

Abstract
An intimate discourse between the blastocyst and uterus is essential for successful implantation. However, the molecular basis of this interaction is not clearly understood. Exploiting genomic Hbegf mutant mice, we show here that maternal deficiency of heparin-binding EGF-like growth factor (HB-EGF) defers on-time implantation, leading to compromised pregnancy outcome. We also demonstrate that amphiregulin, but not epiregulin, partially compensates for the loss of HB-EGF during implantation. In search of the mechanism of this compensation, we found that reduced preimplantation estrogen secretion from ovarian HB-EGF deficiency is a cause of sustained expression of uterine amphiregulin before the initiation of implantation. To explore the significance specifically of uterine HB-EGF in implantation, we examined this event in mice with conditional deletion of uterine HB-EGF and found that this specific loss of HB-EGF in the uterus still defers on-time implantation without altering preimplantation ovarian estrogen secretion. The observation of normal induction of uterine amphiregulin surrounding the blastocyst at the time of attachment in these conditional mutant mice suggests a compensatory role of amphiregulin for uterine loss of HB-EGF, preventing complete failure of pregnancy. Our study provides genetic evidence that HB-EGF is critical for normal implantation. This finding has high clinical relevance, because HB-EGF signaling is known to be important for human implantation.
Keywords: amphiregulin, implantation, uterus, blastocyst
The initiation of implantation is the result of coordinated integration of various signaling pathways between the blastocyst and the uterus. Early studies have provided valuable clues to this process involving a range of endocrine, paracrine, autocrine, and juxtacrine modulators (1). In this regard, signaling initiated by the EGF family of ligands, including EGF itself, transforming growth factor α (TGFα), heparin-binding EGF (HB-EGF), amphiregulin, epiregulin, and betacellulin, has been studied extensively before and during implantation in mice (2–5). However, thus far, no implantation defects have been reported in mice deficient in amphiregulin, epiregulin, or even with compound deficiency of EGF/TGFα/amphiregulin (6–8), challenging the essentiality of amphiregulin, epiregulin, EGF, or TGFα in implantation.
Increasing evidence suggests that HB-EGF participates in a wide range of physiological and pathological processes, including heart development (9, 10), wound healing (11), atherosclerosis and pulmonary hypertension (12, 13), and tumor development and angiogenesis (14, 15). HB-EGF has also been highlighted as an early molecular marker of embryo–uterine cross-talk during implantation (5). It is expressed as both transmembrane and soluble forms in the uterine luminal epithelium at the site of the blastocyst before the attachment reaction, influencing blastocyst activities in a paracrine and/or juxtacrine manner by interacting with ErbB1 and ErbB4 that are displayed on the blastocyst cell surface (16–18). More interestingly, our recent study shows that HB-EGF produced by implantation-competent blastocysts and secreted by ectodomain shedding of proHB-EGF induces its own gene expression in the receptive uterus via an autoinduction loop (19). Although these findings suggest that HB-EGF is a key signaling molecule involved in setting up a hierarchy of events between the blastocyst and uterus for implantation, genetic evidence for a definitive role of HB-EGF in this process remains elusive. In the present investigation, we have combined genetic, pharmacological, and physiological approaches to address whether HB-EGF is critical for normal implantation.
Results and Discussion
Genetic Ablation of Hbegf Compromises Term Pregnancy with Reduced Litter Size in Mice.
To assess the physiological relevance of HB-EGF signaling in early pregnancy events, we first examined pregnancy outcome in Hbegf−/− females crossed with fertile Hbegf−/− males. As illustrated in Fig. 1A, mice missing HB-EGF show compromised term pregnancy with significantly reduced litter size compared with WT littermates, suggesting that HB-EGF signaling is crucial for normal pregnancy. Because a range of physiological functions is ascribed to HB-EGF owing to its cell-specific expression in ovaries (20), early embryos and uteri during early pregnancy (5, 16, 19, 21), it is possible that one or all of these targets are affected, contributing to reduced litter sizes in Hbegf−/− mice. Therefore, to explore underlying causes of subfertility in Hbegf−/− females, we examined early pregnancy events.
Fig. 1.

HB-EGF deficiency defers the window of implantation, leading to compromised pregnancy. (A) Litter sizes from Hbegf−/− mice are smaller than those of WT mice (Student t test, *, P < 0.01). Numbers within bars indicate numbers of mothers examined. Data are presented as mean ± SEM. (B and C) Ovulation and fertilization are comparable between WT and null mice. Numbers within bars in B indicate the number of mice with eggs/total number of mice examined, and those in C are number of mice with two-cell embryos examined. (D) Preimplantation embryo development is normal in Hbegf−/− mice. Numbers within bars indicate number of mice examined. (E) HB-EGF deficiency leads to deferral of implantation as examined on day 4 evening (2000 h and 2400 h) and day 5 morning (0800 h) by the blue dye method. Numbers within bars indicate numbers of mice with implantation sites (IS) per the total number of mice examined. (F) Representative photographs of WT uteri with IS and Hbegf−/− uteri without blue bands on day 4 midnight (2400 h). (G) Representative photomicrograph of unimplanted morphologically normal blastocysts recovered from Hbegf−/− females without blue reaction at 2400 h of day 4.
Ovulation and Fertilization Are Comparable in WT and Hbegf−/− Mice.
Normal ovulation and fertilization was observed in Hbegf−/− females mated with null males when examined on day 2 of pregnancy. As shown in Fig. 1B, all Hbegf−/− mice (n = 15) ovulated with comparable numbers of ova as WT females (n = 10). The yield of two-cell embryos among ovulated eggs was used to assess the fertilization rate. A comparable number of two-cell embryos was recovered in 12 of 15 mutant females and 8 of 10 WT females (Fig. 1C). These results show that Hbegf−/− females have normal ovulation and fertilization, consistent with previous findings that HB-EGF, instead of being induced like amphiregulin and epiregulin, is down-regulated in preovulatory mural granulosa cells in response to the preovulatory gonadotropin surge (20, 22). In fact, genetic evidence points toward essential roles of amphiregulin and epiregulin in oocyte maturation and cumulus expansion (23). This ligand diversity for ErbB signaling during ovulation ensures normal progression of oocyte meiotic maturation and subsequent fertilization. We next analyzed periimplantation events after fertilization to search for potential causes of reduced fertility in Hbegf−/− mice.
Normal “Window” of Implantation Is Altered in Hbegf−/− Mice.
Under normal conditions, embryo implantation occurs within a limited time span termed the window of implantation, which absolutely requires timely synchronization of the blastocyst achieving implantation competency and the uterus reaching the receptive state (1). In mice, fertilized eggs develop into morulae after several rounds of cleavage, travel through the oviduct into the uterus, and differentiate into blastocysts during the early morning of day 4, then escape from their zona pellucidae to ultimately implant into the receptive uterus around midnight of day 4 (5). We have previously shown that mouse oviduct epithelial cells express HB-EGF during the preimplantation period (24), and HB-EGF also enhances embryo development and differentiation in culture and improves implantation success in vivo (25, 26). To explore whether HB-EGF deficiency impairs early embryo development, we examined morula–blastocyst transformation in both WT and mutant females. We found that Hbegf−/− embryos form blastocysts normally in vivo when examined on day 4 morning (Fig. 1D). These results indicate that HB-EGF signaling is not essential for normal preimplantation embryo growth in vivo. Because HB-EGF signaling through ErbB receptors is thought to mediate reciprocal embryo–uterine interactions before the initiation of the attachment reaction (5, 19, 27), we were particularly interested to examine implantation in Hbegf−/− mice. As shown in Fig. 1 E and F, although seven of eight WT mice showed distinct implantation sites at 2000 h on day 4 as examined by the blue dye method, none of the Hbegf−/− mice showed any signs of implantation. Moreover, at 2400 h of day 4, when all WT mice (n = 12) showed blue bands, only 5 of 11 null females had implantation. Unimplanted blastocysts with normal morphology were frequently recovered from mutant females (Fig. 1G), reinforcing our previous observation of normal preimplantation development. The results clearly show deferral of on-time implantation from the loss of HB-EGF, providing genetic evidence that HB-EGF signaling is indispensable for normal implantation. However, the necessity of maternal vs. embryonic HB-EGF in this process needs to be addressed.
Maternal HB-EGF Is Crucial for Implantation.
To obtain genetic evidence for the relative contribution of maternal vs. embryonic HB-EGF in implantation, we performed reciprocal embryo-transfer experiments. Day 4 WT or Hbegf−/− blastocysts were transferred into WT or null recipients on day 4 of pseudopregnancy, and implantation rates were examined 24 h later by the blue dye method. As shown in Table 1, eight of nine WT recipients receiving WT blastocysts showed normal implantation (46%). By contrast, WT blastocysts transferred into Hbegf−/− recipients showed considerably reduced number of implantation sites; only 12% of the transferred blastocysts showed implantation in only two of eight mutant recipients. However, impaired implantation was not observed when Hbegf−/− blastocysts were transferred into WT uteri; ≈43% of null blastocysts transferred implanted in seven of eight WT recipients. Collectively, the results show that maternal HB-EGF is the primary contributor to the on-time initiation of blastocyst implantation.
Table 1.
Reciprocal embryo transfer in WT and Hbegf−/− mice
| Genotypes |
No. of blastocysts transferred | No. of recipients | No. of mice with IS (%) | No. of IS (%) | No. of blastocysts recovered from mice without IS (%) | |
|---|---|---|---|---|---|---|
| Blastocysts | Recipients | |||||
| +/+ | +/+ | 2143 | 9 | 8 (89) | 66 (46) | 0 (0) |
| +/+ | −/− | 156 | 8 | 2 (25) | 19 (12) | 19 (12) |
| −/− | +/+ | 83 | 8 | 7 (88) | 36 (43) | 2 (2) |
Day 4 WT (+/+) or Hbegf null (−/−) blastocysts were transferred into WT or mutant pseudopregnant recipients on day 4 midmorning, and implantation was examined 24 h later by the blue dye method.
Amphiregulin Partially Compensates for the Loss of HB-EGF During Implantation.
Our present findings that the implantation process is not completely abolished with the loss of HB-EGF suggest that there are other signaling molecules replacing HB-EGF's function in implantation. Because the EGF family of ligands and ErbBs show overlapping uterine expression around the time of implantation (2, 4, 5), and because this ligand-receptor signaling network manifests great redundancy at multiple levels, such as diverse ligand selectivity and dimeric ErbB receptor complexes (28), we surmised that one or more of the family members acting via appropriate ErbBs partially compensate for the loss of HB-EGF during implantation.
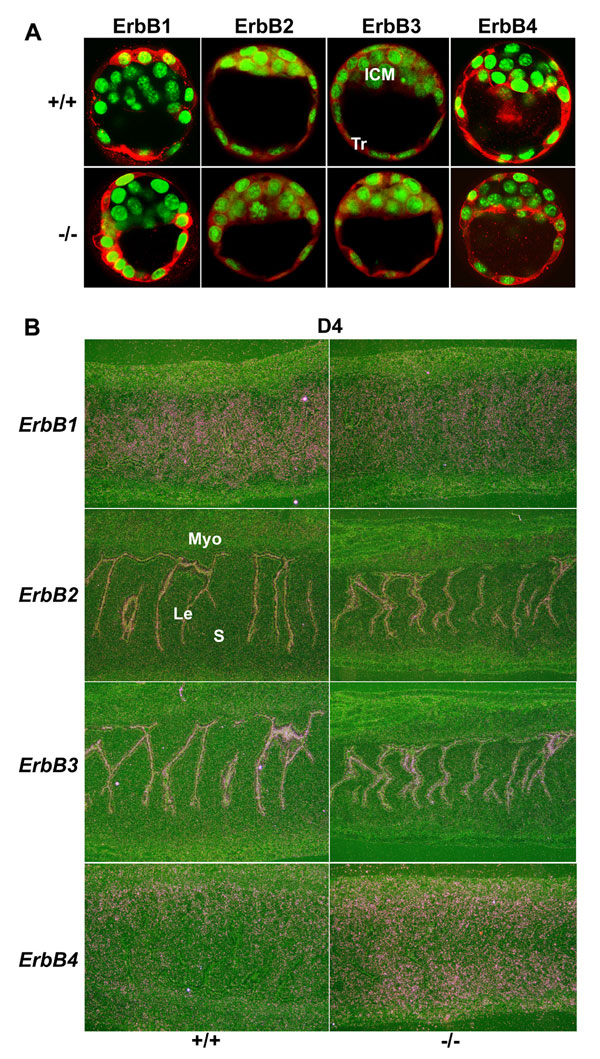
To test this hypothesis, we first examined expression patterns of ErbB receptors in blastocysts and uteri in WT and Hbegf−/− mice. In agreement with earlier studies (16–18), immunofluorescence revealed comparable expression patterns for ErbB receptors in both WT and Hbegf−/− day 4 blastocysts with ErbB1 and ErbB4 primarily being localized in the trophectoderm [supporting information (SI) Fig. 6A]. In addition, in situ hybridization showed unaltered cell-specific expression of these receptors in day 4 receptive uteri in the absence of HB-EGF (SI Fig. 6B). These results suggest that the machinery for the ErbB signaling network is functional in the periimplantation uterus and blastocyst in the absence of HB-EGF. In light of this finding, we next examined whether there is functional redundancy of the EGF family of ligands in Hbegf−/− mice during implantation.
We specifically analyzed the expression of Areg and Ereg, which encode amphiregulin and epiregulin, respectively, by in situ hybridization, because amphiregulin shares the similar heparin-binding feature as HB-EGF (29), and epiregulin has the same ErbB receptor binding selectivity as HB-EGF (30). In addition, they show overlapping expression pattern with HB-EGF in the periimplantation uterus (2, 4, 5). As shown in Fig. 2, we observed a unique compensatory expression of Areg, but not Ereg, in uteri of mice missing HB-EGF. In WT mice, Hbegf expression is first detected in the luminal epithelium surrounding the blastocyst on day 4 of pregnancy at 1800 h, before the initiation of attachment (Fig. 2A), but not at 0800 h (data not shown). In contrast, Areg expression is primarily present in glandular and luminal epithelia on day 4 morning (data not shown), but is largely down-regulated at 1800 h on day 4 regardless of the presence of blastocysts (Fig. 2B). With the onset of the attachment reaction (2400 h), Areg is induced solely in the luminal epithelium surrounding the blastocyst (Fig. 2B). This biphasic expression pattern of Areg suggests its dual role, first in regulating uterine receptivity and later in mediating the embryo–uterine interaction for attachment. However, this biphasic expression profile of Areg is altered in Hbegf null uteri, showing a sustained high level of expression in the luminal and glandular epithelia from day 4 morning through midnight (Fig. 2B). Conversely, Ereg, which was first detected in the luminal epithelium and underlying stroma at the blastocyst attachment site on day 4 midnight in WT mice, was barely detected even at 2400 h of day 4 in Hbegf−/− mice (Fig. 2C). This interesting finding suggests that sustained epithelial expression of Areg partially replaces the function of HB-EGF in initiating implantation in Hbegf−/− females.
Fig. 2.

Amphiregulin compensates for the loss of HB-EGF during implantation. (A) In situ hybridization of Hbegf in the luminal epithelium surrounding the blastocyst before and during the onset of implantation in WT uteri. (B) Areg is expressed much earlier and at higher levels in the absence of HB-EGF. (C) Ereg is first detected in the luminal epithelium at the site of blastocyst attachment in WT mice, but its expression is barely detectable even at 2400 h of day 4 in Hbegf−/− uteri. Arrows indicate location of blastocysts (×40). Le, luminal epithelium; ge, glandular epithelium; s, stroma; Bl, blastocysts; Myo, myometrium.
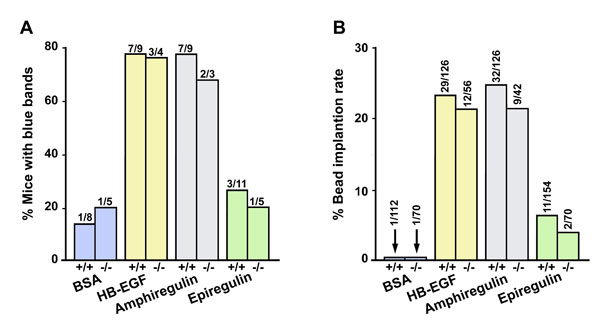
To test the effectiveness of amphiregulin in promoting implantation, we performed bead transfer experiments by introducing beads preabsorbed with purified HB-EGF, amphiregulin or epiregulin into receptive uteri on day 4 of pseudopregnancy. Under these conditions, blastocyst-size beads presoaked with the carrier protein BSA fail to induce implantation-like reactions and decidualization (27, 31). We observed that beads loaded with HB-EGF or amphiregulin, but not epiregulin, induced discrete local responses similar to those elicited by living blastocysts in WT and Hbegf−/− recipients (SI Fig. 7A and B). These results support our contention that amphiregulin has the potential to replace HB-EGF's function in initiating the attachment reaction. However, the underlying mechanism allowing compensatory expression of Areg with the loss of HB-EGF still remained unknown.
Attenuated Preimplantation Ovarian Estrogen Secretion in Hbegf−/− Mice Contributes to Sustained Uterine Areg Expression.
In mice, ovarian P4 and estrogen are the principal hormones that coordinate blastocyst activation and uterine receptivity for normal implantation (1). Because P4 and estrogen exert stimulatory and repressive roles, respectively, in regulating uterine Areg expression (4), we speculated that its compensatory up-regulation in Hbegf−/− uteri could be due to altered preimplantation secretion of ovarian P4 and/or estrogen. In fact, we observed significantly reduced serum levels of both P4 and 17β-estradiol (E2) when examined on day 4 morning (0800 h) in Hbegf null females compared with wild-type littermates (Fig. 3 A and B). However, the question remained as to how HB-EGF regulates ovarian steroidogenesis during the periimplantation period. To address this, we first examined the expression status of HB-EGF and ErbB receptors in the ovary during early pregnancy.
Fig. 3.

Reduced preimplantation ovarian estrogen secretion retains uterine Areg expression in Hbegf null uteri. (A and B) Serum progesterone (P4) and estradiol-17β (E2) levels are significantly lower in Hbegf−/− mice than WT females on day 4 morning (0800 h). Data are means ± SEM (*, P < 0.05; Student t test). Numbers within bars indicate numbers of mice examined. (C) RU486 (400 μg/mouse at 2400 h on day 3) abolishes Areg expression in WT uteri, whereas ICI-182780 (ICI) (125 μg per mouse at 2400 h on day 3) retains Areg expression at 0800 h and 1800 h on day 4 (×40). (D) Whereas P4 (2 mg per mouse at 0800 h on day 4) enhances Areg expression, E2 (3 ng per mouse at 0800 h on day 4) largely neutralizes the retained Areg expression in Hbegf−/− uteri at 1800 h on day 4 (×100). However, Areg is normally reinduced in luminal epithelial cells at the site of blastocyst apposition on day 4 midnight (2400 h) in null females treated with E2 (3 ng per mouse at 0800 h on day 4). Le, luminal epithelium; ge, glandular epithelium; s, stroma; Myo, myometrium.
Immunohistochemistry analysis revealed HB-EGF expression in growing follicles, newly formed corpora lutea and interstitial tissues on days 1 and 4 of pregnancy (SI Fig. 8A). With respect to ErbBs, whereas ErbB1 is primarily localized in corpora lutea, strong signals for ErbB2, ErbB3 and ErbB4 are detected in the developing follicles with lower levels in corpora lutea and interstitial tissues (SI Fig. 8A). These observations suggest that HB-EGF is a potential player in regulating ovarian steroidogenesis during early pregnancy. To search for the cause of reduced P4 and E2 secretion in the absence of HB-EGF, we examined the expression of key steroid biosynthetic enzymes, such as cytochrome P450 cholesterol side-chain cleavage enzyme (P450scc), 3β-hydroxysteroid dehydrogenase (3β-HSD), and cytochrome P450 aromatase (P450Arom), in day 4 ovaries. As shown in SI Fig. 8 B and C, we noted substantially reduced expression of P450scc (the rate-limiting enzyme of P4 and E2 biosynthesis) in corpora lutea and interstitial tissues lacking HB-EGF, whereas expression of 3β-HSD and P450Arom was comparable between WT and null ovaries. Decreased ovarian P450scc expression correlates well with our finding of overall decreased serum P4 and E2 levels in null mice (Fig. 3 A and B). However, reduced P4 levels cannot explain the phenotype of sustained uterine Areg expression before and during implantation in the absence of HB-EGF.
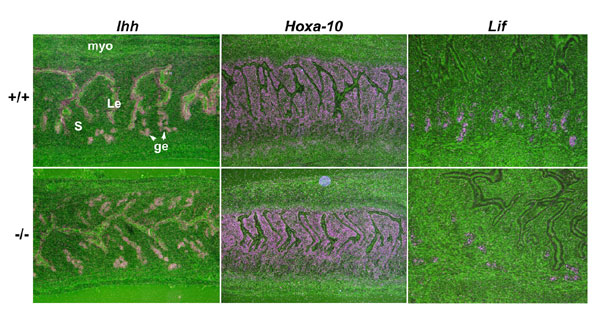
One could argue that P4 even at reduced levels is sufficient to maintain normal P4-responsive gene expression in the uterus. Indeed, expression of Indian hedgehog (Ihh) and Hoxa-10, known P4-target genes (32, 33), was comparable in WT and Hbegf−/− day 4 uterine epithelial and stromal cells, respectively (SI Fig. 9), supporting the view that reduced preimplantation P4 secretion from the loss of HB-EGF doest not disrupt P4's role in uterine receptivity. Because estrogen is also critical for uterine receptivity, we examined the effect of reduced preimplantation E2 secretion on uterine expression of leukemia inhibitory factor (LIF), an E2-target gene (34), and found that Lif was expressed in uterine glands in both WT and Hbegf−/− females on day 4 morning (SI Fig. 9). These results suggest that reduced preimplantation P4 and E2 secretion from the loss of HB-EGF is still capable of governing events central to uterine receptivity.
To distinguish the roles of P4 and E2 in regulating uterine Areg expression, we used selective antagonists to progesterone receptor and estrogen receptor, RU486 and ICI-182780, respectively. In contrast to normal uterine Areg expression in WT females on day 4 morning (control; 0800 h), its expression greatly diminished in uteri of mice treated with RU486 on day 3 midnight (2400 h) (Fig. 3C). This result supports our earlier observation that P4 is the primary inducer of Areg in receptive uteri (4). More interestingly, we observed sustained Areg expression through day 4 afternoon (1800 h) in WT mice receiving ICI-182780 treatment (Fig. 3C vs. Fig. 2B). This induction of Areg by ICI-182780 is similar to its compensatory expression in Hbegf−/− uteri. These findings provide direct evidence that although rising P4 levels induce uterine Areg expression on the morning of day 4, preimplantation ovarian E2 secretion down-regulates uterine Areg expression several hours before the attachment reaction.
To further explore roles of ovarian steroids in regulating Areg expression in the periimplantation uterus, we examined the influence of P4 or E2 treatment on Areg expression in Hbegf−/− uteri. As illustrated in Fig. 3D, the expression of Areg in null uteri on day 4 afternoon (1800 h) was substantially enhanced when mutant females were treated with P4 (2 mg per mouse) on day 4 morning (0800 h). In contrast, the compensatory expression of Areg in mutant uteri at 1800 h on day 4 was neutralized in Hbegf−/− mice receiving an injection of E2 (3 ng per mouse) on day 4 morning. This observation provides additional evidence that the sustained uterine expression of Areg in Hbegf−/− females is due to reduced preimplantation ovarian E2 secretion. Moreover, we observed that Areg expression reappears in luminal epithelial cells at the site of blastocyst apposition on day 4 midnight (2400 h) in Hbegf−/− mice receiving an E2 (3 ng per mouse) injection (Fig. 3D). It is possible that this late phase of Areg expression is induced by implantation-competent blastocysts and contributes to overriding HB-EGF deficiency in initiating attachment reaction.
Amphiregulin Compensates for the Loss of HB-EGF in Delayed Implanting Mice.
We used the delayed implantation model with defined doses of P4 and E2 to further explore amphiregulin's compensatory role in implantation in the absence of HB-EGF. As expected, implantation occurred in delayed implanting WT and Hbegf−/− females when examined 24 h after an E2 administration (25 ng per mouse) (Fig. 4 A and B). In situ hybridization analysis revealed that although Areg was primarily expressed in the glandular epithelium with lower levels in the luminal epithelium in P4-primed delayed uteri of both WT and Hbegf−/− mice, its expression was promptly induced in the luminal epithelium surrounding the blastocyst in WT and null uteri as early as 6 h after E2 injection (Fig. 4C), which is 6–8 h before the initiation of implantation in delayed implanting mice by 25 ng of E2. These results support our view that the late phase of Areg expression coincident with blastocyst activation compensates for the loss of HB-EGF during implantation.
Fig. 4.

Amphiregulin compensates for the loss of HB-EGF in delayed implanting mice. (A) Implantation rates in delayed implanting WT or Hbegf−/− mice after E2 injection (25 ng per mouse). Mice were ovariectomized on the morning of day 4 of pregnancy and administered P4 from days 5 through 7. IS were examined 24 after E2 injection on day 7 by the blue dye method. Numbers within bars indicate the number of mice with IS per total number of mice examined. (B) Average number of IS in WT and Hbegf−/− mice. Data are presented as mean ± SEM. Numbers within the bars indicate the number of mice examined. (C) In situ hybridization showing maintained Areg expression in the glandular (ge) and luminal (Le) epithelia in P4-primed delayed uteri (P4, 6 and 24 h). Moreover, Areg is induced in the luminal epithelium surrounding the blastocyst as early as 6 h after E2 injection (×40). S, stroma; Myo, myometrium. (D and E) Implantation of normal day 4 WT blastocysts transferred into delayed WT and Hbegf−/− recipients on day 7 of pseudopregnancy. Immediately after transfer, the recipient received an E2 injection (3 or 25 ng per mouse, s.c.); implantation was examined 24 h later. Numbers within the bar in D indicate the number of recipients with IS per the total number of mice examined, and those in E are the number of IS per the total number of blastocysts transferred. *, P < 0.05; Student t test.
Because Hbegf and Areg expression in delayed implanting uteri depends on the status of blastocyst activation (Fig. 4C and ref. 5), we surmised that uterine loss of HB-EGF would alter the minimum level of estrogen required for the initiation of the attachment reaction in delayed implanting mice. To address this, we did blastocyst transfer experiments. Day 4 WT blastocysts were transferred into P4-primed delayed WT or Hbegf−/− recipients on day 7 of pseudopregnancy. Immediately after transfer, recipients received an E2 injection (3 or 25 ng per mouse, sc) and implantation rates were examined 24 h later by the blue dye method. As shown in Fig. 4 D and E, 60% of the transferred blastocysts implanted in all WT recipients (n = 5) receiving 3 ng of E2. By contrast, only 26% of the transferred blastocysts implanted in 7 of 12 Hbegf−/− recipients injected with 3 ng of E2. HB-EGF-deficient recipients receiving even 25 ng of E2 had reduced implantation rate. For example, whereas 68% of the transferred blastocysts implanted in WT recipients (n = 5), only 44% of the transferred blastocysts showed implantation in Hbegf−/− recipients (n = 5). Collectively, the results show that uterine HB-EGF deficiency contributes to impaired implantation in Hbegf−/− mice.
Conditional Deletion of Uterine HB-EGF Confers Implantation Defects.
During the course of this investigation, we consistently observed that a large number of Hbegf−/− mice failed to survive to adulthood, perhaps because of severe heart failure as previously reported (9). This raised the concern that implantation defects we observed in Hbegf mutant females could be due to their poor health condition. To address this question, we established a PR-Cre+/−/Hbegflox/lox mouse line by intercrossing Hbegflox/lox females with hemizygous progesterone receptor Cre (PR-Cre) knockin males (35) to achieve conditional deletion of the Hbegf gene in the PR-expressing tissues, such as the uterus and ovary. The deletion of HB-EGF cDNA leads to the expression of the lacZ gene inserted downstream of the HB-EGF cDNA as described (9).
As shown in Fig. 5A, although LacZ-stained blue cells are visualized in both the lung and heart of genomic Hbegf−/− mice, this gene is not deleted in these tissues of PR-Cre+/−/Hbegflox/lox mice, as evident from the absence of LacZ-stained cells. Most importantly, PR-Cre-mediated deletion of the Hbegf gene was observed in the uterine epithelium at the attachment sites on day 5 morning, resembling that in systemic Hbegf−/− females (Fig. 5A). RT-PCR analysis of Hbegf mRNA showed that this gene was almost totally deleted in the uterus, but not in the heart, of PR-Cre+/−/Hbegflox/lox mice (Fig. 5B). In contrast, ovarian Hbegf mRNA levels were still abundant on days 1 and 4, indicating that the efficiency of Hbegf gene deletion is low in PR-Cre+/−/Hbegflox/lox ovaries (Fig. 5B). This is perhaps due to transient expression of PR-Cre in corpora lutea as previously reported (35) and that HB-EGF is also expressed in follicles and interstitial tissues (SI Fig. 8A), which show little or no PR-Cre activity (35). Thus, ovarian cells still expressing HB-EGF in PR-Cre+/−/Hbegflox/lox mice maintained normal steroidogenesis as seen from comparable serum levels of P4 and E2 in Hbegflox/lox and PR-Cre+/−/Hbegflox/lox mice on day 4 (Fig. 5 C and D). These results led us to ask whether the implantation process is still impaired with conditional deletion of uterine HB-EGF.
Fig. 5.

Conditional deletion of uterine HB-EGF confers implantation defects similar to systemic Hbegf deletion. (A) LacZ staining indicates HB-EGF deletion specifically in uteri but not in lung or heart in PR-Cre+/−/Hbegflox/lox mice (×40). (B) RT-PCR analysis of Hbegf mRNA revealing almost total deletion of Hbegf gene in PR-Cre+/−/Hbegflox/lox uteri, but not in the heart. Hbegf mRNA levels were abundant in PR-Cre+/−/Hbegflox/lox ovaries on days 1 and 4 of pregnancy. (C and D) Comparable levels of serum progesterone (P4) and estradiol-17β (E2) in Hbegflox/lox and PR-Cre+/−/Hbegflox/lox mice on day 4 morning (0800 h). Numbers within the bars indicate number of mice examined. Data are means ± SEM. (E) Implantation is severely compromised in PR-Cre+/−/Hbegflox/lox mice examined at 2400 h on day 4. Numbers within the bars indicate number of mice with IS per total number of mice examined. (F) Litter sizes of mice are significantly smaller in PR-Cre+/−/Hbegflox/lox mice (*, P < 0.05; Student t test). Numbers within the bars indicate number of mice with litters per the total number of plug-positive mice. (G) In situ hybridization of uterine Areg in Hbegflox/lox and PR-Cre+/−/Hbegflox/lox mice (×40). Areg expressed surrounding the blastocyst (arrows) during the late phase contributes to replace uterine HB-EGF's function during implantation.
It was exciting to see that uterine-specific deletion of HB-EGF results in similar phenotypic defects of deferred on-time implantation with compromised litter size (Fig. 5 E and F). To further test the compensatory contribution of biphasic expression of Areg, we performed in situ hybridization analysis. Interestingly, although no sustained Areg expression was observed in uterine epithelia at 1800 h on day 4, Areg was induced solely in the luminal epithelium surrounding the blastocyst on day 4 midnight (2400 h) in both Hbegflox/lox and PR-Cre+/−/Hbegflox/lox mice (Fig. 5G). These results reinforce that the second phase of Areg expression surrounding the blastocyst contributes to override HB-EGF deficiency in initiating attachment reaction. These results provide genetic evidence that uterine-derived HB-EGF signaling is also essential for on-time implantation and normal pregnancy outcome.
Emerging evidence suggests that a short deferral of the attachment of blastocysts to the uterine lumen during early pregnancy adversely affects term pregnancy success (1). Our genetic, pharmacological, and physiological evidence show that deferral of on-time implantation from the loss of maternal HB-EGF leads to compromised pregnancy outcome, supporting our contention that implantation is the gateway to pregnancy success. Moreover, it is intriguing that HB-EGF function during implantation is selectively, but partially, replaced by amphiregulin, another heparin-binding growth factor of the EGF family (29), but not by epiregulin, which also utilizes ErbB receptors (30). This ligand redundancy among specific EGF ligands may serve as a safeguard regulatory mechanism ensuring normal progression of early pregnancy under various pathophysiological conditions. In search of the underlying mechanism for the compensatory expression of Areg in Hbegf null uteri, we also show that ovarian HB-EGF deficiency attenuates preimplantation ovarian secretion of P4 and estrogen by limiting the expression of P450scc in the ovary. Using pharmacological approaches and the delayed implantation mouse model, we further show that reduced ovarian E2 secretion, but not P4, leads to sustained Areg expression in Hbegf null uteri during the periimplantation period. This is a remarkable finding that reduced ovarian E2 secretion from systemic HB-EGF deficiency promotes compensatory expression of Areg to override uterine HB-EGF deficiency during implantation. In conclusion, this study provides genetic evidence that maternal HB-EGF is a critical signaling molecule for early pregnancy success in mice. These findings are clinically relevant to humans because there is evidence that HB-EGF signaling is also important for implantation in humans (21, 36, 37).
Methods
Mice.
HB-EGF-deficient mice on a C57BL/6J genetic background were generated as described (9). Mouse HB-EGF cDNA containing the polyadenylation sequence flanked by loxP sequences was fused with the first exon of the mouse Hbegf gene. Cre-mediated recombination causes the deletion of Hbegf cDNA with the expression of the lacZ inserted downstream of Hbegf cDNA. Mice with systemic deletion of Hbegf were generated by breeding Hbegflox/lox mice with CAG-Cre mice. To induce uterine-specific deletion of the Hbegf gene, Hbegflox/lox mice were crossed with PR-Cre mice (35). LacZ staining and RT-PCR analysis of Hbegf mRNA in day 1 uteri of Hbegflox/lox and PR-Cre+/−/Hbegflox/lox mice revealed PR-Cre activity and deletion efficiency. All mice used in this investigation were housed in the Vanderbilt Animal Care Facility according to National Institutes of Health and institutional guidelines for laboratory animals. Female mice were mated with fertile or vasectomized male mice of the same genotype to induce pregnancy or pseudopregnancy, respectively (day 1 = vaginal plug). Experimental procedures to analyze ovulation, fertilization, and implantation are provided in SI Methods.
P4 and E2 Assay.
Mouse blood samples were collected on day 4 (0800 h). Serum P4 and E2 levels were measured by RIA.
In Situ Hybridization.
Frozen sections were hybridized with 35S-labeled cRNA probes to murine Hbegf, Areg, Ereg, ErbB1, ErbB2, ErbB3, ErbB4, Hoxa-10, Lif, or Ihh as described (5).
LacZ Staining.
LacZ staining in frozen sections was performed as we described (38).
Supplementary Material
Acknowledgments
We thank J. Ian Mason (University of Edinburgh, Edinburgh, U.K.) for his generous gift of 3β-HSD antibody. P4 and E2 assays were performed by the University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core (Charlottesville, VA) supported by National Institute of Child Health and Human Development (NICHD) Grant U54-HD28934. This work was supported in part by National Institutes of Health Grants HD12304, HD33994, and DA06668 (to S. K. Dey), ES07814 and HD37830 (to S. K. Das), and HD50315 (to H.W.). S. K. Dey is the recipient of Method to Extend Research in Time (MERIT) Awards from the NICHD and the National Institute on Drug Abuse (NIDA). H.X. is a Lalor Foundation Postdoctoral Fellow. S.T. is supported by National Research Service Award Individual Fellowship F31 DA021062 from NIDA. We also thank the Turner Foundation for generous support (to H.W.).
Abbreviations
- 3β-HSD
3β-hydroxysteroid dehydrogenase
- E2
17β-estrodial
- HB-EGF
heparin-binding EGF-like growth factor
- LIF
leukemia-inhibitory factor
- P4
progesterone
- PR
progesterone receptor
- P450Arom
cytochrome P450 aromatase
- P450scc
cytochrome P450 cholesterol side-chain cleavage enzyme
- TGFα
transforming growth factor α
- IS
implantation site.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0707909104/DC1.
References
- 1.Wang H, Dey SK. Nat Rev Genet. 2006;7:185–199. doi: 10.1038/nrg1808. [DOI] [PubMed] [Google Scholar]
- 2.Das SK, Das N, Wang J, Lim H, Schryver B, Plowman GD, Dey SK. Dev Biol. 1997;190:178–190. doi: 10.1006/dbio.1997.8694. [DOI] [PubMed] [Google Scholar]
- 3.Tamada H, Das SK, Andrews GK, Dey SK. Biol Reprod. 1991;45:365–372. doi: 10.1095/biolreprod45.2.365. [DOI] [PubMed] [Google Scholar]
- 4.Das SK, Chakraborty I, Paria BC, Wang XN, Plowman G, Dey SK. Mol Endocrinol. 1995;9:691–705. doi: 10.1210/mend.9.6.8592515. [DOI] [PubMed] [Google Scholar]
- 5.Das SK, Wang XN, Paria BC, Damm D, Abraham JA, Klagsbrun M, Andrews GK, Dey SK. Development (Cambridge, UK) 1994;120:1071–1083. doi: 10.1242/dev.120.5.1071. [DOI] [PubMed] [Google Scholar]
- 6.Lee D, Pearsall RS, Das S, Dey SK, Godfrey VL, Threadgill DW. Mol Cell Biol. 2004;24:8907–8916. doi: 10.1128/MCB.24.20.8907-8916.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luetteke NC, Qiu TH, Fenton SE, Troyer KL, Riedel RF, Chang A, Lee DC. Development (Cambridge, UK) 1999;126:2739–2750. doi: 10.1242/dev.126.12.2739. [DOI] [PubMed] [Google Scholar]
- 8.Shirasawa S, Sugiyama S, Baba I, Inokuchi J, Sekine S, Ogino K, Kawamura Y, Dohi T, Fujimoto M, Sasazuki T. Proc Natl Acad Sci USA. 2004;101:13921–13926. doi: 10.1073/pnas.0404217101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iwamoto R, Yamazaki S, Asakura M, Takashima S, Hasuwa H, Miyado K, Adachi S, Kitakaze M, Hashimoto K, Raab G, et al. Proc Natl Acad Sci USA. 2003;100:3221–3226. doi: 10.1073/pnas.0537588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson LF, Qiu TH, Sunnarborg SW, Chang A, Zhang C, Patterson C, Lee DC. EMBO J. 2003;22:2704–2716. doi: 10.1093/emboj/cdg264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marikovsky M, Breuing K, Liu PY, Eriksson E, Higashiyama S, Farber P, Abraham J, Klagsbrun M. Proc Natl Acad Sci USA. 1993;90:3889–3893. doi: 10.1073/pnas.90.9.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyagawa J, Higashiyama S, Kawata S, Inui Y, Tamura S, Yamamoto K, Nishida M, Nakamura T, Yamashita S, Matsuzawa Y, et al. J Clin Invest. 1995;95:404–411. doi: 10.1172/JCI117669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lemjabbar H, Basbaum C. Nat Med. 2002;8:41–46. doi: 10.1038/nm0102-41. [DOI] [PubMed] [Google Scholar]
- 14.Ongusaha PP, Kwak JC, Zwible AJ, Macip S, Higashiyama S, Taniguchi N, Fang L, Lee SW. Cancer Res. 2004;64:5283–5290. doi: 10.1158/0008-5472.CAN-04-0925. [DOI] [PubMed] [Google Scholar]
- 15.Miyamoto S, Hirata M, Yamazaki A, Kageyama T, Hasuwa H, Mizushima H, Tanaka Y, Yagi H, Sonoda K, Kai M, et al. Cancer Res. 2004;64:5720–5727. doi: 10.1158/0008-5472.CAN-04-0811. [DOI] [PubMed] [Google Scholar]
- 16.Wang J, Mayernik L, Schultz JF, Armant DR. Development (Cambridge, UK) 2000;127:33–44. doi: 10.1242/dev.127.1.33. [DOI] [PubMed] [Google Scholar]
- 17.Paria BC, Das SK, Andrews GK, Dey SK. Proc Natl Acad Sci USA. 1993;90:55–59. doi: 10.1073/pnas.90.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paria BC, Elenius K, Klagsbrun M, Dey SK. Development (Cambridge, UK) 1999;126:1997–2005. doi: 10.1242/dev.126.9.1997. [DOI] [PubMed] [Google Scholar]
- 19.Hamatani T, Daikoku T, Wang H, Matsumoto H, Carter MG, Ko MS, Dey SK. Proc Natl Acad Sci USA. 2004;101:10326–10331. doi: 10.1073/pnas.0402597101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park JY, Su YQ, Ariga M, Law E, Jin SL, Conti M. Science. 2004;303:682–684. doi: 10.1126/science.1092463. [DOI] [PubMed] [Google Scholar]
- 21.Yoo HJ, Barlow DH, Mardon HJ. Dev Genet (Amsterdam) 1997;21:102–108. doi: 10.1002/(SICI)1520-6408(1997)21:1<102::AID-DVG12>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 22.Shimada M, Hernandez-Gonzalez I, Gonzalez-Robayna I, Richards JS. Mol Endocrinol. 2006;20:1352–1365. doi: 10.1210/me.2005-0504. [DOI] [PubMed] [Google Scholar]
- 23.Hsieh M, Lee D, Panigone S, Horner K, Chen R, Theologis A, Lee DC, Threadgill DW, Conti M. Mol Cell Biol. 2007;27:1914–1924. doi: 10.1128/MCB.01919-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dalton T, Kover K, Dey SK, Andrews GK. Biol Reprod. 1994;51:597–606. doi: 10.1095/biolreprod51.4.597. [DOI] [PubMed] [Google Scholar]
- 25.Lim JJ, Lee DR, Song HS, Kim KS, Yoon TK, Gye MC, Kim MK. J Assist Reprod Genet. 2006;23:111–119. doi: 10.1007/s10815-006-9021-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin KL, Barlow DH, Sargent IL. Hum Reprod. 1998;13:1645–1652. doi: 10.1093/humrep/13.6.1645. [DOI] [PubMed] [Google Scholar]
- 27.Paria BC, Ma W, Tan J, Raja S, Das SK, Dey SK, Hogan BL. Proc Natl Acad Sci USA. 2001;98:1047–1052. doi: 10.1073/pnas.98.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Citri A, Yarden Y. Nat Rev Mol Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 29.Thorne BA, Plowman GD. Mol Cell Biol. 1994;14:1635–1646. doi: 10.1128/mcb.14.3.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones JT, Akita RW, Sliwkowski MX. FEBS Lett. 1999;447:227–231. doi: 10.1016/s0014-5793(99)00283-5. [DOI] [PubMed] [Google Scholar]
- 31.McLaren A. J Reprod Fertil. 1969;19:199–201. doi: 10.1530/jrf.0.0190199. [DOI] [PubMed] [Google Scholar]
- 32.Matsumoto H, Zhao X, Das SK, Hogan BL, Dey SK. Dev Biol. 2002;245:280–290. doi: 10.1006/dbio.2002.0645. [DOI] [PubMed] [Google Scholar]
- 33.Lim H, Ma L, Ma WG, Maas RL, Dey SK. Mol Endocrinol. 1999;13:1005–1017. doi: 10.1210/mend.13.6.0284. [DOI] [PubMed] [Google Scholar]
- 34.Song H, Lim H, Das SK, Paria BC, Dey SK. Mol Endocrinol. 2000;14:1147–1161. doi: 10.1210/mend.14.8.0498. [DOI] [PubMed] [Google Scholar]
- 35.Soyal SM, Mukherjee A, Lee KY, Li J, Li H, DeMayo FJ, Lydon JP. Genesis. 2005;41:58–66. doi: 10.1002/gene.20098. [DOI] [PubMed] [Google Scholar]
- 36.Leach RE, Khalifa R, Ramirez ND, Das SK, Wang J, Dey SK, Romero R, Armant DR. J Clin Endocrinol Metab. 1999;84:3355–3363. doi: 10.1210/jcem.84.9.5980. [DOI] [PubMed] [Google Scholar]
- 37.Lessey BA, Gui Y, Apparao KB, Young SL, Mulholland J. Mol Reprod Dev. 2002;62:446–455. doi: 10.1002/mrd.10129. [DOI] [PubMed] [Google Scholar]
- 38.Ma W, Tan J, Matsumoto H, Robert B, Abrahamson DR, Das SK, Dey SK. Mol Endocrinol. 2001;15:1983–1992. doi: 10.1210/mend.15.11.0734. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}