

Abstract
Animal fatty acid synthase (FAS; EC 2.3.1.85) is a homodimer of a multifunctional subunit protein and catalyzes the synthesis of palmitate from acetyl-CoA, malonyl-CoA, and NADPH. The subunit (Mr ≈270,000) carries seven distinct component activities and a site for the prosthetic group 4′-phosphopantetheine (acyl carrier protein). Based on proteolytic mapping, the organization of the activity domains along the subunit polypeptide from the N terminus is as follows: β-ketoacyl synthase, acetyl and malonyl transacylases, β-hydroxyacyl dehydratase, enoyl reductase, β-ketoacyl reductase, acyl carrier protein, and thioesterase. By comparing the amino acid sequences of the chicken, rat, and human synthases, we found that kallikrein cleavage sites occur in the least conserved regions of the FAS polypeptide subunit. Determining the amino acid sequences of the N-terminal end of the major kallikrein cleavage peptides helped delineate the most likely boundaries of the component activities in the cDNA-derived amino acid sequence. To confirm this organization, we cloned the chicken FAS cDNA coding for domain I and expressed it in Escherichia coli as a maltose-binding fusion protein. The isolated recombinant protein contained the activities of the acetyl and malonyl transacylases and the β-hydroxyacyl dehydratase. Based on the boundaries of the acetyl and malonyl transacylases and the β-hydroxyacyl dehydratase, we also cloned the appropriate cDNA fragments encoding the domains that contain the transacylases and the dehydratase in pET vectors and expressed them in E. coli as thioredoxin-6xHis fusion proteins. The purified recombinant proteins contained, respectively, the activities of the acetyl and malonyl transacylases and the dehydratase. These results not only confirmed the order of the component activities in domain I, but also paved the way for successful expression and characterization of the remaining activities.
Keywords: cloning subdomains
Animal fatty acid synthase (FAS) is a multifunctional enzyme that catalyzes the synthesis of long-chain fatty acids by using acetyl-CoA as a primer, malonyl-CoA as a two-carbon donor, and NADPH as a reductant for the intermediates (1, 2). We previously determined that the enzyme is a homodimer arranged in a head-to-tail manner such that two catalytic centers are generated for palmitate synthesis (1, 2). Limited proteolytic digestion of FAS with various proteases and the isolation of peptides with active component activities (1–7) revealed that the order of the component activities, starting from the N terminus, are as follows: β-ketoacyl synthase (ketosynthase), acetyl and malonyl transacylases (AT/MT), β-hydroxyacyl dehydratase (dehydratase), enoyl reductase, β-ketoacyl reductase (ketoreductase), acyl carrier protein (ACP), and thioesterase. The cDNA-derived amino acid sequences of the chicken, rat, and human FASs (8–14) further confirmed this order of the partial activities. Most of the component activities of FAS could be readily identified in the cDNA-derived amino acid sequences of animal FASs by locating the already known amino acid sequences at the active sites of the ketosynthase, of AT/MT, and of the thioesterase and at the phosphopantetheine-binding site (ACP) (1) and by identifying the putative nucleotide-binding Gly-X-Gly-X-X-Gly motifs of the enoyl reductase and ketoreductase (8, 10–15).
The amino acid sequences of animal FASs are very similar. Based on the least conserved sequences among animal FASs and comparisons of the sequences of animal FASs with those of bacterial and fungal FASs and polyketide synthases, the putative boundaries of each of the individual activities could be identified (8, 10–15). The unavailability of information concerning the amino acid sequences at the active site of the dehydratase and the lack of discernible motifs of the catalytic function hampered, in the past, the exact identification of the active site of the dehydratase activity. The availability of several bacterial dehydratase sequences and their subsequent comparison revealed the presence of a common motif (H-X-X-X-G-X-X-X-X-P) among the dehydratases. This motif is indeed conserved among animal FASs. Modifying His-878, which is located in this sequence motif in rat FAS, to Ala resulted in the loss of dehydratase function, suggesting that the dehydratase activity is located next to the AT/MT partial domain (16). The presence of an equivalent His-877 in chicken FAS (9) and His-876 in human FAS (10) suggested that the dehydratase activity is located in domain I, assuming that this histidine is located at the active site of the dehydratase (16). To confirm the location of the boundaries of the component activities of FAS that were suggested by comparisons of the amino acid sequences of various animal FASs, it is essential to clone and express appropriate segments of the cDNA sequences in heterologous systems and to purify the proteins containing the specified activity. Previously, we accomplished this with the thioesterase activity of chicken FAS by isolating its partial activity, determining its amino acid sequence, comparing the sequence to that predicted from the chicken FAS cDNA sequence, and thereby defining the span of this region of FAS (3, 4, 8, 17). Similarly, isolating an ACP peptide from the same synthase and determining its amino acid sequence defined the boundaries of this functional unit (8, 18). Moreover, cloning and expressing ACP-thioesterase in Escherichia coli and purifying the recombinant protein that contained thioesterase activity and phosphopantetheine conclusively demonstrated the accuracy of this assignment (19, 20).
Earlier we reported the isolation of a highly purified 26-kDa kallikrein-cleaved peptide from chicken FAS that contained dehydratase activity (7). Here we report on the N-terminal sequence of this peptide and the peptides generated from readily accessible kallikrein cleavage sites (1, 6, 7). The amino-terminal sequences of these peptides confirmed the order of the component activities and delineated the most probable boundaries of these activities. We also describe the cloning and expression in E. coli of the chicken FAS cDNA coding for the domain I region and the purification and characterization of its component activities, the transacylases and the dehydratase.
EXPERIMENTAL PROCEDURES
Materials.
Matrix Gel Red A was obtained from Amicon, and the polyvinylidene difluoride membrane was from Millipore. The expression vector pMALc-2 and amylose resin were obtained from New England Biolabs. The TALON resin was obtained from CLONTECH, and the pET32 vectors and the S-tag Western blot kit were from Novagen. The sources of all other reagents were as described previously (10).
Proteolytic Digestion and Fractionation of Peptides.
FAS was isolated from chicken livers as described (21) and was further purified on a Superose 6 column to a specific activity of 1,300 nmol of NADPH oxidized per min/mg of protein. The purified FAS (35 mg) was dialyzed against buffer A (0.1 M Tris⋅HCl/1 mM EDTA/1 mM DTT), and the protein concentration was adjusted to 4 mg/ml and digested with 1.75 mg of kallikrein at room temperature for 2 h. Proteolysis was stopped by adding benzamidine and phenylmethylsulfonyl fluoride to a final concentration of 2 mM and 10 mM, respectively. The reaction mixture was chilled on ice, and all subsequent procedures were carried out at 0–4°C. The kallikrein digestion products were fractionated by adding solid ammonium sulfate to 40% saturation, and the precipitated protein was separated by centrifugation at 12,000 × g for 20 min and saved. Solid ammonium sulfate was added to the supernatant fluid to a concentration of 90% saturation. The precipitated peptides were collected by centrifugation as described above and dissolved in 3 ml of buffer B (50 mM Tris⋅HCl, pH 7.9/1 mM DTT/1 mM EDTA/50 mM NaCl). The peptides were then loaded onto a Matrix red column (1 × 5 cm), and the flowthrough containing the unbound proteins was collected. The column was washed with 10 column volumes of buffer B, and the bound peptides were eluted with an NaCl gradient (0.1–2.0 M) as described (7). The elution profile showed two distinct peaks; the dehydratase activity eluted in peak 1 and the ketoreductase activity eluted in peak 2. The proteins of these and the flowthrough fractions were precipitated by ammonium sulfate as described before (7), dissolved in buffer A, and separated by electrophoresis in a 4–15% SDS/acrylamide gradient gel. The polypeptides were then transferred electrophoretically to a polyvinylidene difluoride membrane as described (22). The membrane was briefly stained with Coomassie blue, and the protein bands corresponding to the various peptides described by Tsukamoto and Wakil (7) were identified based on their molecular weight. The bands were cut from the membrane and their N-terminal sequences were determined as described earlier (9).
Cloning and Expression of the Chicken FAS cDNA Coding for Domain I.
The restriction map of pL35, a full-length chicken FAS cDNA clone, was constructed from the available cDNA clones (8, 9) by using traditional cloning methods. To clone domain I, we amplified 800 bp of the 5′-terminal chicken FAS cDNA sequence by the polymerase chain reaction (PCR) using appropriate primers. The 5′ primer contained an EcoRI site just upstream of the ATG codon for the convenience of cloning into the pMAL-c2 vector (New England Biolabs). The fragment resulting from PCR was blunt ended with the Klenow fragment of DNA polymerase and cut with EcoRI. This fragment was then cloned at the EcoRI site and at the blunt-ended BamHI site in pMAL-c2 vector. The sequence of the first 500 bp of the insert in the resulting clone, pMAL-c2–720, was confirmed (Fig. 1). The sequence also showed that the chicken FAS coding sequence of the cDNA was in-frame with that of the maltose-binding protein. This clone was then cut with HindIII, and a 2.5-kbp HindIII fragment from pL35 was ligated to it in the proper orientation (Fig. 1). The resulting plasmid, pMAL-c2DI, contained the 1,079 amino acid N-terminal coding sequence of chicken FAS fused to the C terminus of maltose-binding protein (Fig. 1). Since there are no translation termination codons at the 3′ end of this fragment, the reading frame continued in one of the three reading frames of lacZ-α DNA, resulting in the addition of 61 unspecified amino acids at the C terminus. Based on the sequence of this construct, we predicted that expression of pMAL-c2DI in E. coli would result in the production of an ≈165-kDa recombinant protein.
Figure 1.
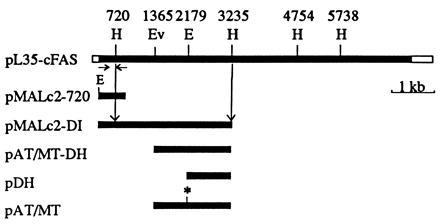
Schematic representation of the construction of plasmids containing various portions of chicken FAS cDNA. Top bar represents the full-length chicken FAS cDNA in pBluescript vector. Relevant restriction sites are indicated as follows: E, EcoRI; Ev, EcoRV; H, HindIII. The span of the coding region being expressed and the sizes of the fusion proteins are also indicated. In pAT/MT, the asterisk denotes the blunt-ended EcoRI site.
Purification of the Chicken FAS Domain I (cFAS-DI).
E. coli strain Top10F′, transformed with the plasmid pMAL-c2DI, was grown and induced as described (23). The cells were harvested, washed once with 10 mM Tris⋅HCl buffer (pH 7.6), which contained 100 mM NaCl and 1 mM EDTA, and resuspended in 200–400 ml of lysis buffer (10 mM Tris⋅HCl, pH 7.6/1 mM EDTA/100 mM NaCl/5 mM 2-mercaptoethanol). The suspension was treated with 8 mg of lysozyme for 10 min on ice with intermittent stirring of the suspension with a glass rod. Lysis was monitored by diluting 0.1 ml of the suspension to 1 ml of lysis buffer and measuring the optical density at 600 nm. The viscous solution was sonicated four times at 60 W for 15 sec, with intermittent chilling on ice for 1 min, to reduce the viscosity by shearing the DNA. The lysate was then centrifuged at 42,000 × g for 30 min. The supernatant fluid was removed and treated with solid ammonium sulfate to 55% saturation. The precipitated protein was separated by centrifugation and dissolved in 50 ml of column buffer (10 mM potassium phosphate, pH 6.8/1 mM EDTA/5 mM 2-mercaptoethanol/200 mM NaCl). The protein solution was passed twice through an amylose resin column (bed volume 50 ml) at a flow rate of 1–2 ml/min. The column was washed with 10 column volumes of column buffer, and the protein was eluted with column buffer containing 10 mM maltose and 5% glycerol. The domain I protein (2–3 mg) was then precipitated with ammonium sulfate at 55% saturation. The precipitated protein was collected by centrifugation, dissolved in 5 ml of column buffer containing 5% glycerol, dialyzed against the same glycerol-containing buffer for 2 h, and stored at −20°C.
Cloning, Expression, and Purification of the AT/MT and Dehydratase Subdomains as Thioredoxin-6xHis Fusion Proteins.
The EcoRV–HindIII fragment [nucleotides 1,365–3,236 (Fig. 1)] coding for the chicken FAS AT/MT and dehydratase sequences was isolated from pMAL-c2DI and ligated with the expression vector pET-32a (Novagen), which had been cut with the same enzymes. This construct (pAT/MT-DH), which expresses the chicken FAS amino acid sequence from residues 455–1,078, yielded a 68-kDa polypeptide that contained the AT/MT and dehydratase activities fused to a 19.5-kDa thioredoxin-S-tag-His-tag sequence at the N terminus and a 1.8-kDa C-terminal vector sequence (Fig. 1). To express the AT/MT component activity independently, pAT/MT-DH was cut with EcoRI (nucleotide 2,268), blunt ended, and recircularized by ligation. This procedure results in a shift of the FAS reading frame to a position past the blunt-ended EcoRI site and in the introduction of a translation termination codon after five unrelated FAS amino acids. This construct (pAT/MT) expressed the 30-kDa AT/MT component fused to a 19.5-kDa vector-encoded sequence as described above. We also constructed a dehydratase expression plasmid by deleting the sequences between EcoRV (nucleotide 1,365) and EcoRI (nucleotide 2,268) in the pAT/MT-DH plasmid and by blunt-end ligating these two ends. The resulting plasmid, pDH (Fig. 1), contained the sequence from nucleotides 2,268 to 3,236, which encodes a 36-kDa peptide that was fused to the vector-coded sequences as described above.
E. coli strain BL21(DE3) (Novagen), which had been transformed with these plasmids, was grown and induced for expression as described above for domain I. The cell suspension was treated with lysozyme, sonicated as described above for domain I, and centrifuged (20,000 × g) for 30 min. The cell-free extracts contained extremely low levels of the recombinant proteins, even though the crude extract obtained before centrifugation showed high levels of expression based on SDS/PAGE analysis. The pellet obtained after centrifugation was suspended in 10–15 ml of 4 M guanidinium-HCl, which contained 200 mM NaCl and 20 mM Tris⋅HCl (pH 8.0), and the viscosity caused by DNA was cleared from the mixture by passing it four times through a 10-ml syringe equipped with an 18-guage needle and centrifuging it at 20,000 × g for 30 min. The clear supernatant fluid was mixed with 5 ml of TALON resin and gently shaken at 0–4°C for 30–60 min. The mixture was then transferred onto a column (3 × 12 cm), and the material that did not bind to the resin was discarded. The column was then washed with 250 ml of 6 M urea containing 200 mM NaCl and 20 mM Tris⋅HCl (pH 8.0). The His-tag protein bound to the resin was processed in one of two ways. In procedure 1, the recombinant protein was eluted from the column with 100 mM imidazole in the same urea buffer. The protein (≈20 ml) was then dialyzed against 2 × 1 liter of phosphate buffer (50 mM phosphate, pH 6.8/1 mM EDTA/5 mM 2-mercaptoethanol) and precipitated with solid ammonium sulfate at 70% saturation. The precipitated protein was collected by centrifugation and dissolved in the dialysis buffer, which contained 5% glycerol. The insoluble material was removed by centrifugation.
In procedure 2, the protein was renatured while still bound to the TALON resin by further washing the urea-washed resin with 250 ml of Tris⋅HCl containing 200 mM NaCl and eluting the protein with imidazole in the same buffer. The eluted protein was then concentrated by using a Centriprep apparatus (Amicon) and simultaneously changing the buffer to buffer C (50 mM phosphate, pH 6.8/1 mM EDTA/5 mM 2-mercaptoethanol/5% glycerol). Beginning with 2 liters of induced culture, these procedures yielded 1–2 mg of protein.
Miscellaneous Procedures.
Recombinant DNA techniques (24), PAGE (7), and DNA and protein sequencing (9) were performed as described. The sequences were compared by using the bestfit program in the package of programs from the Genetics Computer Group.
RESULTS AND DISCUSSION
Determining the Boundaries of the Functional Components of the Chicken FAS.
As reported earlier (7), fractionation of the limited kallikrein digests of chicken FAS with ammonium sulfate and Matrix red chromatography yielded, in the first peak fraction, a dehydratase with a specific activity of about 160 nmol per min/mg, which is about 10 times that of the native FAS (7). The peptides from this fraction and others obtained after ammonium sulfate precipitation were subjected to SDS/PAGE and transferred to a polyvinylidene difluoride membrane (see Experimental Procedures). The major peptides identified by brief staining with Coomassie blue were cut out and their N-terminal sequences were determined. As shown in Table 1, the N-terminal sequence of two of these peptides could not be determined, even though there were sufficient amounts of the proteins available for analyses on the polyvinylidene difluoride membrane based on staining with Coomassie blue. Hence, our conclusion was that these peptides contained the blocked N-terminal residue (N-acetylmethionine) of chicken FAS and represent the N-terminal peptides of the enzyme (9). The N-terminal amino acid sequences of the other protein bands readily matched the amino acid sequence of chicken FAS predicted from the nucleotide sequences of FAS cDNA (8, 9). The locations of the cleavage sites along the derived amino acid sequence of FAS are indicated in the top line of Fig. 2. Based on the sizes and sequences of these peptides (Table 1), we determined the order of the kallikrein cleavage peptides along the FAS subunit as shown in Fig. 2. Based on our previous finding that products of limited kallikrein digestion and those of digestion with other proteases of chicken FAS retained component activities (1–7), we assigned the boundaries of the component activities as indicated in Fig. 2 (bottom line). To determine whether this assignment of boundaries is also valid for rat and human FASs, we compared the amino acid sequences of rat, chicken, and human FASs by using bestfit in the Genetics Computer Group package of programs. The results of this comparison showed that the sequences of rat FAS and human FAS, chicken FAS and rat FAS, and chicken FAS and human FAS have 79%, 78%, and 64% identity, respectively. The peptide segments of FASs with high and low degrees of similarity are shown in Fig. 2; the sites of kallikrein cleavage appear to be targeted to the sequence stretches that have very low similarity. These regions of the chicken FAS subunit that exhibit less similarity (<20%) in amino acid sequences to their counterparts in rat FAS are depicted as thick solid bars in Fig. 2 (top line). These observations are consistent with the presumption that the FAS dimer is a very compact structure and that all the functionally active subdomains are highly structured to carry out their assigned catalytic function. These results also showed that the less conserved sequences are the linker regions between the individual functional units that are outside the compactly folded, catalytically active subcomponents of FAS and, therefore, are readily accessible to proteases. It is noteworthy that the comparison between the sequences of the animal synthases did not highlight the boundaries of the dehydratase or of the enoyl reductase. Based on the analyses of the major kallikrein cleavage sites, however, we were able to divide the amino acid sequence of the chicken FAS subunit into four domains as follows: domain I (residues 1–980) containing the ketosynthase, AT/MT, and the dehydratase; the interdomain region (residues 981–1,629); domain II (residues 1,630–2,230) containing the enoyl reductase, the ketoreductase, and ACP; and domain III (residues 2,231–2,512), which codes for thioesterase (Fig. 2, Table 1). The function of the interdomain region is unknown at this time, but its purpose may be to hold the two FAS subunits together and generate a dimer with two centers of fatty acid synthesis (2, 6, 25, 26). The assignment of AT/MT (residues 360–734) and of the dehydratase (residues 734–980) by proteolytic mapping of domain I (Fig. 2, Table 1) is also consistent with the N-terminal sequence of the transacylase-containing rat FAS tryptic peptide (27) and the identification of His-878 in rat FAS and its location within a putative conserved motif, H-X-X-X-G-X-X-X-X-P (16). The His-877 in chicken FAS is also located in a similar motif (9). Although it is likely that this histidine is located in the active site of the dehydratase (16), it has not been experimentally proven that the dehydratase domain encompasses this motif and is located at this site. We now present experimental evidence to show that this motif is indeed an integral part of a polypeptide domain that is catalytically active as a dehydratase.
Table 1.
N-terminal sequences of major peptides obtained after kallikrein proteolysis of chicken FAS
| Span of amino acid residues* | FAS domain(s) | Component activity | N-terminal sequence | Estimated molecular weight, kDa† | Calculated molecular weight, kDa‡ |
|---|---|---|---|---|---|
| 1–1360 | I | KS, AT/MT, DH | N-terminus blocked | 142 | 140.86 |
| 1–359 | I | KS | N-terminus blocked | 37 | 41.71 |
| 360–733 | I | AT/MT | NPNPDIPAL | 49 | 40.20 |
| 734–980 | I | DH | NVNNLVNPVL | 26 | 29.34 |
| Interdomain | — | 64 | 71.71 | ||
| 981–1293 | Interdomain 1 | — | NVTAKSGLLM | 30 | 34.76 |
| 1294–1630 | Interdomain 2 | — | DAGISFSQWD | 34 | 36.95 |
| 1631–2512 | II–III | — | 95 | 96.17 | |
| 1294–2512 | Interdomain 2/II–III | — | 124 | 133.51 | |
| 1294–2208 | Interdomain-2/II | — | DAGISFSQ | 92 | 99.85 |
| 1631–1876 | II | ER | DCDKRFLWE | 24 | 27.06 |
| 1877–2130 | II | KR | SAISRTSCPP | 23 | 27.35 |
| 2132–2208 | II | ACP | SEGG | 15 | 8.49 |
| 2209–2512 | III | TE | KTGPG | 33 | 33.27 |
KS, β-ketoacyl synthase; DH, β-hydroxyacyl dehydratase; ER, enoyl reductase; KR, β-ketoacyl reductase; TE, thioesterase.
The span of the amino acid residues was based on the N-terminal amino acid sequences of kallikrein cleavage peptides.
Based on SDS/PAGE analyses.
Based on the cDNA-derived amino acid sequences within the indicated sequence span.
Figure 2.
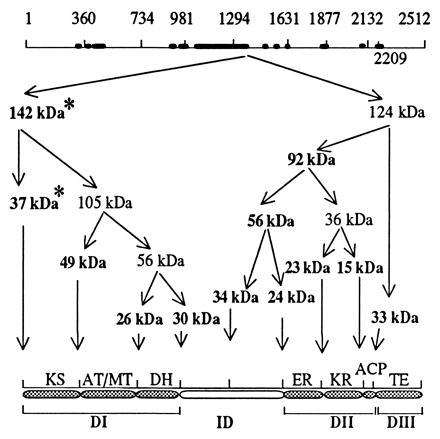
Kallikrein cleavage map of chicken FAS. Top line represents the open reading frame of the chicken FAS cDNA. The positions of the major cleavage sites in the sequence are also indicated. Thick solid bars in the top line indicate the location and span of the least conserved regions of the sequences of the chicken and rat FASs. Arrows depict the origin and the location of the cleavage and point to the possible origin of the major peptides and their sizes. The peptide sizes that are indicated in boldface type are those whose sequence could be obtained, except for the N-terminal blocked peptides, which are denoted by asterisks (see also Table 1). The organization of the component activities and the arrangement of the domain are depicted in the bottom line as follows: domain I (DI), interdomain region (ID), domain II (DII), and domain III (DIII).
Cloning and Expression of Chicken FAS Domain I in E. coli.
To further confirm that domain I contains the ketosynthase, AT/MT, and dehydratase activities, we cloned and expressed a chicken FAS cDNA fragment that codes for domain I in E. coli as a 165-kDa maltose-binding protein fusion protein as described in Experimental Procedures and in Fig. 1. After purifying the fusion protein by affinity chromatography using amylose resin, a protein fraction was obtained that had both the AT/MT and dehydratase (residues 455–1,076) activities. SDS/PAGE analysis revealed the presence of a major protein band (Mr ≈165,000), which in Western blot analysis reacted positively with anti-chicken FAS antibodies (Fig. 3). Assays of the affinity-purified protein showed the presence of AT/MT and dehydratase activities at levels comparable with those of chicken FAS (Table 2). The presence of the dehydratase activity in this domain further confirmed the order of the three activities in domain I that were deduced from the amino acid sequences of the peptides described above. However, assays of this fraction for ketosynthase activity were negative as determined by the incorporation of [14C]bicarbonate into malonyl-CoA by the reverse reaction (26), by the formation of triacetic acid lactone (10), or by the acyl-binding assay (28). These results are consistent with earlier observations that showed that the FAS monomer subunit, which contains all the active partial components of FAS reactions, does not exhibit ketosynthase activity (1, 2, 26). This was expected, based on the model of the FAS mechanism we proposed earlier (1, 2): in the FAS head-to-tail dimer complex, the acetyl moiety from acetyl-CoA binds the Ser—OH of the transacylase of one subunit and then is transferred to the Cys—SH of the ketosynthase partial activity of the second subunit. This organization is not fulfilled by the presence of domain I only. As anticipated, we could detect the acetyl and malonyl binding to the Ser—OH of the recombinant domain I, albeit at low levels, but we could not detect any transfer of either of these groups to the Cys—SH of the ketosynthase (Table 3). These results suggest that this transfer of the acetyl group takes place via ACP, which is not located in this domain.
Figure 3.
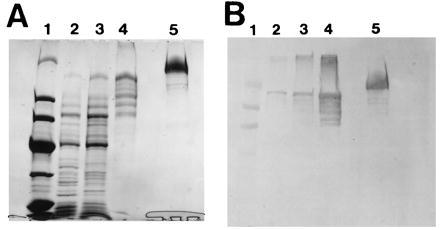
SDS/PAGE and Western blot analyses of a partially purified recombinant maltose-binding protein–domain I fusion protein. (A) The protein was analyzed by SDS/PAGE, and the gel was stained with Coomassie blue. Lanes: 1, protein standards with molecular weights (in kDa) of 200, 97, 68, 43, 29, and 18; 2, cell-free extract of the induced E. coli; 3, 0–55% ammonium sulfate fraction; 4, fraction purified by affinity chromatography; 5, chicken FAS. (B) Western blot analysis with anti-chicken FAS of a gel run as in A, except that lane 1 contained prestained protein markers.
Table 2.
Comparison of the catalytic activities of the recombinant chicken FAS domain I and its constituent subdomains with those of native chicken FAS
| Protein fractions | Specific activity, nmol per/mg
|
||
|---|---|---|---|
| AT | MT | DH | |
| Chicken FAS domain I | |||
| Crude extract | 15 | 90 | ND |
| 0–55% (NH4)SO4 | 58 | 86 | ND |
| Amylose affinity (2.5 mg) | 405 | 200 | 14 |
| Chicken FAS AT/MT-DH | 422 | 195 | 14 |
| Chicken FAS AT/MT | 400 | 220 | ND |
| Chicken FAS DH* | ND | ND | 30 |
| Native chicken FAS | 730 | 300 | 16 |
The partial activities were assayed as described in Experimental Procedures. AT, acetyl transacylase; MT, malonyl transacylase; DH, β-hydroxyacyl dehydratase; ND, not detectable.
This dehydratase preparation was obtained by using procedure 2 as described in Experimental Procedures.
Table 3.
Acyl-binding assay
| Moles of acyl moieties bound/moles of enzyme
|
|||
|---|---|---|---|
| Total | Nonthiol | Thiol | |
| Acetyl-CoA | |||
| Chicken FAS domain I | |||
| Experiment 1 | 0.017 | 0.016 | — |
| Experiment 2 | 0.013 | 0.015 | — |
| Chicken FAS | 0.880 | 0.350 | 0.54 |
| Malonyl-CoA | |||
| Chicken FAS domain I | |||
| Experiment 1 | 0.020 | 0.021 | — |
| Experiment 2 | 0.020 | 0.020 | — |
| Chicken FAS | 1.350 | 0.620 | 0.74 |
Cloning and Expression of the AT/MT and Dehydratase Component Activities.
As described in Experimental Procedures and in Fig. 1, we cloned and expressed the AT/MT and dehydratase component activities together and individually. The thioredoxin-6xHis fusion protein containing the AT/MT and the dehydratase (residues 455–1,076) was found to be soluble and could be enriched to about 30% by affinity chromatography using TALON resin (data not shown). The activities of AT/MT and the dehydratase present in this preparation are about 60% of those exhibited by native chicken FAS (Table 2).
As stated earlier, the copiously expressed recombinant AT/MT protein (residues 455–726) was insoluble and was associated with the pellet fraction after removal of the soluble proteins. An active AT/MT preparation was obtained after the protein was solubilized in guanidinium-HCl solution and purified by affinity chromatography as described. The partially purified recombinant protein showed AT/MT activities of about 60% of the activity of the native chicken FAS and had no detectable dehydratase activity (Table 2). SDS/PAGE and Western blot analyses of the preparation showed that the protein was more than 50% pure and reacted with S-Tag antibodies (Fig. 4A and 4B, lanes 1 and 2).
Figure 4.
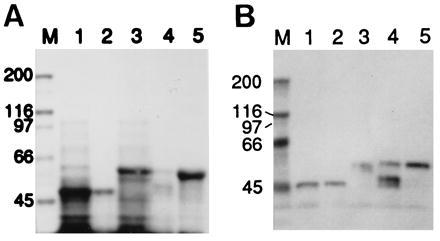
SDS/PAGE and Western blot analyses of partially purified thioredoxin-6xHis fusion proteins of the acetyl and malonyl transacylases (AT/MT) and the dehydratase. (A) The preparations were analyzed by SDS/PAGE, and the gel was stained with Coomassie blue. Lanes: M, protein standards of the indicated molecular weights in kDa; 1, lysate of E. coli expressing AT/MT; 2, AT/MT fraction purified by TALON affinity chromatography after solubilization in guanidinium-HCl and renaturation by dialysis; 3, cell-free extract of E. coli expressing the dehydratase component; 4, affinity-purified dehydratase fraction after solubilization in guanidinium-HCl and renaturation by dialysis; 5, affinity-purified dehydratase as in lane 4, except that the protein was renatured on the column by washing it with buffer containing no denaturant. (B) Western blot analysis with S-protein AP conjugate of a gel run as in A, except that lane M contained prestained markers.
The recombinant protein containing the dehydratase activity (residues 727–1,076) could also be purified by using a protocol similar to the one we used to purify the AT/MT recombinant protein. When the dehydratase preparation was isolated according to procedure 1 (see Experimental Procedures), the specific activity of the dehydratase was about 14 nmol of crotonyl-CoA consumed per min/mg, and the yield was relatively low. However, when the recombinant dehydratase protein was bound to the TALON resin and the column was washed extensively at room temperature with buffer devoid of a denaturing reagent as described in procedure 2, a highly purified protein fraction was obtained (Fig. 4) that exhibited dehydratase activity twice that of the native chicken FAS dehydratase and did not have detectable transacylase activities (Table 2).
The proteolytic mapping of animal FAS defined the amino acid span of the ketosynthase to be between residues 1 and 360 (Table 1). Comparing the amino acid sequence in this region with the sequences of the various bacterial polyketide synthases revealed that the β-ketoacyl synthases are evolutionarily conserved. Although the AT/MT subdomain is next to the ketosynthase, beginning at a point near residue 360, the amino acid sequence between residues 500 and 730 has the highest identity (25%) with bacterial acetyl and malonyl transacylases. Furthermore, when the cDNA clone pAT/MT (Fig. 1), which codes for amino acid residues 455–726 of chicken FAS, was expressed in E. coli as described above, it produced a recombinant protein that had AT/MT activities. These results suggest that the sequence between amino acid residues 360 and 450 would constitute a linker region between the ketosynthase and AT/MT activity domains. Finally, the N-terminal end of the dehydratase subdomain at amino acid residue 734 of chicken FAS, which was derived from amino acid sequencing (Table 2), is reaffirmed by the dehydratase activity of the recombinant protein derived from pDH (Fig. 1, Table 2).
The results described above show conclusively that the ketosynthase, AT/MT, and dehydratase component activities are located in the domain I region of FAS. Our initial mapping of the component activities, as previously determined by limited proteolysis (1–7), and now our N-terminal sequence analysis of the major products of kallikrein cleavage, have further defined the boundaries of these functional units. Although we assigned the dehydratase to domain II in our earlier map, it is the same fragment that we isolated initially as containing dehydratase activity (7) and are now able to assign to domain I on the basis of the N-terminal sequence analysis, cloning and expression of pDH in E. coli, and purification of the recombinant protein. It is gratifying to find that our first functional map of animal FAS (1, 7), which we based solely on unsophisticated proteolytic mapping, is still exceptionally good. Furthermore, knowing the sequences of the boundaries of the component activities will help us in cloning and expressing the component activities in heterologous systems and in determining their characteristics.
Acknowledgments
The computer software programs used for the sequence analyses were accessed through the Molecular Biology Computational Resource at Baylor College of Medicine. We thank Pamela Paradis Powell (Editor in the Life Sciences) for editing the manuscript. This work was supported by a grant from the National Institutes of Health (GM-19091).
ABBREVIATIONS
- FAS
fatty acid synthase
- ACP
acyl carrier protein
- AT/MT
acetyl and malonyl transacylases
Footnotes
To whom reprint requests should be addressed at: Department of Biochemistry, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030. e-mail: swakil@bcm.tmc.edu.
References
- 1.Wakil S J, Stoops J K, Joshi V C. Annu Rev Biochem. 1983;52:537–579. doi: 10.1146/annurev.bi.52.070183.002541. [DOI] [PubMed] [Google Scholar]
- 2.Wakil S J. Biochemistry. 1989;28:4523–4530. doi: 10.1021/bi00437a001. [DOI] [PubMed] [Google Scholar]
- 3.Mattick J S, Tsukamoto Y, Nickless J, Wakil S J. J Biol Chem. 1983;258:15291–15299. [PubMed] [Google Scholar]
- 4.Mattick J S, Nickless J, Mizugaki M, Yang C-Y, Uchiyama S, Wakil S J. J Biol Chem. 1983;258:15300–15304. [PubMed] [Google Scholar]
- 5.Wong H, Mattick J S, Wakil S J. J Biol Chem. 1983;258:13305–13311. [PubMed] [Google Scholar]
- 6.Tsukamoto Y, Wong H, Mattick J S, Wakil S J. J Biol Chem. 1983;258:15312–15322. [PubMed] [Google Scholar]
- 7.Tsukamoto Y, Wakil S J. J Biol Chem. 1988;263:16225–16229. [PubMed] [Google Scholar]
- 8.Chirala S S, Kasturi R, Pazirandeh M, Stolow D T, Huang W-Y, Wakil S J. J Biol Chem. 1989;264:3750–3757. [PubMed] [Google Scholar]
- 9.Huang W-Y, Chirala S S, Wakil S J. Arch Biochem Biophys. 1994;314:45–49. doi: 10.1006/abbi.1994.1410. [DOI] [PubMed] [Google Scholar]
- 10.Jayakumar A, Tai M-H, Huang W-Y, Al-Feel W, Hsu M, Abu-Elheiga L, Chirala S S, Wakil S J. Proc Natl Acad Sci USA. 1995;92:8695–8699. doi: 10.1073/pnas.92.19.8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holzer K P, Liu W, Hammes G G. Proc Natl Acad Sci USA. 1989;85:4387–4391. doi: 10.1073/pnas.86.12.4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amy C M, Witkowski A, Naggert J, Williams B, Randhawa Z, Smith S. Proc Natl Acad Sci USA. 1989;86:3114–3118. doi: 10.1073/pnas.86.9.3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beck K-F, Schreglmann R, Stathopulos I, Klein H, Hoch J, Schweizer M. DNA Sequence. 1992;2:359–386. doi: 10.3109/10425179209020817. [DOI] [PubMed] [Google Scholar]
- 14.Witkowski A, Rangan V S, Randhawa Z I, Amy C M, Smith S. Eur J Biochem. 1991;198:571–579. doi: 10.1111/j.1432-1033.1991.tb16052.x. [DOI] [PubMed] [Google Scholar]
- 15.Smith S. FASEB J. 1994;8:1248–1259. [PubMed] [Google Scholar]
- 16.Joshi A K, Smith S. J Biol Chem. 1993;268:22508–22513. [PubMed] [Google Scholar]
- 17.Yang C-Y, Huang W-Y, Chirala S S, Wakil S J. Biochemistry. 1988;27:7773–7777. doi: 10.1021/bi00420a028. [DOI] [PubMed] [Google Scholar]
- 18.Huang W-Y, Stoops J K, Wakil S J. Arch Biochem Biophys. 1988;270:92–98. doi: 10.1016/0003-9861(89)90011-8. [DOI] [PubMed] [Google Scholar]
- 19.Pazirandeh M, Chirala S S, Wakil S J. J Biol Chem. 1989;264:18195–18201. [PubMed] [Google Scholar]
- 20.Pazirandeh M, Chirala S S, Wakil S J. J Biol Chem. 1991;266:20946–20952. [PubMed] [Google Scholar]
- 21.Arslanian M J, Wakil S J. Methods Enzymol. 1975;35:59–65. doi: 10.1016/0076-6879(75)35138-0. [DOI] [PubMed] [Google Scholar]
- 22.Towbin H, Staehelin T, Gordon J. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jayakumar A, Huang W-Y, Raetz B, Chirala S S, Wakil S J. Proc Natl Acad Sci USA. 1996;93:14509–14514. doi: 10.1073/pnas.93.25.14509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maniatis T, Fritsch E F, Sambrook J. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1982. [Google Scholar]
- 25.Stoops J K, Wakil S J, Uberbacher E C, Bunick G J. J Biol Chem. 1987;262:10246–10251. [PubMed] [Google Scholar]
- 26.Stoops J K, Wakil S J. J Biol Chem. 1981;256:5128–5133. [PubMed] [Google Scholar]
- 27.Rangan V S, Witkowski A, Smith S. J Biol Chem. 1991;266:19180–19185. [PubMed] [Google Scholar]
- 28.Joshi V C, Plate C A, Wakil S J. J Biol Chem. 1970;245:2857–2867. [PubMed] [Google Scholar]