or solvent (−) for 40 min. LSCM was performed in vivo and Fin was calculated. Lower panels: AGS-B cells co-transfected with eGFP-PKD2 (wt-PKD2), a control siRNA (scrambled) or CK1δ/ɛ siRNA for 40 h were treated with solvent (−) or 100 nM gastrin for 40 min. LSCM was performed and Fin was calculated. Knockdown of CK1 δ/ɛ was analyzed by western blotting using antibodies recognizing CK1ɛ (upper panel) or CK1δ (middle panel). (B) Gastrin activates CK1. Kinase assays were performed in the presence and absence of 1.6 μM IC261 using two peak kinase fractions (A, B) of unstimulated or gastrin (G)-stimulated cells and GST-p531−65S4,6,9A as substrate. AGS-B cell lysates from solvent (−) or gastrin (G+)-treated cells were immunoprecipitated with an anti-Erk1/2 or an anti-CK1ɛ-specific antibody and subjected to IVKs using MBP (100 μg/ml) as a substrate. The position of phosphorylated MBP is indicated by arrowheads. Equal expression of CK1ɛ and Erk1/2 in each condition was detected by western analysis (lower panel). (C) Phosphorylation of GST-PKD2-CRD by CK1α, CK1ɛ and CK1δ. IVKs for CK1α, CK1δ or CK1ɛ were performed using a GST-PKD2-CRD fusion protein or casein as substrates. Phosphorylated proteins were separated by SDS–PAGE and visualized by autoradiography and Coomassie staining. (D) Phosphorylation of GST-PKD2-CRD and GST-PKD2-CRD-S244A by the CK1δ kinase domain. GST-PKD2-CRD (red circles) and GST-PKD2-CRD-S244A (blue squares) were phosphorylated by the CK1δ-kinase domain for the times indicated. Means ± s.d. of two independent experiments are shown.
A detailed analysis of the amino-acid sequence around Ser244 in PKD2 revealed that SSSASSYTGRP does not confer the critical substrate motif for a PKC substrate as the corresponding sequence in PKD1. However, this sequence has properties of a potential substrate motif for the CK1 family of acidophilic serine/threonine kinases (Obenauer et al, 2003) that regulate differentiation, proliferation and apoptosis (Knippschild et al, 2005).
First, we examined whether CK1 is activated by gastrin in AGS-B cells. There were major peaks of kinase activities eluting between 100 and 270 mM NaCl in cell extracts that were substantially enhanced upon gastrin stimulation (data not shown). Phosphorylation of the CK1 substrate GST-p531−64 was markedly increased in fractions obtained from lysates of gastrin-treated cells. In contrast, a GST-p531−64-S4/6/9A fusion protein that lacks the major N-terminal serine residues phosphorylated by CK1δ and ɛ in vitro and in vivo (Knippschild et al, 1997) was not phosphorylated by kinases present in these fractions (Supplementary Figure S3B). The analysis of the catalytic activity present in two fractions by in vitro kinase assays (IVKs) performed in the presence and absence of IC261 revealed that gastrin treatment of AGS-B cells induces a 4- to 5-fold increase in the catalytic activity present in the two fractions that was abolished in the presence of IC261. In addition, gastrin increased the catalytic activity of CK1ɛ that was immunopurified from AGS-B cells (Figure 4B). Thus, activation of the CCK2R leads to stimulation of CK1 activity in AGS-B cells.
Next we determined whether CK1 could directly phosphorylate the cystein-rich domain (CRD) of PKD2 containing S244. A fusion protein comprising the entire CRD of PKD2 (GST-PKD2-CRD) was efficiently phosphorylated in vitro by CK1δ and ɛ and to a lesser extent by CK1α (Figure 4C). In contrast, phosphate incorporation into a mutant fusion protein lacking a phosphorylatable Ser at 244 (GST-PKD2-CRD-S244A) was reduced by 70% in the presence of the CK1δ kinase domain (Figure 4D and Supplementary Figure S3C), indicating that Ser244 is a major CK1 phosphorylation site in the CRD of PKD2.
To test whether incubation of cells with gastrin induced phosphorylation of PKD2 at Ser244 also in vivo, we generated a phospho-specific antibody directed against a peptide surrounding pSer244 in PKD2. Phosphorylation of Ser244 in response to gastrin treatment was time-dependent, reaching a maximum after 30 min of incubation, whereas phosphorylation of Ser706/710 peaked already at 5 min after incubation of cells with gastrin (Figure 4E). The band corresponding to PKD2 phosphorylated at Ser244 was not detectable upon competition of the antibody with the immunizing peptide. Overexpression of active CK1δ stimulated phosphorylation at Ser244, whereas selective knockdown of CK1δ/ɛ prevented phosphorylation at Ser244 in response to gastrin (Figure 4E).
Figure 4b.

(E) Gastrin induces phosphorylation of PKD2 at Serine 244 in vivo. Left panels: AGS-B cells were treated with solvent or 100 nM gastrin for 2.5, 5, 15, 30, 45 and 60 min, lysed and subjected to western analysis using phosphorylation-specific antibodies recognizing pSer244 (upper panel, PKD2-pS244) and pSer706/710 (middle panel, pS706/710) or anti-PKD2 antibody (lower panel, PKD2). Relative positions of Ser244- or Ser706/710-phosphorylated PKD2 are indicated by arrowheads. Right panel: WCLs of untreated (−) or gastrin-treated (G) AGS-B cells were analyzed by western blotting using the pSer244 antiserum (upper panel), or the same antibody preincubated with the immunizing pSer244 peptide (middle panel) or an anti-PKD2 antibody. Lower left panel: catalytically active CK1 induces phosphorylation of PKD2 at Ser244. AGS-B cells were transfected with a control plasmid or truncated, catalytically active CK1δ. WCLs were prepared and analyzed by western blotting with the PKD2 pSer244 antibody. Expression of endogenous or the truncated, catalytically active CK1δ was detected with an antibody directed against CK1δ (right panels). Lower right panel: Ser244 phosphorylation can be inhibited by CK1 siRNA. AGS-B cells transfected with scrambled siRNA or CK1δ/ɛ siRNA were incubated with solvent (−) or 100 nM gastrin (G). WCL was prepared and analyzed using the PKD2-pSer244 antiserum (upper panels), an anti-CK1δ antibody (middle panel) or an anti-β actin antibody. Positions of pSer244, CK1δ or β-actin are indicated by arrowheads. (F) Gastrin-induced nuclear accumulation of PKD2 is dependent on CK1δ/ɛ. Left panel: AGS-B cells expressing eGFP-PKD2 (wt-PKD2) were transfected with scrambled siRNA or CK1δ/ɛ siRNA. At 24 h after transfection, cells were retransfected with siRNA and a control plasmid or a catalytically active CK1δ plasmid. After 24 h, cells were incubated with solvent (−) or gastrin and subjected to LSCM and Fin was calculated (right panel). (G) Catalytically active CK1δ and PKCη are sufficient to induce nuclear accumulation of PKD2. AGS-B cells transfected with eGFP-PKD2 (top), eGFP-PKD2-S706/710E (bottom) and constitutively active CK1δ (CK1δ active, +) and/or constitutively active PKCη (+) for 48 h were incubated with solvent (−) or 100 nM gastrin (G+) for 40 min. Fin was calculated (right panels). Size bars indicate 5 μm.
In conclusion, (i) incubation of AGS-B cells with gastrin leads to activation of CK1δ/ɛ, (ii) CK1δ/ɛ phosphorylate PKD2 in the CRD and (iii) Ser244 is the major phosphorylation site in PKD2 targeted by CK1δ/ɛ both in vitro and in vivo.
CK1 and PKCη are sufficient to induce maximal nuclear accumulation of PKD2
It was now important to determine whether activation of PKD2 by PKCs and phosphorylation at Ser244 by CK1δ were required and sufficient to trigger its nuclear accumulation. Selective CK1 siRNA constructs reduced nuclear accumulation of PKD2 in response to gastrin. This inhibition was rescued by overexpression of a constitutively active fragment of rat CK1δ that is not targeted by the siRNA constructs (Figure 4F).
Constitutively active CK1δ alone did not induce nuclear accumulation of wt-PKD2. However, nuclear accumulation of wt-PKD2 was accomplished by co-transfection of CK1δ and active PKCη. Active CK1δ induced maximum nuclear accumulation of active PKD2-S706/710E that was not further increased by co-transfection of PKCη or gastrin (Figure 4G). Thus, CK1δ/ɛ and PKCη are sufficient to confer all the post-translational modifications required for nuclear accumulation of PKD2 in response to activation of GPCRs, that is phosphorylation of PKD2 at Ser244 and Ser706/S710.
PKD2 interacts with and phosphorylates HDAC7 at the N terminus
Recently, we and others demonstrated that PKD1 phosphorylates members of the class IIa family of histone deacetylases such as HDAC5 and HDAC7 (Vega et al, 2004; Dequiedt et al, 2005; Parra et al, 2005). Class IIa HDACs possess a conserved N-terminal catalytic domain that mediates interaction with MEF2 transcription factors via a 17 amino-acid motif leading to the recruitment of class IIa HDACs to DNA-bound MEF2 and transcriptional repression of MEF2-target genes. The repressive activity of class IIa HDACs is controlled by nucleocytoplasmic shuttling and by their association with 14-3-3 proteins in a signal-specific manner (Dequiedt et al, 2003). At present, it is unclear in which subcellular compartment(s) class II HDACs are phosphorylated by members of the PKD family.
Class IIa HDACs are endogenously expressed in various cell types, including AGS-B cells. To determine whether PKD2 interacts with the prototypical class IIa member HDAC7 in vivo, AGS-B cells were transfected with Flag-HDAC7 and eGFP-PKD2. In co-immunoprecipitation (IP) experiments, HDAC7 was associated with both endogenous and eGFP-PKD2. Endogenous PKD2 also interacted with endogenous HDAC7 in AGS-B cells. Interestingly, interaction between HDAC7 and PKD2 was enhanced by gastrin treatment (Figure 5A). To determine the site of HDAC7 that interacted with PKD2, an N-terminal (GST-H7-Nter; aa 1–490) or a C-terminal fragment (GST-H7-Cter; aa 490–915) of HDAC7 was incubated with extracts of AGS-B cells, either untreated or treated with gastrin. Endogenous PKD2 associated with GST-H7-Nter, but not with GST-H7-Cter in response to gastrin (Figure 5B). Thus, the catalytic activity of PKD2 enhances its interaction with the N terminus of HDAC7.
Figure 5a.

(A) PKD2 interacts with HDAC7. Left panels: AGS-B cells transfected with Flag-tagged HDAC7 (Flag-H7, +), eGFP (+) or eGFP-PKD2 (+) for 48 h as indicated were incubated with solvent (−) or 100 nM gastrin for 40 min (G+) followed by anti-Flag IP and anti-PKD2 (top panel) or anti-Flag (bottom panel) western analysis. Positions of endogenous PKD2 (105 kDa), eGFP-PKD2 (133 kDa) and Flag-HDAC7 (115 kDa) are indicated by arrowheads. Right panels: AGS-B cells treated with solvent (−) or gastrin (G, +) were lysed and immunoprecipitated with an antibody against HDAC7 (middle panels, top) or PKD2 (middle panels, bottom) followed by PKD2 and HDAC7 western analysis. (B) PKD2 associates with the N terminus of HDAC7 upon gastrin treatment. Lysates of AGS-B cells treated with solvent (−) or 100 nM gastrin (G, +) for 40 min were incubated with agarose-coupled GST-fusion proteins of the C terminus (GST-H7-Cter) or the N terminus of HDAC7 (GST-H7-Nter). GST pull downs (upper panel) were analyzed by anti-PKD2 western blotting. The position of endogenous PKD2 is indicated by an arrowhead. H7-Cter and H7-Nter fusion proteins are shown by Coomassie staining and their positions are indicated by arrowheads (lower panel). (C) PKD2 phosphorylates HDAC7 in vitro. GST-H7-Nter was immobilized on glutathione sepharose beads and incubated with equal amounts of purified recombinant GST-PKD2 (PKD2wt+) or GST-PKD2-S706/710E (PKD2ca+) followed by IVKs and autoradiography (upper panel) or Coomassie staining (lower panel). (D) Endogenous PKD2 phosphorylates HDAC7 in gastrin-treated cells. AGS-B cells treated with solvent (−) or 100 nM gastrin (G+) for 40 min were lysed and subjected to anti-PKD2 immunoprecipitation followed by IVK analysis using GST-HDAC7-Nter as substrate (top panel). Equal expression of endogenous PKD2 was determined by anti-PKD2 western analysis (middle panel). Coomassie staining of GST-HDAC7-Nter was used as loading control (bottom panel). (E) Gastrin stimulates phosphorylation of endogenous HDAC7 at Ser181 in vivo. Left panels: AGS-B cells were incubated with 100 nM gastrin (G+) or solvent (−) for 40 min, lysed and further analyzed by anti-pHDAC7-Ser181(pH 7) or anti-HDAC7 western analysis. Right panels: AGS-B cells expressing eGFP-HDAC7 (top panels) or eGFP-HDAC7-S181A (bottom panels) were incubated with 100 nM gastrin (G+) or solvent for 40 min. IPs were performed using an anti-GFP antibody and further analyzed by anti-pHDAC7-Ser181 (pH 7). Expression of HDAC7 was detected by an anti-HDAC7 antibody.
To investigate whether HDAC7 is a direct substrate of PKD2, recombinant wt-PKD2 and PKD2-S706/710E were used in IVKs using GST-H7-Nter as substrate. Wt and even more constitutively active PKD2 phosphorylated the N terminus of HDAC7 in vitro (Figure 5C). When endogenous PKD2 was immunoprecipitated from AGS-B cells and used in IVKs, we observed phosphorylation of GST-H7-Nter, which was substantially increased upon incubation of cells with gastrin (Figure 5D). Gastrin treatment also induced phosphorylation of HDAC7 in vivo as demonstrated by a phospho-specific antibody that selectively detects HDAC7 phosphorylated at Ser181 in the N terminus (Dequiedt et al, 2006) (Figure 5E). Phosphorylation of HDAC7 at Ser181 in response to gastrin was completely prevented by incubation of cells with the selective CK1 inhibitor IC261 (data not shown), confirming that PKD2 phosphorylates HDAC7 by a pathway that requires CK1 activation.
PKD2 activity and subcellular localization regulate HDAC7 nucleocytoplasmic shuttling
Next we investigated the biological relevance of HDAC7 phosphorylation by the CCK2R–PKD2 signaling pathway. In untreated AGS-B cells, HDAC7 localizes predominantly in the nucleus, with only a small proportion of the protein found in the cytoplasm. Strikingly, treatment with gastrin resulted in a dramatic relocalization of HDAC7 from the nuclear compartment to the cytoplasm, which was not observed when cells were transfected with an HDAC7-S181A mutant (Figure 5F). This indicates that phosphorylation of HDAC7 at Ser181 is crucial for gastrin-mediated nucleocytoplasmic transport of HDAC7. Of note, co-transfection of the catalytically active, but predominantly cytoplasmic PKD2-S706/710E mutant did not induce a similar relocalization, whereas the nuclear and constitutively active PKD2-S244/706/710E triple mutant induced complete nuclear exclusion of HDAC7 (Figure 5F). Likewise, PKCη or CK1δ alone had no effect on the subcellular distribution of HDAC7. Only co-transfection of both active kinases induced cytoplasmic accumulation of HDAC7 that was comparable to the effect of gastrin or PKD2-S244/706/710E (Figure 5G).
Figure 5b.

(F) Phosphorylation of PKD2 at Ser244 and Ser706/710 induces maximum nuclear exclusion of HDAC7. AGS-B, Cos7 or HeLa cells co-transfected with eGFP-HDAC7 or eGFP-HDAC7-S181A and the CCK2R (in Cos7 and HeLa cells), Flag-PKD2-S706/710E or Flag-PKD2-S244/706/710E for 48 h were incubated with solvent (−, first lane) or 100 nM gastrin (G) as indicated. Images were captured in vivo and nuclear exclusion of HDAC7 was calculated. The bars represent the mean percentage of HDAC7 nuclear exclusion ± s.e. in at least 300 cells per condition. Size bars indicate 5 μm. (G) Nuclear exclusion of HDAC7 requires both active CK1δ and PKCη. AGS-B cells transfected with eGFP-HDAC7 together with active CK1δ (CK1δactive), constitutively active PKCη or both CK1δactive and PKCη for 48 h were analyzed by LSCM and the percentage of HDAC7 excluded from the nuclear compartment was calculated. The bars represent the mean percentage of HDAC7 nuclear exclusion ± s.e. in at least 300 cells per condition. Size bars indicate 5 μm. (H) Left and bottom right: gastrin-induced nuclear exclusion of HDAC7 requires PKD2. Cells were transfected with eGFP-HDAC7 together with solvent (−), scrambled siRNA (scrambled oligo), PKD2-siRNA (PKD2-siRNA) for 24 h and were retransfected with siRNA and a control vector or the PKD2 siRNA-resistant mutant Flag-PKD2ΔPH for 24 h. Cells were then incubated with solvent (−) or 100 nM gastrin for 40 min, LSCM was performed in vivo and Fin was calculated. Bars represent the Fin in each condition. ***P<0.001. Top right: PKD2ΔPH is siRNA resistant. AGS-B cells expressing eGFP-HDAC7 and Flag-PKD2ΔPH were transfected with scrambled siRNA or PKD2-siRNA. WCL was prepared and analyzed with an antibody against PKD2 or HDAC7.
Finally, selective knockdown of PKD2 resulted in a significant reduction of HDAC7 nuclear export in response to gastrin (Figure 5H). The siRNA constructs were selective for PKD2 and did not affect PKD1 or PKD3 (Supplementary Figure S4). The inhibitory effect of the PKD2 siRNA constructs on gastrin-induced nuclear exclusion of HDAC7 was rescued by overexpression of a PKD2 mutant lacking the PH domain that contains the siRNA target sequence (Figure 5H).
These data demonstrate that PKD2 is a crucial mediator of HDAC7 nuclear export upon activation of the CCK2R.
Nuclear, active PKD2 is required for efficient derepression of HDAC7 targets
Phosphorylation-dependent nucleocytoplasmic shuttling of class IIa HDACs controls their activity as transcriptional repressors. We examined the expression of genes regulated by HDAC7 such as the gene coding for the nuclear orphan receptor Nur77 (Dequiedt et al, 2003). Treatment of cells with gastrin resulted in a 15-fold increase in Nur77 promoter activity and a marked increase in Nur77 protein in AGS-B cells (Figure 6A). Inhibition of CK1δ/ɛ by IC261 significantly inhibited gastrin-induced Nur77 promoter activity (Figure 6B). Selective knockdown of PKD2 in AGS-B cells also resulted in a substantial reduction of gastrin-induced Nur77 transcription (Figure 6C). Thus, the CK1-PKD2 signaling pathway is the major mediator of gastrin-induced activation of HDAC7 target genes in AGS-B cells.
Figure 6.

PKD2 activation by gastrin leads to nuclear export of HDAC7 and activation of the Nur77 promoter. (A) AGS-B cells transfected with a luciferase reporter plasmid driven by the Nur77 promoter (pNur77-Luc) were incubated with solvent (−) or 100 nM gastrin (G) for 2 h. For all reporter assays (A–D), luciferase activities were determined and normalized based on Renilla luciferase expression and represented as relative light units (RLUs) of firefly luciferase. Results are representative for at least three independent experiments each performed in triplicate. Insert: lysates (WCL) of untreated (−) or gastrin-stimulated (G) cells were analyzed by anti-Nur77 or anti-β-actin western blotting. (B) Gastrin-induced Nur77 reporter activity requires CK1 activity. AGS-B cells transfected with pNur77-Luc for 40 h were incubated with 1.6 μM IC261 (IC261, +) or solvent (−) for 1 h prior to treatment with 100 nM gastrin (G+) or solvent (−). Luciferase assays were performed as described. Parallel cultures were analyzed by anti-pPKD2-Ser706/710 and anti-PKD2 western analysis. (C) Gastrin stimulation of Nur77 reporter activity requires PKD2. AGS-B cells were transfected with pNur77-Luc for 12 h and then with PKD2-siRNA or a scrambled oligonucleotide for another 24 h prior to incubation with solvent (−) or 100 nM gastrin (G) for 4 h followed by analysis of Nur77 reporter activity. In parallel cultures, efficient and selective knock-down of PKD2 by the specific RNAi oligo was determined (inset). (D) AGS-B cells transfected with pNur77-Luc together with eGFP, eGFP-PKD2-S706/710E or eGFP-PKD2-S244/706/710E for 40 h were lysed and further analyzed by luciferase assays (left panel) or anti-Nur77, anti-PKD2, anti-GFP and anti-β-actin western blotting (right panels). (E) Nuclear action of PKD2: in resting cells, PKD2 continuously shuttles between the nucleus and the cytoplasm by reversible binding of importin and Crm-1. The rate of nuclear export exceeds that of nuclear import. CK1 (basal kinase activity*) and PKCs (low catalytical activity) are localized in the cytoplasm. HDAC7 (red) is localized in the nucleus acting as a transcriptional repressor. Arrow direction and thickness represent the differential rates and directionality of PKD2 transport. Upon binding of gastrin to the CCK2R, PKCs (PKC*) are activated in the cytoplasm phosphorylating PKD2 at Ser706 and Ser710, which leads to activation of PKD2 (*). In parallel, CK1 activity is enhanced by gastrin (***), resulting in CK1 nuclear accumulation. Active PKD2 is trapped in the nucleus by CK1-induced phosphorylation at Ser244 and in turn phosphorylates HDAC7. This leads to nuclear exclusion of HDAC7 and derepression of HDAC7-regulated transcription.
Active, but cytoplasmic PKD2-S706/710E only slightly induced Nur77 transcription and protein expression. In contrast, the nuclear PKD2-S244/706/710E triple mutant markedly stimulated Nur77 promoter activity and expression of Nur77 protein (Figure 6D). Thus, in complete agreement with our model, we observed that nuclear localization of active PKD2 is necessary to phosphorylate nuclear targets such as class IIa HDACs and is required to fully relieve the inhibitory effect of HDAC7 on Nur77 transcriptional activity.
Discussion
To ensure proper cellular function, the spatial distribution of different proteins is tightly coordinated and the elucidation of these mechanisms has attracted considerable interest. PKDs are localized in various cellular compartments including plasma membrane, Golgi apparatus, cytoplasm and nucleus, and are implicated in diverse biological events such as migration, vesicle shedding, proliferation, apoptosis and signal transduction in response to oxidative stress and GPCRs (Rykx et al, 2003; Storz and Toker, 2003; Rozengurt et al, 2005). So far, little is known about the precise regulation of PKD's subcellular trafficking and its impact on the specific functions of the kinase. We have recently demonstrated that PKD2 shuttles between the cytoplasm and the nucleus using functional nuclear export signal and nuclear localization signal in the zinc-finger domain (Auer et al, 2005). Here, we addressed the central question of how activation of the CCK2R leads to nuclear accumulation of PKD2 in epithelial tumor cells and which events are triggered by PKD2 in the nucleus.
Three phosphorylation events are required for nuclear accumulation of PKD2 upon activation of the CCK2R
Our data show that catalytic activity of PKD2 induced by phosphorylation at Ser706 and Ser710 by PKCs (Sturany et al, 2002) is required but not sufficient to target PKD2 to the nucleus. Ser244 is identified as a novel residue in PKD2 phosphorylated upon activation of the CCK2R. pSer244 has no effect on the catalytic activity, but prevents the interaction of PKD2 with the critical components of the nuclear export machinery. Consequently, phosphorylation at Ser706/710 and Ser244 is sufficient to trigger maximum nuclear accumulation of PKD2. The sequence around Ser244 is unique in PKD2. Ser255 in the corresponding sequence in PKD1 is also trans-phosphorylated. In contrast to Ser244 in PKD2, Ser255 in PKD1 is phosphorylated by PKCs and enhances the catalytic activity of PKD1 upon phosphorylation (Vertommen et al, 2000). There are no data on a potential impact of pSer255 on the subcellular localization of PKD1. Also, PKD3 exhibits marked differences in the amino-acid sequence in the corresponding region that exhibits no potential phosphorylation site for CK1. PKD3 is also predominantly localized in the nucleus, even in the absence of an exogenous stimulus. Thus, we conclude that PKD2 is regulated in a distinct way compared to PKD1 and PKD3 and that this mechanism contributes to the fine tuning of PKD isoform activity in cells that express more than one isoform.
CK1 is an upstream regulator of PKD2
Our data establish that CK1δ and ɛ directly phosphorylate PKD2 at Ser244 in vitro and in vivo making PKD2 an—albeit atypical—K1δ/ɛ substrate. PKD2 does not exhibit an FXXXF motif around Ser244. However, the presence of this motif in the substrate is not an absolute requirement for phosphorylation by CK1. This motif is present in both NFAT and β-catenin, two well-established CK1 substrates, and the FXXXF motif is important for phosphorylation of NFAT by CK1 (Okamura et al, 2004). However, it is not required at all for efficient phosphorylation of β-catenin by CK1 (Bustos et al, 2006).
Kinases of the CK1 family can be isolated as active enzymes from many cell lines, but their activity can also be increased by, for example receptor tyrosine kinases (Knippschild et al, 2005). Our data show that CK1δ can be activated via the CCKR2, which results in its nuclear accumulation (data not shown), in contrast to previous reports demonstrating cytoplasmic localization of active CK1δ (Milne et al, 2001). The difference could be due to distinct levels of CK1δ catalytic activity, which is enhanced above baseline in gastrin-treated AGS-B cells. Thus, a novel signaling pathway downstream of the CCK2R could be established that triggers activation and nuclear accumulation of PKD2. CK1δ and PKCη confer all the post-translational modifications required for nuclear accumulation of PKD2 upon activation of GPCRs, that is phosphorylation at Ser244 and Ser706/710. CK1δ and PKD2 colocalize only in the nucleus upon activation. Since active PKD2 shuttles between the cytoplasm and the nucleus, we propose a model in which upon gastrin stimulation PKD2 is activated by PKCs in the cytoplasm and subsequently phosphorylated at Ser244 in the nucleus by active CK1, which blocks nuclear exit of active PKD2. This conclusion is also supported by the kinetics of PKD2 phosphorylation at Ser706/710 and Ser244. Under physiological conditions, dephosphorylation of Ser244 subsequently regulates PKD2 nuclear export (Figure 6E).
Nuclear PKD2 interacts with and phosphorylates HDAC7
PKD1 phosphorylates HDAC7 in thymocytes (Dequiedt et al, 2005; Parra et al, 2005) and B cells (Matthews et al, 2006). The repressive activity of class IIa HDACs is controlled by phosphorylation of specific serine residues in their N terminus, which leads to their cytoplasmic accumulation (Grozinger and Schreiber, 2000; Dressel et al, 2001; Kao et al, 2001). In gastric cancer cells, which mainly express PKD2 and exhibit nuclear HDAC7 under resting conditions, the kinase interacts with the N terminus of HDAC7 in an activation-dependent manner and phosphorylates its N terminus in vitro and in vivo. Binding to and phosphorylation of HDAC7 by PKD2 requires CK1 activity. Furthermore, the coordinated phosphorylation of PKD2 by active PKCη and active CK1 recapitulates the effect of gastrin or the triple S244/706/710E PKD2 mutant on nuclear exclusion of HDAC7. This further supports our model where the three phosphorylation sites that are regulated by CK1 and PKCs are sufficient for the nuclear activity of PKD2 in AGS-B cells. The fact that active PKD2 only efficiently phosphorylates HDAC7 upon nuclear accumulation demonstrates that HDAC7 is indeed phosphorylated/inactivated by PKD2 in the nuclear compartment in gastric cancer cells. Consequently, only active and nuclear PKD2 markedly induced expression of Nur77 in AGS-B cells. However, in other cell types in which PKD1 has been shown to phosphorylate HDACs such as lymphocytes, the compartment in which this phosphorylation event occurs may be different since the site in PKD1 that corresponds to Ser244 in PKD2 is a PKC phosphorylation site and therefore PKCs alone can induce all phosphorylation events at the three serine residues in PKD1 (Vertommen et al, 2000). PKD3 is also expressed in AGS cells. In many cells, PKD3 is detected in the nucleus and this kinase can also phosphorylate HDACs. It has been reported that there is redundancy between PKD1 and PKD3 in chicken B lymphocytes upon knockout of one isoform (Matthews et al, 2006). In AGS gastric cancer cells, we did not observe such a redundancy. However, in our model, expression of PKD2 was only reduced, but not completely abrogated, which might influence the degree by which specific isoforms act in a redundant manner. Furthermore, it is likely that there are cell-type-specific differences in the compartment-specific signaling properties of PKD isoforms.
Nur77 is induced by multiple extracellular signals, including growth factors, apoptotic and inflammatory signals and hormones, in a cell-type-specific manner. Nur4A family members have been found to play a role in T-cell apoptosis, dopaminergic neuron development, pituitary function and glucose metabolism in the liver (Pei et al, 2006). We establish the CCK2R as a novel inducer of Nur77 expression. The fact that gastrin-induced Nur77 expression is critically dependent on PKD2 stresses the role of PKD2 as a mediator of gastrin-induced transcriptional regulation.
In conclusion, we demonstrate a novel signal transduction pathway downstream of the CCK2R that culminates in phosphorylation of class IIa HDACs in the nucleus and derepression of their target genes. PKD2 is a central mediator of this signaling cascade and we identify the three critical modifications of the kinase as well as the upstream regulators that confer these modifications. One regulator, PKCη, controls the catalytic activity of PKD2, whereas the other regulator, CK1δ, defines the subcellular compartment in which the activity occurs. It will be interesting to determine whether similar coregulatory mechanisms of compartment-specific kinase activity are a feature of other signaling pathways.
Materials and methods
Cell culture
HEK293, AGS-B, HeLa and Cos7 cells were maintained as described (Sturany et al, 2001). AGS-B cells overexpressing the CCK2R have been described elsewhere (Sturany et al, 2001).
Plasmids
C-terminal eGFP fusion proteins of human HDAC7 and GST-fusion proteins of the N- (aa 1–490) and C terminus (aa 490–915) of HDAC7 (Dequiedt et al, 2003, 2005), the CK1 and p53 expression plasmids (Knippschild et al, 1997) and constitutively active C-terminal truncated PKCη have been described earlier (Sturany et al, 2002; Winter et al, 2004). CK1δ1–375 was amplified by PCR using pGEX2T-wtCK1δ1−428 (Knippschild et al, 1997) as template and the 5′ primer 5′-CGAATTCCATGGAGCGGGAACGCCG-3′ and 3′ primer 5′-ACTCGAGTCGACACGGTGCAGCCGCA-3′. The PCR product was cloned into the pcDNA-3.1/V5-His-TOPO vector generating pcDNA-3.1-CK1δ1−375 (plasmid 861).
Generation of PKD2 mutants
PKD2 and its mutants were ligated into peGFP-C2 (BD Clontech), pcDNA3 (Invitrogen) containing an N-terminal Flag tag, pGEX4T or pGMEXT3. Mutations within eGFP-PKD2 resulting both in single, double and triple amino-acid substitutions (D695A, S706/710E, S706/710A, S244A, S244E, S244/706/710E, S244A706/710E, S244E/S706/710A) were performed by PCR-based site-directed mutagenesis (Stratagene). To obtain GST-PKD2-CRD or GST-PKD2-S244A, the entire C1a/C1b domain of PKD2 was amplified with PCR using either eGFP-PKD2 or eGFP-PKD2-S244A as template and inserted into the pGEX-4T vector. All mutants were verified by sequencing.
Transient transfections of AGS-B, HEK293, HeLa and Cos7 cells
Cells were transfected using FUGENE (Roche, Mannheim, Germany) according to the manufacturer's instructions.
RNA interference and rescue experiments
Functionally validated Stealth RNAi directed against PKD2 and CK1δ/ɛ and a Stealth™ RNAi negative control were from Invitrogen (CA). eGFP-HDAC7 (for immunofluorescence analysis) or pNur77-Luc (for luciferase assays) were transfected together with PKD2 siRNA (150 nM) or control siRNA using HiPerfect (Qiagen). For rescue experiments, siRNA-transfected cells were retransfected after 24 h with siRNA and respective vectors (Flag-pcDNA3, Flag-PKD2ΔPH lacking the entire PH domain of PKD2 or ratCK1δ).
Western blotting, IPs and IVKs
IPs were performed as described (Sturany et al, 2001) with anti-GFP or anti-PKD2 antibodies and further analyzed by western blotting using various antibodies as indicated in the figure legends. IVKs to determine the catalytic activity of PKD2 or various PKD2 GST-fusion proteins were performed as described (Auer et al, 2005) using histone or HDAC7 as substrates. Quantification of autoradiographs was performed by scanning densitometry using the MacBAS V2.5 software (Fuji Photo Film, Tokyo, Japan).
Bacterial GST-fusion protein expression and purification
Recombinant, active GST-fusion proteins (p53 and p53 mutants, GST-PKD2, GST-PKD2-S706/710E, GST-HDAC7-Nter and Cter, GST-14-3-3η, GST-PKD2-CRD and GST-PKD2-CRD244A) were expressed in Top10 or BL21-RIP bacterial strains and isolated as described in Supplementary data.
GST pull downs
GST-PKD2, GST-HDAC7 or GST-14-3-3η proteins were bound to glutathione sepharose. For pull-down reactions, cells were lysed for 10 min in GST pull-down lysis buffer and further analyzed as described in the figure legends.
NanoLC/ES-MS/MS
The SDS–PAGE separated band of GST-PKD2 fusion proteins was digested as described (Shevchenko et al, 1996) followed by NanoLC/ES-MS/MS of the extracted peptides according to Tolson et al (2006) using a 150 mm × 75 μm (ID) Vydac Everest C18 (5 μm, 300 Å) capillary column (Alltech Grom GmbH, Rottenburg-Hailfingen, Germany). Peptides were gradient eluted (250 nl/min flow rate): 0–5 min 5% B, 5–100 min 5–40% B, 100–130 min 40–60% B, where system A was 0.1% formic acid in water (v/v) and system B was 0.1% formic acid, 80% acetonitrile in water (v/v/v). The data-dependent tandem mass spectrum analyses included the acquisition of a survey spectrum followed by MS/MS spectra of the three most abundant ions in the survey scan (m/z 200–2000). In addition to the data-dependent analyses, the theoretical m/z values of the triply charged precursor ions of the unphosphorylated (m/z 799.9) and mono-phosphorylated (m/z 826.4) peptides Arg233-Lys255 (in full-length PKD2) were used as preferred masses for target MS/MS analyses. Uninterpreted MS/MS data were searched using MASCOT (Matrix Science, London, UK). Assignments of the phosphopeptides were confirmed by manual comparison of the MS/MS mass spectra with the predicted fragmentation generated by the MS-Product component of Protein Prospector.
Immunofluorescence studies
AGS-B, Cos7, HEK293 or HeLa cells were plated onto coverslips for 24 h and transfected with the respective plasmids as described (Auer et al, 2005) for another 24 h at 37°C. Cells were subsequently fixed and primary antibodies in PBS were added overnight at 4°C followed by incubation with Alexa-coupled secondary antibodies (Dianova, Hamburg, Germany) for 1 h at room temperature and embedding in mounting medium (Immunotech, Marseille, France). Imaging was performed with a confocal laser scanning microscope (LSM510; Zeiss, Jena, Germany) with a 25 mW krypton–argon laser and a × 63 objective (NA 1.4). Laser energy and parameters of intensity detection were kept constant for all slides. The images shown represent typical cells from at least three independent experiments.
Live cell imaging, fluorescence intensity evaluation and FLIP
Imaging of live and fixed cells expressing eGFP-tagged proteins was performed as described by Auer et al (2005) using the LSM510. The relative nuclear fluorescence intensity was calculated with the following equation: Finuclear (Fin in arbitrary units a.u.): 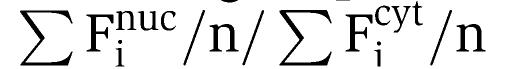 (where Finuc is the fluorescence intensity in the nucleus, Ficyt is the relative fluorescence intensity in the cytoplasm and n is the number of cells examined). To quantify HDAC7 nuclear exclusion (HDAC7 nuc ex), cells with nuclear or cytoplasmic HDAC7 were counted in at least 300 HDAC7-positive cells and the percentage of cytoplasmic HDAC7-positive cells was calculated. FLIP was determined as described (White and Stelzer, 1999; Lippincott-Schwartz et al, 2001). nFi (relative nuclear fluorescence intensity) was obtained by dividing the nuclear fluorescence intensity at various time points (Fit) after bleaching the cytoplasm by the nuclear fluorescence intensity prior to bleaching the cytoplasm (Fiinitial). The background-corrected nuclear fluorescence at the time prior to the initial photobleaching was set as 100%, nFi (%)=(Fit/Fiinitial) × 100.
(where Finuc is the fluorescence intensity in the nucleus, Ficyt is the relative fluorescence intensity in the cytoplasm and n is the number of cells examined). To quantify HDAC7 nuclear exclusion (HDAC7 nuc ex), cells with nuclear or cytoplasmic HDAC7 were counted in at least 300 HDAC7-positive cells and the percentage of cytoplasmic HDAC7-positive cells was calculated. FLIP was determined as described (White and Stelzer, 1999; Lippincott-Schwartz et al, 2001). nFi (relative nuclear fluorescence intensity) was obtained by dividing the nuclear fluorescence intensity at various time points (Fit) after bleaching the cytoplasm by the nuclear fluorescence intensity prior to bleaching the cytoplasm (Fiinitial). The background-corrected nuclear fluorescence at the time prior to the initial photobleaching was set as 100%, nFi (%)=(Fit/Fiinitial) × 100.
Statistical analysis
Statistical significance was tested in an unpaired Student's t-test using the Graph Pad Prism software. Compared data sets were termed as statistically significant (P<0.01 (*)) or very significant (P<0.001 (***)).
Materials
[γ-32P]ATP (5000 Ci/mmol; 37 GBq=1 mCi) was from Amersham. Anti-Crm-1 antibody was from BD Transduction (Lexington, USA) and anti-GFP antibody was from Roche (Mannheim, Germany). CKI-7 was from Seikagaku (Tokyo, Japan), and DD4476 and GF 109203X were supplied by Calbiochem (Nottingham, UK). IC261 was from Merck (Switzerland). Anti-phospho-PKD1-Ser744/748 and anti-HDAC7 antibodies were from Cell Signaling (Beverly, MA, USA). Anti-PKD2 antibody was purchased from Biomol (Hamburg, Germany). Anti-Nur77 and anti-CK1ɛ antibodies were from active motif and anti-CK1δ antibody was from ICOS Corp. (Bothell, WA, USA). Anti-FlagM2 and anti-β-actin antibodies were supplied by Sigma Aldrich (Taufkirchen, Germany). The anti-phospho-pSer244 was generated by Panatecs (Tübingen, Germany). The anti-phospho-HDAC7 (HDAC7 pS-181) antibody was generated by Dr Franck Dequiedt. Anti-GST antibody was from Santa Cruz (CA, USA).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S3 Continued
Supplementary Figure S4
Supplementary Figure Legends
Acknowledgments
JvB was a fellow of the GRK1041. TS is supported by DFG (SFB 518/B3 and GRK1091), UK is supported by DFG (SFB518/B15) and Deutsche Krebshilfe (10-2237-KN3), and JVL by the Belgian Federal Government (grant IAP 5/12) and the FWO-Vlaanderen. FD is Research Associate of the FNRS and was supported by a Televie grant (# 7.4629.07) from le Fonds de la Recherche Scientifique-FNRS. We thank Ulrike Meyr-Beyerle for expert technical assistance.
References
- Auer A, von Blume J, Sturany S, von Wichert G, Van Lint J, Vandenheede J, Adler G, Seufferlein T (2005) Role of the regulatory domain of protein kinase D2 in phorbol ester binding, catalytic activity, and nucleocytoplasmic shuttling. Mol Biol Cell 16: 4375–4385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustos VH, Ferrarese A, Venerando A, Marin O, Allende JE, Pinna LA (2006) The first armadillo repeat is involved in the recognition and regulation of beta-catenin phosphorylation by protein kinase CK1. Proc Natl Acad Sci USA 103: 19725–19730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dequiedt F, Kasler H, Fischle W, Kiermer V, Weinstein M, Herndier BG, Verdin E (2003) HDAC7, a thymus-specific class II histone deacetylase, regulates Nur77 transcription and TCR-mediated apoptosis. Immunity 18: 687–698 [DOI] [PubMed] [Google Scholar]
- Dequiedt F, Martin M, Von Blume J, Vertommen D, Lecomte E, Mari N, Heinen MF, Bachmann M, Twizere JC, Huang MC, Rider MH, Piwnica-Worms H, Seufferlein T, Kettmann R (2006) New role for hPar-1 kinases EMK and C-TAK1 in regulating localization and activity of class IIa histone deacetylases. Mol Cell Biol 26: 7086–7102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dequiedt F, Van Lint J, Lecomte E, Van Duppen V, Seufferlein T, Vandenheede JR, Wattiez R, Kettmann R (2005) Phosphorylation of histone deacetylase 7 by protein kinase D mediates T cell receptor-induced Nur77 expression and apoptosis. J Exp Med 201: 793–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dressel U, Bailey PJ, Wang SC, Downes M, Evans RM, Muscat GE (2001) A dynamic role for HDAC7 in MEF2-mediated muscle differentiation. J Biol Chem 276: 17007–17013 [DOI] [PubMed] [Google Scholar]
- Grozinger CM, Schreiber SL (2000) Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc Natl Acad Sci USA 97: 7835–7840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi A, Seki N, Hattori A, Kozuma S, Saito T (1999) PKCnu, a new member of the protein kinase C family, composes a fourth subfamily with PKCmu. Biochim Biophys Acta 1450: 99–106 [DOI] [PubMed] [Google Scholar]
- Kao HY, Verdel A, Tsai CC, Simon C, Juguilon H, Khochbin S (2001) Mechanism for nucleocytoplasmic shuttling of histone deacetylase 7. J Biol Chem 276: 47496–47507 [DOI] [PubMed] [Google Scholar]
- Knippschild U, Gocht A, Wolff S, Huber N, Lohler J, Stoter M (2005) The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell Signal 17: 675–689 [DOI] [PubMed] [Google Scholar]
- Knippschild U, Milne DM, Campbell LE, DeMaggio AJ, Christenson E, Hoekstra MF, Meek DW (1997) p53 is phosphorylated in vitro and in vivo by the delta and epsilon isoforms of casein kinase 1 and enhances the level of casein kinase 1 delta in response to topoisomerase-directed drugs. Oncogene 15: 1727–1736 [DOI] [PubMed] [Google Scholar]
- Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B, Yoshida M, Horinouchi S (1999) Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci USA 96: 9112–9117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Snapp E, Kenworthy A (2001) Studying protein dynamics in living cells. Nat Rev Mol Cell Biol 2: 444–456 [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S (2002) The protein kinase complement of the human genome. Science 298: 1912–1934 [DOI] [PubMed] [Google Scholar]
- Mashhoon N, DeMaggio AJ, Tereshko V, Bergmeier SC, Egli M, Hoekstra MF, Kuret J (2000) Crystal structure of a conformation-selective casein kinase-1 inhibitor. J Biol Chem 275: 20052–20060 [DOI] [PubMed] [Google Scholar]
- Matthews SA, Liu P, Spitaler M, Olson EN, McKinsey TA, Cantrell DA, Scharenberg AM (2006) Essential role for protein kinase D family kinases in the regulation of class II histone deacetylases in B lymphocytes. Mol Cell Biol 26: 1569–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne DM, Looby P, Meek DW (2001) Catalytic activity of protein kinase CK1 delta (casein kinase 1delta) is essential for its normal subcellular localization. Exp Cell Res 263: 43–54 [DOI] [PubMed] [Google Scholar]
- Oancea E, Bezzerides VJ, Greka A, Clapham DE (2003) Mechanism of persistent protein kinase D1 translocation and activation. Dev Cell 4: 561–574 [DOI] [PubMed] [Google Scholar]
- Obenauer JC, Cantley LC, Yaffe MB (2003) Scansite 2.0 Proteome Protwide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res 31: 3635–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura H, Garcia-Rodriguez C, Martinson H, Qin J, Virshup DM, Rao A (2004) A conserved docking motif for CK1 binding controls the nuclear localization of NFAT1. Mol Cell Biol 24: 4184–4195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra M, Kasler H, McKinsey TA, Olson EN, Verdin E (2005) Protein kinase D1 phosphorylates HDAC7 and induces its nuclear export after T-cell receptor activation. J Biol Chem 280: 13762–13770 [DOI] [PubMed] [Google Scholar]
- Pei L, Waki H, Vaitheesvaran B, Wilpitz DC, Kurland IJ, Tontonoz P (2006) NR4A orphan nuclear receptors are transcriptional regulators of hepatic glucose metabolism. Nat Med 12: 1048–1055 [DOI] [PubMed] [Google Scholar]
- Rittinger K, Budman J, Xu J, Volinia S, Cantley LC, Smerdon SJ, Gamblin SJ, Yaffe MB (1999) Structural analysis of 14-3-3 phosphopeptide complexes identifies a dual role for the nuclear export signal of 14-3-3 in ligand binding. Mol Cell 4: 153–166 [DOI] [PubMed] [Google Scholar]
- Rozengurt E, Rey O, Waldron RT (2005) Protein kinase D signaling. J Biol Chem 280: 13205–13208 [DOI] [PubMed] [Google Scholar]
- Rykx A, De Kimpe L, Mikhalap S, Vantus T, Seufferlein T, Vandenheede JR, Van Lint J (2003) Protein kinase D: a family affair. FEBS Lett 546: 81–86 [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M (1996) Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68: 850–858 [DOI] [PubMed] [Google Scholar]
- Storz P, Toker A (2003) Protein kinase D mediates a stress-induced NF-kappaB activation and survival pathway. EMBO J 22: 109–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturany S, Van Lint J, Gilchrist A, Vandenheede JR, Adler G, Seufferlein T (2002) Mechanism of activation of protein kinase D2(PKD2) by the CCK(B)/gastrin receptor. J Biol Chem 277: 29431–29436 [DOI] [PubMed] [Google Scholar]
- Sturany S, Van Lint J, Muller F, Wilda M, Hameister H, Hocker M, Brey A, Gern U, Vandenheede J, Gress T, Adler G, Seufferlein T (2001) Molecular cloning and characterization of the human protein kinase D2. A novel member of the protein kinase D family of serine threonine kinases. J Biol Chem 276: 3310–3318 [DOI] [PubMed] [Google Scholar]
- Tolson JP, Flad T, Gnau V, Dihazi H, Hennenlotter J, Beck A, Mueller GA, Kuczyk M, Mueller CA (2006) Differential detection of S100A8 in transitional cell carcinoma of the bladder by pair wise tissue proteomic and immunohistochemical analysis. Proteomics 6: 697–708 [DOI] [PubMed] [Google Scholar]
- Van Lint J, Rykx A, Maeda Y, Vantus T, Sturany S, Malhotra V, Vandenheede JR, Seufferlein T (2002) Protein kinase D: an intracellular traffic regulator on the move. Trends Cell Biol 12: 193–200 [DOI] [PubMed] [Google Scholar]
- Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA (2004) Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol 24: 8374–8385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertommen D, Rider M, Ni Y, Waelkens E, Merlevede W, Vandenheede JR, Van Lint J (2000) Regulation of protein kinase D by multisite phosphorylation. Identification of phosphorylation sites by mass spectrometry and characterization by site-directed mutagenesis. J Biol Chem 275: 19567–19576 [DOI] [PubMed] [Google Scholar]
- White J, Stelzer E (1999) Photobleaching GFP reveals protein dynamics inside live cells. Trends Cell Biol 9: 61–65 [DOI] [PubMed] [Google Scholar]
- Winter M, Milne D, Dias S, Kulikov R, Knippschild U, Blattner C, Meek D (2004) Protein kinase CK1delta phosphorylates key sites in the acidic domain of murine double-minute clone 2 protein (MDM2) that regulate p53 turnover. Biochemistry 43: 16356–16364 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S3 Continued
Supplementary Figure S4
Supplementary Figure Legends