

Abstract
We have previously shown that activation of the homologous recombinational repair pathway leads to a block of cell division in corrected cells, possibly through the activity of checkpoint proteins Chk1 and Chk2. In this study, we examine the long term impact of this stalling on the growth of cells that have enabled gene repair events. Using a mutated eGFP gene as an episomal reporter, we show that corrected (eGFP positive) cells contain only a few active replication templates two weeks after electroporation, yet do not display an apoptotic or senescent phenotype. By six weeks after electroporation, cells resume active replication with a cell cycle profile that is comparable to that of the non-corrected (eGFP negative) population. These results indicate that the initial stalling is transient and eGFP positive cells eventually resume a normal phenotypic growth pattern, allowing for passaging and expansion in vitro.
Keywords: gene repair, single-stranded oligonucleotides
Introduction
Gene repair directed by oligonucleotides has been demonstrated in a variety of cell lines, primary cells and animal models [1, 2]. Substantial progress has been made in understanding the mechanism by which the mutant base is corrected and several pathways have been proposed to explain the regulation of the reaction. Recently, numerous reports have lent credence to a route that involves DNA replication as a controlling mechanism in gene repair [3-7]. In this context, most of the data generated from studies on the mechanism of correction indicate that the oligonucleotide may become part of a growing replication fork (summarized in 1). The molecule essentially anneals to the complementary, transient single-stranded regions located most likely on the lagging strand of the replication template [3]. This type of activity has the potential to provide the basis for genetic treatment of inherited diseases, particularly those that arise by single base mutations. In contrast, to viral-based gene therapy approaches, this nonviral approach directs the removal of a mutated base and replaces it with the normal (wild-type) residue.
We have recently shown that activation of a DNA damage response mediated by ATM takes place following introduction of the oligonucleotide into mammalian cells [1]. These results are consistent with previous observations of Nur et al [8], who showed that ATM is activated in cells transfected with single-stranded DNA. Corrected cells contain phosphorylated Chk1 and Chk2 foci, are stalled in S phase and contain few actively replicating templates for up to 72 hours after the introduction of the oligonucleotide [9]. These data provide an explanation for phenomena initially reported by Krauss and colleagues [5, 6] who showed that CHO-K1 cells containing a corrected eGFP gene score positive for cell cycle arrest. Previous work by Gilchrest and colleagues [10-12] demonstrated that specific oligonucleotides bearing sequences resembling 3’ telomeric DNA induce a stalling of DNA replication and cell senescence as a result of a p53-mediated SOS response.
We sought to characterize eGFP positive cells that have stalled in their cell cycle progression. Several cellular responses are possible. First, they may undergo apoptosis and effectively be removed from the overall population. Second, they could adopt a senescent phenotype and remain as a stable part of the population without dividing. Third, they could remain quiescent as recovery takes place and subsequently resume DNA replication and cell division. Should this occur in targeted cells, then the enthusiasm for gene repair as a form of gene therapy will be reduced. Corrected cells should not be altered in their growth characteristics by this process or uncorrected cells will dominate and the effect will be lost.
Our studies reveal that stalled HCT116 cells that have undergone a gene correction event directed by a single-stranded oligonucleotide eventually resume active DNA replication and cell cycle progression. We show that the initial replication and cell division block, induced by the entry of the targeting molecule, is overcome with time.
Results and Discussion
These experiments were designed to evaluate the cellular response to the gene repair protocol and not simply to detail the mechanism of correction at the genomic level. The HCT116 cell line was used as a model system in this study; cells were transfected with the plasmid pEGFP-N3, which contains an eGFP reporter gene with a single mutation at nucleotide position 201 to establish the target site [3, 4, 13] This mutation (C to G) creates a stop codon which leads to the expression of nonfunctional eGFP. Conversion of the TAG to TAC directed by a single-stranded oligonucleotide (EGFP3S/47NT) (Fig. 1a) rescues the expression of the protein [14, 15]. Repair of the mutation can be measured by FACS analysis or by fluorescence microscopy using green fluorescence as a phenotypic marker of corrected cells.
Figure 1. Oligonucleotide-directed gene repair.
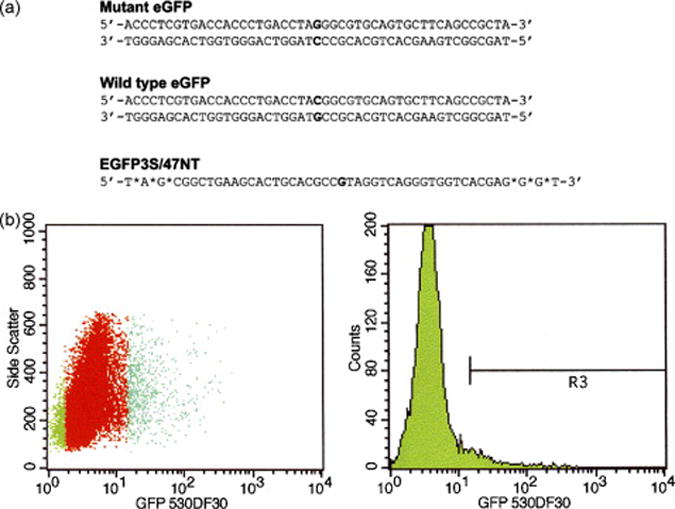
(a) The mutant eGFP gene, containing a mutation at nucleotide position 201 (bold) is depicted together with the wild type (corrected) gene. EGFP3S/47NT indicates the oligonucleotide used to direct gene repair. The sequence of the oligonucleotide is complementary to the non-transcribed strand of the mutant eGFP gene; asterisks indicate phosphorothioate linkages. (b) HCT116 cells were electroporated with 5 μg of mutant pEGFP-N3 plasmid (Clontech) and 4 μM EGFP3S/47NT and recovered for 24 h before sorting. Cells were sorted with FACSVantage SE Sort (Becton Dickinson) into two populations each containing approximately 50,000 cells based on the presence (R3) or absence (R4) of eGFP fluorescence. In the left panel (dot plot) the sorted populations are indicated by a green color.
Over 50% of the cells receive the plasmid and enable expression of the eGFP protein (data not shown) as judged by the transfection of a plasmid containing a wild type copy of the eGFP gene. In this study, we used the mutated pEGFP-N3 plasmid as a target because a high level of correction (5.2% +/- 1.5%) is seen routinely. The high frequency of repair facilitates sorting and the isolation of a larger number of corrected cells for further study. Episomal targeting is also useful in order to measure a response that is not dependent on the sequence context of the chromosomal target site.
The experimental strategy was to carry out gene repair of the mutated eGFP using a 47-base oligonucleotide and to sort eGFP positive cells. We compared the eGFP-negative and positive cell populations for the presence of active replication forks and the onset of apoptosis and/or senescence. Then, we followed the growth of these isolated populations for six weeks, analyzing DNA replication activity, cell cycle progression and the capacity to establish new colonies.
Non-corrected and corrected cell populations contain different levels of DNA replication activity but do not exhibit apoptotic or senescent character
The 47-mer, EGFP3S/47NT, was co-electroporated into HCT116 cells with the plasmid pEGFP-N3 and gene repair was allowed to take place for 24 h, before the entire population of cells was sorted by FACS (Figure 1b). The specific area indicated as R4 reflects the position of eGFP-positive cells, gated at a fluorescence value of 101 on FACS. Approximately 3.7% of the cell population was found to be eGFP-positive and isolated for further analyses (a comparable number of eGFP-negative cells were also isolated). Each population (about 50,000 cells) was plated and allowed to grow for 14 days before being analyzed for DNA replication activity by measuring BrdU incorporation.
We have recently demonstrated that the vast majority of eGFP-positive cells contain phosphorylated Chk1 and Chk2 [9]. The reduced level of replication activity in this population can be explained by a higher level of activation of a DNA damage response. Activated Chk1 and Chk2 are responsible for arresting/delaying cell cycle progression following DNA damage [16] and, if this arrest is permanent, then the cells would enter either an apoptotic [5] or senescent pathway. To examine each of these possibilities, we first stained the two cultures with Annexin V, testing for the presence of apoptotic activity. As shown in Figure 2a, both cell populations are positive for 6-carboxyfluorescein diacetate (6-CFDA; cells appear green), which is used as a measure of cell viability (see Materials and Methods), but neither sample stains positive for Annexin V. Camptothecin, a known inducer of DNA damage [17-19], was used here as a positive control as it causes DNA damage and initiates the apoptotic response. Thus, corrected HCT116 cells, stalled in their cell cycle progression and containing a low level of DNA replication activity, do not contain detectable apoptotic cells when analyzed two weeks after the introduction of the oligonucleotide. This response is not unique to HCT116 as the same phenomenon is observed in DLD-1 cells [9].
Figure 2. Corrected cells have a reduced number of replication templates but do not display an apoptotic or senescent phenotype.
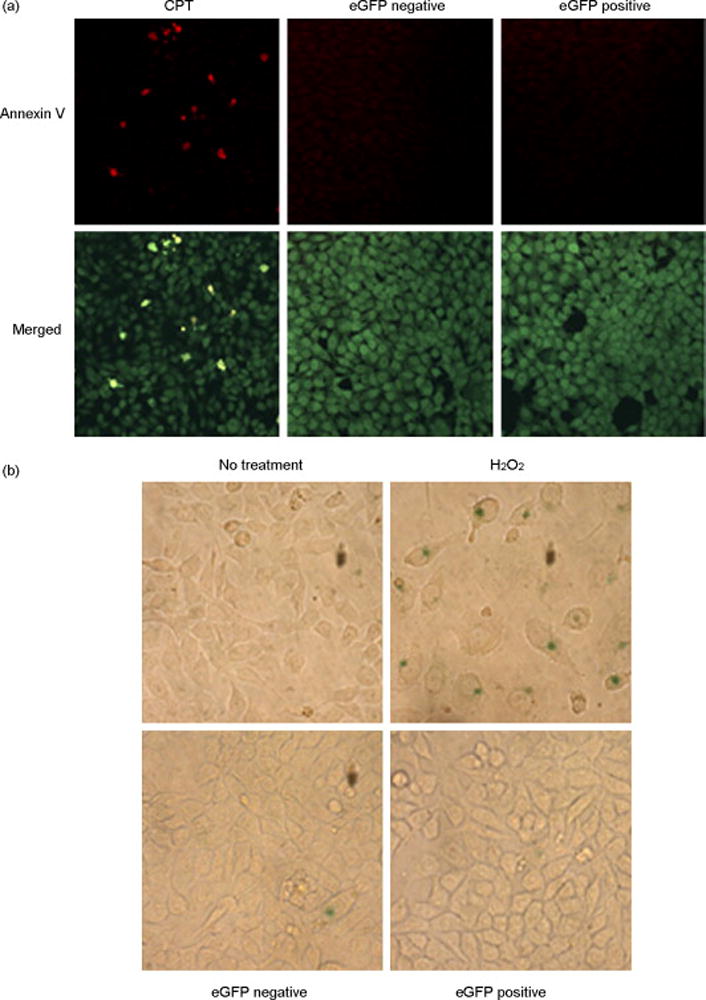
(a) Annexin V staining of sorted cells. Sorted cells and cells treated with 1 μM camptothecin (CPT) were stained with fluorescein-conjugated Annexin V (Apoptosis Detection Kit, Sigma), following manufacturer’s instructions. Apoptotic cells appear yellow in the merged image. The green staining (6-carboxyfluorescein diacetate) is a measure of cell viability. (b) β-galactosidase staining. Cells were treated with 200 μM H2O2 for 2 h and then cultured for 5 days before fixation or, for the sorted eGFP-positive and negative cells, directly fixed with 4% paraformaldehyde for 10 min. Cells were washed twice in PBS and then incubated with the following solution: 40 mM citric acid/sodium phosphate pH 6.0, 150 mM NaCl, 2 mM MgCl2, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 1 mg/ml X-Gal. After 16 h incubation at 37°C (without CO2), cells were washed in PBS and then analyzed under the microscope. The blue color in the cytoplasm suggests that these cells are senescent.
Unlike apoptosis, no unique molecular marker or unequivocal cellular feature can assess the transition to cellular senescence. However, one assay has been traditionally used to identify senescent cells and it is based on the observation that cells in a senescent state express β-galactosidase [20]. As a positive control, we treated cells with H2O2, a known inducer of cellular senescence [21]. As seen in Figure 2b, cells treated with H2O2 exhibit a blue color after staining, indicating that HCT116 have the capacity to develop a senescent phenotype. In contrast, neither eGFP-negative nor eGFP-positive populations contain a significant number of cells that stain positive for β-galactosidase activity. Thus, we suggest that corrected cells are not entering the senescent pathway when analyzed two weeks after the electroporation of the oligonucleotide.
DNA replication and cell cycle recovery in the corrected cell population
To analyze the long term growth capacity of each population, the isolated eGFP-negative and eGFP-positive cells were cultured beyond the two-week time point described above and tested for the potential to form new colonies. Two hundred cells from each culture were plated at two and four weeks after electroporation and the number of colonies determined one week later (at three and five weeks, respectively). As shown in Table 1, a wide disparity between the two populations is observed at the three-week time point, with the non-corrected population (eGFP negative) having a much higher colony forming capacity than the corrected cells (eGFP positive). These results are consistent with the data presented in Figure 3a, wherein the eGFP-positive cell population is found to have a low level of replication activity. The gap between the eGFP-positive and eGFP-negative cells shortens substantially, however, when the plating efficiency is measured after five weeks. Here, we observe a difference of only 1.2 fold, indicating that the corrected cells have recovered from the initial replication block and are now capable of forming colonies.
Table 1. Plating efficiency of sorted cells.
Two hundred cells were plated at 2 and 4 weeks after sorting and the number of colonies was determined after one week, at 3 and 5 weeks, respectively. The colonies (total of 200) were detected by staining with 0.5% methylene blue in 50% methanol. The difference between the number of colonies in the eGFP-positive and eGFP-negative populations is statistically significant (t-test, p<0.001) at both the three-week and five-week time point.
| eGFP-positive | eGFP-negative | |
|---|---|---|
| 3 weeks | 13.5% | 43.85% |
| 5 weeks | 64.25% | 76.65% |
Figure 3. Corrected cells resume active replication.
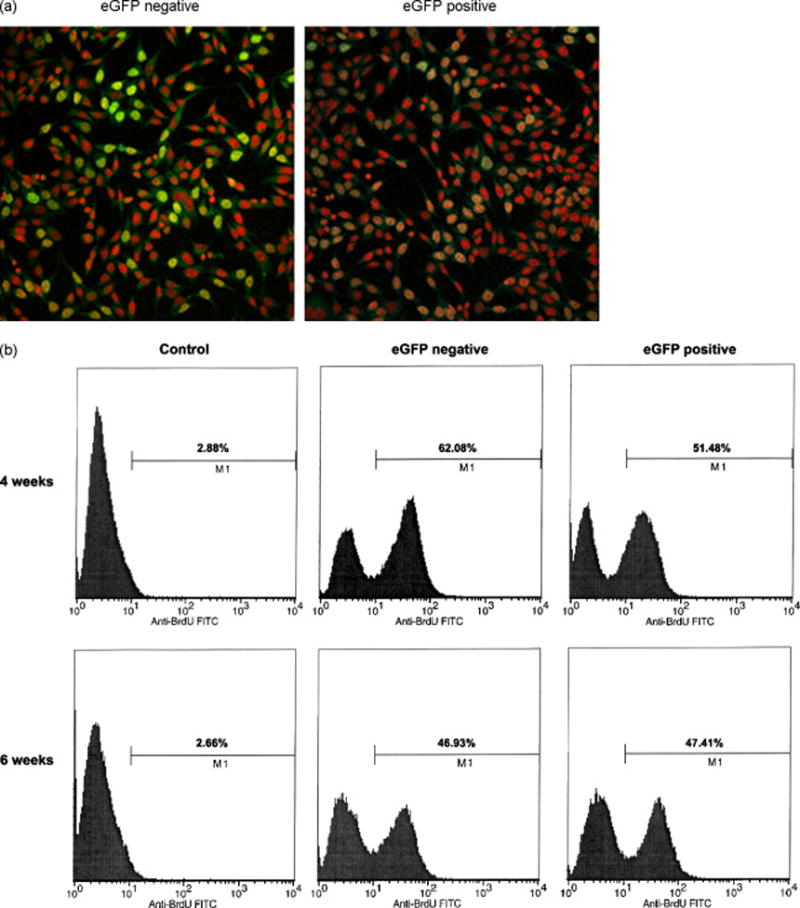
(a) BrdU incorporation in sorted cells. Fourteen days after electroporation, cells were incubated with 10 μM BrdU for 1 h before fixation in 100% cold methanol. Cells were then washed in PBS and DNA was denatured in 0.2 M HCl for 1 h at 37°C. After two washes in 0.1 M sodium borate and three washes in PBS, cells were blocked in PBS-0.5%BSA-0.1% Tween-20 for 20 min. Cells were then incubated for 1 h at room temperature with Anti-BrdU-fluoresceine antibody (Roche Diagnostics) diluted in PBS-0.1% BSA at a final concentration of 25 μg/ml. Red, nuclear staining (DAPI); green, BrdU staining. (b) Sorted HCT116 cells were analyzed for BrdU incorporation at 4 and 6 weeks after electroporation of the oligonucleotide. Cells were incubated with 10 μM BrdU for 30 min before harvesting, fixed in 70% cold ethanol and incubated with a fluorescin-anti BrdU antibody following manufacturer’s instructions (In Situ Cell Proliferation Kit, FLUOS, Roche Diagnostics). The sample labeled as control shows cells that were processed without prior incubation with BrdU and are used here to detect the background fluorescence of the antibody. The numbers indicate the percentage of BrdU-positive cells in each population.
Next, we examined the replication activity in both populations by measuring BrdU incorporation at four and six weeks after electroporation. Figure 3a shows that the eGFP-negative population (uncorrected), stained with an anti-BrdU antibody and analyzed by confocal microscopy, contains an abundance of cells bearing BrdU-positive cells (green). In contrast, replication-positive cells are barely visible in the eGFP-positive population. Consistent with the observations of Olsen et al [6] and our own previous data [9], eGFP-positive cells initially contain fewer actively replicating templates than their eGFP-negative counterparts. Thus, we suggest that the two populations of cells have different growth characteristics depending on whether they have received enough oligonucleotides to undergo a gene repair event or not (Figure 3b). After four weeks, DNA replication activity is clearly higher in the eGFP-negative population, but after six weeks, the replication activity in the corrected cell population has reached the same level as the non-corrected sample.
This recovery is also observed when the cell cycle profile from each population is measured at the two-week time point (Figure 4a), at which time both samples exhibit a normal profile with approximately an equal number of cells in each phase. The same effect is observed with DLD-1 cells containing an integrated mutant eGFP gene when analyzed at 6 weeks [13]. We showed previously that the percentage of eGFP-positive cells in the targeted population decreases steadily as a function of time [9]. Using this system and sorting eGFP-positive cells, we followed their cells cycle progression for 28 days. Figure 4c illustrates cell cycle profile of the eGFP-positive and eGFP-negative cells 28 days after targeting. The profiles are very similar, both resembling untreated cells and exhibiting the normal cycling pattern of DLD-1 cells [see 13] We suggest that multiple cell lines from different sources are similarly affected by the gene repair protocol both in the stalling phenotype and their subsequent recovery. Thus, a population of targeted cells can recover after an initial block in replication and resume normal DNA replication activity and cell cycle progression.
Figure 4. Cell cycle recovery of corrected cells.
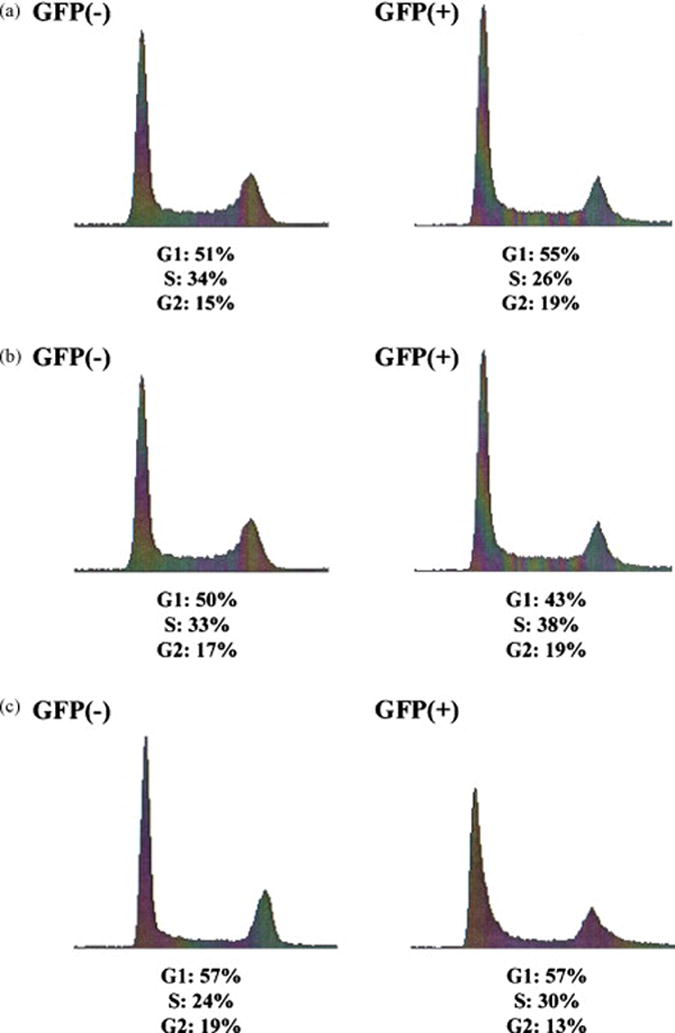
(a) Sorted eGFP-positive and eGFP-negative cells were analyzed for DNA content 2 weeks after electroporation of the oligonucleotide. Asynchronously growing cultures were fixed in 70% cold ethanol, incubated at 4°C overnight, then washed with PBS and resuspended in 0.5 ml of PBS containing 50 μg/ml RNaseA, 1% FBS and 2.5 μg/ml propidium iodide. Cells were incubated at 37°C for 1 h and then overnight at 4°C before analysis of DNA content by Becton Dickinson FACScalibur flow cytometer (Becton Dickinson). (b) Sorted eGFP-positive and eGFP-negative cells were analyzed for DNA content 6 weeks after electroporation of the oligonucleotide. (c) Cells were synchronized utilizing a double thymidine block [see 14] and released for 3 hours prior to targeting. About 2×106 cells were resuspended in 100 ul of serum free medium, transferred to a 4 mm gap cuvette, 4 μM 72NT oligonucleotide was added and cells electroporated at standard settings. Twenty four hours post electroporation, cells were collected via trypsinization and resuspended in PBS + 0.5% BSA at 1×106 cells/ml. Cells were sorted on ice and a total of 4×105 GFP(+) cells were obtained, equivalent to roughly 3.65% of the entire population. These cells were grown for 28 days, at which point the cultured cells were pooled together and frozen. Cells were thawed and allowed to grow for 3 and 6 days before taken for cell cycle analysis. Image depicts a culture of DLD-1 integrated mutant eGFP cells, non-corrected [GFP(-)], and the recovered GFP(+) cells after thawing.
Our data support the observations of Olsen et al [5, 6], in which a high percentage of cells in the corrected population exhibit a different growth phenotype than non-corrected cells. These workers convincingly showed that addition of the Chk1 inhibitor, Go6976, enabled 30% of the corrected cells to resume cell division. This observation aligns with our previous data [9], which showed that the presence of activated Chk1 may be responsible for the stalling of corrected cells in S or G2. Thus, this stalling phenomenon has now been reported in multiple cell lines and more importantly the capacity for the cells to recover has now been validated in several systems.
We also find that the vast majority of corrected HCT116 cells return to a normal growth pattern six weeks after receiving the oligonucleotide and undergoing gene repair. In this study, we have used an episomal gene as our target for repair, but preliminary data (not shown) indicate a similar effect when chromosomal genes serve as targets. We believe that the effect on cell cycle is due to the presence of high levels of oligonucleotide, rather than the gene repair reaction itself. Control reactions in which the same level of wild type plasmid expressing eGFP was transfected resulted in a profile resembling that of the uncorrected, eGFP-negative cells. Thus, the repair reaction is clearly dependent on the level of oligonucleotide received by the cell. We hypothesize that correction events require high levels of oligonucleotide and those cells bearing a repaired gene are likely to have received enough vector to only generate eGFP-positive cells and impact cell cycle progression in a negative fashion. These observations indicate that the gene repair reaction has characteristics of a mass action event only when high levels of oligonucleotide are present at a time that is prime for gene correction. This “time” is most likely to be S phase as cells in S phase have been found to be most amenable to correction [3, 4, 6, 13, 22].
Considering the harsh effect of oligonucleotide entry into targeted cells and the subsequent response by the cells, the observation that eGFP-positive cells (corrected) cells eventually resume a normal growth pattern provides some hope for gene repair as an ex vivo approach. If the cells had not been able to proliferate, then the use of gene repair as an in vitro treatment, followed by expansion of cells explanted from patients, would be dealt a serious blow.
Supplementary Material
Acknowledgments
We wish to thank Dr. Kirk Czymmek (University of Delaware) for help with confocal microscopy, Dr. Stuart Linn (University of California, Berkeley) for critical advice on the work and members of the Kmiec laboratory for extensive discussions. This work was supported by a grant from NIH (R01 CA89325-01A1).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Parekh-Olmedo H, Ferrara L, Brachman E, Kmiec EB. Gene therapy progress and prospects: targeted gene repair. Gene Ther. 2005;12:639–646. doi: 10.1038/sj.gt.3302511. [DOI] [PubMed] [Google Scholar]
- 2.Igoucheva O, Alexeev V, Yoon K. Mechanism of gene repair open for discussion. Oligonucleotides. 2004;14:311–321. doi: 10.1089/oli.2004.14.311. [DOI] [PubMed] [Google Scholar]
- 3.Brachman EE, Kmiec EB. DNA replication and transcription direct a DNA strand bias in the process of targeted gene repair in mammalian cells. J Cell Sci. 2004;117:3867–3874. doi: 10.1242/jcs.01250. [DOI] [PubMed] [Google Scholar]
- 4.Brachman EE, Kmiec EB. Gene repair in mammalian cells is stimulated by the elongation of S phase and transient stalling of replication forks. DNA Repair (Amst) 2005;4:445–457. doi: 10.1016/j.dnarep.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Olsen PA, McKeen C, Krauss S. Branched oligonucleotides induce in vivo gene conversion of a mutated EGFP reporter. Gene Ther. 2003;10:1830–1840. doi: 10.1038/sj.gt.3302079. [DOI] [PubMed] [Google Scholar]
- 6.Olsen PA, Randol M, Krauss S. Implications of cell cycle progression on functional sequence correction by short single-stranded DNA oligonucleotides. Gene Ther. 2005;12:546–551. doi: 10.1038/sj.gt.3302454. [DOI] [PubMed] [Google Scholar]
- 7.Radecke S, Radecke F, Peter I, Schwarz K. Physical Incorporation of a single-stranded oligodeoxynucleotide during targeted repair of a human chromosomal locus. The Journal of Gene Medicine. 2005 doi: 10.1002/jgm.828. [DOI] [PubMed] [Google Scholar]
- 8.Nur-E-Kamal, Li TK, Zhang A, Qi H, Hars ES, Liu LF. Single-stranded DNA induces ataxia telangiectasia mutant (ATM)/p53-dependent DNA damage and apoptotic signals. J Biol Chem. 2003;278:12475–12481. doi: 10.1074/jbc.M212915200. [DOI] [PubMed] [Google Scholar]
- 9.Ferrara L, Kmiec EB. Targeted gene repair activates Chk1 and Chk2 and stalls replication in corrected cells. DNA Repair (Amst) 2006;5:422–431. doi: 10.1016/j.dnarep.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 10.Eller MS, Puri N, Hadshiew IM, Venna SS, Gilchrest BA. Induction of apoptosis by telomere 3’ overhang-specific DNA. Exp Cell Res. 2002;276:185–193. doi: 10.1006/excr.2002.5531. [DOI] [PubMed] [Google Scholar]
- 11.Li GZ, Eller MS, Firoozabadi R, Gilchrest BA. Evidence that exposure of the telomere 3’ overhang sequence induces senescence. Proc Natl Acad Sci U S A. 2003;100:527–531. doi: 10.1073/pnas.0235444100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eller MS, Maeda T, Magnoni C, Atwal D, Gilchrest BA. Enhancement of DNA repair in human skin cells by thymidine dinucleotides: evidence for a p53-mediated mammalian SOS response. Proc Natl Acad Sci U S A. 1997;94:12627–12632. doi: 10.1073/pnas.94.23.12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu Y, Parekh-Olmedo H, Drury M, Skogen M, Kmiec EB. Reaction parameters of targeted gene repair in Mammalian cells. Mol Biotechnol. 2005;29:197–210. doi: 10.1385/MB:29:3:197. [DOI] [PubMed] [Google Scholar]
- 14.Ferrara L, Parekh-Olmedo H, Kmiec E. Enhanced oligonucleotide-directed gene targeting in mammalian cells following treatment with DNA damaging agents. Exp Cell Res. 2004;300:170–179. doi: 10.1016/j.yexcr.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 15.Ferrara L, Kmiec EB. Camptothecin enhances the frequency of oligonucleotide-directed gene repair in mammalian cells by inducing DNA damage and activating homologous recombination. Nucleic Acids Res. 2004;32:5239–5248. doi: 10.1093/nar/gkh822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 17.Kaufmann SH. Cell death induced by topoisomerase-targeted drugs: more questions than answers. Biochim Biophys Acta. 1998;1400:195–211. doi: 10.1016/s0167-4781(98)00136-5. [DOI] [PubMed] [Google Scholar]
- 18.Pourquier P, Pommier Y. Topoisomerase I-mediated DNA damage. Adv Cancer Res. 2001;80:189–216. doi: 10.1016/s0065-230x(01)80016-6. [DOI] [PubMed] [Google Scholar]
- 19.Ryan AJ, Squires S, Strutt HL, Johnson RT. Camptothecin cytotoxicity in mammalian cells is associated with the induction of persistent double strand breaks in replicating DNA. Nucleic Acids Res. 1991;19:3295–3300. doi: 10.1093/nar/19.12.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen QM, Tu VC, Liu J. Measurements of hydrogen peroxide induced premature senescence: senescence-associated beta-galactosidase and DNA synthesis index in human diploid fibroblasts with down-regulated p53 or Rb. Biogerontology. 2000;1:335–339. doi: 10.1023/a:1026590501344. [DOI] [PubMed] [Google Scholar]
- 22.Radecke S, Radecke F, Peter I, Schwarz K. Physical Incorporation of a single-stranded oligodeoxynucleotide during targeted repair of a human chromosomal locus. The Journal of Gene Medicine. 2005 doi: 10.1002/jgm.828. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.