

Abstract
We propose a mechanism for the role of the bacterial chaperonin GroEL in folding proteins. The principal assumptions of the mechanism are (i) that many unfolded proteins bind to GroEL because GroEL preferentially binds small unstructured regions of the substrate protein, (ii) that substrate protein within the cavity of GroEL folds by the same kinetic mechanism and rate processes as in bulk solution, (iii) that stable or transient complexes with GroEL during the folding process are defined by a kinetic partitioning between formation and dissociation of the complex and the rate of folding and unfolding of the protein, and (iv) that dissociation from the complex in early stages of folding may lead to aggregation but dissociation at a late stage leads to correct folding. The experimental conditions for refolding may play a role in defining the function of GroEL in the folding pathway. We propose that the role of GroES and MgATP, either binding or hydrolysis, is to regulate the association and dissociation processes rather than affecting the rate of folding.
Keywords: folding intermediates, aggregation, dihydrofolate reductase
The bacterial chaperonin GroEL is probably the best studied of all the proteins that are involved as accessory proteins for correct substrate protein folding. A member of the Hsp60 class of proteins, it is composed of 14 identical subunits arranged as two stacked heptameric rings. As determined from electron microscopy (1) and crystallographic (2, 3) studies, each ring contains a large cavity into which a substrate protein binds and subsequently folds. Even though the crystal structure is known and numerous site-directed mutants have been made (4), there is not much information about the molecular processes involved in GroEL-mediated folding. Thus, a series of questions remain unanswered. (i) Why do so many unfolded proteins bind to GroEL? A survey of the literature (as exemplified by the chaperonin web page†) indicates at least 40 proteins that form stable complexes with GroEL. Many of these proteins are not even from bacterial systems. For example, of those substrate proteins that have been examined most closely, several, like rhodanese and α-lactalbumin, are mammalian proteins whereas others, like barnase, are not from Escherichia coli. (ii) What is the mechanism of protein folding in the presence of GroEL? Many studies examining folding do so on the basis of recovery of enzymatic activity. Although these results show that the efficiency of activity recovery is increased in the presence of GroEL or that stable complexes may form with GroEL, they give little information about the mechanism of the folding process. A few studies (5–7) have examined the kinetics of refolding in the presence and absence of GroEL, but the conditions or proteins used were not always conducive to determining mechanism. (iii) What is the nature of complexes formed between GroEL and substrate protein? Most studies discuss the formation of stable complexes, and although, for example, murine dihydrofolate reductase (DHFR) does form a stable complex, we have shown that (under the same solution conditions) the E. coli DHFR forms only a transient complex with GroEL (8). When stable complexes are formed they may be relatively unstructured or structured depending on which substrate protein is being studied. Recent results (9–11), for example, suggest that a substrate protein in such a complex can be significantly protected against exchange of amide protons when compared with other protein substrates such as cyclophilin (12), barnase (13), or α-lactalbumin (14) that show extensive amide proton exchange when bound to GroEL. (iv) Does GroEL interact with native protein or with some preexisting isoform in equilibrium with the native form? Some controversy exists in the literature with Fersht and coworkers (e.g., ref. 15) suggesting the former mechanism while others propose the latter (e.g., ref. 16).
We have recently been studying the folding of several structurally homologous DHFRs both in the presence and absence of GroEL. DHFR has been used as a model protein for complex formation with the bacterial chaperonin GroEL (17), but an examination of the literature showed that it was the mammalian enzyme, rather than the E. coli enzyme that was used in these studies. Indeed, we found that the E. coli DHFR does not form a stable complex with GroEL at temperatures below 25°C (8). This observation led to investigation of the differences between the E. coli and mammalian forms of DHFR. Superimposition of the E. coli (18) and human (19) DHFR–folate binary complex structures suggested that although most of the structure was similar between the two enzymes, three external loops existed in the mammalian protein that were not present in the E. coli protein. By site-directed mutagenesis, we inserted each of two loops into the E. coli protein (20). This allowed the examination of four structurally similar proteins: the wild-type E. coli DHFR, the murine DHFR (which has essentially the same sequence as the human enzyme), and two mutant proteins, each containing a loop similar to that found in the mammalian protein but not in the E. coli protein. Although our comments may be biased because of the studies with DHFR, the results from these and other studies now allow us to make specific proposals about the mechanism of protein folding in the presence of GroEL.
Promiscuity
As mentioned above, numerous proteins, both prokaryotic and eukaryotic, form stable complexes with GroEL. A seemingly unrelated result may explain this observation. GroEL contains no tryptophan residues (21) yet the usual preparations show significant fluorescence emission typical of tryptophan. This suggests that tryptophan-containing peptides are tightly bound to GroEL and are not removed during protein preparation. We have examined, by electrospray ionization mass spectrometry, the molecular weight distribution of species in a sample of protein that shows substantial fluorescence. Although there are species whose molecular weights are below 350, surprisingly, there are no observed masses between these low molecular weight species and that of the GroEL itself (∼57,200) (22). This result shows the contaminating, presumably tightly bound, material to be very low molecular weight, certainly no larger than tripeptides‡. Although larger peptides (23, 24) and small amounts of numerous proteins (P. Horowitz, personal communication) have been shown to interact with GroEL, the most tightly bound species could be very small and unstructured segments of the substrate protein. Indeed, single amino acids have been shown to bind, although somewhat weakly (25), to GroEL. In this regard, it is of interest to calculate the concentration of a single molecule in the GroEL cavity. From crystallographic studies, the cavity within a single ring-like structure may be about 125,000 Å3 (2). Within a cavity this size, a single molecule may have a concentration of more than 10 mM. We propose, therefore, that the reason GroEL binds to many different proteins is that the binding site(s), which appears to contain many hydrophobic residues (26), recognize unstructured regions, but with some specificity toward hydrophobic residues such as tryptophan. It has been proposed on the basis of site-directed mutagenesis (4) and equilibrium binding studies (27) that hydrophobicity is an important factor in protein binding to GroEL. Many possible intermediates of a single protein substrate may form stable complexes because the apparent concentration within the cavity is so high. Conclusions from such experiments, however, may be misleading. In our studies of structurally homologous DHFRs, it is clear that it is the complex formed just prior to the final folding step that defines complex stability (8).
The Mechanism of Folding
Much of the literature refers to the action of GroEL as assisting or increasing the efficiency of folding and even catalyzing folding. We believe none of these concepts is strictly correct. Our studies on the refolding of the four structurally similar monomeric DHFRs show that the kinetics of the refolding process is the same in the presence or absence of GroEL (8). Thus the large cavity that exists in GroEL must be similar to bulk solution. Of course, since only a few (one to two) molecules are contained within this cavity, the chance of aggregation is slight. Consequently, the chaperonin increases the probability of the protein refolding correctly by decreasing the probability of aggregation that may occur in the bulk solution. The observation with respect to the DHFRs is not an isolated one: similar kinetics with and without GroEL have been observed by others (28). We propose, therefore that this is the general case, i.e., that folding in the cavity of GroEL occurs by the same mechanism as in bulk solution and that it occurs at the same rate until a stable complex is formed. Ellis (29), in describing the concept of the Anfinsen cage, has also suggested that folding within the GroEL cavity is the same as in bulk solution.
The Nature of the Stable Complex
As mentioned above, there are conflicting data in the literature concerning the extent of protein substrate structure that is present in the GroEL–protein complex. Those results indicating a highly structured intermediate are consistent with our observations made with DHFR (8). In this case, since the kinetic behavior in the presence of GroEL is the same as in its absence, it is unlikely that a stable complex is formed early in refolding. That the protein substrate in the stable complex is an intermediate directly preceding the formation of the native structure is suggested both from the kinetic data and from the observation that the same complex is formed when starting with unfolded or native protein (8). In the latter case, the kinetic data show that complex formation is a consequence of GroEL binding not to the native structure itself but to a species that interconverts to the native structure (8). This indicates that at least one conformation or a region within the native structure may be similar to a partially unfolded form. However, in general, the nature of the substrate protein within the stable complex is really a function of a kinetic partitioning between the association and dissociation of the substrate protein with GroEL and the forward and reverse rate constants for formation of a specific intermediate. Thus the stability of a particular complex may be simply a matter of defining a slow step in the folding process as well as the affinity of an intermediate for GroEL. For protein substrates that are oligomeric in solution, but not within the GroEL cavity, the slow step in the formation of an active enzyme may well be the association of competently folded subunits and in such cases any stable complex would be with an intermediate close to the final folded form of the subunit.
Stability of the Complex and What it Means
With these thoughts in mind we need to consider only a portion of the folding process to define the nature of the stable complex. We can write a general scheme as follows.
The unfolded protein, U, may fold through the formation of one or more intermediates prior to forming the native state. In this mechanism the species Ii is the last intermediate state prior to forming the native structure. The species ELI1 is written as if there is only a single molecule of protein substrate in the cavity, but it is certainly possible that the stoichiometry of substrate protein to GroEL may be greater than one. This will not affect the discussion, although the rate constants described in this case may be more complex than indicated in the above scheme. Additionally, the scheme does not include the possibility of conformational changes within GroEL that could affect association or dissociation rates without changing the folding mechanism. According to the scheme, intermediates may dissociate from GroEL at any point along the folding pathway, but if dissociation occurs early, the dissociated intermediates may aggregate, presumably due to exposed hydrophobic regions of the protein, rather than refold to a correct structure. This may be the case for a protein like rhodanese (30) or citrate synthase (31). In any case, the stability of any of the complexes is clearly defined by the relative magnitude of rate constants leading into and out of the complex. Transient formation of the final complex may occur if binding of Ii to GroEL is weak and/or if the formation of the native structure from Ii is fast. This is what is observed for wild-type E. coli DHFR at temperatures below 25°C (8). Indeed, this is what one would want to happen for E. coli proteins under normal growth conditions. For if, under such conditions, a stable complex were formed, the cells would not continue to grow. This, in fact, is the heat shock response: an overproduction of GroE protects the viability of the cell by forming stable complexes with proteins.
The formation of a stable complex can also be defined in terms of these rate constants. If the binding of Ii to GroEL is reasonably tight and/or if the rate constant for the process N → Ii is large, the complex formed will be stable. The finding that many stable complexes are formed with nonbacterial proteins may simply be a question of the relative values of rate constants leading into or out of that complex, as shown in Scheme SI. In this regard, it is of interest that mouse DHFR does not appear to form a stable complex with yeast Hsp60 at 25°C (32), whereas it does with GroEL at the same temperature (8). Wild-type E. coli DHFR does form at stable complex with GroEL as the temperature is raised above 30°C (unpublished data). This temperature dependence is exactly what would be expected for a heat shock response and has to do with the relative values of the rate constants shown in Scheme SI.
Scheme I.
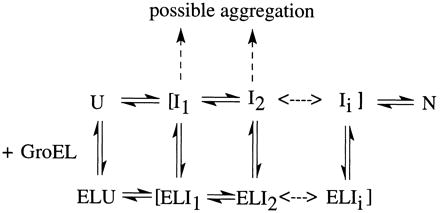
The role of the kinetic partitioning between binding to GroEL and folding rates may play a part in the different results obtained for different protein substrates. Typical refolding experiments are, of course, performed by dilution of the chemical denaturant, either urea or guanidine hydrochloride. The rate of refolding is found to be dependent on the final denaturant concentration relative to the midpoint of the denaturation curve, increasing at lower final denaturant concentrations. On the other hand, the binding or dissociation of the substrate protein (or any intermediate) to GroEL is probably not very sensitive to the final denaturant concentration under refolding conditions. Thus the issue of the nature of the stable complex or the tendency of the substrate protein to aggregate may be a function of the conditions of the procedure used in the refolding experiment.
The Role of GroES and ATP
Some proteins require the presence of GroEL, GroES, and MgATP to fold properly (e.g., refs. 33 and 34) but others may require only MgATP in the presence of GroEL to form the final folded structure (17, 35–37). Since folding occurs within the GroEL cavity obviously neither GroES nor MgATP alone has chaperone activity. Rather, these components serve only to regulate the rate constants involved in the formation or dissociation of the GroEL–substrate protein complexes. This regulation may be quite complex (38–40) involving cooperativity between the two heptameric rings of GroEL and certainly related not only to MgATP binding but also to MgATP hydrolysis (34) as well as capping of the GroEL cavity (either cis or trans to the substrate protein) by GroES (41, 42). Even though formation of the substrate protein–GroEL complex may involve conformational changes within GroEL, the mechanism of the folding process is probably unchanged. We would expect that the observation of a temperature dependence of stable complex formation with wild-type E. coli DHFR reflects what would happen in vivo but that the temperature dependence would be regulated by the presence of GroES and ATP.
Conclusion
We propose that the mechanism and rate constants for folding of a protein are the same in the presence or absence of GroEL. Preferential binding of small unfolded structural units is responsible for the lack of specificity in forming the initial complex with GroEL. At any point along the folding pathway the protein may dissociate from the GroEL, but if this occurs early, aggregation may result. The ability of GroEL to fold proteins correctly without aggregation is a consequence of the stability of the complex formed between GroEL and the protein conformation just prior to the final folding step. The stability of this complex is defined by rate constants involved in complex formation or dissociation compared with the rate constants for folding to or unfolding from the native protein as well as the experimental conditions used for the refolding experiment. Thus, rather than ask whether a given protein binds to GroEL, the relevant question would appear to be at what point in the folding pathway a given protein associates and dissociates from GroEL.
Acknowledgments
This work was supported by National Institutes of Health Grant DK13332.
ABBREVIATION
- DHFR
dihydrofolate reductase
Footnotes
The URL for the GroE chaperonin web page is http://bioc09.uthscsa.edu/~seale/Chap/chap.html.
We note that many of these contaminating species, including essentially all the fluorescent ones, may be removed from GroEL by use of a Reactive Red 120-agarose column (20) and that this is important in the studies described herein with the DHFRs. The Reactive Red column or a similar procedure that removes low molecular weight contaminants should always be used in any quantitative study involving binding of proteins or peptides to GroEL.
References
- 1.Harris J R, Zahn R, Pluckthun A. J Struct Biol. 1995;115:68–77. [Google Scholar]
- 2.Braig K, Otwinowski Z, Hegde R, Boisvert D C, Joachimiak A, Horwich A L, Sigler P B. Nature (London) 1994;371:578–586. doi: 10.1038/371578a0. [DOI] [PubMed] [Google Scholar]
- 3.Hunt J F, Weaver A J, Landry S J, Gierasch L, Deisenhofer J. Nature (London) 1996;379:37–45. doi: 10.1038/379037a0. [DOI] [PubMed] [Google Scholar]
- 4.Fenton W A, Kashi Y, Furtak K, Horwich A L. Nature (London) 1994;371:614–619. doi: 10.1038/371614a0. [DOI] [PubMed] [Google Scholar]
- 5.Katsumata K, Okazaki A, Kuwajima K. J Mol Biol. 1996;258:827–838. doi: 10.1006/jmbi.1996.0290. [DOI] [PubMed] [Google Scholar]
- 6.Ayling A, Baneyx F. Protein Sci. 1996;5:478–487. doi: 10.1002/pro.5560050309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sparrer H, Lilie H, Buchner J. J Mol Biol. 1996;258:74–87. doi: 10.1006/jmbi.1996.0235. [DOI] [PubMed] [Google Scholar]
- 8.Clark A C, Frieden C. J Mol Biol. 1997;268:512–525. doi: 10.1006/jmbi.1997.0969. [DOI] [PubMed] [Google Scholar]
- 9.Gervasoni P, Staudenmann W, James P, Gehrig P, Pluckthun A. Proc Natl Acad Sci USA. 1996;93:12189–12194. doi: 10.1073/pnas.93.22.12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Groβ M, Robinson C V, Mayhew M, Hartl F U, Radford S E. Protein Sci. 1996;5:2506–2513. doi: 10.1002/pro.5560051213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldberg M S, Zhang J, Sonder S, Matthews C R, Fox R O, Horwich A L. Proc Natl Acad Sci USA. 1997;94:1080–1085. doi: 10.1073/pnas.94.4.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zahn R, Spitzfaden C, Ottiger M, Wuthrich K, Pluckthun A. Nature (London) 1994;368:261–265. doi: 10.1038/368261a0. [DOI] [PubMed] [Google Scholar]
- 13.Zahn R, Perrett S, Stenberg G, Fersht A R. Science. 1996;271:642–645. doi: 10.1126/science.271.5249.642. [DOI] [PubMed] [Google Scholar]
- 14.Robinson C V, Groβ M, Eyles S J, Ewbank J J, Mayhew M, Hartl F U, Dobson C M, Radford S E. Nature (London) 1994;372:646–651. doi: 10.1038/372646a0. [DOI] [PubMed] [Google Scholar]
- 15.Corrales F J, Fersht A R. Proc Natl Acad Sci USA. 1995;92:5326–5330. doi: 10.1073/pnas.92.12.5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walter S, Lorimer G H, Schmid F X. Proc Natl Acad Sci USA. 1996;93:9425–9430. doi: 10.1073/pnas.93.18.9425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Viitanen P V, Donaldson G K, Lorimer G H, Lubben T H, Gatenby A A. Biochemistry. 1991;30:9716–9723. doi: 10.1021/bi00104a021. [DOI] [PubMed] [Google Scholar]
- 18.Bolin J T, Filman D J, Matthews D A, Hamlin R C, Kraut J. J Biol Chem. 1982;257:13650–13662. [PubMed] [Google Scholar]
- 19.Davies J D, Delcamp T J, Prendergast N J, Ashford V A, Freisheim J H, Kraut J. Biochemistry. 1990;29:9467–9479. doi: 10.1021/bi00492a021. [DOI] [PubMed] [Google Scholar]
- 20.Clark A C, Hugo E, Frieden C. Biochemistry. 1996;35:5893–5901. doi: 10.1021/bi953051v. [DOI] [PubMed] [Google Scholar]
- 21.Hemmingsen S M, Woolford C, van der Vies S M, Tilly K, Dennis D T, Georgopoulos C P, Hendrix R W, Ellis R J. Nature (London) 1988;333:330–334. doi: 10.1038/333330a0. [DOI] [PubMed] [Google Scholar]
- 22.Clark, A. C., Ramanathan, R. & Frieden, C. (1997) Methods Enzymol., in press. [DOI] [PubMed]
- 23.Landry S J, Jordan R, Mcmacken R, Gierasch L M. Nature (London) 1992;355:455–457. doi: 10.1038/355455a0. [DOI] [PubMed] [Google Scholar]
- 24.Rosenberg H F, Ackerman S J, Tenen D G. J Biol Chem. 1993;268:4499–4503. [PubMed] [Google Scholar]
- 25.Richarme G, Kohiyama M. J Biol Chem. 1994;269:7095–7098. [PubMed] [Google Scholar]
- 26.Fenton W A, Weissman J S, Horwich A L. Chem Biol. 1996;3:157–161. doi: 10.1016/s1074-5521(96)90257-4. [DOI] [PubMed] [Google Scholar]
- 27.Lin Z, Schwartz F P, Eisenstein E. J Biol Chem. 1995;270:1011–1014. doi: 10.1074/jbc.270.3.1011. [DOI] [PubMed] [Google Scholar]
- 28.Laminet A A, Ziegelhoffer T, Georgopoulos C, Pluckthun A. EMBO J. 1990;9:2315–2319. doi: 10.1002/j.1460-2075.1990.tb07403.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ellis R. Folding Design. 1996;1:R9–R15. [PubMed] [Google Scholar]
- 30.Mendoza J A, Rogers E, Lorimer G H, Horowitz P M. J Biol Chem. 1991;266:13044–130449. [PubMed] [Google Scholar]
- 31.Buchner J, Schmidt M, Fuchs M, Jaenicke R, Rudolph R, Schmid F X, Kiefhaber T. Biochemistry. 1991;30:1586–1591. doi: 10.1021/bi00220a020. [DOI] [PubMed] [Google Scholar]
- 32.Rospert S, Looser R, Dubaquie Y, Matouschek A, Glick B S, Schatz G. EMBO J. 1996;15:764–774. [PMC free article] [PubMed] [Google Scholar]
- 33.Martin J, Langer T, Boteva R, Schramel A, Horwich A L, Hartl F U. Nature (London) 1991;352:36–42. doi: 10.1038/352036a0. [DOI] [PubMed] [Google Scholar]
- 34.Goloubinoff P, Christeller J T, Gatenby A A, Lorimer G H. Nature (London) 1989;342:884–889. doi: 10.1038/342884a0. [DOI] [PubMed] [Google Scholar]
- 35.Badcoe I G, Smith C J, Wood S, Halsall D J, Holbrook J J, Lund P, Clarke A R. Biochemistry. 1991;30:9195–9200. doi: 10.1021/bi00102a010. [DOI] [PubMed] [Google Scholar]
- 36.Fisher M T. Biochemistry. 1992;31:3955–3963. doi: 10.1021/bi00131a010. [DOI] [PubMed] [Google Scholar]
- 37.Murai N, Taguchi H, Yoshida M. J Biol Chem. 1995;270:19957–19963. doi: 10.1074/jbc.270.34.19957. [DOI] [PubMed] [Google Scholar]
- 38.Todd M J, Viitanen P V, Lorimer G H. Science. 1994;265:659–666. doi: 10.1126/science.7913555. [DOI] [PubMed] [Google Scholar]
- 39.Yifrach O, Horovitz A. J Mol Biol. 1996;255:356–361. doi: 10.1006/jmbi.1996.0028. [DOI] [PubMed] [Google Scholar]
- 40.Hayer-Hartl M K, Weber F, Hartl F-U. EMBO J. 1996;15:6111–6121. [PMC free article] [PubMed] [Google Scholar]
- 41.Weissman J S, Hohl C M, Kovalenko O, Kashi Y, Chen S X, Braig K, Saibil H R, Fenton W A, Horwich A L. Cell. 1995;83:577–587. doi: 10.1016/0092-8674(95)90098-5. [DOI] [PubMed] [Google Scholar]
- 42.Mayhew M, Dasilva A C R, Martin J, Erdjumentbromage H, Tempst P, Hartl F U. Nature (London) 1996;379:420–426. doi: 10.1038/379420a0. [DOI] [PubMed] [Google Scholar]