

Abstract
Phosphorylation of human CTP synthetase 1 by mammalian protein kinase C was examined. Using purified Escherichia coli-expressed CTP synthetase 1 as a substrate, protein kinase C activity was time-and dose-dependent, and dependent on the concentrations of ATP and CTP synthetase 1. The protein kinase C phosphorylation of the recombinant enzyme was accompanied by a 95-fold increase in CTP synthetase 1 activity. Phosphopeptide mapping and phosphoamino acid analyses showed that CTP synthetase 1 was phosphorylated on multiple serine and threonine residues. The induction of PKC1R398A-encoded protein kinase C resulted in a 50% increase for human CTP synthetase 1 phosphorylation in the Saccharomyces cerevisiae ura7Δ ura8Δ mutant lacking yeast CTP synthetase activity. Synthetic peptides that contain the protein kinase C motif for Ser462 and Thr455 were substrates for mammalian protein kinase C, and S462A and T455A mutations resulted in decreases in the extent of CTP synthetase 1 phosphorylation that occurred in vivo. Phosphopeptide mapping analysis of S. cerevisiae-expressed CTP synthetase 1 mutant enzymes phosphorylated with mammalian protein kinase C confirmed that Ser462 and Thr455 were phosphorylation sites. The S. cerevisiae-expressed and purified S462A mutant enzyme exhibited a 2-fold reduction in CTP synthetase 1 activity, whereas the purified T455A mutant enzyme exhibits a 2-fold elevation in CTP synthetase 1 activity (Choi, M.-G., and Carman, G.M. (2006) J. Biol. Chem. 282, 5367–5377). These data indicated that protein kinase C phosphorylation at Ser462 stimulates human CTP synthetase 1 activity, whereas phosphorylation at Thr455 inhibits activity.
CTP synthetase (EC 6.3.4.2, UTP: ammonia ligase (ADP-forming)) is an allosterically regulated enzyme that is essential for the growth and metabolism of all organisms (1). The essential nature of the enzyme stems from the fact that its reaction product CTP is required for the synthesis of RNA, DNA, and membrane phospholipids (1). CTP synthetase activity controls the balance of nucleotide pools (2–8) and regulates the pathways by which phospholipids are synthesized (8–10). Moreover, an unregulated level of CTP synthetase activity is a common property of several cancers (11–16), and thus, the enzyme is a target of antiproliferative drug development for cancer therapy (17–22). The enzyme is also a target for parasite (23, 24) and viral (25) diseases.
The CTP synthetase enzyme has been isolated from both prokaryotic and eukaryotic organisms (7, 24, 26–30), and structures for bacterial (31, 32) and human (33) enzymes have been solved. CTP synthetase enzymes contain conserved synthetase and glutamine amide transfer domains that are involved in catalysis (31–33) (Fig. 1). The enzymological and kinetic properties of the enzyme from various organisms are similar, although some differences have been noted (24, 28). The enzyme catalyzes a complex set of reactions that include the ATP-dependent transfer of the amide nitrogen from glutamine (i.e., glutaminase reaction) to the C-4 position of UTP to generate CTP (26, 34). GTP stimulates the glutaminase reaction by accelerating the formation of a covalent glutaminyl enzyme intermediate (26, 35). The enzyme exhibits positive cooperative kinetics with respect to UTP and ATP, and negative cooperative kinetics with respect to glutamine and GTP (7, 26, 28, 29, 35–39). The cooperativity with respect to UTP and ATP is ascribed to CTP synthetase oligomerization resulting in the active tetrameric form of the enzyme (7, 26, 28, 29, 35–41).
FIGURE 1. Domain structure of human CTP synthetase 1 and putative protein kinase C phosphorylation sites.
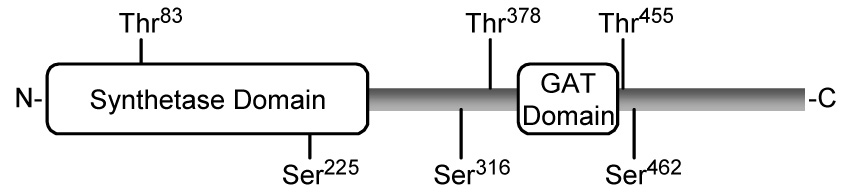
The diagram shows the positions of the synthetase domain (residues 1-272) and the glutamine amide transfer (GAT) domain (residues 394–405) in the CTP synthetase 1 protein sequence. The putative serine (S) and threonine (T) residues for protein kinase C phosphorylation are indicated.
CTP synthetase enzymes are allosterically regulated by CTP product inhibition (7, 26, 28–30). CTP inhibits activity by increasing the positive cooperativity of the enzyme for UTP (7, 26, 29, 30). Mutagenesis and structural studies have shown that CTP inhibits CTP synthetase activity through interaction with sites other than the UTP binding site of the enzyme (8, 42). dCTP does not substitute for CTP as a feedback inhibitor of CTP synthetase activity using dUTP or UTP as a substrate (28, 43). CTP synthetase mutations that alleviate CTP feedback inhibition result in abnormally high intracellular levels of CTP and dCTP (3, 8, 44), resistance to nucleotide analog drugs used in cancer chemotherapy (45–48), and an increased rate of spontaneous mutations (4, 46, 48).
Covalent modification by phosphorylation is another mechanism by which CTP synthetase activity is regulated (49–51). Studies with the Saccharomyces cerevisiae URA7-encoded enzyme have shown that phosphorylation by protein kinase A (49) or by protein kinase C (50, 51) results in the stimulation of CTP synthetase activity. Because the regulation of CTP synthetase activity is so important to normal cell growth and development, we initiated studies to examine the phosphorylation of the human CTPS1-encoded enzyme (52, 53). The functional expression of the human CTPS1 gene in the S. cerevisiae ura7Δ ura8Δ double mutant devoid of CTP synthetase activity has shown that human CTP synthetase 1 is phosphorylated in vivo (52). Some of this phosphorylation is mediated by protein kinase A (52), and that phosphorylation at Thr455 results in the inhibition of CTP synthetase 1 activity (53).
In the present work, we addressed the hypothesis that phosphorylation of human CTP synthetase 1 was also mediated by protein kinase C. This hypothesis is based on the presence of potential protein kinase C target sites in the CTP synthetase 1 enzyme (Fig. 1). We showed here that mammalian protein kinase C does in fact phosphorylate purified human CTP synthetase 1. Ser462 and Thr455 were identified as major protein kinase C phosphorylation sites. The phosphorylation at Ser462 results in the stimulation of CTP synthetase 1 activity, whereas phosphorylation at Thr455 results in the inhibition of activity. This work identifies an important human target of protein kinase C and advances the understanding of human CTP synthetase 1 regulation by phosphorylation.
EXPERIMENTAL PROCEDURES
Materials
All chemicals were reagent grade. Growth medium supplies were obtained from Difco Laboratories. New England Biolabs was the source of modifying enzymes, recombinant Vent DNA polymerase, and restriction endonucleases. DNA gel extraction kit, plasmid DNA purification kit, and Ni2+-NTA agarose resins were purchased from Qiagen. Oligonucleotides were synthesized by Genosys Biotechnologies, Inc. The QuikChange site-directed mutagenesis kit was purchased from Stratagene. Carrier DNA for yeast transformation was from Clontech. Peptides were synthesized and purified by Bio-Synthesis, Inc. Radiochemicals were purchased from Perkin-Elmer Life Sciences. Nucleotides, 5-fluoroorotic acid, aprotinin, benzamidine, bovine serum albumin, leupeptin, pepstatin, phenylmethylsulfonyl fluoride, phosphoamino acids, and L-1-tosylamido-2-phenylethyl chloromethyl ketone-trypsin were purchased from Sigma. Bio-Rad was the source of DNA size ladders, electrophoresis reagents, immunochemical reagents, molecular mass protein standards, and protein assay reagents. Mouse monoclonal anti-His6 antibodies were from Cell Signaling Technology. Mouse monoclonal anti-HA antibodies (12CA5) and goat anti-mouse IgG alkaline phosphatase conjugates were from Roche Molecular Biochemicals and Pierce, respectively. Conventional mammalian protein kinase C (rat brain) was purchased from Promega. Lipids were obtained from Avanti Polar Lipids. Protein A-Sepharose CL-4B beads, Hybond-P polyvinylidene difluoride paper, and the enhanced chemifluorescence Western blotting detection kit were purchased from GE Healthcare. Cellulose thin layer glass plates were from EMD Chemicals. Scintillation counting supplies and acrylamide solutions were purchased from National Diagnostics.
Strains and Growth Media
The strains used in this work are listed in Table 1. E. coli strain DH5α was used for the propagation of plasmids. Cells were grown in LB medium (1% tryptone, 0.5% yeast extract, 1% NaCl, pH 7.4) at 37 °C. Ampicillin (100 µg/ml) was added to cultures of DH5α cells that carried plasmids. Human CTP synthetase 1 was expressed in E. coli BL21(DE3) cells bearing plasmid pET-28b(+)-hCTPS1 as described by Han et al. (52). Methods for yeast growth were performed as described previously (54, 55). Yeast cultures were grown in YEPD medium (1% yeast extract, 2% peptone, 2% glucose) or in synthetic complete medium (56) containing 2 % glucose at 30°C. For selection of cells carrying plasmids, appropriate amino acids were omitted from the growth medium. Cell numbers in liquid medium were determined spectrophotometrically at an absorbance of 600 nm. The growth medium was supplemented with 2% (for yeast) or 1.5% (for E. coli) agar for growth on plates.
TABLE 1.
Strains used in this work
| Strain | Relevant characteristics | Source or Ref. |
|---|---|---|
| E. coli | ||
| DH5α | F− ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rk− mk+) phoA supE44 λ−thi-1 gyrA96 relA1 | (55) |
| S. cerevisiae | ||
| SDO195 | MATa leu2–3,112 trp1-289 ura3–52 ura7Δ::TRP1 ura8Δ:: hisG [pDO134] | (8) |
| GHY52 | MATa leu2–3,112 trp1-289 ura3–52 ura7Δ::TRP1 ura8Δ::hisG [pDO134][pDO105] | (52) |
| GHY53 | MATa leu2–3,112 trp1-289 ura3–52 ura7Δ::TRP1 ura8Δ::hisG [pDO134][pDO105-hCTPS1] | (52) |
| GHY55 | MATa leu2–3,112 trp1-289 ura3–52 ura7Δ::TRP1 ura8Δ::hisG [pDO105-hCTPS1] | (52) |
| MCY20 | MATa leu2–3,112 trp1-289 ura3–52 ura7Δ::TRP1 ura8Δ::hisG [pDO105-hCTPS1(T455A)] | (53) |
| YCY1 | MATa leu2–3,112 trp1-289 ura3–52 ura7Δ::TRP1 ura8Δ:: hisG [pDO105-hCTPS1(S462A)] | This work |
| YCY2 | MATa leu2–3,112 trp1-289 ura3–52 ura7Δ::TRP1 ura8Δ:: hisG [pDO105-hCTPS1(S462A,T455A)] | This work |
| YCY3 | MATa leu2–3,112 trp1-289 ura3–52 ura7Δ::TRP1 ura8Δ:: hisG [pDO105-hCTPS1][pBM743] | This work |
| DL470 | MATa leu2–3,112 ura3-52 trp1-1 his4 can1r pkc1Δ::LEU2 [pGAL1::PKC1(R398A::HA)] | (61) |
| YCY4 | MATa leu2–3,112 trp1-289 ura3–52 ura7Δ::TRP1 ura8Δ:: hisG [pDO105-hCTPS1][pGAL1::PKC1(R398A::HA)] | This work |
| YCY5 | MATa leu2–3,112 trp1-289 ura3–52 ura7Δ::TRP1 ura8Δ:: hisG [pDO105-hCTPS1(S462A)][pGAL1::PKC1(R398A::HA)] | This work |
| YCY6 | MATa leu2–3,112 trp1-289 ura3–52 ura7Δ::TRP1 ura8Δ:: hisG [pDO105-hCTPS1(T455A)][pGAL1::PKC1(R398A::HA)] | This work |
| YCY7 | MATa leu2–3,112 trp1-289 ura3–52 ura7Δ::TRP1 ura8Δ:: hisG [pDO105-hCTPS1(S462A,T455A)][pGAL1::PKC1(R398A::HA)] | This work |
DNA Manipulations, Amplification of DNA by PCR, and DNA Sequencing
Standard methods were used to prepare genomic and plasmid DNA, to digest DNA with restriction enzymes, and to ligate DNA (55). Transformation of E. coli (55) and yeast (57, 58) was performed according to standard methods. The methods of Innis and Gelfand (59) were used to optimize PCR reactions. DNA sequencing reactions were performed by the dideoxy method using Taq DNA polymerase (55) and analyzed by automated DNA sequencing.
Construction of Plasmids and Expression of Human CTPS1 in S. cerevisiae
The plasmids used in this work are listed in Table 2. Plasmid pDO105-hCTPS1 directs the overexpression of full-length His6-tagged (C terminus) human CTP synthetase 1 in S. cerevisiae (52). The CTPS1S462A (primer 5’-CAGACCAAGAACGCAGTCATGAGGAAACTCTATGG-3’ and its complement) mutant allele was constructed by PCR with the QuikChange site-directed mutagenesis kit using plasmid pDO105-hCTPS1 as the template. The CTPS1S462A,T455A mutant allele was constructed with the primers for the S462A mutation using plasmid pDO105-hCTPS1(T455A) as the template. All mutations were confirmed by DNA sequencing.
Table 2.
Plasmids used in this work
| Plasmid | Relevant characteristics | Source or Ref. |
|---|---|---|
| YEpLac181 | Multicopy E. coli/yeast shuttle vector with LEU2 | (76) |
| YEpLac195 | Multicopy E. coli/yeast shuttle vector with URA3 | (76) |
| pDO105 | Derivative of YEpLac181 with the ADH1 promoter and multiple cloning sites | (8) |
| pDO134 | URA7 derivative of YEpLac195 | (8) |
| pDO105-hCTPS1 | CTPS1 derivative of pDO105 | (52) |
| pDO105-hCTPS1(T455A) | CTPS1T455A derivative of pDO105-hCTPS1 | (53) |
| pDO105-hCTPS1(S462A) | CTPS1S462A derivative of pDO105-hCTPS1 | This work |
| pDO105-hCTPS1(S462A,T455A) | CTPS1S462A,T455A derivative of pDO105-hCTPS1(T455A) | This work |
| pBM743 | Single copy vector containing the GAL1 promoter with URA3 | (77) |
| pGAL1::PKC1(R398A::HA) | PKC1(R398A::HA) derivative of pBM743 | (61) |
Strain SDO195 was transformed to leucine prototrophy with plasmids containing the wild type and mutant alleles of the CTPS1 gene. Strain SDO195 is an ura7Δ ura8Δ double mutant carrying plasmid pDO134. This plasmid, which bears a wild type yeast URA7 allele, rescues the lethal phenotype of the ura7Δ ura8Δ mutant (6), and at the same time, confers uracil prototrophy to strain SDO195 because it contains the URA3 gene (8). The yeast transformants, which contained URA3- and LEU2-based plasmids bearing the URA7 and CTPS1 genes, respectively, were streaked onto synthetic complete-leucine plates containing 5-fluoroorotic acid to evict (60) the plasmid bearing the URA3 gene. 5-Fluoroorotic acid-resistant cells were further confirmed by their uracil auxotrophy. Immunoblot analysis using anti-His6 antibodies confirmed that the 5-fluoroorotic acid-resistant ura7Δ ura8Δ mutant cells expressed the wild type and the individual CTPS1 mutant alleles.
To examine protein kinase C phosphorylation of human CTP synthetase 1 in vivo, the CTPS1 gene was expressed in S. cerevisiae cells (strain YCY4) that also expressed a galactose-inducible activated PKC1R398A allele. Plasmid pGAL1::PKC1(R398A::HA), which directs the expression of yeast protein kinase C activity (61), was isolated from strain DL470 (61) and transformed into strain GHY55 to construct strain YCY4. For galactose induction, cells were first grown to A600 nm = 0.7 in the synthetic complete medium with 2% glucose. The cells were then washed and resuspended in synthetic complete medium with 2% raffinose followed by incubation at 30°C for 10–12 hr to deplete the internal stores of glucose. Cells were then harvested and resuspended in synthetic complete medium with 2% raffinose and labeled with 32Pi (0.25 mCi/ml) for 1.5 h. Galactose was then added to a final concentration of 2% to induce the expression of the activated PKC1R398A gene (61). After a 1.5-h induction, the cells were treated with 20 mM NaN3 and harvested by centrifugation.
Purification of Human CTP Synthetase 1 Enzymes from E. coli and from S. cerevisiae
His6-tagged wild type human CTP synthetase 1 was expressed and purified from E. coli by Ni2+-NTA chromatography and with Poros HQ ion-exchange chromatography as described by Han et al. (52). His6-tagged wild type and mutant human CTP synthetase 1 enzymes were expressed and purified from yeast cell extracts with Ni2+-NTA resin as described by Choi and Carman (53). These enzyme preparations were used for in vitro phosphorylation experiments. Alternatively, the overexpressed wild type and S462A mutant CTP synthetase 1 enzymes were purified from the cytosolic fraction of yeast as described by Park et al. (62). These preparations were used for enzyme activity measurements. As described previously (53, 62), SDS-PAGE analysis indicated that the enzyme preparations were highly purified.
Enzyme Assays
CTP synthetase activity was determined by measuring the conversion of UTP to CTP (molar extinction coefficients of 182 and 1,520 M−1 cm−1, respectively) by following the increase in absorbance at 291 nm on a recording spectrophotometer (26). The standard reaction mixture contained 50 mM Tris-HCl (pH 8.0), 10 mM MgCl2, 10 mM β-mercaptoethanol, 2 mM L-glutamine, 0.1 mM GTP, 2 mM ATP, 2 mM UTP, and an appropriate dilution of enzyme protein in a total volume of 0.1 ml. Enzyme assays were performed in triplicate with an average standard deviation of ± 3%. All assays were linear with time and protein concentration. A unit of CTP synthetase activity was defined as the amount of enzyme that catalyzed the formation of 1 nmol of product/min.
SDS-PAGE and Immunoblot Analysis
SDS-PAGE (63) and immunoblotting (64) with polyvinylidene difluoride membrane were performed by standard procedures. Mouse monoclonal anti-His6 antibodies and alkaline phosphatase-conjugated goat anti-mouse antibodies were used at dilutions of 1:1000 and 1:5000, respectively. Proteins were detected on immunoblots using the enhanced chemifluorescence Western blotting detection kit. FluorImaging was used to acquire images from immunoblots, and the relative densities of the images were analyzed using ImageQuant software. Signals were in the linear range of detectability.
Phosphorylation Reactions
Phosphorylation reactions were measured for 10 min at 30 °C in a total volume of 25 µl. The protein kinase C reaction mixture contained 50 mM Tris-HCl buffer (pH 8.0), 10 mM MgCl2, 10 mM 2-mercaptoethanol, 0.375 mM EDTA, 0.375 mM EGTA, 1.7 mM CaCl2, 20 µM diacylglycerol, 50 µM phosphatidylserine, 50 µM [γ-32P]ATP (11,000 cpm/pmol), protein kinase C (0.5 unit/ml) and the indicated preparation of CTP synthetase 1. At the end of the phosphorylation reactions, samples were treated with 4x Laemmli's sample buffer (63), subjected to SDS-PAGE, and transferred to polyvinylidene difluoride membranes. Phosphorimaging was used to visualize phosphorylated enzyme, and the extent of phosphorylation was quantified with ImageQuant software. The data were normalized to amount of human CTP synthetase 1 protein on the membranes as determined by immunoblot analysis. In some experiments, the extent of enzyme phosphorylation was quantified directly from dried polyacrylamide gels. Reactions using human CTP synthetase 1 synthetic peptides were performed in a total volume of 25 µl, and were terminated by spotting the reaction mixtures onto P81 phosphocellulose paper. The papers were washed with 75 mM phosphoric acid and then subjected to scintillation counting. Phosphorylation reactions were performed in triplicate. A unit of protein kinase C activity was defined as the amount of enzyme that catalyzed the formation of 1 nmol of phosphorylated product/min.
In Vivo Labeling of Human CTP synthetase 1
The S. cerevisiae ura7Δ ura8Δ mutant carrying the human CTPS1 alleles was used to examine the in vivo phosphorylation of the human CTP synthetase 1 enzyme as described by Choi and Carman (53). Cultures (50 ml) in synthetic complete medium were grown to the exponential phase of growth; cells were harvested by centrifugation, resuspended in 5 ml of fresh medium containing 32Pi (0.25 mCi/ml), and incubated for 3 h. The 32P-labeled cells were harvested by centrifugation, suspended in 0.25 ml of spheroplast buffer (25 mM Tri-HCl (pH 7.4), 1 M sorbitol, 5 mM dithiothreitol, 10 mM NaF, 10 mM NaN3, and 60 units of lyticase), and incubated for 30 min at 30 °C. The spheroplasts were lysed by boiling for 3 min in 2% SDS. His6-tagged human CTP synthetase 1 enzymes were isolated from the lysates with Ni2+-NTA resin, subjected to SDS-PAGE, and transferred to polyvinylidene difluoride membrane. The membrane was used for phosphorimaging analysis of 32P-labeled human CTP synthetase 1 enzymes. ImageQuant software was used to quantify the extent of enzyme phosphorylation. The data were normalized to amount of human CTP synthetase 1 protein on the membranes as determined by immunoblot analysis using anti-His6 antibodies.
Phosphoamino Acid and Phosphopeptide Mapping Analyses
A portion of a polyvinylidene difluoride membrane containing 32P-labeled human CTP synthetase 1 protein was subjected to acid hydrolysis with 6 N HCl (49), followed by the analysis of phosphoamino acids by two-dimensional electrophoresis on cellulose thin-layer chromatography plates (65). Following electrophoresis, the cellulose thin-layer plates were dried and subjected to phosphorimaging analysis. Standard phosphoamino acids (2.5 µg phosphoserine, 2.5 µg phosphothreonine, and 5 µg phosphotyrosine) were visualized by spraying the plate with 0.25% ninhydrin in acetone. For phosphopeptide mapping analysis, polyvinylidene difluoride membrane slices containing 32P-labeled CTP synthetase 1 protein were subjected to proteolytic digestion with L-1-tosylamido-2-phenylethyl chloromethyl ketonetrypsin, followed by two-dimensional peptide mapping analysis using cellulose thin-layer chromatography plates (66). In the first dimension, the peptides were separated by electrophoresis, and in the second dimension, they were separated by ascending chromatography (66). Phosphorimaging was used to analyze phosphoamino acids and phosphopeptides on the cellulose thin-layer plates.
Analysis of Protein and CTP
Protein concentration was estimated by the method of Bradford (67) using bovine serum albumin as the standard. Nucleotides were extracted from S. cerevisiae cells (5) and analyzed for CTP by high performance liquid chromatography (43).
Analyses of Data
Kinetic data were analyzed according to the Michaelis-Menten equation using the EZ-FIT enzyme kinetic model-fitting program (68). Statistical analyses were performed with SigmaPlot 7 software. P values < 0.05 were taken as a significant difference.
RESULTS
Phosphorylation of E. coli-expressed Human CTP Synthetase 1 by Protein Kinase C
Analysis of human CTP synthetase 1 with the NetPhosK 2.0 server (69) indicated that the enzyme has putative protein kinase C target sites at three serine (Ser225, Ser316, and Ser462) and three threonine (Thr83, Thr378, and Thr455) residues with a relatively high score of prediction (Fig. 1). To examine the hypothesis that human CTP synthetase 1 is a substrate for mammalian protein kinase C, we utilized a homogeneous preparation of the enzyme that was expressed and purified from E. coli (52). The purified enzyme was incubated with mammalian protein kinase C in the presence of [γ-32P]ATP, and the phosphorylation of human CTP synthetase 1 was monitored by the incorporation of the radioactive γ phosphate into the protein. Phosphorimaging analysis of a polyvinylidene difluoride membrane containing the reaction product showed that human CTP synthetase 1 was a substrate for protein kinase C (Fig. 2A). Phosphoamino acid analysis of the 32P-labeled protein showed that protein kinase C phosphorylated human CTP synthetase 1 at serine and threonine residues (Fig. 2B). The relative proportion of phosphoserine to phosphothreonine was 2:1.
FIGURE 2. Phosphorylation and phosphoamino acid analysis of E. coli-expressed human CTP synthetase 1 phosphorylated by protein kinase C.
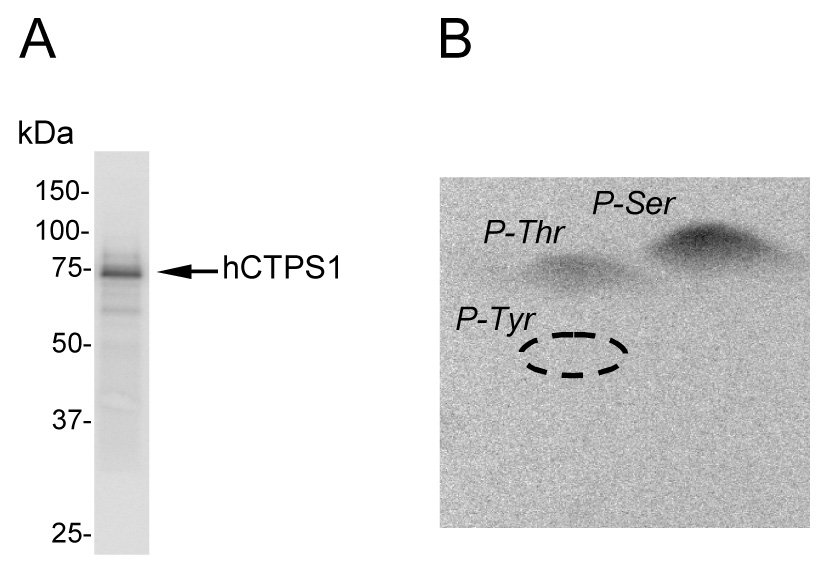
A, human CTP synthetase 1 (50 µg/ml) was phosphorylated with mammalian protein kinase C (0.5 unit/ml) and 50 µM [γ-32P]ATP (11,000 cpm/pmol) for 20 min followed by SDS-PAGE, transfer to polyvinylidene difluoride membrane, and phosphorimaging analysis. B, the PVDF membrane slice containing 32P-labeled enzyme was hydrolyzed with 6N HCl for 90 min at 110 °C, and the hydrolysate was separated by 2-dimensional electrophoresis. The positions of the standard phosphoamino acids phosphoserine (P-Ser), phosphothreonine (P-Thr), and phosphotyrosine (P-Tyr) are indicated in the figure. The data shown are representative of three independent experiments.
The phosphorylation of human CTP synthetase 1 by protein kinase C was examined in more detail. Mammalian protein kinase C activity was dependent on the time of the reaction (Fig. 3A), and on the concentration of the kinase (Fig. 3B). In addition, the dependence of protein kinase C activity on the concentrations of ATP (Fig. 4A) and human CTP synthetase 1 (Fig. 4B) followed saturation kinetics. Analyses of the data according to the Michaelis-Menten equation yielded Km values for ATP and human CTP synthetase 1 of 2 µM and 30 µg/ml, respectively. The stoichiometry of the phosphorylation was determined by calculating the amount of radioactive phosphate incorporated into the enzyme after the reaction was carried out to completion. At the point of maximum phosphorylation, protein kinase C catalyzed the incorporation of 0.2 mol of phosphate per mol of native CTP synthetase 1.
FIGURE 3. Time- and dose-dependent phosphorylation of E. coli-expressed human CTP synthetase 1 by protein kinase C.
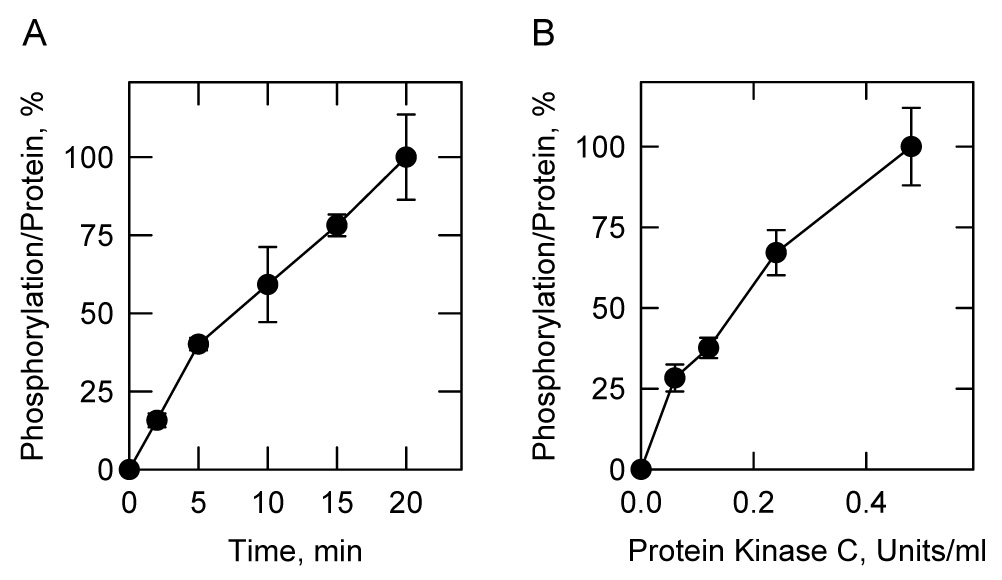
A, human CTP synthetase 1 (50 µg/ml) was incubated with mammalian protein kinase C (0.5 unit/ml) and 50 µM [γ-32P]ATP (11,000 cpm/pmol) for indicated time intervals. B. human CTP synthetase 1 (50 µg/ml) was incubated with indicated concentrations of mammalian protein kinase C and 50 µM [γ-32P]ATP (11,000 cpm/pmol) for 10 min. After the phosphorylation reactions, the samples were subjected to SDS-PAGE. The SDS-polyacrylamide gels were dried and the phosphorylated proteins were subjected to phosphorimaging analysis. The relative amounts of phosphate incorporated into CTP synthetase 1 were quantified using ImageQuant software. Each data point represents the average of duplicate determinations.
FIGURE 4. Dependence of protein kinase C activity on the concentrations of ATP and E. coli-expressed human CTP synthetase 1.

A, mammalian protein kinase C (0.5 unit/ml) was incubated with human CTP synthetase 1 (50 µg/ml) and the indicated concentrations of [γ-32P]ATP (11,000 cpm/pmol) for 10 min. B, mammalian protein kinase C (0.5 unit/ml) was incubated with 50 µM [γ-32P]ATP (11,000 cpm/pmol) and the indicated concentrations of human CTP synthetase 1 for 10 min. After the phosphorylation reactions, the samples were subjected to SDS-PAGE. The SDS-polyacrylamide gels were dried and the phosphorylated proteins were subjected to phosphorimaging analysis. The relative amounts of phosphate incorporated into CTP synthetase 1 were quantified using ImageQuant software. Each data point represents the average of duplicate determinations.
The effect of protein kinase C phosphorylation on human CTP synthetase 1 activity was examined. As described previously (52), the E. coli-expressed and purified enzyme had a very low level (2.5 nmol/min/mg) of CTP synthetase 1 activity (Fig. 5). The phosphorylation of the enzyme with protein kinase C resulted in a time-dependent stimulation of CTP synthetase 1 activity (Fig. 5). After a 20-min incubation period with protein kinase C, the activity of the synthetase was stimulated 95-fold.
FIGURE 5. Effect of protein kinase C phosphorylation on E. coli-expressed human CTP synthetase 1 activity.
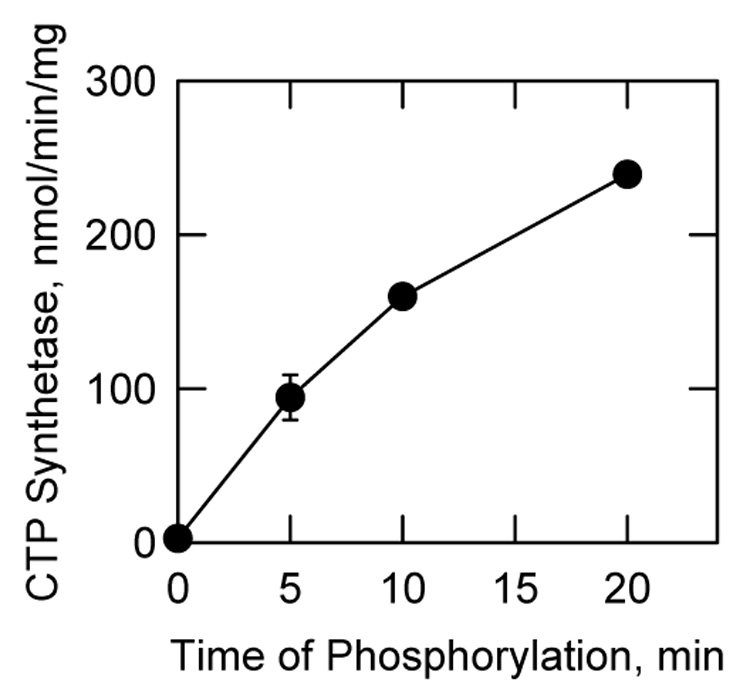
Human CTP synthetase 1 (160 µg/ml) was phosphorylated with mammalian protein kinase C (0.5 unit/ml) for the indicated time intervals using unlabeled ATP. Following the phosphorylations, samples were diluted 4-fold into the standard CTP synthetase 1 reaction mixture and activity was measured by following the conversion of UTP to CTP at 291 nm using a recording spectrophotometer. Each data point represents the average of triplicate determinations ± S.D.
Phosphorylation of S. cerevisiae-expressed Human CTP Synthetase 1 by Protein Kinase C
Previous work has shown that the human CTP synthetase 1 enzyme is functionally expressed in the S. cerevisiae ura7Δ ura8Δ double mutant that lacks yeast CTP synthetase activity (52). The enzyme expressed in yeast is phosphorylated on serine and threonine residues, and some of this phosphorylation has been attributed to protein kinase A (52, 53). We questioned whether human CTP synthetase 1 was phosphorylated in yeast by protein kinase C. To address this question, human CTP synthetase 1 was expressed in a S. cerevisiae ura7Δ ura8Δ double mutant that also expressed the galactose-inducible activated PKC1R398A allele encoding yeast protein kinase C. The extent of enzyme phosphorylation was determined by 32P-labeling following the induction of yeast protein kinase C. The amount of 32Pi incorporated into human CTP synthetase 1 in yeast cells expressing the activated PKC1R398A allele was 50% greater when compared with the control cells that did not express the activated allele (Fig. 6A).
FIGURE 6. Protein kinase C phosphorylation of S. cerevisiae-expressed human CTP synthetase 1.

A, Cultures (50 ml) of S. cerevisiae ura7Δ ura8Δ mutant cells containing at plasmids for the expression of human CTP synthetase 1 (pDO105-hCTPS1) and for the galactose-inducible expression of yeast protein kinase C (pGAL1::PKC1(R398A::HA)) (strain YCY4) were grown to the exponential phase of growth in synthetic complete medium with 2% glucose. The cells were then washed, resuspended in synthetic complete medium with 2% raffinose, and incubated overnight to deplete internal stores of glucose. The cells were then collected by centrifugation, resuspended in 2 ml of the same growth medium, and labeled with 32Pi (0.25 mCi/ml) for 1.5 h. The expression of yeast protein kinase C was induced by the addition of 2% galactose. Following a 1.5-h induction period, human CTP synthetase 1 was isolated from cell extracts with Ni2+-NTA resin, subjected to SDS-PAGE, and transferred to the polyvinylidene difluoride membrane. The 32P-labeled human CTP synthetase 1 proteins were visualized by phosphorimaging analysis, and their relative intensity was quantified using ImageQuant software. The control contains plasmid pBM743, which lacks the galactose-inducible PKC1(R398A::HA) allele (strain YCY3). The extent of CTP synthetase 1 phosphorylation in cells lacking the plasmid that directed protein kinase C activated expression was set at 100%. The data were normalized to the amount of human CTP synthetase 1 proteins as determined by immunoblot analysis using anti-His6 antibodies. The induction of yeast protein kinase C was confirmed by immunoblot analysis using anti-HA antibodies. Each data points represents the average of three experiments ± S.D. B, the human CTP synthetase 1 that was expressed and purified from S. cerevisiae ura7Δ ura8Δ cells was incubated with [γ- 32P]ATP and the indicated amounts of mammalian protein kinase C for 20 min. After incubations, samples were subjected to SDS-PAGE and then transferred to polyvinylidene difluoride membrane. The membrane was subjected to phosphorimaging analysis, and the relative amounts of phosphate incorporated were quantified using ImageQuant software. The maximum extent of CTP synthetase 1 phosphorylation was set at 100%. The data were normalized to the amount of human CTP synthetase 1 protein as determined by immunoblot analysis using anti-His6 antibodies. The values reported were average of three separate experiments ± S.D. The inset shows the results of a phosphoamino acid analysis of the protein kinase C-phosphorylated enzyme. The positions of the standard phosphoamino acids phosphoserine (P-Ser) and phosphothreonine (P-Thr) are indicated in the figure.
We also examined the protein kinase C phosphorylation of the human CTP synthetase 1 enzyme that was expressed and purified from the S. cerevisiae ura7Δ ura8Δ double mutant. The purified enzyme was a substrate for mammalian protein kinase C in vitro. As described above for the E. coli-expressed enzyme, this phosphorylation occurred on serine and threonine residues, and was dependent on the concentration of protein kinase C (Fig. 6B). The concentration of mammalian protein kinase C that was needed to phosphorylate the S. cerevisiae-expressed human CTP synthetase 1 enzyme was higher than that needed to phosphorylate the E. coli-expressed enzyme. That the enzyme purified from yeast was already phosphorylated in vivo (52) may account for this difference. We did not attempt to dephosphorylate the S. cerevisiae-expressed enzyme prior to its phosphorylation with protein kinase C in vitro.
Identification of Ser462 and Thr455 as Protein Kinase C Phosphorylation Sites in Human CTP Synthetase 1
Peptides containing the six putative protein kinase C phosphorylation sites (Fig. 1) in the human CTP synthetase 1 were synthesized and tested for their ability to serve as substrates for mammalian protein kinase C (Table 3). The peptides S462 and T455, which contained the putative Ser462 and Thr455 phosphorylation sites, respectively, were the best substrates for the kinase. At the concentration of 1 mM, the protein kinase C activity using the peptide S462 was 5.6-fold greater when compared with peptide T455 (Table 3). The dependence of protein kinase C activity on peptide S462 followed Michaelis-Menten kinetics (Fig. 7A). The kinetic analysis of the data yielded Vmax and Km values for peptide S462 of 2,150 nmol/min/mg and 0.27 mM, respectively. The phosphorylation of peptide T455 by protein kinase C was also dependent on the concentration of the peptide (Fig. 7B). However, kinetic constants for peptide T455 could not be determined because the reaction did not reach saturation at the highest concentration of substrate available. Peptide T455 contains a second threonine residue that corresponds to Thr459. To address whether this threonine residue was phosphorylated, the A455 peptide (Thr455 was changed to an alanine) was tested as a substrate (Table 3). That peptide A455 did not serve as a protein kinase C substrate confirmed that Thr455 was the site of phosphorylation in peptide T455.
Table 3.
Protein kinase C activity using human CTP synthetase 1 synthetic peptides
| Peptidea | Sequenceb | Protein kinase C activity |
|---|---|---|
| nmol/min/mg | ||
| T83 | LDIRLTKDNN | NAc |
| S225 | NPLDTSVKEK | 5.63 ± 1.0 |
| S316 | SDSYASVIKA | 26.30 ± 1.4 |
| T378 | FGVRGTEGKI | 0.6 ± 0.1 |
| T455 | LGKRRTLFQT | 271.02 ± 37 |
| A455 | LGKRRALFQT | NA |
| S462 | KNSVMRKLYG | 1518.53 ± 89 |
Protein kinase C activity measured with 1 mM peptide.
Putative phosphorylation site is underlined.
No activity.
FIGURE 7. Phosphorylation of human CTP synthetase 1 synthetic peptides by protein kinase C.
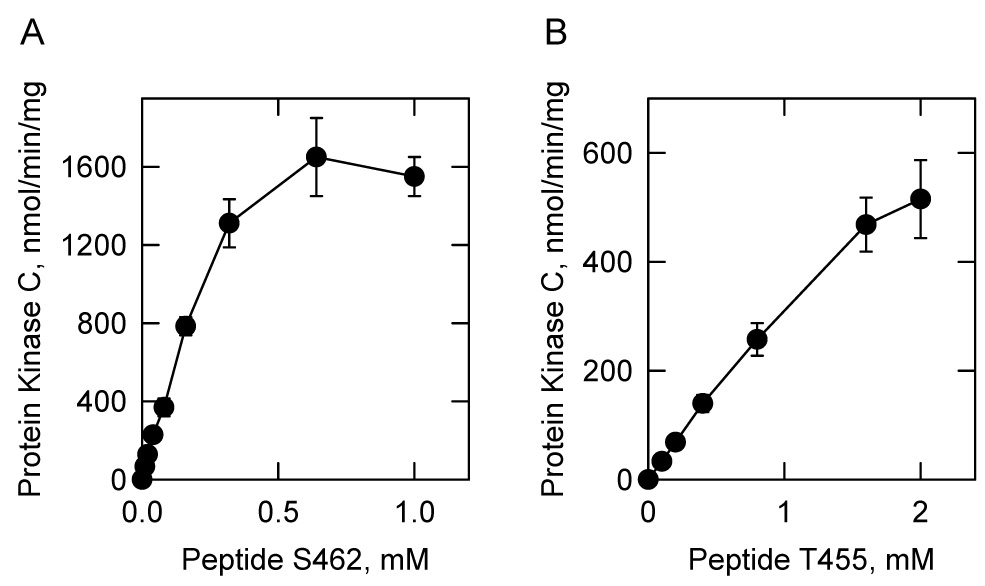
Mammalian protein kinase C activity was measured as a function of the concentration of the indicated synthetic peptides S462 and T455. The values reported were the average of three experiments ± S.D.
The codons for Ser462 and Thr455 within the human CTPS1 gene were changed to alanine codons by site-directed mutagenesis. The mutations were made individually, and in combination to further test the hypothesis that Ser462 and Thr455 might be targets for protein kinase C phosphorylation. His6-tagged versions of the S462A, T455A, and S462A, T455A mutant CTP synthetase 1 enzymes were expressed on a multicopy plasmid in the S. cerevisiae ura7Δ ura8Δ double mutant. Immunoblot analysis using anti-His6 antibodies confirmed the expression of the mutant enzymes. The S462A,T455A mutant enzyme however, was highly susceptible to proteolytic degradation. Cells bearing the S462A and T455A mutant enzymes grew at similar growth rates when compared with cells bearing the wild type human enzyme. However, cells bearing the S462A,T455A double mutation grew at a much slower growth rate.
The wild type and mutant His6-tagged CTP synthetase 1 enzymes were purified from yeast cell extracts using Ni2+-NTA-agarose resin, and the resin bound proteins were incubated with mammalian protein kinase C and [γ-32P]ATP. Following the phosphorylation, the samples were subjected to trypsin digestion and two-dimensional phosphopeptide mapping analysis (Fig. 8). This analysis showed that mammalian protein kinase C phosphorylated the S. cerevisiae-expressed wild type human CTP synthetase 1 on multiple residues. Two major phosphopeptides present in the phosphopeptide map of the wild type enzyme were missing in the map of the S462A mutant enzyme (Fig. 8, indicated by the ellipses). This indicated that Ser462 was contained in these phosphopeptides. Likewise, a major phosphopeptide that was present in the wild type enzyme was absent in the T455A mutant enzyme (Fig. 8, indicated by the square). This indicated that Thr455 was contained in this phosphopeptide. All three phosphopeptides were missing in the phosphopeptide map of the S462A,T455A mutant enzyme (Fig. 8).
FIGURE 8. Effects of the S462A, T455A, and S462A,T455A mutations on the phosphopeptide map of human CTP synthetase 1 phosphorylated by protein kinase C.

Wild type (WT) and the S462A, T455A, and S462A,T455A mutant human CTP synthetase 1 enzymes were expressed and purified from S. cerevisiae ura7Δ ura8Δ cells. The purified enzymes were phosphorylated with mammalian protein kinase C and [γ-32P]ATP for 20 min. After phosphorylation, the samples were subjected to SDS-PAGE and transferred to the polyvinylidene difluoride membrane. The 32P-labeled proteins were digested with L-1-tosylamido-2-phenylethyl chloromethyl ketone-trypsin. The resulting peptides were separated on cellulose thin layer plates by electrophoresis (from left to right) in the first dimension and by chromatography (from bottom to top) in the second dimension. The positions of the phosphopeptides that were absent in the S462A (indicated by an ellipses) and absent in the T455A (indicated by a square) mutant enzymes but were present in the wild type enzyme are indicated in the figure. The data are representative of two independent experiments.
Effects of the S462A and T455A Mutations on the Phosphorylation of Human CTP Synthetase 1 in S. cerevisiae
The effects of the S462A and T455A mutations on the phosphorylation of human CTP synthetase 1 in yeast were examined. For this study, the His6-tagged wild type enzyme and the S462 and T455A mutant enzymes were expressed in S. cerevisiae ura7Δ ura8Δ cells that also expressed the galactose-inducible activated PKC1R398A allele encoding yeast protein kinase C. Following induction, the cells were incubated with 32Pi to label the CTP synthetase 1 enzymes. The phosphorylated enzymes were then isolated from cell extracts with Ni2+-NTA resin, subjected to SDS-PAGE, and transferred to polyvinylidene difluoride membrane. The analysis of the polyvinylidene difluoride membrane by phosphorimaging showed that the extent of phosphorylation of the S462A mutant and T455A mutant enzymes were reduced by 61% and 58%, respectively, when compared with the phosphorylation of the wild type enzyme (Fig. 9A). The identity of the phosphorylated proteins as CTP synthetase 1 enzymes was confirmed by immunoblot analysis using anti-His6 antibodies. The phosphorylation state of the S462A,T455A mutant enzyme appeared to be much less than that of the individual mutant enzymes. However, the extent of phosphorylation could not be determined with accuracy because the protein was susceptible to proteolytic degradation.
FIGURE 9. Effects of the S462A and T455A mutations on the phosphorylation of human CTP synthetase 1 in vivo, and the effect of the S462A mutation on CTP synthetase 1 activity in vitro.
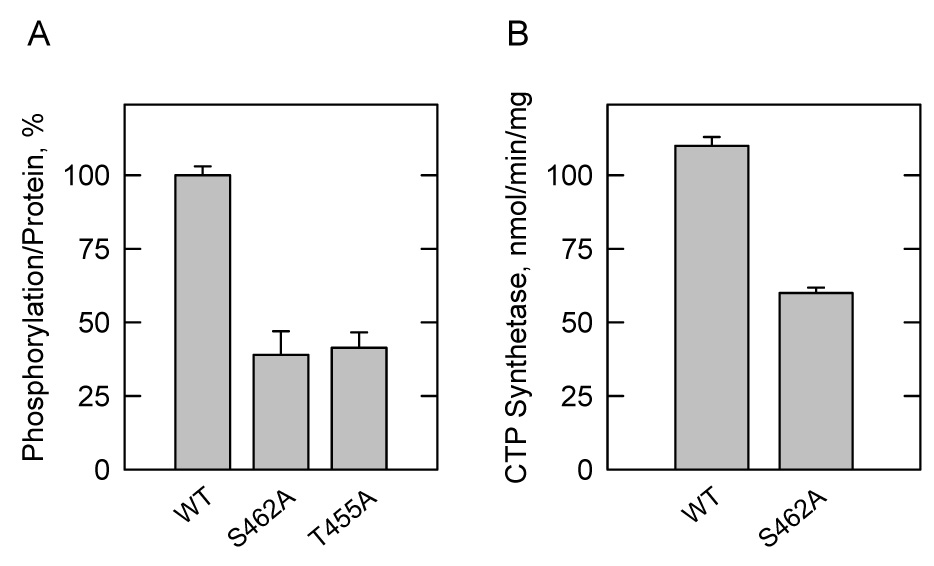
A, Cultures (50 ml) of S. cerevisiae ura7Δ ura8Δ cells containing plasmids for the expression of wild type (WT) (pDO105-hCTPS1) (strain YCY4), S462A mutant (pDO105-hCTPS1(S462A)) (strain YCY5), and T455A mutant (pDO105-hCTPS1(T455A)) (strain YCY6) human CTP synthetase 1 enzymes and for the galactose-inducible expression of yeast protein kinase C (pGAL1::PKC1(R398A::HA)) were grown and labeled with 32Pi (0.25 mCi/ml) as described in the legend to Fig. 6B. The wild type and mutant enzymes were isolated from cell extracts with Ni2+-NTA resin, subjected to SDS-PAGE, and transferred to the polyvinylidene difluoride membrane. The 32P-labeled human CTP synthetase 1 proteins were visualized by phosphorimaging analysis, and their relative intensity was quantified using ImageQuant software. The maximum extent of wild type CTP synthetase 1 phosphorylation was set at 100%. The data were normalized to the amount of human CTP synthetase 1 proteins as determined by immunoblot analysis using anti-His6 antibodies. B, the wild type and S462A mutant human CTP synthetase 1 enzymes were expressed and purified from S. cerevisiae ura7Δ ura8Δ cells. CTP synthetase 1 activity was measured by following the conversion of UTP to CTP at 291 nm using a recording spectrophotometer. The values reported were the average of three experiments ± S.D.
Effects of the S462A Mutation on CTP Synthetase 1 Activity
The effect of the S462A mutation on purified human CTP synthetase 1 activity was examined. The specific activity of the S462A mutant enzyme was 46% lower than the specific activity of the purified wild type enzyme (Fig. 9B). This result supported the conclusion that phosphorylation at Ser462 by protein kinase C contributed to the stimulation of CTP synthetase 1 activity. We questioned whether the reduced level of CTP synthetase 1 activity exhibited by the S462A mutant enzyme correlated with a change in the cellular concentration of CTP in S. cerevisiae. The S462A mutant enzyme was expressed in S. cerevisiae ura7Δ ura8Δ cells; nucleotides were extracted from exponential phase cells and analyzed for CTP by high performance liquid chromatography. We did not detect a significant difference in the CTP concentration of cells expressing the S462A mutant enzyme when compared with cells expressing the wild type enzyme. It was difficult to obtain reproducible results for the CTP concentration of S. cerevisiae ura7Δ ura8Δ cells expressing the S462A,T455A double mutant enzyme because the cells were difficult to grow.
DISCUSSION
CTP synthetase is an essential enzyme because it catalyzes the formation of the nucleotide precursor CTP for the synthesis of nucleic acids and membrane phospholipids (1). Studies on the regulation of human CTP synthetase are important because the misregulation of the enzyme is a common characteristic of several forms of cancer (11–16). Phosphorylation is a common mechanism by which enzymes are regulated, and CTP synthetase activity is regulated by this mechanism (49–51, 53). In this work, we initiated studies on the phosphorylation of human CTP synthetase 1 by protein kinase C. The specificity of mammalian protein kinase C phosphorylation of human CTP synthetase 1 was examined using enzyme that was expressed and purified from E. coli. Using the E. coli-expressed enzyme as a substrate, protein kinase C activity was time- and dose-dependent, and dependent on the concentrations of ATP and the human CTP synthetase 1 protein. Phosphoamino acid and phosphopeptide mapping analyses showed that mammalian protein kinase C phosphorylated CTP synthetase 1 on multiple serine and threonine residues.
The E. coli-expressed human CTP synthetase 1 had negligible activity when measured in vitro (52). This has been attributed to a lack of post-translational modifications in E. coli that are required for maximum expression of CTP synthetase 1 activity (52). Upon phosphorylation with mammalian protein kinase C, CTP synthetase 1 activity increased by 95-fold. This indicated that protein kinase C phosphorylation was an important posttranslational modification for the expression of activity. CTP synthetase 1 is also phosphorylated by protein kinase A (52, 53). However, the consequence of this phosphorylation is the inhibition of activity (53). Thus, the effects of phosphorylation are complex with protein kinases A and C having opposite effects on the regulation of CTP synthetase 1 activity.
We sought evidence that the phosphorylation of CTP synthetase 1 by protein kinase C occurred in vivo. To address this question, the human enzyme was expressed in the S. cerevisiae ura7Δ ura8Δ mutant that also expressed a galactose-inducible activated PKC1R398A allele encoding yeast protein kinase C. The yeast protein kinase C has substrate specificity similar to that of the α, β, and γ isoforms of the conventional mammalian protein kinase C (61, 70). As described previously (52, 53), human CTP synthetase 1 was phosphorylated in the S. cerevisiae ura7Δ ura8Δ mutant. The induction of the yeast protein kinase C enzyme resulted in a 50% increase for CTP synthetase 1 phosphorylation. In addition, the purified S. cerevisiae-expressed human CTP synthetase 1 was a substrate for mammalian protein kinase C.
Through a computer analysis, we identified six potential protein kinase C target sites that have a relatively high score of prediction (Fig. 1). Synthetic peptides with sequences for these sites were tested as substrates for mammalian protein kinase C. The peptides containing a protein kinase C phosphorylation motif at Ser462 (peptide S462) and Thr455 (peptide T455) were the best substrates. The preference for peptide S462 over peptide T455 mimicked the preference for serine phosphorylation over threonine phosphorylation when the purified E. coli-expressed and purified S. cerevisiae-expressed CTP synthetase 1 enzymes were tested as substrates for mammalian protein kinase C. This assay provided confidence that Ser462 and Thr455 might be bona fide sites for protein kinase C phosphorylation. To provide support for this hypothesis, S462A and T455A mutant CTP synthetase 1 enzymes were expressed and purified from S. cerevisiae ura7Δ ura8Δ cells, and then tested as substrates for mammalian protein kinase C. Phosphopeptide-mapping analysis of the protein kinase C-phosphorylated enzymes showed that two phosphopeptides present in the wild type enzyme were missing in the S462A mutant enzyme, and that a distinct phosphopeptide present in the wild type enzyme was missing in the T455A mutant enzyme. These results provided strong evidence that Ser462 and Thr455 were sites for mammalian protein kinase C phosphorylation. Moreover, the S462A and T455A mutations resulted in decreases (61% and 58%, respectively) in the extent of CTP synthetase 1 phosphorylation that occurred in S. cerevisiae ura7Δ ura8Δ cells when compared with the phosphorylation of the wild type enzyme. The phosphopeptide mapping experiments indicated that protein kinase C phosphorylated the enzyme on sites other than Ser462 and Thr455. However, the identification of those sites will require additional studies.
The S. cerevisiae-expressed and purified S462A mutant enzyme exhibited reduced CTP synthetase 1 activity. Thus, phosphorylation at Ser462 contributed to the stimulation of enzyme activity. This interpretation correlated with the stimulation of CTP synthetase 1 activity upon protein kinase C phosphorylation in vitro. In contrast, the T455A mutation results in an increase in purified CTP synthetase 1 activity, and an increase in the cellular content of CTP in the S. cerevisiae ura7Δ ura8Δ mutant (53). This effect has been attributed to the lack of phosphorylation of Thr455 by protein kinase A (53). As shown in this work, the effect of the T455A mutation on activity may also be attributed to the lack of phosphorylation of Thr455 by protein kinase C. Taken together, these results indicated that phosphorylation at Thr455 by protein kinase A or by protein kinase C contributed to the inhibition of CTP synthetase 1 activity. It is not too surprising that Thr455 was phosphorylated by protein kinases A and C. The major protein kinase A target site (i.e., Ser424) in the S. cerevisiae URA7-encoded CTP synthetase enzyme is also phosphorylated by protein kinase C (71, 72). Protein kinase A does not utilize peptide S462 as a substrate nor does the S462A mutation affect the protein kinase A phosphorylation of human CTP synthetase 12. Thus, Ser462 is not a target site for protein kinase A phosphorylation.
Clearly, the phosphorylation of CTP synthetase 1 by protein kinase C is complex because the effect of phosphorylation on activity depends on which site is phosphorylated (i.e., Ser462 versus Thr455). The opposing effects of the S462A and T455A mutations on CTP synthetase 1 activity may account for the lack of a significant effect of the S462A mutation on the cellular concentration of CTP in yeast. The phosphorylation of Thr455 by protein kinase A or by protein kinase C in the S462A mutant enzyme might balance any negative effect the S462A mutation might have on the cellular CTP concentration. We were unable to assess the effects of the S462A,T455A double mutant enzyme on the cellular levels of CTP. S. cerevisiae ura7β ura8β cells expressing the mutant enzyme grew slowly, and the mutant enzyme was highly susceptible to protein degradation during the purification. Whether the S462A,T455A mutant enzyme was less stable in vivo was unclear, and additional studies are required to address this issue. Nonetheless, these results indicated that phosphorylation at Ser462 and Thr455 are important for CTP synthetase 1 function and cell growth.
Protein kinase C is a transducer of second messengers (e.g., diacylglycerol and calcium) that are generated by receptor-mediated hydrolysis of membrane phospholipids (e.g., phosphatidylinositol 4,5-bisphosphate and phosphatidylcholine) (73–75). The enzyme is also the receptor for phorbol ester and other tumor promoters (73–75). Protein kinase C activity plays a central role in the regulation of a myriad of cellular functions (e.g., cell growth and proliferation) through its activation by growth factors, hormones, and other agonists (73–75). While there is a great deal known about the structure, function, and regulation of protein kinase C (73–75), there is much less known about the targets of its phosphorylation. The work reported here identifies a very important human target of protein kinase C phosphorylation, and indicates a mechanism by which lipid signal transduction is linked to CTP synthetase 1 regulation and cell growth. The knowledge that protein kinase C phosphorylates CTP synthetase 1 at Ser462 and at Thr455 will facilitate studies to examine the consequences of protein kinase C phosphorylation of the enzyme in human cells.
Acknowledgments
We are grateful to David E. Levin for providing us with the strains and plasmids for the expression of yeast protein kinase C. We also thank Mal-Gi Choi, Gil-Soo Han, and Avula Sreenivas for helpful discussions during the course of this work.
Footnotes
This work was supported in part by United States Public Health Service, National Institutes of Health Grants GM-50679 (to G.M.C.) and GM-63109 (to E.P.B).
M.-G. Choi and G.M. Carman, unpublished data.
REFERENCES
- 1.Stryer L. Biochemistry. Fourth Ed. New York: W. H. Freeman and Company; 1995. [Google Scholar]
- 2.Aronow B, Ullman B. J. Biol. Chem. 1987;262:5106–5112. [PubMed] [Google Scholar]
- 3.Robert de Saint Vincent B, Buttin G. Biochim. Biophys.Acta. 1980;610:352–359. [Google Scholar]
- 4.Meuth M, L'Heureux-Huard N, Trudel M. Proc.Nat.Acad.Sci.USA. 1979;76:6505–6509. doi: 10.1073/pnas.76.12.6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ozier-Kalogeropoulos O, Fasiolo F, Adeline M-T, Collin J, Lacroute F. Mol.Gen.Genet. 1991;231:7–16. doi: 10.1007/BF00293815. [DOI] [PubMed] [Google Scholar]
- 6.Ozier-Kalogeropoulos O, Adeline M-T, Yang WL, Carman GM, Lacroute F. Mol.Gen.Genet. 1994;242:431–439. doi: 10.1007/BF00281793. [DOI] [PubMed] [Google Scholar]
- 7.Yang W-L, McDonough VM, Ozier-Kalogeropoulos O, Adeline M-T, Flocco MT, Carman GM. Biochemistry. 1994;33:10785–10793. doi: 10.1021/bi00201a028. [DOI] [PubMed] [Google Scholar]
- 8.Ostrander DB, O'Brien DJ, Gorman JA, Carman GM. J.Biol.Chem. 1998;273:18992–19001. doi: 10.1074/jbc.273.30.18992. [DOI] [PubMed] [Google Scholar]
- 9.Hatch GM, McClarty G. J.Biol.Chem. 1996;271:25810–25816. doi: 10.1074/jbc.271.42.25810. [DOI] [PubMed] [Google Scholar]
- 10.McDonough VM, Buxeda RJ, Bruno MEC, Ozier-Kalogeropoulos O, Adeline MT, McMaster CR, Bell RM, Carman GM. J.Biol.Chem. 1995;270:18774–18780. doi: 10.1074/jbc.270.32.18774. [DOI] [PubMed] [Google Scholar]
- 11.van den Berg AA, van Lenthe H, Busch S, de Korte D, Roos D, van Kuilenburg ABP, van Gennip AH. Eur.J.Biochem. 1993;216:161–167. doi: 10.1111/j.1432-1033.1993.tb18128.x. [DOI] [PubMed] [Google Scholar]
- 12.van den Berg AA, van Lenthe H, Kipp JB, de Korte D, Van Kuilenburg AB, van Gennip AH. Eur J Cancer. 1995;31A:108–112. doi: 10.1016/0959-8049(94)00442-8. [DOI] [PubMed] [Google Scholar]
- 13.Verschuur AC, van Gennip AH, Muller EJ, Voute PA, Van Kuilenburg AB. Adv Exp Med Biol. 1998;431:667–671. doi: 10.1007/978-1-4615-5381-6_129. [DOI] [PubMed] [Google Scholar]
- 14.Kizaki H, Williams JC, Morris HP, Weber G. Cancer Res. 1980;40:3921–3927. [PubMed] [Google Scholar]
- 15.Weber G, Lui MS, Takeda E, Denton JE. Life Sci. 1980;27:793–799. doi: 10.1016/0024-3205(80)90333-1. [DOI] [PubMed] [Google Scholar]
- 16.Weber G, Olah E, Lui MS, Tzeng D. Adv.Enzyme Regul. 1979;17:1–21. doi: 10.1016/0065-2571(79)90005-0. [DOI] [PubMed] [Google Scholar]
- 17.Hindenburg AA, Taub RN, Grant S, Chang G, Baker MA. Cancer Res. 1985;45:3048–3052. [PubMed] [Google Scholar]
- 18.Kang GJ, Cooney DA, Moyer JD, Kelley JA, Kim H-Y, Marquez VE, Johns DG. J.Biol.Chem. 1989;264:713–718. [PubMed] [Google Scholar]
- 19.Politi PM, Xie F, Dahut W, Ford H, Jr., Kelley JA, Bastian A, Setser A, Allegra CJ, Chen AP, Hamilton JM. Cancer Chemother.Pharmacol. 1995;36:513–523. doi: 10.1007/BF00685802. [DOI] [PubMed] [Google Scholar]
- 20.Zhang H, Cooney DA, Zhang MH, Ahluwalia G, Ford H, Jr., Johns DG. Cancer Res. 1993;53:5714–5720. [PubMed] [Google Scholar]
- 21.Verschuur AC, van Gennip AH, Leen R, Meinsma R, Voute PA, van Kuilenburg ABP. Br.J.Haematol. 2000;110:161–169. doi: 10.1046/j.1365-2141.2000.02136.x. [DOI] [PubMed] [Google Scholar]
- 22.Verschuur AC, van Gennip AH, Leen R, Muller EJ, Elzinga L, Voute PA, Van Kuilenburg AB. Eur.J.Cancer. 2000;36:627–635. doi: 10.1016/s0959-8049(00)00021-6. [DOI] [PubMed] [Google Scholar]
- 23.Hofer A, Steverding D, Chabes A, Brun R, Thelander L. Proc.Natl.Acad.Sci.U.S.A. 2001;98:6412–6416. doi: 10.1073/pnas.111139498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fijolek A, Hofer A, Thelander L. J Biol.Chem. 2007;282 doi: 10.1074/jbc.M611580200. in press. [DOI] [PubMed] [Google Scholar]
- 25.Dereuddre-Bosquet N, Roy B, Routledge K, Clayette P, Foucault G, Lepoivre M. Antiviral Res. 2004;61:67–70. doi: 10.1016/j.antiviral.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 26.Long CW, Pardee AB. J.Biol.Chem. 1967;242:4715–4721. [PubMed] [Google Scholar]
- 27.Anderson PM. Biochemistry. 1983;22:3285–3292. doi: 10.1021/bi00282a038. [DOI] [PubMed] [Google Scholar]
- 28.Wadskov-Hansen SL, Willemoes M, Martinussen J, Hammer K, Neuhard J, Larsen S. J.Biol.Chem. 2001;276:38002–38009. doi: 10.1074/jbc.M100531200. [DOI] [PubMed] [Google Scholar]
- 29.Nadkarni AK, McDonough VM, Yang W-L, Stukey JE, Ozier-Kalogeropoulos O, Carman GM. J.Biol.Chem. 1995;270:24982–24988. doi: 10.1074/jbc.270.42.24982. [DOI] [PubMed] [Google Scholar]
- 30.Thomas PE, Lamb BJ, Chu EHY. Biochim.Biophys.Acta. 1988;953:334–344. doi: 10.1016/0167-4838(88)90042-8. [DOI] [PubMed] [Google Scholar]
- 31.Endrizzi JA, Kim H, Anderson PM, Baldwin EP. Biochemistry. 2004;43:6447–6463. doi: 10.1021/bi0496945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goto M, Omi R, Nakagawa N, Miyahara I, Hirotsu K. Structure.(Camb.) 2004;12:1413–1423. doi: 10.1016/j.str.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 33.Kursula P, Flodin S, Ehn M, Hammarstrom M, Schuler H, Nordlund P, Stenmark P. Acta Crystallograph.Sect.F.Struct.Biol.Cryst.Commun. 2006;62:613–617. doi: 10.1107/S1744309106018136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liberman I. J.Biol.Chem. 1956;222:765–775. [PubMed] [Google Scholar]
- 35.Levitzki A, Koshland DE., Jr. Biochemistry. 1972;11:241–246. doi: 10.1021/bi00752a015. [DOI] [PubMed] [Google Scholar]
- 36.Levitzki A, Koshland DE., Jr. Proc.Nat.Acad.Sci.USA. 1969;62:1121–1128. doi: 10.1073/pnas.62.4.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levitzki A, Koshland DE., Jr. Biochemistry. 1971;10:3365–3371. doi: 10.1021/bi00794a008. [DOI] [PubMed] [Google Scholar]
- 38.Lewis DA, Villafranca JJ. Biochemistry. 1989;28:8454–8459. doi: 10.1021/bi00447a027. [DOI] [PubMed] [Google Scholar]
- 39.von der Saal W, Anderson PM, Villafranca JJ. J.Biol.Chem. 1985;260:14993–14997. [PubMed] [Google Scholar]
- 40.Levitzki A, Koshland DE., Jr. Biochemistry. 1972;11:247–252. doi: 10.1021/bi00752a016. [DOI] [PubMed] [Google Scholar]
- 41.Pappas A, Yang W-L, Park T-S, Carman GM. J.Biol.Chem. 1998;273:15954–15960. doi: 10.1074/jbc.273.26.15954. [DOI] [PubMed] [Google Scholar]
- 42.Endrizzi JA, Kim H, Anderson PM, Baldwin EP. Biochemistry. 2005;44:13491–13499. doi: 10.1021/bi051282o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pappas A, Park T-S, Carman GM. Biochemistry. 1999;38:16671–16677. doi: 10.1021/bi9920127. [DOI] [PubMed] [Google Scholar]
- 44.Trudel M, van Genechten T, Meuth M. J.Biol.Chem. 1984;259:2355–2359. [PubMed] [Google Scholar]
- 45.Meuth M, Goncalves O, Thom P. Somat.Cell Genet. 1982;8:423–432. doi: 10.1007/BF01538705. [DOI] [PubMed] [Google Scholar]
- 46.Aronow B, Watts T, Lassetter J, Washtien W, Ullman B. J.Biol.Chem. 1984;259:9035–9043. [PubMed] [Google Scholar]
- 47.Kaufman ER. Muta.Res. 1986;161:19–27. doi: 10.1016/0027-5107(86)90096-5. [DOI] [PubMed] [Google Scholar]
- 48.Chu EHY, McLaren JD, Li I-C, Lamb B. Biochem.Genet. 1984;22:701–715. doi: 10.1007/BF00485854. [DOI] [PubMed] [Google Scholar]
- 49.Yang W-L, Carman GM. J.Biol.Chem. 1996;271:28777–28783. doi: 10.1074/jbc.271.46.28777. [DOI] [PubMed] [Google Scholar]
- 50.Yang W-L, Carman GM. J.Biol.Chem. 1995;270:14983–14988. doi: 10.1074/jbc.270.25.14983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang W-L, Bruno MEC, Carman GM. J.Biol.Chem. 1996;271:11113–11119. doi: 10.1074/jbc.271.19.11113. [DOI] [PubMed] [Google Scholar]
- 52.Han G-S, Sreenivas A, Choi MG, Chang YF, Martin SS, Baldwin EP, Carman GM. J Biol.Chem. 2005;280:38328–38336. doi: 10.1074/jbc.M509622200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choi MG, Carman GM. J Biol.Chem. 2007;282:5367–5377. doi: 10.1074/jbc.M610993200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rose MD, Winston F, Heiter P. Methods in Yeast Genetics: A Laboratory Course Manual. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press; 1990. [Google Scholar]
- 55.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning, A Laboratory Manual. 2nd Ed. Cold Spring Harbor, N.Y.: Cols Spring Harbor Laboratory; 1989. [Google Scholar]
- 56.Culbertson MR, Henry SA. Genetics. 1975;80:23–40. doi: 10.1093/genetics/80.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ito H, Yasuki F, Murata K, Kimura A. J.Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schiestl RH, Gietz RD. Curr.Genet. 1989;16:339–346. doi: 10.1007/BF00340712. [DOI] [PubMed] [Google Scholar]
- 59.Innis MA, Gelfand DH. In: PCR Protocols. A Guide to Methods and Applications. Innis MA, Gelfand DH, Sninsky JJ, White TJ, editors. San Diego: Academic Press, Inc.; 1990. pp. 3–12. [Google Scholar]
- 60.Sikorski RS, Boeke JD. Methods Enzymol. 1991;194:302–318. doi: 10.1016/0076-6879(91)94023-6. [DOI] [PubMed] [Google Scholar]
- 61.Watanabe M, Chen C-Y, Levin DE. J.Biol.Chem. 1994;269:16829–16836. [PubMed] [Google Scholar]
- 62.Park T-S, O'Brien DJ, Carman GM. J.Biol.Chem. 2003;278:20785–20794. doi: 10.1074/jbc.M301394200. [DOI] [PubMed] [Google Scholar]
- 63.Laemmli UK. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 64.Haid A, Suissa M. Methods Enzymol. 1983;96:192–205. doi: 10.1016/s0076-6879(83)96017-2. [DOI] [PubMed] [Google Scholar]
- 65.Boyle WJ, Van der Geer P, Hunter T. Methods Enzymol. 1991;201:110–149. doi: 10.1016/0076-6879(91)01013-r. [DOI] [PubMed] [Google Scholar]
- 66.MacDonald JIS, Kent C. J.Biol.Chem. 1994;269:10529–10537. [PubMed] [Google Scholar]
- 67.Bradford MM. Anal.Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 68.Perrella F. Anal.Biochem. 1988;174:437–447. doi: 10.1016/0003-2697(88)90042-5. [DOI] [PubMed] [Google Scholar]
- 69.Blom N, Sicheritz-Ponten T, Gupta R, Gammeltoft S, Brunak S. Proteomics. 2004;4:1633–1649. doi: 10.1002/pmic.200300771. [DOI] [PubMed] [Google Scholar]
- 70.Antonsson B, Montessuit S, Friedli L, Payton MA, Paravicini G. J.Biol.Chem. 1994;269:16821–16828. [PubMed] [Google Scholar]
- 71.Park T-S, Ostrander DB, Pappas A, Carman GM. Biochemistry. 1999;38:8839–8848. doi: 10.1021/bi990784x. [DOI] [PubMed] [Google Scholar]
- 72.Choi M-G, Park TS, Carman GM. J.Biol.Chem. 2003;278:23610–23616. doi: 10.1074/jbc.M303337200. [DOI] [PubMed] [Google Scholar]
- 73.Newton AC. J.Biol.Chem. 1995;270:28495–28498. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 74.Newton AC. Chem.Rev. 2001;101:2353–2364. doi: 10.1021/cr0002801. [DOI] [PubMed] [Google Scholar]
- 75.Mackay HJ, Twelves CJ. Endocr.Relat Cancer. 2003;10:389–396. doi: 10.1677/erc.0.0100389. [DOI] [PubMed] [Google Scholar]
- 76.Gietz RD, Sugino A. Gene. 1988;74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- 77.Johnston M, Davis RW. Mol.Cell Biol. 1984;4:1440–1448. doi: 10.1128/mcb.4.8.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]