

Abstract
The use of a flow-injection surface plasmon resonance (FI-SPR) spectrometer equipped with a bicell detector or a position-sensitive device for determining coordination of heavy metal ions (Cd2+ and Hg2+) by surface-confined apo-metallothionein (apo-MT) molecules is described. To facilitate the formation of a compact MT adsorbate layer with a uniform surface orientation, MT molecules were attached onto a preformed alkanethiol self-assembled monolayer. The method resorts to the generation of apo-MT at the surface by treating the MT-covered sensor chip with glycine–HCl and the measurement of the apo-MT conformation changes upon metal ion incorporation. Domain-specific metal ion binding processes by the apo-MT molecules were observed. Competitive replacement of one metal ion by another can be monitored in real time by FI-SPR. The tandem use of an immobilization scheme for forming a sub-monolayer of MT molecules at the sensor surface and the highly sensitive FI-SPR instrument affords a low concentration detection level. The detection level for Cd2+ (0.1 μM or 15 ppb) compares favorably with similar studies and the methodology complements to other well-established sensitive analytical techniques. The extent of metal incorporation by apo-MT molecules was also determined.
Keywords: Surface plasmon resonance, Flow injection, Metallothionein, Self-assembled monolayer, Conformation changes, Metal replacement
1. Introduction
Development of highly sensitive and selective techniques for detecting heavy metal ions is at the forefront of environmental monitoring and protection. Many techniques have been developed for trace metal analysis, which include, but are not limited to, atomic spectroscopy (e.g., atomic absorption, emission, and fluorescence spectrometry [1]), mass spectrometry (e.g., inductively coupled plasma-mass spectrometry, ICP-MS [2]), and electrochemical techniques (e.g., ion-selective potentiometry and anodic stripping voltammetry [3, 4]). Although these techniques possess many attractive analytical “figures of merit”, each of them has one or several undesirable limitations [1]. For example, ICP-MS, arguably the most sensitive and multi-elemental detection technique, is bulky (i.e., not suitable for in-situ field monitoring [2]), expensive, and not selective to different charge states and chemical forms of an element. While voltammetric methods are simple, inexpensive, and portable, they are only amenable to select metal species (e.g., in anodic stripping voltammetry only amalgam-forming metal species can be measured) and could be subjected to interferences inherent in complex sample matrices. Thus, it is important to develop alternative techniques that are complementary to the existing techniques and can be tailored for specific applications.
Surface plasmon resonance (SPR) [5–8] has emerged as a powerful optical technique for measuring the thickness and structures of ultrathin adsorbate layers and for evaluating affinity of biomolecules at metal films [9–24]. At a specific resonance angle, the absorption of incident light by a thin metal film causes collective oscillation of electrons (surface plasmons) in the film, which launches an evanescent wave into the dielectric layer adjacent to the metal film. The propagation of the evanescent wave decays exponentially away from the metal film and is thus significantly perturbed by the adsorption of a species at the metal film or changes in the adlayer structure. An attractive aspect of SPR in affinity binding assays and kinetic studies is that analytes of interest do not require labeling [5]. A major inherent limitation is that the selectivity of SPR is relatively low (i.e., any molecules that cause a change in the refractive index in the vicinity of the metal film or nonspecifically adsorb onto the surface can generate a SPR signal). For example, Tao and coworkers [25] and Zare and coworkers [26] used SPR for measuring heavy metals ions and achieved rather low detection levels. However, selective determination of a given metal ion cannot be achieved because the detection methods relied on the nonspecific, electrostatic interaction between metal cations and the carboxylic acid groups at the termini of preformed alkanethiol self-assembled monolayers (SAMs).
One of the strategies to afford selectivity to SPR sensing is to rely on biomolecular recognition. Along this line, biomolecules such as bacteria, enzymes, proteins, and DNA have been implemented for selective determinations of a variety of analytes [27, 28]. Among these biomolecules, metallothionein (MT), an intracellular metalloprotein that plays an important role in regulation of essential metals and detoxification of toxic metal ions, has been used as a probe for heavy metal ion detection. MT has the shape of a dumbbell and envelops metals in two separate domains. In the N-terminal β-domain (residues 1–30) of MT there are three-metal clusters and in the C-terminal α-domain (residues 31–61) there are four-metal clusters. One MT molecule can bind to 7 Cd ions or 7, 11 or 18 Hg ions [29–31]. The two domains can ligate metal ions strongly, with formation constants exceeding 1013 M−1 [32]. Moreover, the two domains have been reported to interact with metal ions differently [33] and there exists a binding hierarchy of MT towards different metal ions (e.g., Hg(II) >Ag(I) ≫ Cu(I) > Cd(II) >Zn(II) [34]). As a consequence, metal ions in MT molecules can be competitively displaced by other metal ions that have stronger affinities to MT. Therefore, these unique properties of MT provide one level of selectivity. Reconstitutions of unstructured metal-free MT or apo-MT molecules with metal ions result in protein folding or substantial conformation changes.
Detection of metal ion binding by MT or related molecules immobilized onto a sensor chip has been reported recently [35]. Saber and Piskin attached a hexapeptide (FT) onto a Au-coated quartz crystal and monitored metal ion binding using a quartz crystal microbalance [36]. Because the measurement was not conducted in real time, the assessment of the MT binding affinities toward different metal ions was not straightforward. Phytochelatin, the MT analog in plants, was used by Bontidean et al. for metal sensing [37]. Wu and Lin were the first to construct a MT-based SPR sensor to realize heavy metal ion determination [35]. In their work, MT molecules were covalently linked onto carboxymethylated dextran thin films and a commercial instrument (Biacore X) was successfully used to measure the signals associated with metal ion incorporation. Kinetic parameters of the metal ion binding by apo-MT were also determined. Although the dextran film is known to be effective in eliminating nonspecific protein adsorption, there are no inherent advantages of using such a film for trace metal analysis since metal ions in general do not adsorb non-discriminately onto sensor surface. Compared to preformed SAMs, the dextran film is relatively thick and non-uniform. Consequently could cause the metal ion incorporation into MT molecules to be measured at distances further away from the metal film where the evanescent field is weaker. This could partially explain why the limit of detection (LOD) reported (~2 μM) [35] was not particularly low. Furthermore, the work described in reference [35] displayed three separate calibration curves in different metal ion concentration ranges and did not reveal the domain-specific binding of MT molecules towards metal ions. We also noticed that the MT immobilization step was conducted at pH = 4.0. Although the surface density of immobilized MT is higher at a lower pH, an acidic environment is known to cause partial loss of the pristine metals in MT molecules [35, 38]. The loss of metal ions changes the MT structure and exposes different amino acid residues (e.g., the several lysine residues) that could interact with the sensor surface. It is possible that the relatively random MT surface orientations could be produced and would consequently complicate the SPR differentiation of metal binding between the two domains.
Recently, we have described the use of a flow injection highly sensitive SPR instrument [8, 25] (FI-SPR) for bioaffinity studies [23, 39] and its combination with electrochemistry for the investigation of redox-induced changes of molecular conformations [24, 40]. The high sensitivity of such a SPR instrument (down to femtomolar for DNA assays [39]) is resulted from the use of a bicell detector, which sensitively measures the movement of the SPR resonance angle at the sensor chip. In our more recent work based on coupling scanning electrochemical microscopy to SPR [40], we showed that a conformation change of immobilized cytochrome c as tiny as 0.27 ± 0.03 Å can be measured. Such a resolution is almost two orders of magnitude lower than the size of rabbit MT (3 nm in length and 1.5 nm in width [46]). We thus envision that such a highly sensitive SPR, used in tandem with a sensor surface at which the MT molecules adopt a uniform orientation, may allow us to determine metal ions by measuring the conformation changes of MT molecules during metal ion binding and to follow the domain-specific metal incorporation by immobilized MT molecules. In this work, we used a FI-SPR instrument equipped with a bicell detector and a commercial instrument based on a position-sensitive detector to achieve a metal ion detection level lower than obtained by similar studies [35, 36]. We demonstrate that the methodology can provide a complementary approach to well-established cost-effective techniques for trace metal analysis (e.g., anodic stripping voltammetry). We also monitored competitive metal binding/replacement in real-time to assess the MT binding affinity towards a specific metal ion.
2. Experimental
2.1 Materials and reagents
Metallothionein-2 (MT) isolated from rabbit liver, ethanolamine hydrochloride (AE), the pentapeptide (Ala-Ala-Ala-Ala-Ala, Ala5), and glycine were purchased from Sigma (St. Louis, MO). 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) and 11-mercaptoundecanoic acid (MUA) were obtained from Aldrich Chemicals (Milwaukee, WI). N-hydroxysulfosuccinimide (NHS) was acquired from Pierce (Rockford, IL) and high-purity HCl (99.999%) was obtained from Alfa Aesar (Ward Hill, MA). Dimethylsulfoxide (DMSO), K2HPO4·3H2O, KH2PO4, NaCl, CdCl2 and HgCl2 were all of AR grade and were purchased from either Aldrich Chemicals or Beijing Chemical Reagent Co. (Beijing, China). Water with resistivity of more than 18.2 MΩ·cm was collected from a Millipore Simplicity 185 system (Millipore Co.). MT (10.0 μM), EDC (0.4 M) and NHS (0.1 M) were freshly prepared in pH 7.4 PBS buffer (10.0 mM of K2HPO4·3H2O and KH2PO4 each in 1 mM NaCl), and MUA (4 mM) was prepared in DMSO. The stock solutions of HCl (0.1 M) and AE (1 M) were prepared in deionized H2O. Stock solutions of 5.0 mM CdCl2 or 5.0 mM HgCl2 were prepared in water and adjusted to pH 6.0 using 0.1 M HCl and then diluted to the desired concentrations. Glycine–HCl buffer (50 mM, pH 2.0) were used to release metal ions from MT. All experiments were performed at room temperature.
2.2 Procedure
BK7 glass cover slides (Fisher Scientific, Tustin, CA) were cleaned by exposing to ammonia solution (H2O2 (30%): NH4OH·HCl (saturated): H2O = 1:1:5 (V: V: V)) at 80 °C for 30 min. This step was followed by rinsing the surface with deionized water. After drying with N2, each glass slide was coated with a 2-nm thick chromium adhesive layer and then covered with a 50-nm thick gold film using a sputtering coater (Model 108 SE/auto, Kurt J. Lester Co., Clairton, PA).
Prior to use, the gold film was briefly annealed under a hydrogen flame to reduce surface contamination and to generate a smooth Au surface. The annealed gold film was immediately immerged in a 4 mM MUA solution overnight, followed by rinsing with DMSO and H2O and drying with N2. The gold film was then covered with a mixture of 0.4 M EDC, 0.1 M NHS, and 10 μM MT (30 μL each, pH ≅ 7.0) and kept in a humidified chamber for 3 h. Upon the MT attachment step, the surface was rinsed with PBS buffer and H2O and dried with N2. To block the unreacted sites, 40 μL of 0.1 M AE were spread onto the surface. The resultant film was either used immediately or stored 4°C for future use.
2.3 Flow injection-SPR apparatus
SPR measurements were conducted either with a home-built flow injection-SPR equipped with a bicell detector [8, 23] or a BI-1000 SPR system (Biosensing Instrument, Tempe, AZ) that uses a position-sensitive device for high-resolution SPR measurements. The sensitivity of the BI-1000 SPR system is about 3–4 times less sensitive than the bicell counterpart but was found to be sufficient for the measurements in this work. The inlet of the flow cell is connected to a six-port Cheminert valve (VICI, Houston, TX). For each measurement, the sample solution was preloaded into a 50-μL loop with a microsyringe (Fisher) and delivered to the flow cell by a Genie Plus syringe pump (Kent Scientific, Torrington, CT). Degassed water was used as the carrier solution. Once a stable baseline signal had been obtained, the glycine–HCl buffer (50 mM, pH 2.0) was injected to remove metal ions coordinated by MT. In our work, all the SPR dip shifts have been converted to degrees using the ethanol calibration method [8, 41].
3. Results and discussion
3.1 MT immobilization
MT molecules can bind to the Au surface through either weak, nonspecific adsorption via their N-termini [42] or covalent attachment of the thiol groups on any free cysteine residues. However, these processes do not allow MT to attach to the Au surface in a predictable and well-organized fashion [7]. Figure 1 depicts the MT conformation change induced by metal ion binding. Briefly, MT molecules are immobilized onto preformed MUA SAMs. The immobilization is realized through the widely used EDC/NHS cross-linking reaction [43], which connects the N-terminus of the MT molecule to the carboxyl end group at the SAM. We should point out that it is also possible that the MT attachment might occur at the lysine residues. Among the lysine residues, Lys 30 and Lys 31 have been shown to be more effective for surface attachment [44]. However, given the steric hindrance (Lys 30 and Lys 31 are close to the cleft of the MT molecule), it is more likely that the MT molecule is anchored at its N-terminus. Advantages of this immobilization scheme include simplicity of the procedure, compactness of the preformed MUA layer, and attachment of MT molecules without a serious loss of their inherent metals. These advantages lead to the formation of MT films with a more uniform orientation. The variation of hydration in the compact MUA film, which affects the refractive index of the film, should also be less than the dextran film. We should mention that the scheme given in Figure 1 leads to sub-monolayer coverage, whereas at the dextran film multilayers of MT might be deposited. As mentioned in the Introduction, the dextran film is more extended, providing a greater accessibility to the MT attachment and possessing a larger number of reactive sites. While multilayered apo-MT films are expected to incorporate more metal ions, we will show that the combination of our SPR and the uniformity of the MT film actually resulted in a lower detection level (vide infra). Perhaps the most interesting result obtained at a uniform and sub-monolayered MT film is that metal ion incorporation by the two MT domains can be differentiated.
Fig. 1.
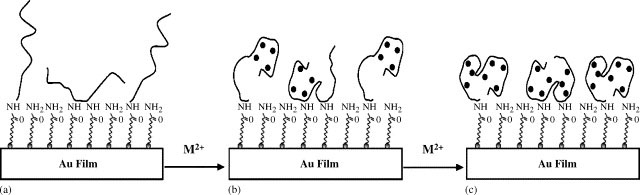
Schematic representation of the conformation changes of apo-MT brought by metal binding. The filled circles in (b) and (c) represent the metal ions (M2+). Notice that the predominant configuration is the attachment of MT molecules via their N-termini, although the linkage via the lysine residues cannot be excluded. Conformation changes associated with metal ion incorporations by the α (b) and β (c) domains of MT are represented in a stepwise fashion.
To evaluate the MT surface coverage, we injected a mixture solution of MT, NHS, and EDC into the flow cell housing a MUA-covered Au sensor chip. The SPR dip shift was determined to be about 0.05 degree. It was reported that the SPR dip shift by 1 degree corresponds to 10 ng/mm2 of protein and is independent of the protein size [10]. We thus deduced the MT surface density (Preal) to be about 0.5 ng/mm2 or 7.7 pmol/cm2. The maximal surface density (Pmax, in ng/mm2) for a close-packed monolayer can be calculated by the following equation [45]:
where MW is the molecular weight, NA is the Avogadro’s number, and A is the surface area occupied by a protein molecule. The surface coverage can be obtained by the following relationship [46]:
The MT surface area is about 4.5 nm2 [47]. Thus, the surface density and surface coverage of MT are 3.0 ng/mm2 and 16%, respectively. The actual films should have a higher coverage since the Au film was exposed to the MT solution for a time (3 h) that is much longer than that for a typical injection (3 min). Nevertheless, the coverage is most likely less than a monolayer, given the steric hindrance and electrostatic repulsion imposed by the adjacent protein molecules [34, 48].
3.2 Real-time SPR monitoring of Cd2+ and Hg2+ incorporation by apo-MT
As mentioned in the Introduction, many metal ions can interact with MT molecules to form metal-thiolate bonds. Metals inherent in MT molecules can also be released under certain conditions. For example, metal ions can be replaced by protons at a low pH value [42] and be transferred to an apo-metalloenzyme under an oxidized condition [49]. The binding between MT and metal ions was demonstrated to be reversible [38, 50].
It is reported that glycine can chelate metal ions and replace the metal-coordination sites in proteins with protons [51]. Because trace metals in a solution can readily bind to apo-MT and the solution pH affects the apo-MT binding to metal ions, deionized and boiled H2O was used as the carrier solution. Similarly, the metal standards were prepared with boiled, deionized H2O. We chose neutral pH because metal-MT complexes might be dissociated below pH 6.0 and above pH 7.7 [35, 38]. Curve a in Figure 2 shows a typical SPR sensorgram. In acquiring this sensorgram, glycine–HCl buffer was first injected to release the pristine metal ions in the immobilized MT molecules. The difference in the baseline SPR angles before and after the injection of the Cd2+ solution (termed as the SPR dip shift,Δθ) is about 0.00028°. To confirm that the net change is indeed due to the folding of the apo-MT back to the MT structure, we conducted a control experiment in which a pentapeptide, Ala5, was immobilized onto a sensor chip. As can be seen in curve b, essentially little SPR dip shift was observed after injecting a Cd2+-containing solution into the flow cell, suggesting that there was no significant surface reorientation of the Ala5 film. Furthermore, since alkanethiol SAM bearing carboxylic acid end groups have been used for SPR measurements of heavy metal ions such as Pb2+ and Hg2+ [36,39], the recovery of the original baseline upon the elution of Cd2+ in curve b also suggests that the negatively charged carboxyl groups on the MUA SAM had likely been converted to neutral amide groups during the peptide or protein immobilization. Such a conversion annihilates the negative charges (i.e., deprotonated carboxylic acid groups) at the surface and reduces electrostatic attraction to metal ions in solution.
Fig. 2.
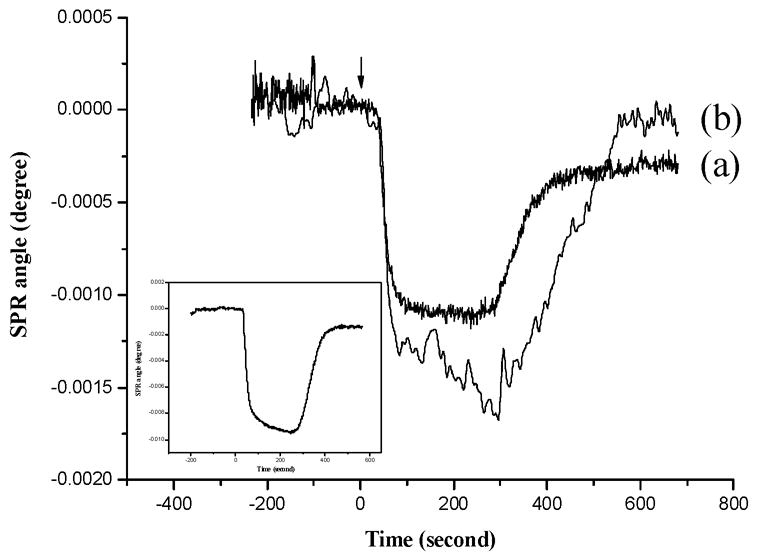
A representative SPR sensorgram corresponding to the incorporation of Cd2+ (0.13 μM) by immobilized apo-MT (curve a). Prior to the incorporation of Cd2+, the pristine metals in the immobilized MT molecules were first released by injecting a glycine–HCl buffer solution (sensorgram shown in the inset). Curve b was acquired in a similar fashion as curve a except that an Ala5-modified Au sensor chip was used. Degassed, boiled water was used as the carrier solution. A flow rate of 1 mL/h and a 50-μL sample loop were used. The arrow indicates the time when the injection was made.
Consistent with other reports [28, 51], we found that glycine–HCl can effectively remove metals from surface-confined MT molecules. The exposure of a MT-covered sensor to glycine–HCl caused a net change by 0.0014° (inset of Figure 2). Such a change can be ascribed to the metal loss and the conformation change in the MT molecules. The second injection of glycine –HCl did not result in any observable SPR dip shift, suggesting that one injection was sufficient to remove all the metals in the MT molecules. Therefore, to avoid excessive disruption of the MT adsorbate layer, the glycine–HCl buffer was injected only once for all the experiments described in this work.
The SPR dip shift is directly correlated with the Cd2+ concentration ([Cd2+]). The calibration plot in Figure 3a contains two regions, viz., from 0.13 to 10 μM (Region 1) and from 10 to 200 μM (Region 2). Linear regression analysis of both regions yielded the following equations with both correlation coefficients R2 greater than 0.97.
Fig. 3.
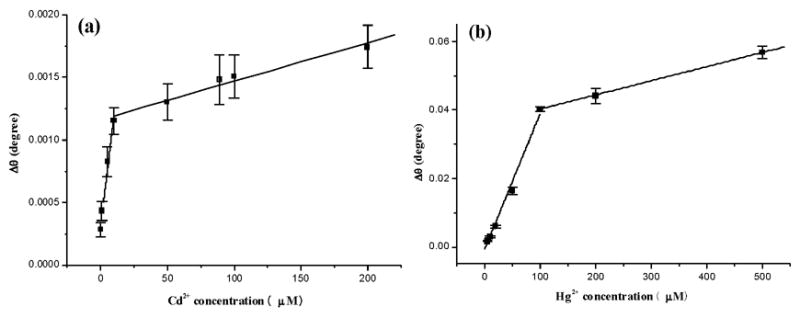
Calibration curves for the measurements of Cd2+ (panel a) and Hg2+ (panel b) using MT-covered Au sensor chips. Concentrations of metal ions injected were 0.13, 1.0, 5.0, 10, 50, 89, 100, 200 μM for Cd2+ and 5, 10, 20, 50, 100, 200, 500 μM for Hg2+. Each concentration was repeated at least three times and the standard deviations are between 9.6% and 18.6% for the Cd2+ calibration curve and range between 1.9% and 11.6% for the Hg2+ calibration curve.
| (Region 1) |
| (Region 2) |
The slope of the plot in the first region is much higher than that in the second region. This reveals that, as the metal ion concentration increases, the extent of conformation change becomes less. A plausible explanation is that the greater binding affinity of the MT α domain is responsible for the ligation of metal ions present at low concentrations (cf. Figure 1). When the metal ion concentration is sufficiently high, the excess metal ions begin to interact with the β domain, producing a smaller slope. Our contention is supported by the observation of Stillman et al. who showed that metals such as Cd2+ bind preferentially to the MT α domain [52]. Moreover, the α domain can coordinate four Cd2+ ions, whereas the β domain holds only three divalent metal ions. Thus, the conformation change brought by metal coordination in the α domain should be greater. Such a preferential binding and the higher number of metal ions coordinated jointly produced the steeper slop in the first region of the calibration curve. We should note that, in the work by Wu and Lin [35], there are three plots constructed for different concentration regions. Although the slopes of these three plots are all different, the discrepancies do not appear to be significant and these researchers concluded that the SPR signal was proportional to the metal ion concentration for all the concentrations studied. We think that the variations in the slopes of the three plots are probably originated from the metal ion incorporation into the two domains of MTs, as well as into apo-MT molecules with different surface orientations. These complications make the differential binding of metal ions to the two MT domains less pronounced. As a consequence, the binding characteristics of the two domains were observed as averaged results.
Another noteworthy point is that, in spite of the smaller number of immobilized MT molecules, our SPR experiments actually measured lower metal concentrations (for Cd2+, approximately 20 times less than that reported in Ref [35]). The detection level (0.1 μM or 15 ppb) is sufficiently low for most applications and compares well with many conventional trace metal analysis (e.g., atomic absorption [1] and anodic stripping voltammetry [3]). Although the SPR instruments used in this work are 1–2 orders of magnitude more sensitive than that used in Ref [35], we think that the predominant reason for the lower detection level is the immobilization procedure. The thinner SAM for anchoring MT molecules keeps the most pronounced portion of the evanescent wave closer to the metal film, allowing the MT conformation to be measured sensitively.
Similar trends were also found for the incorporation of Hg2+ by apo-MT molecules. The two regions in the calibration curve range from 5 to 100 μM and 100 to 500 μM, respectively. The following two equations, with correlation coefficients R2 > 0.98, again suggest good linearity for the relationship between the SPR dip shift and the Hg2+ concentration.
| (Region 1) |
| (Region 2) |
Both slopes are higher than their Cd2+ counterparts. This is understandable given that Hg2+ binds to MT most strongly (vide supra). The higher slopes also indicate that the sensitivity of the SPR measurements for Hg2+ should be greater than that for Cd2+. However, injecting solutions containing [Hg2+] lower than that shown in Figure 3b did not produce a linear relation. It is not clear what factors contributed to this deviation.
The difference between Cd2+ and Hg2+ in their interactions with apo-MT molecules is more obvious in a competitive binding assay. In such an assay, Cd2+ and Hg2+ solutions were alternatively injected (Figure 4). When a solution containing Hg2+ was first injected, the follow-up injection of a Cd2+-containing solution did not cause any appreciable SPR dip shift. This suggests that Hg2+ ions incorporated by the apo-MT molecules are strongly retained and cannot be replaced by Cd2+. On the other hand, when a Cd2+ solution was first injected, Hg2+ ions introduced to the SPR flow cell in the subsequent injection appeared to be able to replace Cd2+ and produced a rather pronounced SPR dip shift (inset). It is known that an apo-MT molecule is capable of binding 7 Cd2+ or 7, 11 or 18 Hg2+ [29–31]. All binding sites in the two domains may be occupied, and each domain in a single MT molecule may be filled by different metal ions (e.g., Zn2Cd5-MT). Despite the large thermodynamic stability of metal-MT complexes, metal ions present in MT molecules are kinetically labile. The intramolecular and intermolecular metal exchange could be fast [53, 54] and intrasite and intersite metal exchanges of Zn and Cd in MT have been reported [55–57]. Note that the SPR dip shift corresponding to the exposure of the Cd-reconstituted MT to 100 μM Hg2+ (Figure 4) was smaller than that corresponding to the interaction of Hg2+ with apo-MT (approximately 27% less. Data not shown). This suggests that Hg2+ only partially replaced the Cd2+ ions and is consistent with results reported by Jaw and Jeffrey [58]. Our data are consistent with the findings that MT molecules can selectively coordinate Cd2+ in the presence of Ni2+ and Zn2+ [35]. We therefore did not further measure binding of Hg2+ by apo-MT in the presence of Cd2+. Our experimental results demonstrate that FI-SPR can be used to gauge binding affinities of MT towards different metal ions in real time.
Fig. 4.
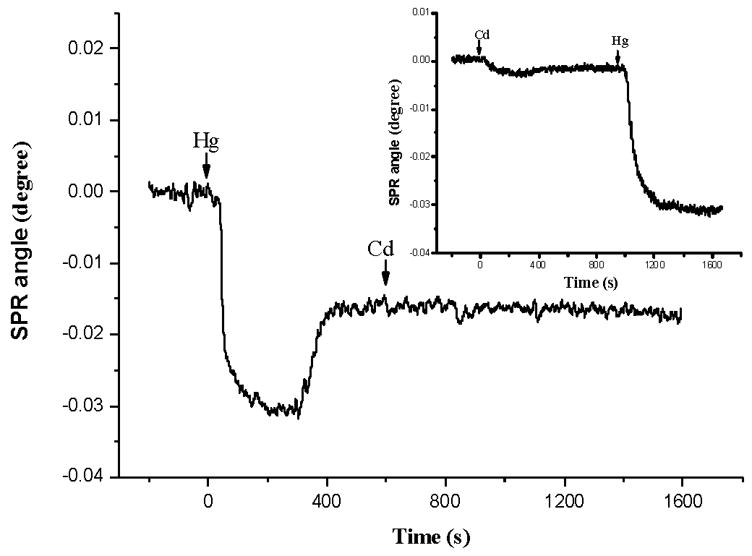
A SPR sensorgram corresponding to two consecutive injections of a 100 μM Hg2+ solution (apo-MT reconstitution) and a 100 μM Cd2+ solution (metal ion replacement). Glycine–HCl buffer was first injected to remove the metal ions in the MT molecules. Inset: upon metal release by the glycine–HCl treatment, the apo-MT was first reconstituted with Cd2+ by injecting 50 μM Cd2+. This was then followed by injecting 50 μM Hg2+ for the replacement study. Arrows indicate the times when injections were made.
In an effort to evaluate the extent of metal incorporation by surface-confined MT molecules, we conducted continuous injections of a given metal ion solution into a flow cell housing an apo-MT-covered sensor chip. Figure 5 displays the time-resolved SPR responses upon consecutive injections of Hg2+ solutions of two different concentrations. As can be seen, the response is dependent on the Hg2+ concentration, with a higher Hg2+ concentration saturating the binding sites more rapidly than the lower one. When the concentration is relatively high (e.g., 200 μM), two consecutive injections appear to be sufficient to coordinate metal ions with all the free sulphydryl groups in the MT molecules. Wu and Lin reported similar findings, though the concentration for saturating the binding sites was higher (~10 mM for Cd2+ [35]).
Fig. 5.
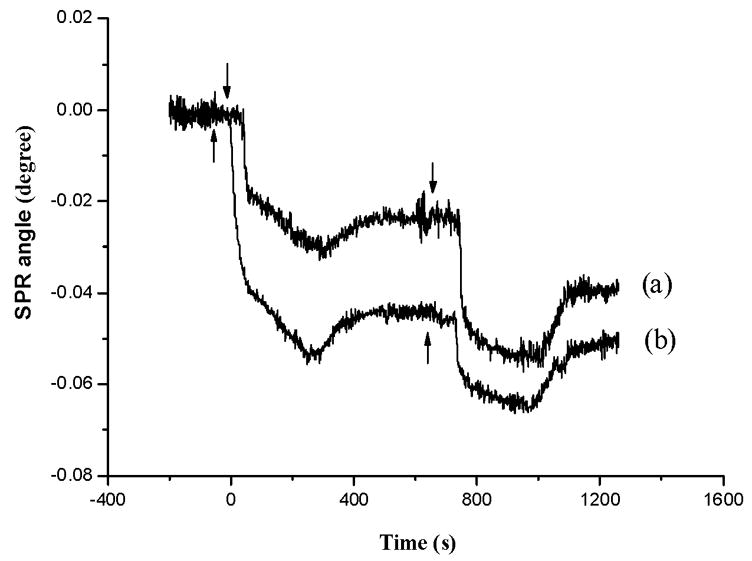
SPR sensorgrams corresponding to continuous injections of solutions containing (a) 50 μM Hg2+ and (b) 200 μM Hg2+. Arrows indicate the times when injections were made.
3.3 Determination of apparent affinity constants for Cd2+ and Hg2+ binding
The plot of the SPR dip shift vs. Cd2+ or Hg2+ concentration is analogous to a Langmuir adsorption isotherm. Therefore, the binding process can be considered as an adsorption equilibrium at the MT-coated sensor chip. Affinity constants (K) for Cd2+ and Hg2+ binding to MT can be estimated with the following equation [25]:
where Δθsat is the SPR dip shift corresponding to the maximal conformation change (or surface coverage), and [M2+] is the metal ion concentration. The binding equilibrium is given by:
It must be noted that, instead of at equilibrium, the process monitored by SPR could be still under kinetic control. So the affinity constant (K) is the “apparent stability constant” [25]. The value of K for Cd2+ binding by MT was estimated to be 4.2×105 M−1, which is in excellent agreement with that reported in reference [35] (4.7×105 M−1) but significantly lower than the apparent stability constant for rabbit Cd–MT2 in solution (6–12.5×1014 M−1 [59, 60]). Wu and Lin suggested that this deviation may be due to the difference in conformations between immobilized MT molecules and MT molecules in solution [35].
The stability constant for the Hg-MT complex was estimated to be 2.7×103 M−1. This is somewhat surprising since it is about two orders of magnitude lower than that of Cd-MT. Possible reasons are that the Hg-MT complex formation is a much more complicated process [61] and the binding stoichiometry is dependent on the metal ion concentration and the specific MT isoform [62]. At issue is whether the Hg-MT follows a pseudo first-order chemical reaction as assumed by the commonly used Langmuir isotherm. We should stress that this value is not entirely unreasonable, since a constant of 4.0×105 M−1 had been measured electrochemically [63].
4. Conclusions
In this work, MT molecules were cross-linked onto preformed self-assembled monolayers and the sensor chips were treated with glycine–HCl right before heavy metal ion analyses. Our work demonstrates that the conformation changes of apo-MT brought by the incorporation of heavy metal ions can be readily detected by highly sensitive FI–SPR equipped with a bicell detector or a position-sensitive detector. Due in large to the formation of compact MT films with a uniform MT surface orientation, a low concentration detection level (~ 0.1 μM or 15 ppb for Cd2+ and 5 μM or 1 ppm for Hg2+) was obtained and compares well with those offered by conventional trace metal analysis techniques. The calibration curves exhibited two linear regions with different slopes, suggesting that FI-SPR could resolve the separate binding processes involving the two MT domains. Continuous injections of solutions containing the same metal ion allowed us to gauge the extent of metal ion incorporation by surface-confined apo-MT molecules, whereas consecutive injections of solutions comprising different metal ions confirmed the binding hierarchy of MT towards heavy metals. Good linearity and reproducibility were obtained for the determinations of Cd2+ and Hg2+. Owing to the compact and versatile design of the instrument and the simplicity of the immobilization scheme, our approach should be complementary to other existing methods for trace metal analysis.
Acknowledgments
We gratefully acknowledge partial support of this work by the National Natural Science Foundation of China (No. 20225517), the Cultivation Fund of the Ministry of Education of China (No. 704036), a NIH-SCORE Subproject (GM 08101), and a NSF-RUI grant (0555224).
Biographies
Yintang Zhang received his M. S. degree in nonferrous metal metallurgy at Changsha Research Institute of Mining and Metallurgy (Changsha, P. R. China) in 2004. He is pursing a Ph. D. degree in Chemistry under the supervision of Professors Feimeng Zhou and Maotian Xu.
Maotian Xu obtained his PhD at Northwestern University (Xi’an, P. R. China) in 2004. He is currently a Professor in the College of Chemistry and Chemical Engineering at Central South University (Changsha, P. R. China). His research interests are on chemically modified electrodes and studies of protein/drug interactions.
Yanju Wang obtained her PhD in Polymer Physics & Chemistry at Changchun Institute of Applied Chemistry (Changchun, P. R. China) in 2002. She is currently a postdoctoral research associate at California State University, Los Angeles (CSULA). Her research interests include SPR, conducting polymers, and optical spectroscopy.
Freddy Toledo obtained his B. S. in Physics at the University of California-Berkeley in 2002 and conducted his graduate studies under the supervision of Dr. Feimeng Zhou. He is currently pursuing his Ph. D. degree at the University of California-Irvine. His research interests are on SPR, scanning tunneling microscopy, and surface chemistry.
Feimeng Zhou obtained his PhD in Chemistry at University of Texas at Austin (USA) in 1993. He is currently a full professor in the Department of Chemistry at CSULA and an adjunct professor at Central South University. He is a recipient of the Dreyfus Teacher-Scholar Award and serves as the director of the Biomedical Research and Infrastructure Program at CSULA. Prof. Zhou has published more than 60 papers. He is conducting research on bioanalytical chemistry, surface analysis, protein structure/function studies, and analytical instrumentation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Skoog DA, Holler FJ, Nieman TA. Principles of instrumental analysis. 5. Saunders College Publishing; Philadelphia: 1992. pp. 206–229. [Google Scholar]
- 2.Taylor HE. Inductively coupled plasma mass spectrometry: Practices and techniques. Academic Press; San Diego: 2001. [Google Scholar]
- 3.Bard AJ, Faulkner LR. Electrochemical methods: Fundamentals and applications. 2. John Wiley & Sons; New York: 2000. pp. 458–464. [Google Scholar]
- 4.Wang J. Stripping analysis: Principles, instrumentation, and applications. VCH; Deerfield: 1985. [Google Scholar]
- 5.Rothenhausler B, Knoll W. Surface-plasmon microscopy. Nature. 1988;332:615–617. [Google Scholar]
- 6.Hanken DG, Jordan CE, Frey BL, Corn RM. In: Electroanalytical Chemistry: A Series of Advances. Bard AJ, Rubenstein I, editors. Vol. 20. Marcel Dekker; New York: 1998. pp. 141–225. [Google Scholar]
- 7.Homola J, Yee SS, Gauglitz G. Surface plasmon resonance sensors: Review. Sens Actuators, B, Chem. 1999;54:3–15. [Google Scholar]
- 8.Tao NJ, Boussaad S, Huang WL, Arechabaleta RA, J DA. High resolution surface plasmon resonance spectroscopy. Rev Sci Instrum. 1999;70:4656–4660. [Google Scholar]
- 9.Piscevic D, Lawall R, Vieth M, Liley M, Okahata Y, Knoll W. Oligonucleotide hybridization observed by surface plasmon optical techniques. Appl Surf Sci. 1995;90:425–436. [Google Scholar]
- 10.Su X, Wu YJ, Knoll W. Comparison of surface plasmon resonance spectroscopy and quartz crystal microbalance techniques for studying DNA assembly and hybridization. Biosens Bioelectron. 2005;21:719–726. doi: 10.1016/j.bios.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 11.Nelson BP, Grimsrud TE, Liles MR, Goodman RM, Corn RM. Surface plasmon resonance imaging measurements of DNA and RNA hybridization adsorption onto DNA microarrays. Anal Chem. 2001;73:1–7. doi: 10.1021/ac0010431. [DOI] [PubMed] [Google Scholar]
- 12.Goodrich TT, Lee HJ, Corn RM. Direct detection of genomic DNA by enzymatically amplified SPR imaging measurements of RNA microarrays. J Am Chem Soc. 2004;126:4086–4087. doi: 10.1021/ja039823p. [DOI] [PubMed] [Google Scholar]
- 13.Peterlinz KA, Georgiadis RM, Herne TM, Tarlov MJ. Observation of hybridization and dehybridization of thiol-tethered DNA using two-color surface plasmon resonance spectroscopy. J Am Chem Soc. 1997;119:3401–3402. [Google Scholar]
- 14.Wolf LK, Fullenkamp DE, Georgiadis RM. Quantitative angle-resolved SPR imaging of DNA-DNA and DNA-drug kinetics. J Am Chem Soc. 2005;127:17453–17459. doi: 10.1021/ja056422w. [DOI] [PubMed] [Google Scholar]
- 15.Zayats M, Kharitonov AB, Pogorelova SP, Lioubashevski O, Katz E, Willner I. Probing photoelectrochemical processes in Au-CdS nanoparticle arrays by surface plasmon resonance: Application for the detection of acetylcholine esterase inhibitors. J Am Chem Soc. 2003;125:16006–16014. doi: 10.1021/ja0379215. [DOI] [PubMed] [Google Scholar]
- 16.He L, Musick MD, Nicewarner SR, Salinas FG, Benkovic SJ, Natan MJ, Keating CD. Colloidal Au-enhanced surface plasmon resonance for ultrasensitive detection of DNA hybridization. J Am Chem Soc. 2000;122:9071–9077. [Google Scholar]
- 17.Bates PJ, Dosanjh HS, Kumar S, Jenkins TC, Laughton CA, Neidle S. Detection and kinetic studies of triplex formation by oligodeoxynucleotides using real-time biomolecular interaction analysis (BIA) Nucleic Acids Res. 1995;23:3627–3632. doi: 10.1093/nar/23.18.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Phillips KS, Han JH, Martinez M, Wang Z, Carter D, Cheng Q. Nanoscale glassification of gold substrates for surface plasmon resonance analysis of protein toxins with supported lipid membrane. Anal Chem. 2006;78:596–603. doi: 10.1021/ac051644y. [DOI] [PubMed] [Google Scholar]
- 19.Wilkop T, Wang Z, Cheng Q. Analysis of μ-contact printed protein patterns by SPR imaging with a led light source. Langmuir. 2004;20:11141–11148. doi: 10.1021/la048177k. [DOI] [PubMed] [Google Scholar]
- 20.Kang X, Jin Y, Cheng G, Dong S. Surface plasmon resonance studies on the electrochemical doping/dedoping processes of anions on polyaniline-modified electrode. Langmuir. 2002;18:10305–10310. [Google Scholar]
- 21.Kai E, Sawata S, Ikebukuro K, Iida T, Honda T, Karube I. Detection of PCR products in solution using surface plasmon resonance. Anal Chem. 1999;71:796–800. doi: 10.1021/ac9807161. [DOI] [PubMed] [Google Scholar]
- 22.Fagerstam LG, Frostell-Karlsson A, Karlsson R, Persson B, Ronnberg I. Biospecific interaction analysis using surface plasmon resonance detection applied to kinetic, binding site and concentration analysis. J Chromatogr. 1992;597:397–410. doi: 10.1016/0021-9673(92)80137-j. [DOI] [PubMed] [Google Scholar]
- 23.Song F, Zhou F, Wang J, Tao N, Lin J, Vellanoweth RL, Morquecho Y, Wheeler-Laidman J. Detection of oligonucleotide hybridization at femtomolar level and sequence-specific gene analysis of the Arabidopsis thaliana leaf extract with an ultrasensitive surface plasmon resonance spectrometer. Nucleic Acids Res. 2002;30:e72. doi: 10.1093/nar/gnf072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yao X, Wang J, Zhou F, Wang J, Tao N. Quantification of redox-induced thickness changes of 11-ferrocenylundecanethiol self-assembled monolayers by electrochemical surface plasmon resonance. J Phys Chem B. 2004;108:7206–7212. [Google Scholar]
- 25.Forzani ES, Zhang H, Chen W, Tao N. Detection of heavy metal ions in drinking water using a high-resolution differential surface plasmon resonance sensor. Environ Sci Technol. 2005;39:1257–1262. doi: 10.1021/es049234z. [DOI] [PubMed] [Google Scholar]
- 26.Chah S, Yi J, Zare RN. Surface plasmon resonance analysis of aqueous mercuric ions. Sens Actuators, B, Chem. 2004;99:216–222. [Google Scholar]
- 27.Gooding JJ, Chow E, Finlayson R. Biosensor for detecting metal ions: New trends. Aust J Chem. 2003;56:159–162. [Google Scholar]
- 28.Gooding JJ, Hibbert DB, Yang W. Electrochemical metal ion sensors. Exploiting amino acids and peptides as recognition elements. Sensors. 2001;1:75–90. [Google Scholar]
- 29.Vallee BL. In: Methods in Enzymology. Riordan JF, Vallee BL, editors. Vol. 205. Academic Press; New York: 1991. pp. 3–7. [Google Scholar]
- 30.Kagi JH. In: Methods in Enzymology. Riordan JF, Vallee BL, editors. Vol. 205. Academic Press; New York: 1991. pp. 613–626. [Google Scholar]
- 31.Stillman MJ. Metallothioneins. Coordin Chem Rev. 1995;144:461–511. [Google Scholar]
- 32.Otvos JD, Petering DH, Shaw CF. Structure-reactivity relationships of metallothionein, a unique metal-binding protein. Comments Inorg Chem. 1989;1:1–35. [Google Scholar]
- 33.Stillman MJ, Zelazowski AJ. Domain specificity in metal binding to metallothionein. A circular dichroism and magnetic circular dichroism study of cadmium and zinc binding at temperature extremes. J Biol Chem. 1988;263:6128–6133. [PubMed] [Google Scholar]
- 34.Hamer DH. Metallothionein. Ann Rev Biochem. 1986;55:913–951. doi: 10.1146/annurev.bi.55.070186.004405. [DOI] [PubMed] [Google Scholar]
- 35.Wu CM, Lin LY. Immobilization of metallothionein as a sensitive biosensor chip for the detection of metal ions by surface plasmon resonance. Biosens Bioelectron. 2004;20:864–871. doi: 10.1016/j.bios.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 36.Saber R, Piskin E. Investigation of complexation of immobilized metallothionein with Zn(II) and Cd(II) ions using piezoelectric crystals. Biosens Bioelectron. 2003;18:1039–1046. doi: 10.1016/s0956-5663(02)00217-8. [DOI] [PubMed] [Google Scholar]
- 37.Bontidean I, Ahlqvist J, Mulchandani A, Chen W, Bae W, Mehra RK, Mortari A, Csoregi E. Novel synthetic phytochelatin-based capacitive biosensor for heavy metal ion detection. Biosens Bioelectron. 2003;18:547–553. doi: 10.1016/s0956-5663(03)00026-5. [DOI] [PubMed] [Google Scholar]
- 38.Muñoz J, Baena JR, Gallego M, Valcárcel M. Development of a method for the determination of inorganic cadmium and cadmium metallothioneins in fish liver by continuous preconcentration on fullerene and flame atomic absorption spectrometry. J Anal At Spectrom. 2002;17:716–720. [Google Scholar]
- 39.Yao X, Li X, Toledo F, Zurita-Lopez C, Gutova M, Momand J, Zhou F. Sub-attomole oligonucleotide and p53 cDNA determinations via a high-resolution surface plasmon resonance combined with oligonucleotide-capped gold nanoparticle signal amplification. Anal Biochem. 2006;354:220–228. doi: 10.1016/j.ab.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 40.Xiang J, Guo J, Zhou F. Scanning electrochemical microscopy combined with surface plasmon resonance: Studies of localized film thickness variations and molecular conformation changes. Anal Chem. 2006;78:1418–1424. doi: 10.1021/ac051601h. [DOI] [PubMed] [Google Scholar]
- 41.Kolomenskii AA, Gershon PD, Schuessler HA. Sensitivity and detection limit of concentration and adsorption measurements by laser-induced surface-plasmon resonance. Appl Optics. 1997;36:6539–6547. doi: 10.1364/ao.36.006539. [DOI] [PubMed] [Google Scholar]
- 42.Chan J, Huang Z, Merrifield ME, Salgado MT, Stillman MJ. Studies of metal binding reactions in metallothioneins by spectroscopic, molecular biology, and molecular modeling techniques. Coordin Chem Rev. 2002;233–234:319–339. [Google Scholar]
- 43.Huang E, Zhou F, Deng L. Studies of surface coverage and orientation of DNA molecules immobilized onto preformed alkanethiol self-assembled monolayers. Langmuir. 2000;16:3272–3280. [Google Scholar]
- 44.Casero E, Vázquez L, Martín-Benito J, Morcillo MA, Lorenzo E, Pariente F. Immobilization of metallothionein on gold/mica surfaces: Relationship between surface morphology and protein-substrate interaction. Langmuir. 2002;18:5909–5920. [Google Scholar]
- 45.Lahiri J, Isaacs L, Grzybowski B, Carbeck JD, Whitesides GM. Biospecific binding of carbonic anhydrase to mixed SAMs presenting benzenesulfonamide ligands: A model system for studying lateral steric effects. Langmuir. 1999;15:7186–7198. [Google Scholar]
- 46.Simonian AL, Revzin A, Wild JR, Elkind J, Pishko MV. Characterization of oxidoreductase-redox polymer electrostatic film assembly on gold by surface plasmon resonance spectroscopy and Fourier transform infrared-external reflection spectroscopy. Anal Chim Acta. 2002;466:201–212. [Google Scholar]
- 47.Davis JJ, Hill HAO, Kurz A, Jacob C, Maret W, Vallee BL. A scanning tunneling microscopy study of rabbit metallothionein. PhysChemComm. 1998;1:12–22. [Google Scholar]
- 48.Yuan Y, Oberholzer MR, Lenhoff AM. Size does matter: Electrostatically determined surface coverage trends in protein and colloid adsorption. Coll Surf A. 2000;165:125–141. [Google Scholar]
- 49.Jacob C, Maret W, Vallee BL. Control of zinc transfer between thionein, metallothionein, and zinc proteins. Proc Nati Acad Sci USA. 1998;95:3489–3494. doi: 10.1073/pnas.95.7.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.CanoGauci DF, Sarkar B. Reversible zinc exchange between metallothionein and the estrogen receptor zinc finger. FEBS Lett. 1996;386:1–4. doi: 10.1016/0014-5793(96)00356-0. [DOI] [PubMed] [Google Scholar]
- 51.Bontidean I, Berggren C, Johansson G, Csoregi E, Mattiasson B, Lloyd JR, Jakeman KJ, Brown NL. Detection of heavy metal ions at femtomolar levels using protein-based biosensors. Anal Chem. 1998;70:4162–4169. doi: 10.1021/ac9803636. [DOI] [PubMed] [Google Scholar]
- 52.Stillman MJ, Cai W, Zelazowski AJ. Cadmium binding to metallothioneins. Domain specificity in reactions of alpha and beta fragments, apometallothionein, and zinc metallothionein with Cd2+ J Biol Chem. 1987;262:4538–4548. [PubMed] [Google Scholar]
- 53.Zangger K, Shen G, Oz G, Otvos JD, Armitage IM. Oxidative dimerization in metallothionein is a result of intermolecular disulphide bonds between cysteines in the alpha-domain. Biochem J. 2001;359:353–360. doi: 10.1042/0264-6021:3590353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vallee BL, Maret W. In: Metallothionein III Birkhauser. Suzuki KT, Imuira N, Kimura M, editors. Verlag; Basel: 1993. pp. 1–27. [Google Scholar]
- 55.Otvos JD, Engeseth HR, Nettesheim DG, Hilt CR. Interprotein metal exchange reactions of metallothionein. Experientia Suppl. 1987;52:171–178. doi: 10.1007/978-3-0348-6784-9_10. [DOI] [PubMed] [Google Scholar]
- 56.Romero-Isart N, Jensen LT, Zerbe O, Winge DR, Vasak M. Engineering of metallothionein-3 neuroinhibitory activity into the inactive isoform metallothionein-1. J Biol Chem. 2002;277:37023–37028. doi: 10.1074/jbc.M205730200. [DOI] [PubMed] [Google Scholar]
- 57.Jiang LJ, Vasak M, Vallee BL, Maret W. Zinc transfer potentials of the α- and β-clusters of metallothionein are affected by domain interactions in the whole molecule. Proc Natl Acad Sci USA. 2000;97:2503–2508. doi: 10.1073/pnas.97.6.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jaw S, Jeffery EH. Role of metallothionein in biliary metal excretion. J Toxicol Env Health. 1989;28:39–51. doi: 10.1080/15287398909531327. [DOI] [PubMed] [Google Scholar]
- 59.Muñoz A, Rodríguez AR. Electrochemical behavior of metallothioneins and related molecules. Part III: metallothionein. Electroanalysis. 1995;7:674–680. [Google Scholar]
- 60.Wang Y, Mackay EA, Kurasaki M, Kagi JHR. Purification and characterisation of recombinant sea urchin metallothionein expressed in Escherichia coli. Eur J Biochem. 1994;225:449–457. doi: 10.1111/j.1432-1033.1994.00449.x. [DOI] [PubMed] [Google Scholar]
- 61.Jiang DT, Heald SM, Sham TK, Stillman MJ. Structures of the cadmium, mercury, and zinc thiolate clusters in metallothionein: XAFS Study of Zn7-MT, Cd 7-MT, Hg7-MT, and Hg18-MT formed from rabbit liver metallothionein 2. J Am Chem Soc. 1994;116:11004–11013. [Google Scholar]
- 62.Cai W, Stillman MJ. Hg18-Metallothionein. J Am Chem Soc. 1988;110:7872–7873. [Google Scholar]
- 63.Ju H, Leech D. Electrochemical study of a metallothionein modified gold disk electrode and its action on Hg2+ cations. J Electroanal Chem. 2000;484:150–156. [Google Scholar]