

Abstract
HIV encephalitis (HIVE) is a neurodegenerative disease seen in approximately one in four terminally infected patients. Macaques infected with the simian immunodeficiency virus develop encephalitis (SIVE) very similar to the human disease. Neurodegeneration in both these conditions occurs from the effects of toxic viral proteins and neurotoxins derived from activated brain macrophages. Activated macrophages in the brain of macaques with SIVE can be labeled in vivo using positron emission tomography (PET) using PK11195, a ligand that binds the peripheral benzodiazepine receptor (PBR). However, the functional significance and mechanisms mediating increased PK11195 binding in activated brain macrophages are not known. Using post mortem tissues from macaques with SIVE and macrophages cell cultures activated with lipopolysaccaride (LPS), we show that [3H](R)-PK11195 binding is increased in activated macrophages. Increased [3H](R)-PK11195 binding in LPS activated macrophages was reversed by pharmacologically inhibiting class III phosphatidylinositol-3 kinase (PI3-kinase), but was not altered by inhibiting the mitogen-activated protein kinase (MAP-kinase) pathway. Our results suggest that activated macrophages in lentiviral encephalitis show increased [3H](R)-PK11195 binding in a PI3-kinase dependent fashion which may help elucidate the function of PBR in activated brain macrophages in HIVE and other neuroinflammatory diseases.
Keywords: Peripheral benzodiazepine receptor, Macrophages, PK11195, PI3-kinase, HIV encephalitis, SIV encephalitis
Introduction
About 25% of immunosuppressed patients infected with HIV develop neurological deficits ranging form cognitive impairments, motor abnormalities, behavioral symptoms to frank dementia (HIV-associated dementia) [6, 20]. HIV encephalitis (HIVE) is considered to be the pathological substrate of HIV-associated dementia and is characterized by the presence of microglial nodules, multinucleated giants cells formed by fusion of HIV infected macrophages, and HIV-infected and activated macrophages [3]. Brain macrophages (including resident microglia and infiltrating macrophages) constitute the cell type productively infected with HIV in the brain and are hypothesized to be central to neurodegeneration as sources of viral proteins and toxic substances that initiate synaptic damage and neuronal death [7].
The macaque model for HIVE closely resembles the human disease. Like humans, only a percentage of macaques infected with Simian Immunodeficiency Virus (SIV) develop encephalitis [14]. We have previously shown that PK11195, a ligand that binds the peripheral benzodiazepine receptor (PBR) expressed normally in low levels in the brain, show higher binding in vivo in SIVE using positron emission tomography (PET) compared to SIV infected macaques without encephalitis [32]. These data are in agreement with a large body of work that report increased PK11195 binding corresponding to activated brain macrophages, as determined using PET, in several acute and chronic neuroinflammatory conditions such as stroke [27], multiple sclerosis [2], Alzheimer’s disease [4], Parkinson’s disease [22], and Huntington’s disease [24].
Increased PK11195 binding to PBR in these diseases has been extensively characterized mainly as a diagnostic tool to detect activated brain macrophages using PET. However, the functional significance of increased PK11195 binding to PBR sites as well as the cellular mechanisms mediating increased PK11195 binding in activated brain macrophages have not been elucidated. Using the macaque model of HIVE along with cell culture systems, we show that [3H](R)-PK11195 binding is increased in activated brain macrophages and is reversed by pharmacologically inhibiting the PI3-kinase pathway. These results may help elucidate the functional significance of increased PBR binding sites in activated brain macrophages in neuroinflammatory disorders.
Methods
Macaque model of HIVE
Archival brain tissues from the frontal cortex were obtained from pigtailed macaques infected with SIV (SIVDeltaB670) without encephalitis (n=3) and with SIVE (n=4). Encephalitis was defined by the presence of microglial nodules; multinucleated giant cells and SIV infected macrophages determined by staining for the viral protein SIV gp120 [14]. Frontal cortical tissues from 2 uninfected macaques were used as an additional control.
Autoradiography
Autoradiography was performed as described earlier [32]. Briefly, 15 μm thick frozen sections obtained from the frontal cortex of SIVE macaques were placed on SuperfrostTm glass slides (Sigma) and incubated in ice-cold 50 mM HEPES (pH 7.4) containing 1 nM [3H](R)-PK11195 for 30 min. Specificity of binding was ensured by the inclusion of 1 μM PK11195 in parallel sections. The sections were mounted with a layer of autoradiographic LM-1 emulsion (Amersham, UK), following which they were developed after 4 weeks and imaged on the confocal microscope.
Immunohistochemistry and quantification
Cellular localization of [3H](R)-PK11195 in frozen brain sections from SIVE macaques was evaluated by combining immunostaining and autoradiography. Sections were first immunostained with GFAP (1:1000, mouse monoclonal, DAKO, Carpinteria, CA) or against a lysosomal-associated marker for activated macrophages CD68 (1:100, mouse monoclonal, Serotec, Raleigh, NC), incubated with Cy5-conjugated anti-mouse IgG at a concentration of 1:200 (Jackson Immunoresearch Laboratories Inc., West Grove, PA) and then processed for autoradiography with [3H](R)-PK11195 following which they were imaged on the confocal microscope.
Quantification of CD68 was performed on the confocal microscope on paraffin embedded sections obtained from the same animals used for filtration binding analyses in a manner similar to that described previously [32]. Sections were scanned and quantified on a laser confocal microscope equipped with an argon laser with 458 nm, 477 nm, 488 nm and 514 nm primary emission lines. (LSM 510, Zeiss, Heidelberg, Germany). Each section was scanned along the z-axis to define the middle optical plane used in quantification (262,144 pixels/plane; 1 pixel= 0.25 μm2). Scanning parameters such as laser power aperture, gain, and photomultiplier tube settings were kept constant for each wavelength. An individual blinded to the experimental design imaged 10 areas (40X) encompassing 106,100 μm2. For each cell phenotype scanned, contribution to signal intensity from autofluorescence was minimized using a threshold that was kept constant. In each area the average pixel fluorescence, along with the pixel counts for a given cell phenotype marker that exceeded the threshold, were enumerated. The average pixel fluorescence was multiplied by the total number of pixels to represent the total florescence for in that area. The total fluorescence values determined from the 10 scanned areas were averaged to represent a measure of the cell phenotype.
Brain sections were imaged on the confocal microscope after combined immunostaining and autoradiography. The autoradiographic silver grains were visualized by transmitted light and then pseudocolored on the confocal microscope for better visualization.
Tissue culture
Human peripheral blood mononuclear cells (PBMC) were isolated from buffy coats obtained from the blood bank (Central Blood Bank, Pittsburgh, PA). PBMC were grown for 5 days in complete medium consisting of AIM-V medium (Gibco BRL, Grand Island, NY) and 10% heat inactivated-fetal calf serum (Gibco). Non-adherent cells were washed thoroughly after 5 days and adherent macrophages were cultured for an additional two days in complete medium. Macrophage cultures were activated with 1μg/ml LPS (Sigma) for 48hrs. Parallel macrophage cultures were pretreated with either 10μM/L of PI3-kinase inhibitor LY294002 (Sigma) or the mitogen-activated protein kinase (MAP-kinase) inhibitor UO126 (Cell Signaling, Beverly, MA) for two hours after which they were activated with 1 μM LPS for 48 hrs in the presence of either inhibitor.
Filtration radioligand binding assays
Brain tissues were weighed and homogenized in ice-cold 50 mM HEPES (Sigma) buffer (pH 7.4). LPS activated and control macrophage cultures were harvested and homogenized in ice-cold 50 mM HEPES. Saturation binding experiments were performed by incubating tissues (total protein concentration ranging from 150 to 200 μg) with 0.5–64 nM [3H](R)-PK11195 (sp. Act., 89.9 Ci/mmol; NEN Life Sciences Products, Boston, MA) at 4°C for 2 hr in a final volume of 250 μl of HEPES. Nonspecific binding was determined by the inclusion of 10 μM PK11195 (Sigma). The reaction was terminated by the addition of ice-cold buffer in a vacuum cell harvester (Brandel, Gaithersburg, MD). All samples were run in duplicate. Bmax in fmoles per mg protein, reflective of the total number of binding sites and KD in nM reflective of ligand binding affinity were determined using PRISM software (Graphpad, San Diego, CA).
Statistical analysis
Data were analyzed using PRISM software (Graphpad, San Diego, CA). Student’s t tests or one-way ANOVA tests with post-test Bonferroni correction and 95% confidence intervals were used to analyze data. Nonparametric correlational analyses using 95% confidence intervals were performed to quantify the relationship between [3H](R)-PK11195 binding and activated macrophages assessed by CD68 staining. Results from correlational analyses are represented by r, the Spearman’s coefficient.
Results
[3H](R)-PK11195 is higher in SIVE compared to controls
[3H](R)-PK11195 Bmax values, reflective of the number of binding sites was significantly higher in SIVE (n=4) compared to SIV infected without encephalitis (n=3) and non-infected animals (n=2) (p=0.0097, Figure 1 A–C). The KD reflective of ligand binding affinity did not differ between the three conditions (p=0.5731, Figure 1D). These results were confirmed in brain sections as [3H](R)-PK11195 autoradiography was greater in SIVE compared to SIV infected macaques without encephalitis (Figure 1F & E). [3H](R)-PK11195 binding in SIVE corresponded to the distribution of microglial nodules (Figure 1F) in contrast to the diffuse binding seen in SIV infected macaques without encephalitis (Figure 1E).
Figure 1. [3H](R)-PK11195 binding is significantly higher in SIVE compared to controls.
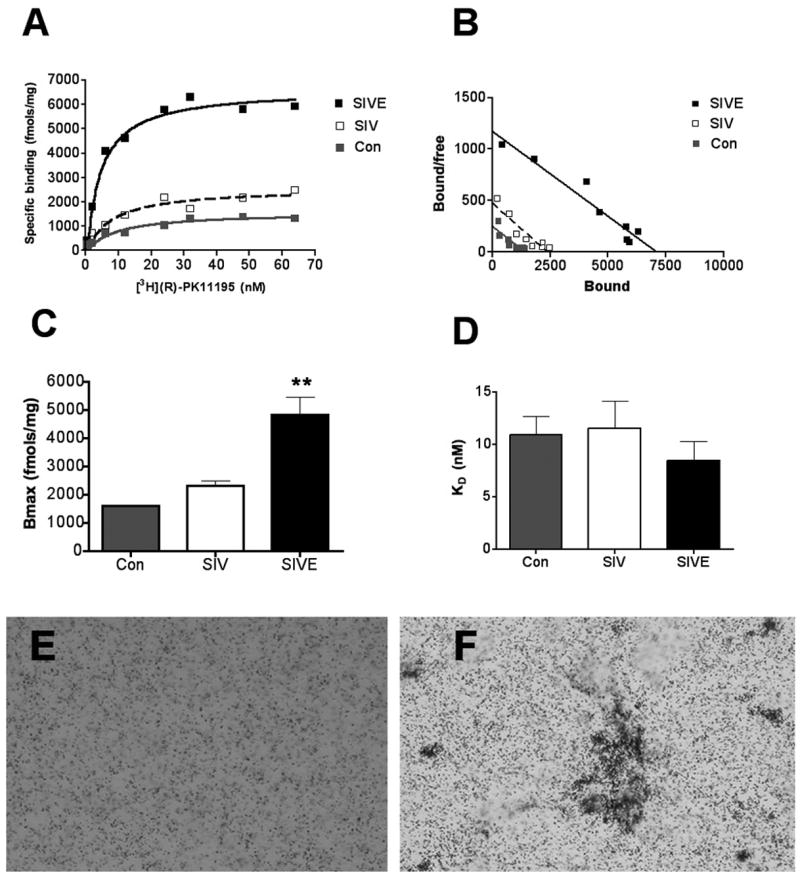
(A & B) Filtration binding (representative saturation binding curves, A and scatchard plot, B) with [3H](R)-PK11195 was higher in frontal cortical tissues from SIVE (n=4, black squares) compared to SIV infected, non-encephalitic (SIV, n=3, clear squares) and non-infected controls (n=2, gray squares).
(C & D) The Bmax (fmols/mg), reflective of the total number of binding sites was significantly higher in SIVE (black bars) compared to SIV infected non-encephalitic (SIV, n=3, clear bars) and non-infected controls (n=2, gray bars). (p=0.0038, C). The KD (nM) reflective of the binding affinity of the ligand to PBR did not significantly differ amongst the three conditions (p=0.2492, D). Data was analyzed using one-sided ANOVA.
(E & F) [3H](R)-PK11195 autoradiograms (black grains) of sections from frontal cortex show higher specific binding in SIVE (F) compared to SIV infected, non-encephalitic macaques (E).
[3H](R)-PK11195 binding in SIVE correlates with activated brain macrophages
[3H](R)-PK11195 autoradiography combined with immunostaining showed that [3H](R)-PK11195 overlapped with regions rich in CD68 labeled activated macrophages (Figure 2A), but not with reactive astrocytosis labeled with GFAP (Figure 2B). We next quantified the degree of microglial activation using CD68 in same regions in the same macaque tissue used for filtration binding analyses and correlated these values with [3H](R)-PK11195 Bmax values. The abundance of CD68 staining activated macrophages correlated with [3H](R)-PK11195 binding (Figure 2C, r=0.8684, p=0.0112).
Figure 2. [3H](R)-PK11195 binding in SIVE corresponds to activated brain macrophages.
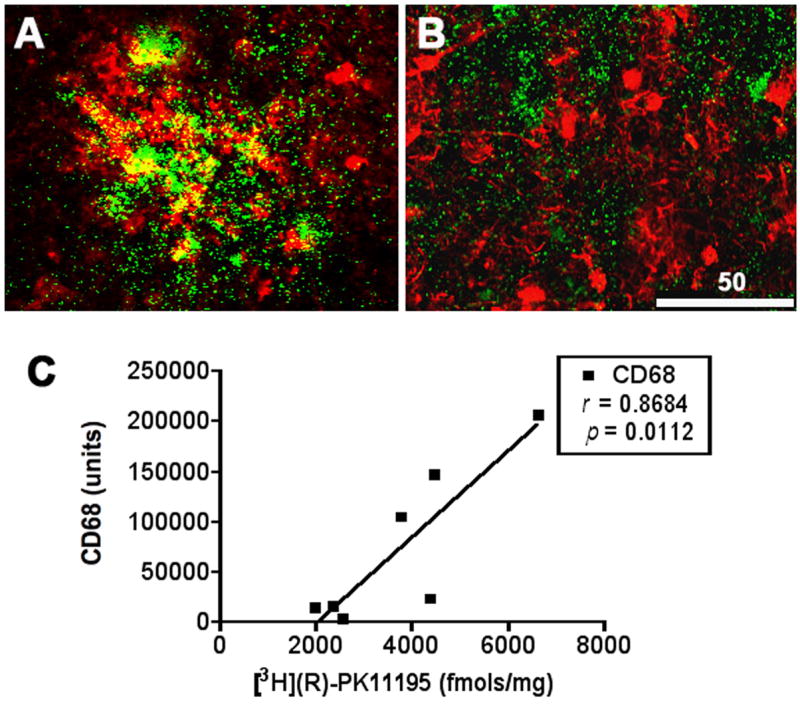
(A, B) Sections obtained from the frontal cortex processed for autoradiography with [3H](R)-PK11195 (green) were immunostained to label activated macrophages with CD68 (A, red) or astrocytes with GFAP (B, red). [3H](R)-PK11195 specific binding overlaped with CD68 but not with GFAP immunostaining.
(C) [3H](R)-PK11195 Bmax values (X-axis, fmols/mg) correlated significantly with the abundance of activated macrophages assessed by quantification of CD68 staining (Y-axis)
LPS activated macrophages show increased [3H](R)-PK11195 binding compared to controls
To determine if [3H](R)-PK11195 binding is increased in activated macrophages, primary human macrophages were cultured and activated with 1μM LPS for 48 hrs (Figure 3A & B). [3H](R)-PK11195 Bmax values, reflective of the number of binding sites, was significantly higher in macrophages activated with LPS compared to non-activated macrophages (p=0.0017, Figure 3C–D and Figure 4A). The KD reflective of ligand binding affinity did not differ significantly between activated and control macrophage cultures (p=0.0896, Figure 4B).
Figure 3. Macrophages activated with show higher binding with [3H](R)-PK11195.
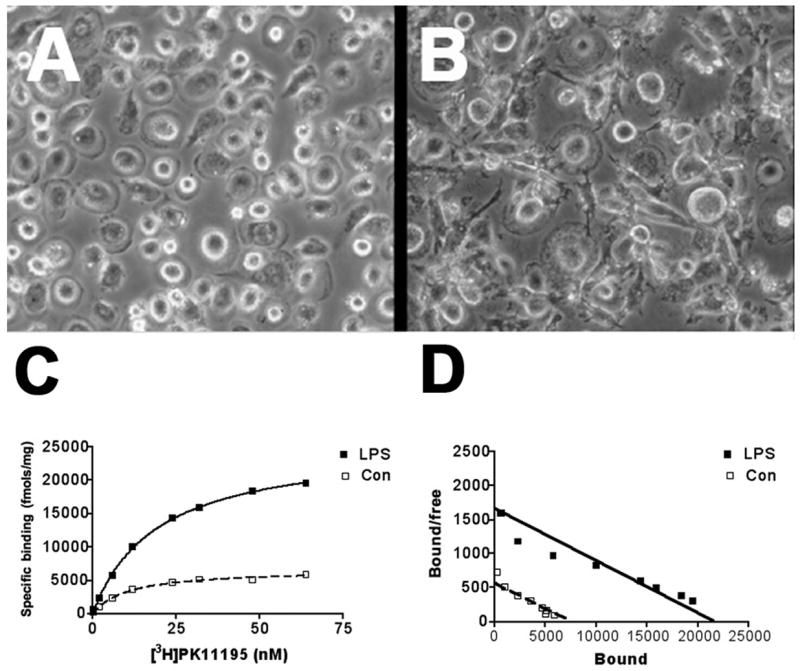
Primary human macrophage cultures (n=3) were activated with (B) or without (A) LPS. Filtration binding (representative saturation binding curves, C and scatchard plots, D) with [3H](R)-PK11195 was higher in LPS activated macrophages (black squares) compared with untreated cultures (white squares).
Figure 4. Pharmacologic inhibition on the PI3-kinase pathway with LY29002 reverses increase in [3H](R)-PK11195 binding in LPS activated macrophages.
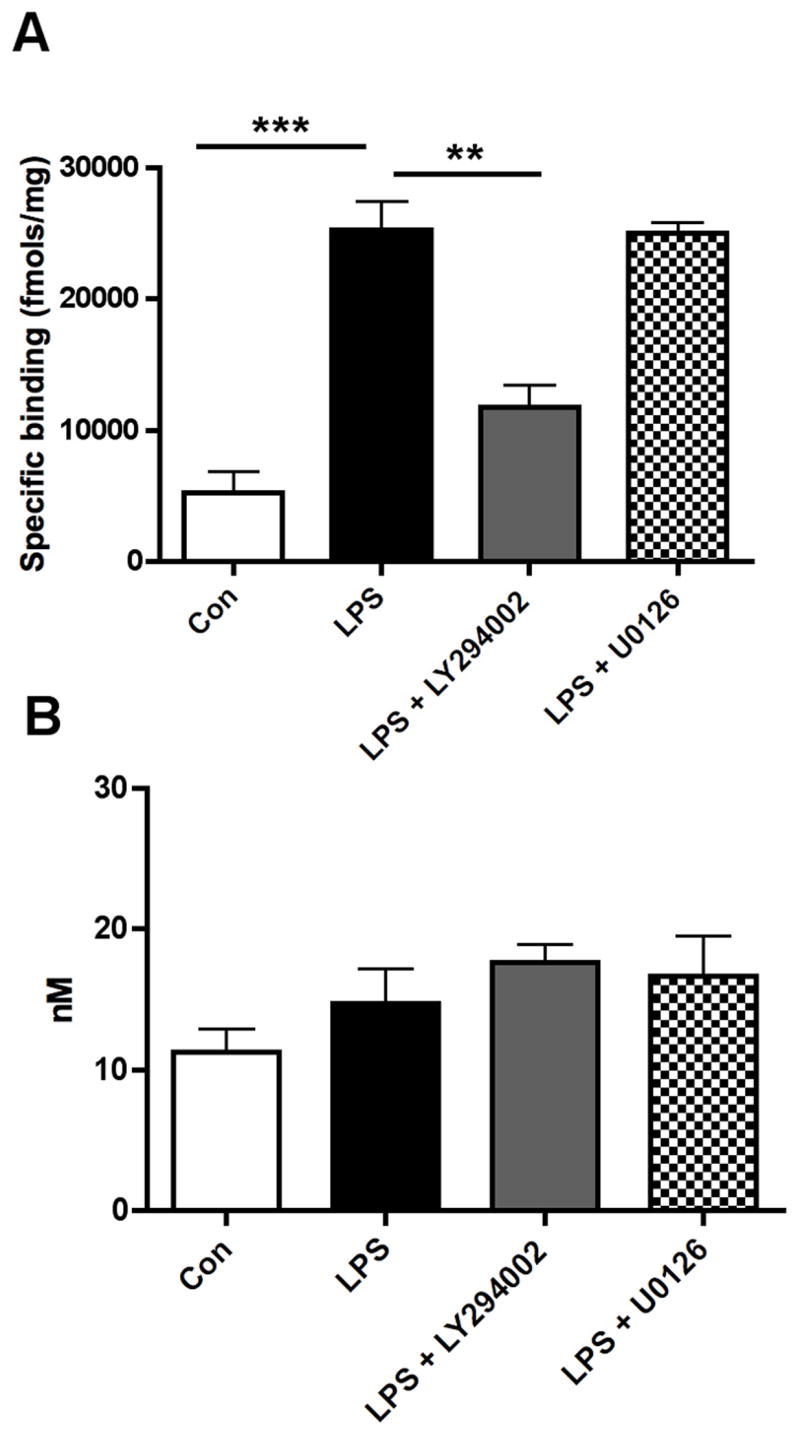
(A) Bmax, reflective of the total number of binding sites, with [3H](R)-PK11195 was higher in primary human macrophages activated with LPS (black bars) compared to untreated controls (clear bars). Treatment with the PI3-kinase pathway inhibitor LY294002 (grey bars) but not MAP-kinase inhibitor U0126 (hatched bars) reversed increase in Bmax values in LPS activated macrophages (n=3, each, p<0.001).
(B) The KD (reflective of the binding affinity) was not significantly different in all the conditions (p=0.2284). Data analyzed using one-sided ANOVA.
Increased [3H](R)-PK11195 binding in LPS activated macrophages is reversed by the PI3-kinase inhibitor LY294002
We sought to determine the cellular signaling pathways that mediate increased [3H](R)-PK11195 binding in activated macrophages. Macrophages treated with the PI3-kinase inhibitor LY294002, but not the MAPK inhibitor U0126 reversed LPS induced increase in [3H](R)-PK11195 Bmax (p<0.0001, Figure 4A). The KD did not significantly differ with either treatment from controls (p=0.5166, Figure 4B).
Discussion
We examined [3H](R)-PK11195 binding in a macaque model of HIVE and cell culture systems and found that [3H](R)-PK11195 binding corresponded to activated brain macrophages. SIVE tissue showed increased [3H](R)-PK11195 binding compared to SIV infected, non-encephalitic and non-infected brain tissues (Figure 1). Increased [3H](R)-PK11195 binding overlapped with and correlated with the abundance of CD68 labeled brain macrophages (Figure 2). In tissue culture experiments, we found increased [3H](R)-PK11195 binding in macrophages activated with LPS compared to control macrophages (Figure 3). Finally, increased [3H](R)-PK11195 binding in LPS treated macrophages was reversed by a pharmacological inhibitor of PI3-kinase, but not of by a pharmacological inhibitor of MAP-kinase (Figure 4). These results suggest that [3H](R)-PK11195 binding in activated brain macrophages may be regulated by the PI3-kinase pathway.
Our results showing increased [3H](R)-PK11195 binding corresponding to brain macrophages are similar to others in rat models of stroke [19], ischemia [28] and facial nerve axotomy [1], multiple sclerosis [2, 33], SIVE [16], and hippocampal lesions in mice [25]. While the functional significance and mechanisms that mediate increased [3H](R)-PK11195 binding to PBR in brain macrophages are not known, it is thought that PBR is part of a hetero-oligomeric complex comprised of the voltage-dependent anion channel and an adenine nucleotide carrier forming the putative mitochondrial permeability transition pore [17]. In tissues synthesizing steroids, PBR is involved in transport of cholesterol from the outer to the inner mitochondrial membranes [23]. As a constituent of the mitochondrial permeability transition pore, it is thought to regulate cell death [17] and mitochondrial respiration [11]. PBR overexpression in many cell types has been shown to protect against various apoptotic insults including the cytopathic effects of sindbis and myxoma viral infections [8, 12]. Forced PBR expression in neurons in vivo and jurkat cells in vitro protects these cells from apoptosis [12, 29]. PBR upregulation in testicular leydig cells protects them from cytokine-induced toxicity [30]. PBR also protects peripheral phagocytes against oxidant induced cell death [5]. Several proteins involved in apoptosis including Bcl-2, Bcl-Xl and Bax physically interact with the voltage dependent anion channel and the adenine nucleotide carrier [10]. PBR can thus influence cell death processes either by directly affecting the molecular components of the pore or indirectly by interfering with interactions with apoptotic proteins.
The PI3-kinase pathway is a key regulator of cell survival [21]. Further, PI3-kinase/Akt activation is an important survival-regulation pathway in brain macrophages [13], peripheral macrophages and other hemopoietic cell [18, 31]. Moreover, inhibition of the PI3-kinase pathway in peripheral macrophages causes these cells to die due to loss of mitochondrial transmembrane potential [15]. Our data suggests that PBR is directly regulated by PI3-kinase, and since PBR is a constituent of the permeability transition pore and may play a role in maintaining mitochondrial transmembrane potential, it is possible that PBR is an essential player in regulating macrophage cell survival in the CNS in HIVE. Very little is known about how macrophages survive in the brain in HIVE in vivo. In the SCID mouse model of HIV encephalitis, macrophages survive for several months in the brain [26]. HIV infected macrophages also survive in culture for as long as 60 days [9]. Since macrophages mediate the pathology of HIV encephalitis, it is conceivable that a longer life span of macrophages would lead to exacerbation of the neurodegenerative process seen in HIVE. PBR expression in activated brain macrophages may prolong their life span by influencing mitochondrial permeability transition and preventing apoptosis of these cells in the CNS. The full functional significance of increased PBR binding sites in brain macrophages in HIVE and other neuroinflammatory conditions remains to be determined.
Acknowledgments
We thank James Kasenchak and Jason Nugyen for technical help and Dr. Dafna Bonneh-Barkay for banking macaque brain tissues. This work was supported by the National Institutes of Health RO1 MH64921 (CAW), K24 MH01717 (CAW).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Banati RB, Myers R, Kreutzberg GW. PK (‘peripheral benzodiazepine’)--binding sites in the CNS indicate early and discrete brain lesions: microautoradiographic detection of [3H]PK11195 binding to activated microglia. J Neurocytol. 1997;26:77–82. doi: 10.1023/a:1018567510105. [DOI] [PubMed] [Google Scholar]
- 2.Banati RB, Newcombe J, Gunn RN, Cagnin A, Turkheimer F, Heppner F, Price G, Wegner F, Giovannoni G, Miller DH, Perkin GD, Smith T, Hewson AK, Bydder G, Kreutzberg GW, Jones T, Cuzner ML, Myers R. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain. 2000;123(Pt 11):2321–37. doi: 10.1093/brain/123.11.2321. [DOI] [PubMed] [Google Scholar]
- 3.Budka H. Neuropathology of human immunodeficiency virus infection. Brain Pathol. 1991;1:163–75. doi: 10.1111/j.1750-3639.1991.tb00656.x. [DOI] [PubMed] [Google Scholar]
- 4.Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358:461–7. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- 5.Carayon P, Portier M, Dussossoy D, Bord A, Petitpretre G, Canat X, Le Fur G, Casellas P. Involvement of peripheral benzodiazepine receptors in the protection of hematopoietic cells against oxygen radical damage. Blood. 1996;87:3170–8. [PubMed] [Google Scholar]
- 6.Dore GJ, Correll PK, Li Y, Kaldor JM, Cooper DA, Brew BJ. Changes to AIDS dementia complex in the era of highly active antiretroviral therapy. Aids. 1999;13:1249–53. doi: 10.1097/00002030-199907090-00015. [DOI] [PubMed] [Google Scholar]
- 7.Ellis R, Langford D, Masliah E. HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci. 2007;8:33–44. doi: 10.1038/nrn2040. [DOI] [PubMed] [Google Scholar]
- 8.Everett H, Barry M, Sun X, Lee SF, Frantz C, Berthiaume LG, McFadden G, Bleackley RC. The myxoma poxvirus protein, M11L, prevents apoptosis by direct interaction with the mitochondrial permeability transition pore. J Exp Med. 2002;196:1127–39. doi: 10.1084/jem.20011247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garaci E, Caroleo MC, Aloe L, Aquaro S, Piacentini M, Costa N, Amendola A, Micera A, Calio R, Perno CF, Levi-Montalcini R. Nerve growth factor is an autocrine factor essential for the survival of macrophages infected with HIV. Proc Natl Acad Sci U S A. 1999;96:14013–8. doi: 10.1073/pnas.96.24.14013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harris MH, Thompson CB. The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ. 2000;7:1182–91. doi: 10.1038/sj.cdd.4400781. [DOI] [PubMed] [Google Scholar]
- 11.Hirsch JD, Beyer CF, Malkowitz L, Beer B, Blume AJ. Mitochondrial benzodiazepine receptors mediate inhibition of mitochondrial respiratory control. Mol Pharmacol. 1989;35:157–63. [PubMed] [Google Scholar]
- 12.Johnston C, Jiang W, Chu T, Levine B. Identification of genes involved in the host response to neurovirulent alphavirus infection. J Virol. 2001;75:10431–45. doi: 10.1128/JVI.75.21.10431-10445.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim WK, Hwang SY, Oh ES, Piao HZ, Kim KW, Han IO. TGF-beta1 represses activation and resultant death of microglia via inhibition of phosphatidylinositol 3-kinase activity. J Immunol. 2004;172:7015–23. doi: 10.4049/jimmunol.172.11.7015. [DOI] [PubMed] [Google Scholar]
- 14.Lackner AA, Smith MO, Munn RJ, Martfeld DJ, Gardner MB, Marx PA, Dandekar S. Localization of simian immunodeficiency virus in the central nervous system of rhesus monkeys. Am J Pathol. 1991;139:609–21. [PMC free article] [PubMed] [Google Scholar]
- 15.Liu H, Perlman H, Pagliari LJ, Pope RM. Constitutively activated Akt-1 is vital for the survival of human monocyte-differentiated macrophages. Role of Mcl-1, independent of nuclear factor (NF)-kappaB, Bad, or caspase activation. J Exp Med. 2001;194:113–26. doi: 10.1084/jem.194.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mankowski JL, Queen SE, Tarwater PJ, Adams RJ, Guilarte TR. Elevated peripheral benzodiazepine receptor expression in simian immunodeficiency virus encephalitis. J Neurovirol. 2003;9:94–100. doi: 10.1080/13550280390173283. [DOI] [PubMed] [Google Scholar]
- 17.McEnery MW, Snowman AM, Trifiletti RR, Snyder SH. Isolation of the mitochondrial benzodiazepine receptor: association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc Natl Acad Sci U S A. 1992;89:3170–4. doi: 10.1073/pnas.89.8.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minshall C, Arkins S, Freund GG, Kelley KW. Requirement for phosphatidylinositol 3’-kinase to protect hemopoietic progenitors against apoptosis depends upon the extracellular survival factor. J Immunol. 1996;156:939–47. [PubMed] [Google Scholar]
- 19.Myers R, Manjil LG, Cullen BM, Price GW, Frackowiak RS, Cremer JE. Macrophage and astrocyte populations in relation to [3H]PK 11195 binding in rat cerebral cortex following a local ischaemic lesion. J Cereb Blood Flow Metab. 1991;11:314–22. doi: 10.1038/jcbfm.1991.64. [DOI] [PubMed] [Google Scholar]
- 20.Nath A, Sacktor N. Influence of highly active antiretroviral therapy on persistence of HIV in the central nervous system. Curr Opin Neurol. 2006;19:358–61. doi: 10.1097/01.wco.0000236614.51592.ca. [DOI] [PubMed] [Google Scholar]
- 21.Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis. 2004;9:667–76. doi: 10.1023/B:APPT.0000045801.15585.dd. [DOI] [PubMed] [Google Scholar]
- 22.Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, Torizuka T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann Neurol. 2005;57:168–75. doi: 10.1002/ana.20338. [DOI] [PubMed] [Google Scholar]
- 23.Papadopoulos V, Amri H, Li H, Boujrad N, Vidic B, Garnier M. Targeted disruption of the peripheral-type benzodiazepine receptor gene inhibits steroidogenesis in the R2C Leydig tumor cell line. J Biol Chem. 1997;272:32129–35. doi: 10.1074/jbc.272.51.32129. [DOI] [PubMed] [Google Scholar]
- 24.Pavese N, Gerhard A, Tai YF, Ho AK, Turkheimer F, Barker RA, Brooks DJ, Piccini P. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology. 2006;66:1638–43. doi: 10.1212/01.wnl.0000222734.56412.17. [DOI] [PubMed] [Google Scholar]
- 25.Pedersen MD, Minuzzi L, Wirenfeldt M, Meldgaard M, Slidsborg C, Cumming P, Finsen B. Up-regulation of PK11195 binding in areas of axonal degeneration coincides with early microglial activation in mouse brain. Eur J Neurosci. 2006;24:991–1000. doi: 10.1111/j.1460-9568.2006.04975.x. [DOI] [PubMed] [Google Scholar]
- 26.Persidsky Y, Buttini M, Limoges J, Bock P, Gendelman HE. An analysis of HIV-1-associated inflammatory products in brain tissue of humans and SCID mice with HIV-1 encephalitis. J Neurovirol. 1997;3:401–16. doi: 10.3109/13550289709031186. [DOI] [PubMed] [Google Scholar]
- 27.Price CJ, Wang D, Menon DK, Guadagno JV, Cleij M, Fryer T, Aigbirhio F, Baron JC, Warburton EA. Intrinsic activated microglia map to the peri-infarct zone in the subacute phase of ischemic stroke. Stroke. 2006;37:1749–53. doi: 10.1161/01.STR.0000226980.95389.0b. [DOI] [PubMed] [Google Scholar]
- 28.Stephenson DT, Schober DA, Smalstig EB, Mincy RE, Gehlert DR, Clemens JA. Peripheral benzodiazepine receptors are colocalized with activated microglia following transient global forebrain ischemia in the rat. J Neurosci. 1995;15:5263–74. doi: 10.1523/JNEUROSCI.15-07-05263.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stoebner PE, Carayon P, Casellas P, Portier M, Lavabre-Bertrand T, Cuq P, Cano JP, Meynadier J, Meunier L. Transient protection by peripheral benzodiazepine receptors during the early events of ultraviolet light-induced apoptosis. Cell Death Differ. 2001;8:747–53. doi: 10.1038/sj.cdd.4400861. [DOI] [PubMed] [Google Scholar]
- 30.Trincavelli ML, Marselli L, Falleni A, Gremigni V, Ragge E, Dotta F, Santangelo C, Marchetti P, Lucacchini A, Martini C. Upregulation of mitochondrial peripheral benzodiazepine receptor expression by cytokine-induced damage of human pancreatic islets. J Cell Biochem. 2002;84:636–44. [PubMed] [Google Scholar]
- 31.Tyner JW, Uchida O, Kajiwara N, Kim EY, Patel AC, O’Sullivan MP, Walter MJ, Schwendener RA, Cook DN, Danoff TM, Holtzman MJ. CCL5-CCR5 interaction provides antiapoptotic signals for macrophage survival during viral infection. Nat Med. 2005;11:1180–7. doi: 10.1038/nm1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Venneti S, Lopresti BJ, Wang G, Bissel SJ, Mathis CA, Meltzer CC, Boada F, Capuano S, 3rd, Kress GJ, Davis DK, Ruszkiewicz J, Reynolds IJ, Murphey-Corb M, Trichel AM, Wisniewski SR, Wiley CA. PET imaging of brain macrophages using the peripheral benzodiazepine receptor in a macaque model of neuroAIDS. J Clin Invest. 2004;113:981–9. doi: 10.1172/JCI20227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vowinckel E, Reutens D, Becher B, Verge G, Evans A, Owens T, Antel JP. PK11195 binding to the peripheral benzodiazepine receptor as a marker of microglia activation in multiple sclerosis and experimental autoimmune encephalomyelitis. J Neurosci Res. 1997;50:345–53. doi: 10.1002/(SICI)1097-4547(19971015)50:2<345::AID-JNR22>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]