

Abstract
Since the tyrosine phosphatase SHP-1 plays a major role in regulating T-cell signaling, we investigated regulation thereof by Ser/Thr phosphorylation. We found that TCR stimulation induced fast (≤1min) and transient phosphorylation of SHP-1 S591 in both Jurkat and human peripheral blood T-cells (PBT). Phosphorylation of S591 in T-cells could be mediated artificially by a constitutive active PKC-theta construct, but the dose dependence of inhibition by PKC inhibitors indicated that PKCs were not the relevant basophilic kinase in the physiologic response. S591 phosphorylation inhibited phosphatase function since a S591D mutant had lower activity than the S591A mutant. Additional evidence that S591 phosphorylation alters SHP-1 function was provided by studies of Jurkat cells stably expressing SHP-1 wildtype or mutants. In those cells, S591D mutation reduced the capacity of transfected SHP-1 to inhibit TCR-induced phosphorylation of PLC-γ1. Interestingly, SHP-1 Y536 phosphorylation (previously shown to augment phosphatase activity) was also induced in PBT by TCR signal but at a much later time compared to S591 (~30 min). S591 phosphorylation also altered cellular distribution of SHP-1 because: 1) SHP-1 in lipid rafts and a sheared membrane fraction was hypo-phosphorylated; 2) In stably transfected Jurkat cell lines, S591D mutant protein had reduced presence in both lipid raft and the sheared membrane fraction; 3) S591 phosphorylation prevented nuclear localization of a C-terminal GFP tagged SHP-1 construct. Our studies also shed light on an additional mechanism regulating SHP-1 nuclear localization, namely conformational autoinhibition. These findings highlight elegant regulation of SHP-1 by sequential phosporylation of serine then tyrosine.
Keywords: T lymphocyte, signal transduction, lipid raft, nuclear localization
Introduction
Phosphorylation and dephosphorylation are among the most important post-translational protein modifications, which often result in major changes in protein function. Protein kinases and phosphatases play counterbalancing roles regulating protein function in a temporally and spatially controlled fashion. SHP-1 (SH2 domain containing phosphatase 1) is a tyrosine phosphatase expressed mainly in hematopoietic cells[1, 2]. The importance of SHP-1 is highlighted by the characteristic phenotype of the SHP-1 deficient moth-eaten mice[3]. Those mice die at several weeks of age from inflammation of the lung, resulting from uncontrolled granulocytes proliferation and infiltration. They also have lymphocyte abnormalities, specifically hyperactivation of lymphocytes after antigen receptor engagement.
In T lymphocytes, SHP-1 is essential to attenuation of TCR signaling after initiation of the response[4]. Indeed, SHP-1 plays a role in the differential response to agonist/antagonist peptides[5]. Many signaling molecules downstream of TCR are targets of SHP-1 mediated dephosphorylation; each of them has to be dephosphorylated at the right time and place, to allow both efficient and controlled signaling. To achieve this, SHP-1 is likely to be regulated precisely by changes of both its phosphatase activity and cellular distribution. Its phosphatase activity is known to be powerfully increased by phosphorylation of Y536[6]. The structural basis for this upregulation of activity has been suggested by mutational and structural studies showing that the SH2 domains of SHP-1 form extensive interaction with the catalytic domain, inhibiting its activity[7]. It has been proposed that binding of SHP-1 SH2 domains with phospho-tyrosine, either intra-or inter-molecularly, releases it from auto-inhibition[8]. Binding of SHP-1 SH2 domain with phospho-tyrosine motifs (ITIM: immunereceptor tyrosine-based inhibitory motif; ITSM: immunereceptor tyrosine-based switch motif ) is also an important strategy of SHP-1 recruitment [9–12], which brings SHP-1 close to its effector molecules. On the contrary, binding of phosphorylated Y564 of SHP-1 with the SH2 domain of Lck has been shown to recruit SHP-1 to its intended target, which can be inhibited by ERK phosphorylation of lck[5]. Lipid rafts are important membrane micro-domains where signaling process take place. SHP-1 is present in lipid rafts and its lipid raft presence is critical for its function[13]. SHP-1 lipid rafts localization depends on its C-terminal tail but not on the SH2 domains[13, 14], but nothing is known about what can change SHP-1 lipid rafts localization.
Ser/Thr phosphorylation of SHP-1 was identified [15, 16] shortly after the identification of SHP-1[17]. This was associated with decreased phosphatase activity and PKC was proposed to be the relevant kinase [18] but the exact site had not been mapped. Using algorithms developed from our studies on substrate specificity of PKC[19], we found that S591 close the very end of SHP-1 C-terminal tail was potentially a very good phosphorylation site for PKC or other basophilic kinase. The PKC family is a member of a much larger group of “basophilic kinases”, which constitute a large group of Ser/Thr kinases that phosphorylate S/T residues in the context of a cluster of basophilic residues[20, 21]. Such clusters of basophilic residues have important functions including membrane-binding motifs or nuclear localization signals. In fact, previous studies already identified the stretch of positively charged residues around S591 in SHP-1 as a functional nuclear localization signal[22]. Phosphorylation, by neutralizing the net positive charge, is an important way of regulating those biological processes. The present study confirms our in silico prediction, and demonstrates that S591 of SHP-1 is phosphorylated following TCR stimulation. This confirms and extends a report, published during our studies, of S591 phosphorylation in platelets in response to thrombin[23]; but the TCR-induced response appears not to be mediated by PKC. Our studies demonstrate that S591 phosphorylation inhibits phosphatase activity and that the early and transient inhibitory phase of S591 phosphorylation after TCR stimulation is followed by Y536 phosphorylation, previously shown to augment phosphatase activity. Moreover, we demonstrate that SHP-1 nuclear localization is inhibited not only by phosphorylation at this site, but also by intramolecular auto-inhibition.
Material and methods
General
BIM I and III were purchase from EMD Biosciences (San Diego, CA). 4G10 anti-phosphotyrosine mAb and polyclonal anti-SHP-1 antibody made by immunization with whole SHP-1 protein were purchased from Upstate Biotechnology Inc. Monoclonal anti-SHP-1 Ab (clone 52, BD Biosciences) is the product of immunization with a fusion protein containing the SHP-1 C-terminal 106 residues. Anti-SHP-1 anti-peptide Ab (sc-287 from Santa Cruz Biotechnology) is the product of immunization with a peptide containing the C-terminal 19 residues. Antibodies against pY319 of ZAP70 and T202/Y204 of MAP kinase were purchased from Cell Signaling Technologies. Anti-SHP-1 pS591 antibody was made by immunization of rabbits with a phospho-peptide CDKEKSKGpSLKRKOH coupled to KLH. SHP-1 anti-pY536 is a monoclonal antibody raised in mice against pY536 by BD Biosciences. Goat anti-Mouse IRDye 680, Goat anti-rabbit IRDye 800CW were purchase from LI-COR (Lincoln, NE). All figures represent results of at least two independent experiments.
Constructs and expression
mouse SHP-1 cDNA was cloned into Gateway expression vector with N-terminal GFP tag (pcDNA-Dest53) or C-terminal GFP tag (pDest472). Mutations (S591A, S591D, S582D and S588D) were made with Stratagen QuikChange mutagenesis kit. All constructs made are fully sequence-validated.
Cell culture, transfection, stimulation and imaging
HEK293T cells were grown in DMEM medium (Life technologies, Inc) with 10% FCS. Transfection of HEK293T cell is mediated by calcium phosphate as described previously [24]. Jurkat cells (E6-1) were grown in 10% FCS RPMI 1640 (Life Technologies, Inc). For transfection, 1 × 107 Jurkat cells in 0.4ml RPMI 1640 with 20mM HEPES were mixed with appropriate plasmids and electroporated at 310v and 10ms in a BTX ECM 830 electroporator. For stable transfection, the transfected cells were cultured in selection media containing 1mg/ml G418. For stimulation, Jurkat cells were stimulated or not with CD3 mAb (clone 38.1 ascites, 1:1000) plus CD28 mAb (clone 9.3 ascites, 1:1000). For imaging, 20hr after transfection the cells were observed using a Zeiss LSM510 confocal microscope (Carl Zeiss MicroImaging Inc., Thornwood NY) equipped with a 40x C-apochromat (N.A. 1.2) water immersion lens. Confocal images were collected with 0.11 μm × 0.11 μm X-Y pixel sampling and optical slice thickness of 2.0 μm.
SHP-1 phosphatase assay
SHP-1 protein (WT or S591 mutants) was immunoprecipitated from transiently transfected HEK293 cell lysate by mAb against HA tag. The procedure was done without the presence of phosphatase inhibitors and the final wash was done with phosphatase assay buffer, which contained 20mM HEPES, 50mM NaCl, 5mM DTT. The phosphatase assay on phosphotyrosine peptide was done using the phosphatase assay kit from Upstate Biotechnology and the manufacturer’s recommended protocol. A fraction of the immunoprecipitated SHP-1 was blotted with HA mAb for quantification, and phosphatase assay result was adjusted based on the quantification. A series of 2 fold dilution of the immunoprecipitated SHP-1 was used in phosphatase assay and western blot to make sure the amount of different constructs were comparable.
Immunoprecipitation and Western blot
For detection of S591 phosphorylation of SHP-1, Jurkat TAg cells or HEK293T cells (transfected or not) were harvested and centrifuged. The cell pellet was loosened by vortexing and 1xSDS sample buffer was directly added to the pellet, mixed. After sonication to fragment DNA, the cell lysate was boiled for 5min. For immunoprecipitation, the cells were lysed with lysis buffer: 1% Triton X-100, 150mM NaCl, 50mM Tris-HCl pH7.4, 2mM Na3VO4, 5mM Na4P2O7, protease inhibitor cocktail tablet. The lysate was cleared by centrifugation at 14000rpm for 10 min. Immunoprecipitation was done with the indicated Ab and protein G agarose (Invitrogen). For western blot, immunoprecipitate or cell lysate in SDS sample buffer were separated by SDS-PAGE and transferred to nitrocellulose membrane. Detection of some of the Western blot was done using the Amersham ECL kit following the manufacturer’s protocol. Exposure was done either with film or with Fuji LAS-3000 imaging system. Other Western blots were analyzed using an Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, Nebraska).
Isolation of membrane fractions
For lipid raft isolation, 100 × 10@6 Jurkat cells were lysed with 1ml lysis buffer (1%Triton,10mM Tris-HCl pH7.5, 150mM NaCl, 5mM EDTA, protease inhibitor cocktail tablet, 2mM sodium orthovanadate, 5mM sodium pyrophosphate). The lysate was put on ice for 30min and then transferred to a Dounce homogenizer, stroked 10 times with the loose fitting pestle. The lysate was centrifuged at 500g for 10min. The recovered supernatant was mixed with equal volume 80% sucrose and transferred to an ultracentrifuge tube, and then overlaid on top with 5ml of 30% Sucrose and 3ml of 5% sucrose. After centrifuge at 40000rpm, at 4c for 18h, 12 even fractions (each about 0.8ml) were harvested. Lipid raft distribution was detected by blotting with Cholera toxin for the presence of GM1 Ganglioside. Fractions 3, 4, 5 and 6 were pooled as lipid raft fraction. Fractions 11 and 12 were pooled as cytosolic fraction. Isolation of a sheared membrane fraction by a protocol modeled after procedures used for separation of placental microvilli1. In brief, cells were passed repeatedly through a 27G needle. Cells remained viable were removed by sedimentation at 2500g for 5 min. Membrane enriched fragments including microvilli were recovered by sedimentation at 16000g for 40 min.
Results
S591 of SHP-1 is phosphorylated in T lymphocytes in response to TCR stimulation
Predictive algorithms can be very useful in identifying potential phosphorylation sites. But such predictions are most reliable in predicting what class of kinase might phosphorylate a site (e.g. basophilic) rather than exactly which kinase of that class would mediate the phosphorylation (e.g. PKC). During our in silico analysis of molecules involved in TCR signal transduction, we identified a promising site for PKC or another basophilic kinase near the C-terminus of SHP-1. As an initial test of that prediction, we used a “PKC substrate antibody” (pPKC) from Cell Signaling Technology Inc. because the sequence surrounding S591 fits perfectly with the sequence pattern recognized by this antibody. We blotted SHP-1 immunoprecipitated from resting or CD3/28 stimulated Jurkat cell with the pPKC Ab. There is already some signal recognized by this antibody in SHP-1 from resting cells, but the signal in SHP-1 from CD3/28 stimulated cells were stronger, indicating CD3/28 stimulation induced phosphorylation of SHP-1, possibly at S591 (Fig 1A).
Fig 1. SHP-1 is phosphorylated on S591 in Jurkat cells in response to TCR stimulation.
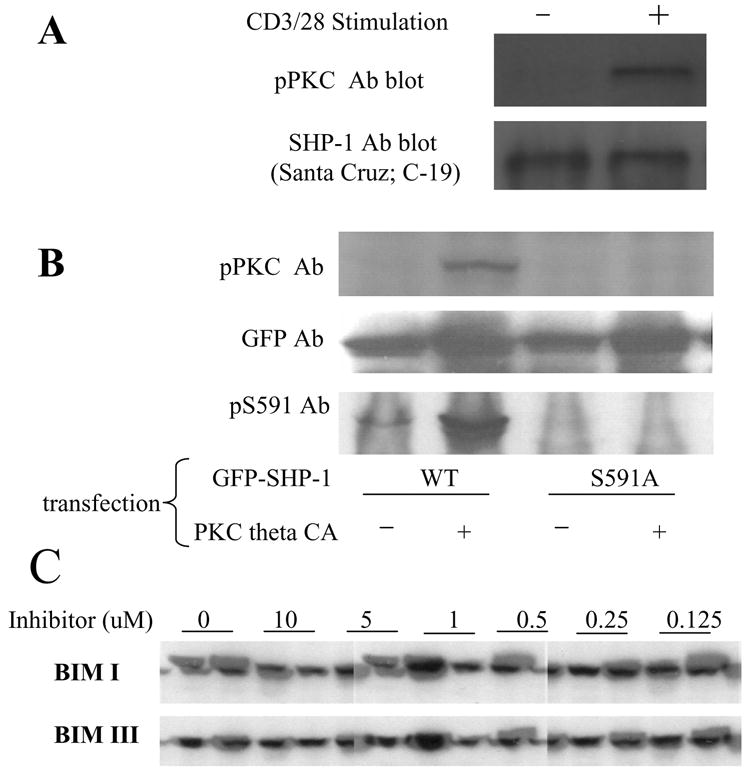
(A): Jurkat T cells were stimulated (or not) with CD3/28 Abs for 1min. Cell lysate was immunoprecipitated with SHP-1 Ab and the precipitated SHP-1 was blotted with pPKC Ab. (B): N-terminal GFP tagged SHP-1 WT or S591A mutant were transfected into Jurkat cells with or without co-transfection of PKC theta CA. The cell lysate was blotted with pPKC Ab and anti-pS591 phospho specific Ab. GFP Ab blot showed comparable expression of SHP-1 in all samples (C): Jurkat cells were pre-treated (or not) with various concentrations of BIM I or BIM III for 15min. Then the cells were stimulated (or not) with CD3/28 Ab for 1min. The cell lysate was blotted with pS591 phospho-specific Ab.
To confirm that S591 is the phosphorylation site on SHP-1 which is recognized by the pPKC Ab, we made SHP-1 constructs, wildtype or S591 mutated to A to prevent phosphorylation, tagged at their N-terminus with GFP (N-GFP_SHP-1 and N-GFP_SHP-1 S591A). Since the sequence surrounding S591 fits the amino acid pattern recognized by PKC (or other basophilic kinase), we expected that overexpression of PKC together with the SHP-1 construct would result in phosphorylation of S591. As predicted, transfected SHP-1 was phosphorylated when cotransfected with constitutively active PKC theta (PKC theta CA) (Fig 1B). In contrast, the phosphorylation recognized by pPKC Ab was not observed when the S591A mutant of SHP-1 was transfected into Jurkat cells together with PKC theta CA. Control conditions show that SHP-1 transfected alone into Jurkat cells alone was not detected well by the pPKC Ab. Thus, the phosphorylation in SHP-1 that is recognized by pPKC Ab is on S591, and that site can be phosphorylated by over-expressed PKC theta.
To facilitate further study of the newly identified phosphorylation site, we made a phospho-specific Ab against S591 of SHP-1. WB with this antibody showed a pattern similar to that observed with the pPKC antibody (Fig 1B). Phosphorylation of transfected SHP-1 occurred only when co-transfected with PKC theta CA and was lost on the S591A mutant, indicating the specificity of the antibody. Thus SHP-1 can be phosphorylated by over-expressed constitutively active PKC theta, and the pS591 antibody specifically recognizes that phosphorylated site.
We tested the possibility that PKC was involved in TCR-induced SHP-1 S591 phosphorylation in Jurkat cells by using PKC inhibitors BIM I and BIM III. As expected, the pS591 Ab detects TCR induced phosphorylation previously detected with the pPKC Ab (Fig 1C). At concentrations of 5uM or above, both BIMs inhibited S591 phosphorylation. But below 1uM neither inhibited effectively. Since the IC50 of both inhibitors are in the low nanomolar range for PKC, the result suggest that some basophilic kinase other than PKC mediates this S591 phosphorylation.
We investigated the kinetics of S591 phosphorylation in both Jurkat cells and primary T cells (Fig 2A). Anti-CD3/28 stimulation of Jurkat cells induced rapid and transient phosphorylation on S591 of SHP-1, which was strong by 1 min after stimulation and decreasing by 10 min. CD3/28-induced S591 phosphorylation in human peripheral blood T-cells (PBT) had similar kinetics to that observed in Jurkat. To study the kinetics of S591 phosphorylation more accurately, we looked at S591 phosphorylation in the time range of 5 to 60 seconds after stimulation of Jurkat cells, and compared it with other phosphorylation events in the cells induced by TCR (Fig 2B). By 15 seconds after stimulation, S591 phosphorylation already had reached its maximum and maintained the level throughout the time range. The kinetics of S591 phosphorylation closely paralleled the early burst of tyrosine phosphorylations in T lymphocyte in response to TCR stimulation as revealed by generic anti-phospho tyrosine Ab blot. In contrast, phosphorylation of ERK was a much later event which only began to appear at 30s after stimulation and was still increasing at 60s after stimulation. The result indicates S591 phosphorylation is a very early event in TCR signal transduction.
Fig 2. TCR induced SHP-1 S591 phosphorylation is fast and transient.
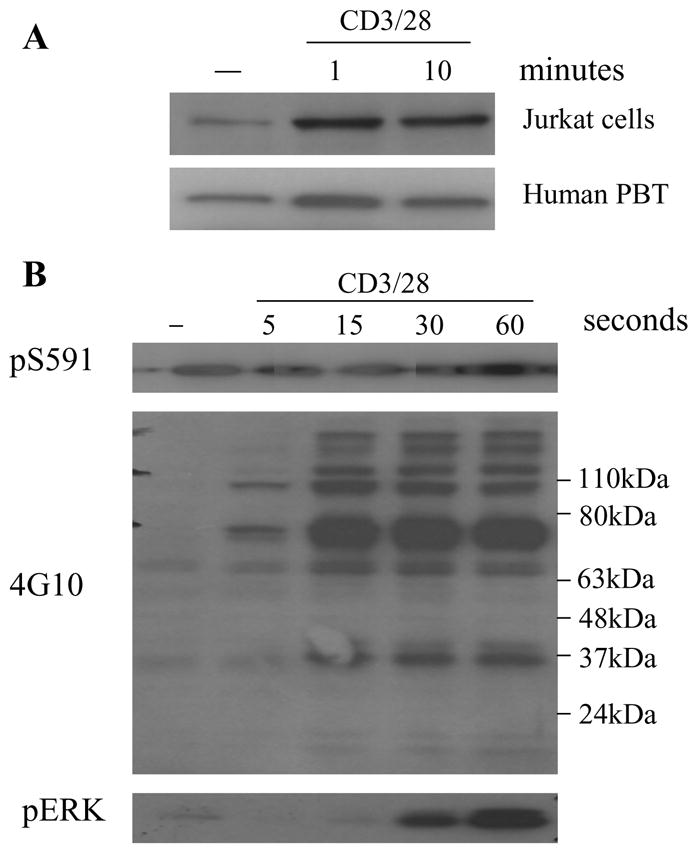
(A) Jurkat cells or human PBT were stimulated (or not) with CD3/28 Abs for 1min or 10min. The cell lysate was blotted with pS591 phospho-specific Ab. (B) Jurkat cells were added with CD3/28 Ab for stimulation. At 5s, 15s, 30s or 60s after stimulation, the stimulation was stopped by cell lysis. The cell lysate was blotted with pS591 phospho-specific Ab, 4G10 Ab, or pERK Ab.
Genetic evidence that PLC-γ1 tyrosine phosphorylation is influenced by S591 phosphorylation
To determine whether S591 phosphorylation changes SHP-1 cellular function, we made stably transfected Jurkat cell lines expressing SHP-1 wildtype, S591A and S591D mutants and assessed tyrosine phosphorylation in those cells following TCR stimulation. After 1 min of CD3/28 stimulation, multiple bands were tyrosine phosphorylated Most of the activation-induced changes in tyrosine phosphorylation were similar between transfected and untransfected cells (Fig 3A). The notable exception was a band at ~150kd, suggestive of PLC-γ1. In Jurkat transfected with wildtype N-GFP_SHP-1 and its S591A mutant, phosphorylation of this band was significantly decreased, but in S591D mutant-transfected Jurkat cells phosphorylation of this band was as strong as in control Jurkat cells. Reprobing of the same cell lysate with PLC-γ1 Ab showed that PLC-γ1 expression were equivalent in all cells. To confirm the finding, PLC-γ1 was immunoprecipitated from equal amounts of cell lysate of control Jurkat cells and SHP-1 constructs stably transfected Jurkat cells stimulated or not with CD3/28 for 1min (Fig 3C) and blotted to detect phospho-tyrosine. The results confirm that PLC-γ1 tyrosine phosphorylation was decreased significantly in wildtype and S591A mutant stably transfected Jurkat cells, but in S591D mutant stably transfected Jurkat cells it was as strong as in control Jurkat cells. To confirm that the change in PLC-γ 1 phosphorylation was selective, two site-specific phospho antibodies were also tested on the same cell lysates (Fig 3B). Results for pY319 of ZAP70 and pT202/Y204 of ERK, showed these phosphorylation events were unaffected by SHP-1 transfection. Quantitative analysis of PLC-γ 1 phosphorylation results from five experiments (Fig 3D) clearly indicated that PLC-γ 1 phosphorylation level in SHP-1 S591D stably transfected Jurkat cells was comparable with wildtype control Jurkat Cells, but higher than SHP-1 WT- or S591Amutant-transfected Jurkat cells. Thus, SHP-1 S591 phosphorylation, mimicked by S591D mutation, can change its function.
Fig 3. SHP-1 S591D mutation interferes with its regulation of PLC-γ1 dephosphorylation in Jurkat cells.
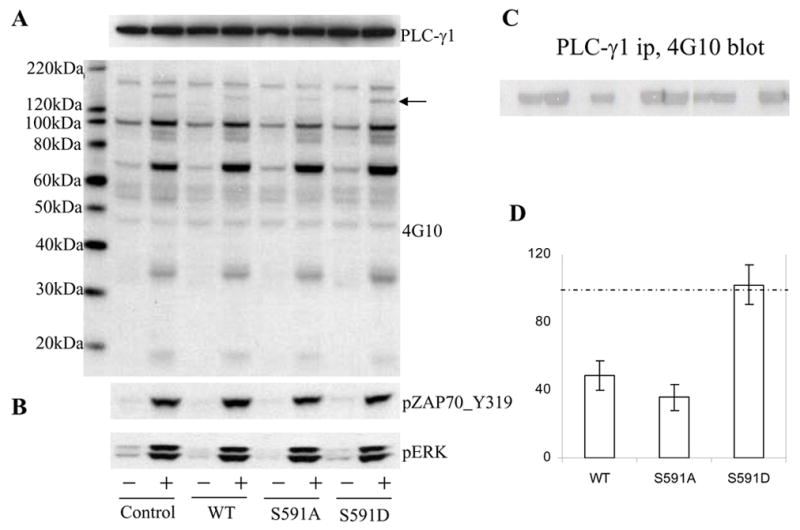
(A) The Jurkat cell parental line (E6-1) or its sublines stably transfected with SHP-1 WT, S591A or S591D mutants were stimulated with CD3/28 Ab for 1min. A fraction of the cell lysate was blotted with either PLC-γ Ab or 4G10 mAb. B) The same lysate was also blotted with pERK and anti-ZAP70 pY319. C) The remaining cell lysate was immunoprecipitated with PLC-γAb and the immunoprecipitated was blotted with 4G10 mAb. D) Statistical analysis (mean ± SEM) of PLC-γ phosphorylation determined by Western blot results in five experiments using an Odyssey Infrared Imaging System for optimal quantitation. Results shown reflect comparisons of each stable line to untransfected Jurkat cells (taken as 100%, indicated by the dashed line)
S591 phosphorylation decreases SHP-1-mediated dephosphorylation
We investigated whether S591 phosphorylation regulates SHP-1 phosphatase activity. In the course of our study, Jones ML et al[23] reported that SHP-1 S591 was phosphorylated in platelets. Their results suggested that S591 phosphorylation reduced phosphatase activity, but complexities of their assay (see discussion) prompted us to investigate the question in alternative ways. We made SHP-1 constructs (N-terminal HA tagged), WT and S591 to A or D mutants, and compared their activity after purification of the protein from transfected HEK293T cells lysate in a time course study with pY peptide as substrate. The results showed that while activity of SHP-1 wildtype and S591A mutant was similar, S591D mutant had about 30% lower activity (Fig 4). These data indicate that phosphorylation of S591 per se regulates its ability to dephosphorylate proteins
Fig 4. SHP-1 S591D mutant has lower phosphatase activity on peptide substrate.
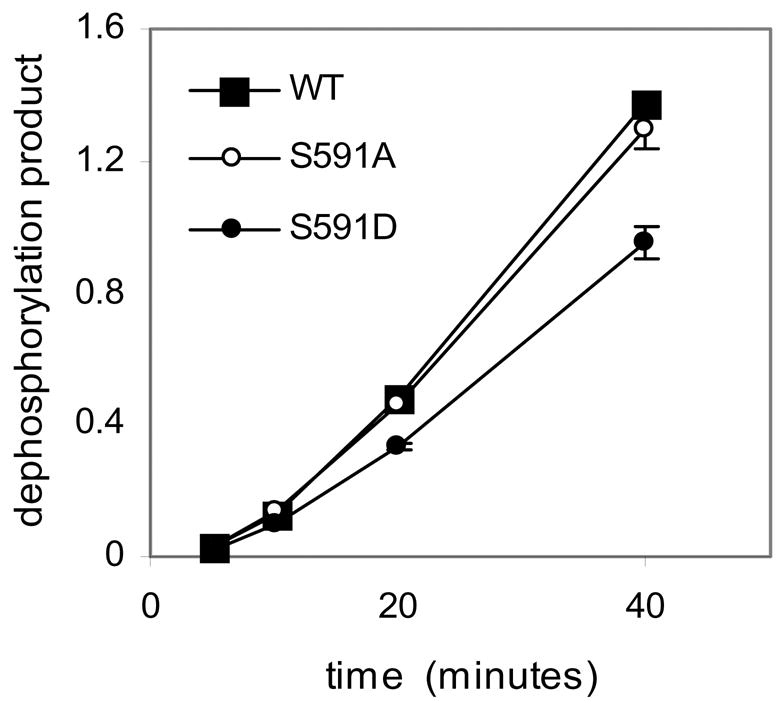
HA tagged SHP-1 WT, S591A or S591D mutant were immunoprecipitated from cell lysate of transiently transfected HEK293T cells. Phosphatase activity was measured by using tyrosine phosphorylated peptide (right panel) as substrate at the indicated time points.
TCR-induced sequential phosphorylation of SHP-1 S591 and Y536 in human PBT
Since it has been shown previously that Y536 phosphorylation up-regulates SHP-1 phosphatase activity [6] in contrast to S591 which decreases its activity, we investigated the temporal relationship between S591 phosphorylation and Y536 phosphorylation of SHP-1 in human PBT. S591 phosphorylation peaked at 1 min after stimulation and then started to decline (Fig 5 and Fig 1). In contrast, Y536 phosphorylation was evident only at 3min of stimulation and keep on increasing until at least 30min after stimulation. The results indicated that the peak of S591 and Y536 phosphorylation are temporally separated which serves to first decrease phosphatase activity and subsequently increase it.
Fig 5. TCR induces sequential SHP-1 S591 and Y536 phosphorylation.
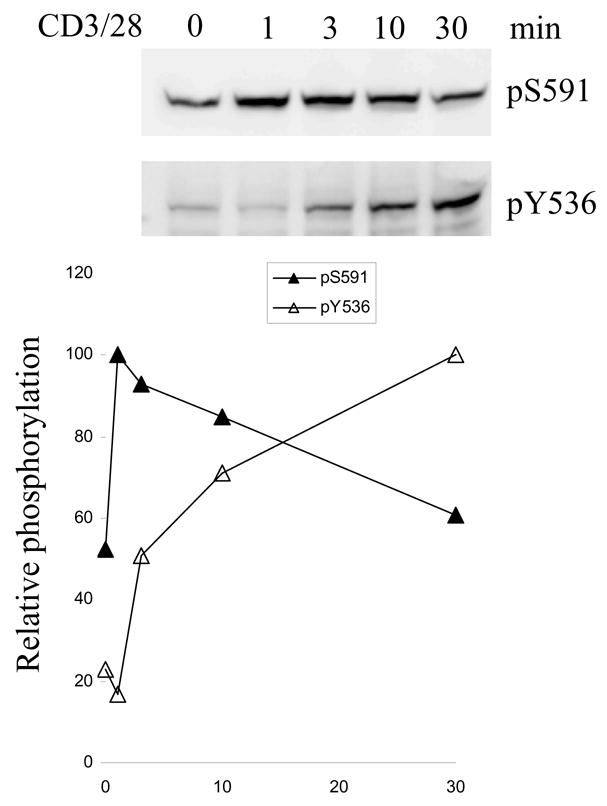
Human PBT was stimulated or not with CD3/28 Abs for various time. The cell lysate was blotted with pS591 or pY535 phospho-specific Abs. Quantitation of the optical density of the immunoblot is also shown, in which the y-axis shows phosphorylation relative to maximum observed.
S591 mutation in N-GFP_SHP-1 changed its cellular distribution in Jurkat cells
We also investigated the possibility that S591 phosphorylation could change SHP-1 cellular distribution as another mechanism to control SHP-1 cellular function. We first investigated SHP-1 localization to lipid raft fractions of Jurkat cells. Fractions were blotted with both the pS591 Ab and a pan-SHP-1 antibody (whose binding is unaffected by the phosphorylation state of S591). To optimize comparison of phosphorylation, amounts of protein in each lane were made more comparable by loading twice as much lipid fraction as cytosolic fraction. Despite comparable loading (Fig 6A) phosphorylation of S591 of SHP-1 in the lipid raft fraction was much lower than that of the cytosolic fraction.
Fig 6. SHP-1 S591 phosphorylation impairs its lipid rafts and membrane localization.
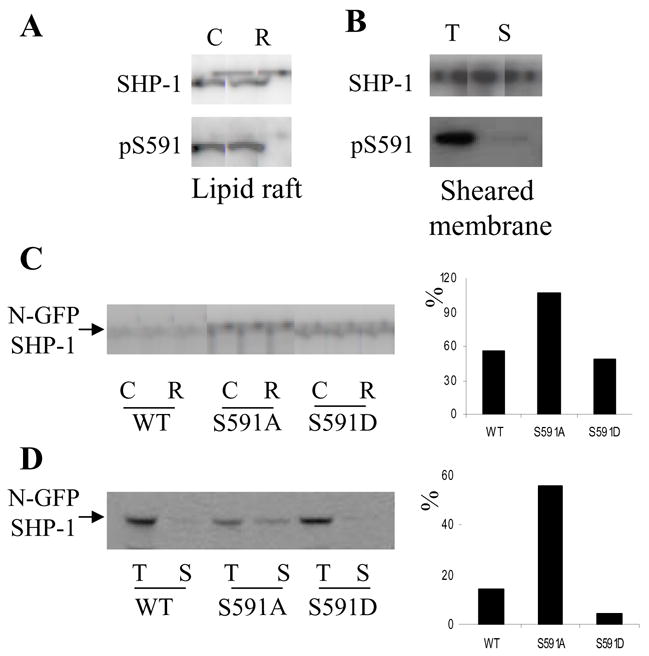
(A): lipid rafts (“R”) and cytosolic fractions (“C”) were separated from Jurkat cells and blotted with SHP-1 Ab or pS591 phospho-specific Ab. B) sheared membrane fraction (“S”) and total cell lysate (“T”) were made from human PBT, and blotted with SHP-1 Ab or pS591 phospho-specific Ab. (C): Lipid rafts and cytosolic fractions were isolated from Jurkat cells stably transfected with SHP-1 WT, S591A or S591D mutants. The fractions were blotted with SHP-1 Ab and pS591 phospho-specific Ab. (D): sheared membrane fraction and total cell lysate were made from Jurkat cells stably transfected with SHP-1 WT, S591A or S591D mutants. The fractions were blotted with SHP-1 Ab and pS591 phospho-specific Ab. In C and D, the result of the western blots were quantified. The percentage of SHP-1 in lipid raft fraction relative to cytosol fraction in the blot was calculated and plotted as a bar graph.
To verify that S591 phosphorylation changes SHP-1 cellular distribution, we used another strategy to enrich for plasma membrane, namely by cell-shearing. This procedure, modeled after published procedures for enrichment of placental microvilli, leaves viable cells and generates a fraction containing about 1% of non-nuclear cell protein and enriched in plasma membrane plus microvilli1. A sheared membrane fraction was made from human PBT, blotted with both SHP-1 Ab and anti-pS591 Ab, and compared with total lysate (Fig 6B). SHP-1 blot showed that the amount of SHP-1 in the total lysate and sheared membrane fraction were about the same, but S591 phosphorylation of SHP-1 in total lysate was much stronger than in the sheared membrane fraction.
The foregoing two approaches demonstrated SHP-1 S591 hypophosphorylation in lipid raft and sheared membrane fraction. It was important to determine whether S591 phosphorylation prevented SHP-1 lipid raft/sheared membrane distribution or lipid raft/sheared membrane distribution caused SHP-1 dephosphorylation on S591. Therefore, we studied the distribution of SHP-1 wildtype and its S591A or S591D mutant in lipid raft and sheared membrane fraction using the stably transfected Jurkat cells described above. The results indicate that the S591A mutant of SHP-1 was more abundant both in lipid rafts (Fig 6C) and in the sheared membrane fraction (Fig 6D) compared with wildtype and S591D mutant of SHP-1. As an internal control we investigated the distribution of endogenous SHP-1 in all three cell lines and found that they are very similar (data not shown). The results clearly indicate that SHP-1 S591 phosphorylation impedes its localization in the lipid and sheared membrane fractions.
Regulation of SHP-1 nuclear localization
S591 is located close to the C-terminus of SHP-1, in the middle of the previously identified nuclear localization signal (NLS) of SHP-1. Since phosphorylation in the proximity of NLS is a common strategy in regulating the function of the NLS, we hypothesized that S591 phosphorylation may regulate SHP-1 nuclear localization. Pilot studies to assess localization demonstrated a dramatic effect of GFP tag position on SHP-1 cellular distribution, which has also been reported by others during this investigation[25]. When the GFP tag was fused to the N-terminus of SHP-1 (N-GFP_SHP-1), the transfected fusion protein was almost exclusively cytoplasmic (Fig 7A). In dramatic contrast, when the GFP tag was fused to the C-terminus of SHP-1 (SHP-1_C-GFP), it was almost exclusively nuclear. By western blot, we found that N-GFP_SHP-1 was phosphorylated on S591. But phosphorylation of S591 was totally absent in SHP-1_C-GFP. So SHP-1 nuclear localization correlated with dephosphorylation of S591.
Fig 7. SHP-1 S591 phosphorylation regulate its nuclear localization.
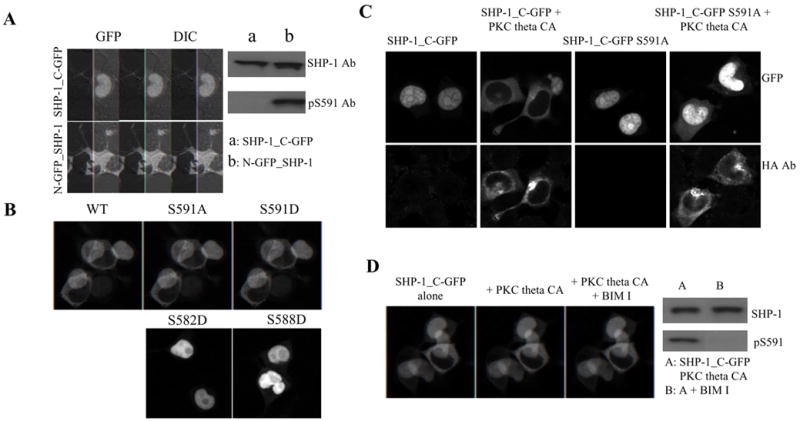
(A): N-GFP_SHP-1 or SHP-1_C-GFP constructs were transfected into HEK293T cells and confocal images cells were acquired at about mid-plane of the cell. Cell lysates were also made from the transfected cells and blotted with SHP-1 Ab or pS591 phospho-specific Ab. (B): SHP-1_C-GFP WT, S591A or S591D mutants were transfected into HEK293T cells and imaged (upper panel); as controls, SHP-1_C-GFP S582D and S588D mutants were also transfected and imaged (lower panel). (C): SHP-1_C-GFP, WT or S591A mutant, were transfected into HEK293T cells, alone or together with PKC theta CA. Cells were stained with HA mAb (red) to monitor expression of PKC theta CA (in addition to the GFP tag on the transfected SHP-1) and imaged with confocal microscope. (D): SHP-1_C-GFP was transfected into HEK293T cells, alone or together with PKC theta CA. The cells were imaged with confocal microscope. Then the PKC theta CA co-transfected cells were treated 10uM BIM I at 37C for 30 min and re-observed. Cell lysates before and after BIM I treatment was also made and blotted with SHP-1 and pS591 phospho-specific Ab.
To analyze genetically the effect of S591 phosphorylation on nuclear localization, we investigated the distribution of three different SHP-1_C-GFP constructs in HEK293T cells: wildtype, S591A or S591D (Fig 7B). Although the wildtype and the S591A mutant distributed mainly in the nucleus of the cells the S591D mutant distributed mainly in the cytoplasm of the cells. Analysis of constructs expressing aspartic acid mutations of other serines nearby (i.e. S582D and S588D), showed that only S591D prevented nuclear localization of SHP-1_C-GFP (Fig 7B).
We investigated phosphorylation per se of S591 to exclude the possibility that the effect on nuclear exclusion of the S591D mutation might not exactly mimic phosphorylation at that site. For this purpose, we took advantage of the fact that PKC theta CA can phosphorylate S591 of SHP-1 when co-transfected. As described above, HEK293T cells transfected SHP-1_C-GFP distributed almost exclusively in the nucleus of the cells. But when PKC theta CA was co-transfected, the localization of SHP-1_C-GFP became almost exclusively cytoplasmic (Fig 7C). Western blot showed that co-transfection of PKC theta CA resulted in S591 phosphorylation of SHP-1_C-GFP (c.f. Fig 1B). S591 phosphorylation per se contributed to the change in localization, because the change caused by PKC-theta was not observed with the S591A mutant. The effect of S591 phosphorylation on SHP-1 nuclear localization was further confirmed by the observation that treatment with a high concentration of the PKC inhibitor, BIM I, blocked S591 phosphorylation by co-transfected PKC theta CA and eliminated its nuclear exclusion (Fig 7D). Although endogenous SHP-1 localized almost exclusively in the cytoplasm, transfected SHP-1_C-GFP distributed mostly in the nucleus in Jurkat T cells as in HEK293T cells (data not shown). Thus, S591 phosphorylation blocks nuclear localization of SHP-1.
We considered the possibility that mechanisms other than S591 phosphorylation might regulate nuclear localization. We reasoned that if the nuclear localization of SHP-1 is solely regulated by S591 phosphorylation, mutating S591 to A to prevent phosphorylation in the N-GFP_SHP-1 should change its localization to nuclear. But the S591A mutant remained cytoplasmic (Fig 8A). The exact structure of the C-terminal tail is still unknown since the C-terminal tail is no present in the solved structure of SHP-1. We hypothesized that the C-terminal tail of SHP-1 normally interacts with another part of the phosphatase, thereby masking the NLS. In our conceptual model the bulky GFP tag in the SHP-1_C-GFP construct prevents that intramolecular interaction of the C-terminus, thereby unmasking the NLS. To provide experimental evidence for the hypothesis, we did limited trypsin digestion experiment, a commonly used strategy in studying the accessibility of molecular regions. The more exposed a region of the molecule is, the easier it will be to digest by trypsin. N-GFP_SHP-1 and SHP-1_C-GFP were transfected into HEK293T cells and the cell lysate was treated with various concentration of trypsin and blotted with two anti-SHP-1 antibodies, a monoclonal antibody (“proximal”) raised against the C-terminal 106 residues and a polyclonal anti-peptide antibody (“distal’) raised against the last 19 amino acids in C-terminal of SHP-1. SHP-1_C-GFP is especially sensitive to proteolytic loss of its GFP moiety based on mw shift (Fig 8B) and loss of reactivity with anti-GFP (not shown). The proximal and distal Abs both recognized a 65kDa fragment, from both N-GFP_SHP-1 and SHP-1_C-GFP. That 65kd fragment is the size of full length SHP-1 and has the intact C-terminal tail, so it appears to be full length SHP-1 molecule after the GFP tag has been cleaved. In addition, there is a 60kd fragment that reacts only with the proximal antibody but not the distal antibody, indicating loss of the C-terminus. This 60kd fragment is already obvious at 0.375ug/ml of trypsin treatment for C-N-GFP_SHP-1, but only appears at 0.75ug/ml of trypsin treatment for N-GFP_SHP-1. The results suggest that the C-terminal tail of SHP-1 in SHP-1_C-GFP is more exposed because of the GFP tag, but somewhat hidden in N-GFP_SHP-1, which would explain why its NLS is not functional.
Fig 8. NLS on the C-terminal tail of SHP-1 is concealed by molecular interaction.
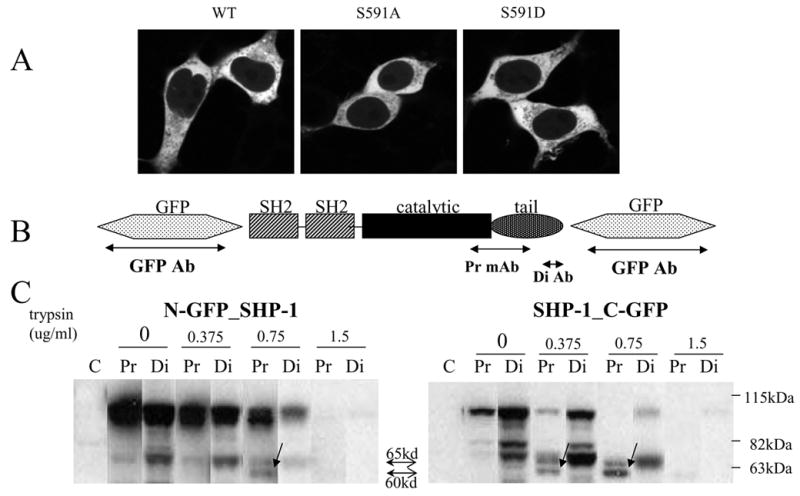
(A): N-GFP_SHP-1 WT, S591A or S591D mutants was transfected into HEK293T cells. Midplane confocal images are shown.
(B) Domain architecture of SHP-1 is shown. Constructs used for limited trypsin digestion had GFP either at N-term or at C-term.
(C): N-GFP_SHP-1 or SHP-1_C-GFP was transfected into HEK293T cells. The cell lysate was treated with graded concentrations of trypsin and then blotted with two different SHP-1 antibodies: a monoclonal antibody (“proximal”) raised against the C-terminal 106 residues and a polyclonal anti-peptide antibody (“distal’) raised against the last 19 amino acids in C-terminal of SHP-1
Discussion
The present study demonstrates that TCR-activation of lymphocytes results in rapid phosphorylation of SHP-1 S591 (Fig 1). This S591 phosphorylation inhibits SHP-1 phosphatase activity (measured on a phosphotyrosine peptide substrate, Fig 4). Notably however, this phosphorylation is transient and is followed by a wave of tyrosine phosphorylation on Y536 (Fig 5). Thus, SHP-1 undergoes a rapid decrease in phosphatase activity in the first several minutes and thereafter an increase in activity. Moreover, phosphorylation of SHP-1 S591 regulates its localization, decreasing membrane localization and preventing nuclear localization. This discussion focuses on the following issues: 1) the basophilic site and candidate kinases; 2) regulation of phosphatase activity; 3) regulation of localization; 4) a conceptual model of the role of S591 phosphorylation for TCR activation
S591 is an ideal site for phosphorylation by a basophilic kinase. First, by virtue of being close to the terminus of the molecule, it is likely to be relatively accessible conformationally. This conformational accessibility is also facilitated by its hydrophilic composition (KEKSKG-S-LKRK-Cterm). Second, the surrounding sequence includes six basic residues in a stretch of 11 amino acids. This gives the region a very strong net positive charge at physiological pH, which is the major prerequisite for recognition by basophilic kinases. Our in silico analysis of SHP-1 (data not shown) indicates that although there are two other potential basophilic phosphorylation sites in SHP-1 (S26 and S32), S591 is the most favorable and likely to be the primary one responsible for the early reports of SHP-1 serine phosphorylation [15, 18, 26]. What is the relevant kinase(s)? Two reports link SHP-1 phosphorylation to PKC: an early report in which the site was not identified [18] and the recent report of S591 phosphorylation in platelets[23]. In contrast, two of our findings in T-cells do not support a physiological role for PKC phosphorylation following TCR stimulation. First, inhibition by BIMs is not as efficient as would be expected to PKCs. Second, constitutive active PKC-theta phosphorylated transfected GFP-tagged SHP-1 but not endogenous SHP-1 (data not shown). Specifically, the inhibitors BIM I and BIM III are both inhibitors for PKC at low nanomolar concentration in vitro (IC50, 10–30nM). In PKC-mediated cellular process concentrations as low as 100-500nM are sufficient to achieve strong inhibition (see Supplementary Fig 1). But instead concentrations of 5–10uM of BIM I and BIM III were needed to completely inhibit TCR-induced S591 phosphorylation (Fig 1). Moreover, kinases other than PKC could very well be inhibited too at those concentrations (for example, PKA, IC50, 500nM; GSK-3, IC50, 360nM per EMD/Calbiochem; see also [27]). Based on those results, PKCs at least are not the only kinase(s) phosphorylating S591 in T lymphocytes. It should be noted that the previous reports of PKC involvement showed partial inhibition at relatively high concentrations of BIM I in intact cells {2 uM in [18] and 5uM in[23]}. It should also be noted that although PKC can phosphorylate SHP-1 in vitro [18] such an in vitro assay is no basis for concluding the in vivo relevance since many basophilic kinases phosphorylate substrates in vitro that are not in vivo substrates. These complexities leave open the possibility that even in the cell systems previously studied, PKC may not be the relevant kinase family.
Our studies and two others support the conclusion that serine phosphorylation of SHP-1 inhibits its phosphatase activity. The most direct evidence for S591 involvement in phosphatase inhibition is provided by the present study in which the S581D mutation was demonstrated to reduce SHP-1 phosphatase activity by about 30% (Fig 4). Since replacement of serine with aspartic acid is only a partial mimic of phosphorylation, it is quite likely that phosphorylation of this residue will result in a much larger inhibition. Downey and co-workers showed that phosphorylation of SHP-1 by PKC inhibited its phosphatase activity[18]. These results do not directly implicate S591 (since the residue was not identified in that study) but are consistent with its involvement. Moreover, both Poole and Downey used another approach in which they demonstrated that SHP-1 in vitro phosphatase activity was increased by 2–5uM BIM inhibition in intact cells: [23]. Again these results do not directly implicate S591, but are consistent with its involvement.
The present studies also demonstrate that phosphorylation of S591 profoundly inhibits nuclear translocation. This is demonstrated both genetically, in the S591D mutant (Fig 7B), and by intracellular phosphorylation by a basophilic kinase (Fig 7D). S591 localizes right in the middle of the identified SHP-1 NLS, which contains a series of positively charged residues ([22, 28]. The regulation of the NLS by S591 phosphorylation is consistent both with the concept that phosphorylation changes the positive charge required in a NLS, and with other molecules in which phosphorylation impairs NLS-based nuclear localization (e.g.[29, 30]). In addition to this regulation, another powerful mechanism inhibits nuclear localization in T-cells; our studies suggest that this mechanism relates to conformational autoinhibition (Fig 8). Our findings that C-terminal GFP tagging results in nuclear translocation in T-cells are consistent with recent findings in fibroblast-derived cell lines[25]. Our studies with endogenous SHP-1 in Jurkat cells and primary T-cells indicate that it is localized in cytoplasm both before and after TCR stimulation (consistent with the localization observed for SHP-1 tagged N-terminally with either HA peptide or GFP), Thus we have no evidence that phosphorylation is important in excluding SHP-1 from the nucleus in early T-cell signal transduction. However, SHP-1 has been clearly demonstrated in the nucleus in numerous contexts with other cell types. For example SHP-1 is found in the nucleus in epithelial cells where it appears to regulate expression of several genes[31]; it is also in the nucleus in ES cells[32]. Further studies will be required to elucidate the cell types and signaling pathway in which phosphorylation-mediated nuclear inhibition of SHP-1 plays an important physiological role.
How does S591 phosphorylation alter TCR-mediated signaling? Our studies demonstrate that S591D is specifically unable to regulate PLC-γ1 phosphorylation (Fig 3). Although such overexpression studies clearly demonstrate regulation of SHP-1 function by a genetic mimic of phosphorylation, they cannot address the fine regulation necessary in receptor-mediated phosphorylation. We estimate that rapid TCR-induced serine phosphorylation of SHP-1 (Fig 2) occurs on ~5-20% of the total SHP-1 pool (based on comparisons with EL-4 cells which have much stronger S591 phosphorylation, data not shown). The SHP-1 which is phosphorylated on S591 will have diminished phosphatase activity (Fig 4) and decreased capacity to localize to membrane (Fig 6). SHP-1 exists in many distinct pools complexed to other molecules [5, 32–34]. Differential phosphorylation of S591 in such pools, which may not be in rapid equilibrium with each other, would confer different functional properties on SHP-1 in such pools. Moreover, our studies demonstrate that early S591 phosphorylation (which decreases the phosphatase activity in that pool) is followed temporally by Y536 phosphorylation, which would be expected to increase its phosphatase activity. It is plausible that this Y536 phosphorylation is likewise in a distinct pool that contributes to the termination of acute TCR signaling. This interplay of positive and negative regulation by phosphorylation illustrates the elegant combinatorial control of SHP-1 function.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the National Cancer Institute, National Institute of Health. We thank Dr. Taolin Yi for providing us the mouse SHP-1 construct; Dr. Guofang Lao for making the mutant constructs of SHP-1; BD Biosciences for the collaborative generation of mAb; and the FACS lab of the Experimental Immunology Branch, for sorting SHP-1 transfected cells.
Footnotes
Hao JJ, Shaw S manuscript in preparation
References
- 1.Veillette A, Latour S, Davidson D. Negative regulation of immunoreceptor signaling. Annu Rev Immunol. 2002;20:669–707. doi: 10.1146/annurev.immunol.20.081501.130710. [DOI] [PubMed] [Google Scholar]
- 2.Ulyanova T, Blasioli J, Thomas ML. Regulation of cell signaling by the protein tyrosine phosphatases, CD45 and SHP-1. Immunol Res. 1997;16:101–13. doi: 10.1007/BF02786326. [DOI] [PubMed] [Google Scholar]
- 3.Shultz LD, Rajan TV, Greiner DL. Severe defects in immunity and hematopoiesis caused by SHP-1 protein-tyrosine-phosphatase deficiency. Trends Biotechnol. 1997;15:302–7. doi: 10.1016/S0167-7799(97)01060-3. [DOI] [PubMed] [Google Scholar]
- 4.Lorenz U, Ravichandran KS, Burakoff SJ, Neel BG. Lack of SHPTP1 results in src-family kinase hyperactivation and thymocyte hyperresponsiveness. Proc Natl Acad Sci U S A. 1996;93:9624–9. doi: 10.1073/pnas.93.18.9624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stefanova I, Hemmer B, Vergelli M, Martin R, Biddison WE, Germain RN. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat Immunol. 2003;4:248–54. doi: 10.1038/ni895. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Z, Shen K, Lu W, Cole PA. The role of C-terminal tyrosine phosphorylation in the regulation of SHP-1 explored via expressed protein ligation. J Biol Chem. 2003;278:4668–4674. doi: 10.1074/jbc.M210028200. [DOI] [PubMed] [Google Scholar]
- 7.Pei D, Lorenz U, Klingmuller U, Neel BG, Walsh CT. Intramolecular regulation of protein tyrosine phosphatase SH-PTP1: a new function for Src homology 2 domains. Biochemistry. 1994;33:15483–93. doi: 10.1021/bi00255a030. [DOI] [PubMed] [Google Scholar]
- 8.Yang J, Liu L, He D, Song X, Liang X, Zhao ZJ, Zhou GW. Crystal structure of human protein-tyrosine phosphatase SHP-1. J Biol Chem. 2003;278:6516–20. doi: 10.1074/jbc.M210430200. [DOI] [PubMed] [Google Scholar]
- 9.Law CL, Sidorenko SP, Chandran KA, Zhao Z, Shen SH, Fischer EH, Clark EA. CD22 associates with protein tyrosine phosphatase 1C, Syk, and phospholipase C-gamma(1) upon B cell activation. J Exp Med. 1996;183:547–60. doi: 10.1084/jem.183.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D’Ambrosio D, Hippen KL, Minskoff SA, Mellman I, Pani G, Siminovitch KA, Cambier JC. Recruitment and activation of PTP1C in negative regulation of antigen receptor signaling by Fc gamma RIIB1. Science. 1995;268:293–7. doi: 10.1126/science.7716523. [DOI] [PubMed] [Google Scholar]
- 11.Otipoby KL, Draves KE, Clark EA. CD22 regulates B cell receptor-mediated signals via two domains that independently recruit Grb2 and SHP-1. J Biol Chem. 2001;276:44315–22. doi: 10.1074/jbc.M105446200. [DOI] [PubMed] [Google Scholar]
- 12.Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173:945–54. doi: 10.4049/jimmunol.173.2.945. [DOI] [PubMed] [Google Scholar]
- 13.Fawcett VC, Lorenz U. Localization of Src homology 2 domain-containing phosphatase 1 (SHP-1) to lipid rafts in T lymphocytes: functional implications and a role for the SHP-1 carboxyl terminus. J Immunol. 2005;174:2849–59. doi: 10.4049/jimmunol.174.5.2849. [DOI] [PubMed] [Google Scholar]
- 14.Kosugi A, Sakakura J, Yasuda K, Ogata M, Hamaoka T. Involvement of SHP-1 tyrosine phosphatase in TCR-mediated signaling pathways in lipid rafts. Immunity. 2001;14:669–80. doi: 10.1016/s1074-7613(01)00146-7. [DOI] [PubMed] [Google Scholar]
- 15.Zhao Z, Shen SH, Fischer EH. Phorbol ester-induced expression, phosphorylation, and translocation of protein-tyrosine-phosphatase 1C in HL-60 cells. Proc Natl Acad Sci U S A. 1994;91:5007–11. doi: 10.1073/pnas.91.11.5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li RY, Gaits F, Ragab A, Ragab-Thomas JM, Chap H. Tyrosine phosphorylation of an SH2-containing protein tyrosine phosphatase is coupled to platelet thrombin receptor via a pertussis toxin-sensitive heterotrimeric G-protein. Embo J. 1995;14:2519–26. doi: 10.1002/j.1460-2075.1995.tb07249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen SH, Bastien L, Posner BI, Chretien P. A protein-tyrosine phosphatase with sequence similarity to the SH2 domain of the protein-tyrosine kinases. Nature. 1991;352:736–9. doi: 10.1038/352736a0. [DOI] [PubMed] [Google Scholar]
- 18.Brumell JH, Chan CK, Butler J, Borregaard N, Siminovitch KA, Grinstein S, Downey GP. Regulation of Src homology 2-containing tyrosine phosphatase 1 during activation of human neutrophils. Role of protein kinase C. Journal of Biological Chemistry. 1997;272:875–882. doi: 10.1074/jbc.272.2.875. [DOI] [PubMed] [Google Scholar]
- 19.Fujii K, Zhu G, Liu Y, Hallam J, Chen L, Herrero J, Shaw S. Kinase peptide specificity: improved determination and relevance to protein phosphorylation. Proc Natl Acad Sci U S A. 2004;101:13744–9. doi: 10.1073/pnas.0401881101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pinna LA, Ruzzene M. How do protein kinases recognize their substrates? Biochim Biophys Acta. 1996;1314:191–225. doi: 10.1016/s0167-4889(96)00083-3. [DOI] [PubMed] [Google Scholar]
- 21.Zhu G, Fujii K, Liu Y, Codrea V, Herrero J, Shaw S. A single pair of acidic residues in the kinase major groove mediates strong substrate preference for P-2 or P-5 Arginine in the AGC, CAMK and STE kinase families. J Biol Chem. 2005;280:36372–9. doi: 10.1074/jbc.M505031200. [DOI] [PubMed] [Google Scholar]
- 22.Craggs G, Kellie S. A functional nuclear localization sequence in the C-terminal domain of SHP-1. J Biol Chem. 2001;276:23719–25. doi: 10.1074/jbc.M102846200. [DOI] [PubMed] [Google Scholar]
- 23.Jones ML, Craik JD, Gibbins JM, Poole AW. Regulation of SHP-1 tyrosine phosphatase in human platelets by serine phosphorylation at its C terminus. J Biol Chem. 2004;279:40475–83. doi: 10.1074/jbc.M402970200. [DOI] [PubMed] [Google Scholar]
- 24.Liu Y, Graham C, Li A, Fisher R, Shaw S. Phosphorylation of PKC-theta activation loop and hydrophobic motif regulate its kinase activity but only activation loop phosphorylation is critical to in vivo NFkB induction. Biochemical Journal. 2002;361:255–265. doi: 10.1042/bj3610255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He D, Song X, Liu L, Burk DH, Zhou GW. EGF-stimulation activates the nuclear localization signal of SHP-1. J Cell Biochem. 2005;94:944–53. doi: 10.1002/jcb.20307. [DOI] [PubMed] [Google Scholar]
- 26.Lorenz U, Ravichandran KS, Pei D, Walsh CT, Burakoff SJ, Neel BG. Lck-dependent tyrosyl phosphorylation of the phosphotyrosine phosphatase SH-PTP1 in murine T cells. Mol Cell Biol. 1994;14:1824–34. doi: 10.1128/mcb.14.3.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang W, Tabrizi M, Yi T. A bipartite NLS at the SHP-1 C-terminus mediates cytokine-induced SHP-1 nuclear localization in cell growth control. Blood Cells Mol Dis. 2002;28:63–74. doi: 10.1006/bcmd.2002.0485. [DOI] [PubMed] [Google Scholar]
- 29.Valovka T, Verdier F, Cramer R, Zhyvoloup A, Fenton T, Rebholz H, Wang ML, Gzhegotsky M, Lutsyk A, Matsuka G, Filonenko V, Wang L, Proud CG, Parker PJ, Gout IT. Protein kinase C phosphorylates ribosomal protein S6 kinase betaII and regulates its subcellular localization. Mol Cell Biol. 2003;23:852–863. doi: 10.1128/MCB.23.3.852-863.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sheng T, Chi S, Zhang X, Xie J. Regulation of Gli1 localization by the cAMP/protein kinase A signaling axis through a site near the nuclear localization signal. J Biol Chem. 2006;281:9–12. doi: 10.1074/jbc.C500300200. [DOI] [PubMed] [Google Scholar]
- 31.Duchesne C, Charland S, Asselin C, Nahmias C, Rivard N. Negative regulation of beta-catenin signaling by tyrosine phosphatase SHP-1 in intestinal epithelial cells. J Biol Chem. 2003;278:14274–83. doi: 10.1074/jbc.M300425200. [DOI] [PubMed] [Google Scholar]
- 32.Paling NR, Welham MJ. Tyrosine phosphatase SHP-1 acts at different stages of development to regulate hematopoiesis. Blood. 2005;105:4290–7. doi: 10.1182/blood-2004-08-3271. [DOI] [PubMed] [Google Scholar]
- 33.Kanagasundaram V, Jaworowski A, Byrne R, Hamilton JA. Separation and characterization of the activated pool of colony-stimulating factor 1 receptor forming distinct multimeric complexes with signalling molecules in macrophages. Mol Cell Biol. 1999;19:4079–92. doi: 10.1128/mcb.19.6.4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perez-Villar JJ, Whitney GS, Bowen MA, Hewgill DH, Aruffo AA, Kanner SB. CD5 negatively regulates the T-cell antigen receptor signal transduction pathway: involvement of SH2-containing phosphotyrosine phosphatase SHP-1. Mol Cell Biol. 1999;19:2903–12. doi: 10.1128/mcb.19.4.2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.