

Abstract
Numbers of substitutions per site for 15 protein-coding genes and six introns of the plant mitochondria were estimated to compare modes and tempos of evolution between exons and introns, and numbers of insertions–deletions per site also were investigated in introns. Intra-gene homogeneity of numbers of substitutions per site was assessed further among different taxa and between mitochondrial and nuclear paralogs translocated from the mitochondrial genome. Gene-to-gene differences in numbers of substitutions per site were found to be higher for nonsynonymous than synonymous sites, and this could be due to differential selection if mutation rate is assumed constant for the genome. Some mitochondrial genes have evolved as fast as chloroplast genes, thus faster than previously thought. For coxI, relative rate tests showed that woody taxa evolved slower than annuals at synonymous sites. Generation time, population size, and speciation rate are likely factors involved in this rate heterogeneity. Introns were less constrained than their adjacent exons for both overall numbers of substitutions per site and indels, but, on average, overall numbers of substitutions per site for introns were similar to numbers of synonymous substitutions per site for exons. Correlations were generally high between numbers of substitutions and numbers of indels per site for the same intron. Mitochondrial genes transferred to the nucleus had an accelerated rate of substitution per site, which was most significant at synonymous sites. These differences between paralogs in two different genomes are likely the result of different mutation rates.
Keywords: substitutions, insertions–deletions, generation time, transferred genes, relative rate test
Genes from the three plant genomes show contrasting differences in their evolutionary rates. Nuclear genes evolve the fastest, followed by chloroplast genes and lastly mitochondrial genes (1), despite the fact that the mitochondrial genome shows extensive rearrangement of its structural organization (2). This slow rate of evolution has made mitochondrial gene sequences unappealing to plant phylogenetic studies at the suborder and subfamily levels but has proved to be useful in inferring ancient phylogenetic relationships (3) and in estimating the time of early diversification events in seed plants (4).
In the most extensive survey of substitution rates of plant mitochondrial gene sequences, the number of genes analyzed was restricted to six, and gene sequences were available from a maximum of nine taxa (1). Now, many angiosperm sequences are available for a variety of mitochondrial genes in addition to the complete mitochondrial sequence of liverwort, Marchantia polymorpha (5). Moreover, many mitochondrial intron sequences have been determined, opening up the possibility to estimate with accuracy their evolutionary rates and compare them with those of coding sequences (exons).
Similarly, the issue of substitution rate heterogeneity of mitochondrial genes among diverse plant taxa may now be addressed more efficiently. There is a growing use of DNA sequences to estimate divergence times (4), so one must assess a priori that the candidate genes behave as molecular clocks. Heterogeneity of substitution rates has been reported between lineages for several plant nuclear, chloroplastic, and mitochondrial genes (6–13). Particularly, the annual angiosperms have been shown to have evolved significantly faster than perennial angiosperms, gymnosperms, ferns, and liverwort for the chloroplastic gene rbcL (8, 14). Also, five different mitochondrial genes have been found to evolve significantly faster at nonsynonymous sites in annual angiosperms than in liverwort (4), paralleling results found with the gene rbcL (8). Thus, it would be of interest to know if there is any rate heterogeneity among angiosperm taxa for mitochondrial genes and if it follows any trend related to life history or taxonomic status.
A third potential level of heterogeneity is between mitochondrial genes and their paralogs transferred to the nuclear genome. Transfer of nucleic acids from the mitochondria to the nucleus has been widely reported (15–19). It is generally accepted that gene transfer leads to the formation of pseudogenes in the donor genome and that these pseudogenes have higher substitution rates than normally expressed genes because all mutations are selectively neutral (20, 21). Furthermore, it is likely that genes transferred to the nuclear genome are subject to a higher mutation rate than their mitochondrial paralogs. However, heterogeneity of substitution rates between mitochondrial and nuclear paralogs has not been formally demonstrated in plants. This hypothesis can now be specifically addressed, and rate differences, if any, can be quantified by using the sequences of the mitochondrial genes coxII and rps12. In several species, one or the other of these genes has been shown to be transferred from the mitochondria to the nucleus (17, 18).
MATERIALS AND METHODS
Retrieving DNA Sequences from the GenBank Database.
Published sequences of plant mitochondrial genes were obtained from GenBank database. cDNA sequences, when available, also were retrieved to compare with genomic sequences to identify edited sites (22, 23). Genomic DNA sequences from a total of 15 protein-coding genes belonging to 29 species for a total of 168 sequences were used in the present study (see Tables 1, 3 and 4). The protein-coding genes sampled were: cytochrome oxydase subunits 1, 2, and 3 (coxI, coxII, and coxIII), adenosine–triphosphatase subunits 1, 6, and 9 (atp1, atp6, and atp9), cytochrome b (cob), NADH dehydrogenase subunits 1, 3, 4, and 5 (nad1, nad3, nad4, and nad5), ribosomal protein S3, S12, and S13 (rps3, rps12, and rps13), and ORF 25 (orf25). The intron sequences sampled were from genes coxII (intron between exons 1 and 2), rps3 (intron between exons 1 and 2), nad1 (intron between exons 2 and 3), nad4 (intron between exons 1 and 2), nad5–1,2 (intron between exons 1 and 2), and nad5–4,5 (intron between exons 4 and 5). For each gene, the sequence from the liverwort M. polymorpha also was obtained.
Table 1.
Average (±) numbers of synonymous (KS) and nonsynonymous (Ka) substitutions per site and transition/transversion (ts/tv) ratios between angiosperms and outgroup Marchantia polymorpha for 15 mitochondrial genes
| Gene* | La | Ka | Test† | Ls | Ks | Test† | ts/tv |
|---|---|---|---|---|---|---|---|
| orf25 | 345 | 0.442 ± 0.048 | a | 90 | 1.163 ± 0.356 | a | 0.71 |
| rps3 | 722 | 0.274 ± 0.023 | ab | 193 | 0.526 ± 0.076 | bcdef | 0.70 |
| rps13 | 251 | 0.253 ± 0.038 | ab | 67 | 0.481 ± 0.120 | cdef | 0.84 |
| rps12 | 258 | 0.107 ± 0.022 | ab | 81 | 0.661 ± 0.130 | abcd | 1.21 |
| atp1 | 1127 | 0.098 ± 0.010 | ab | 337 | 0.640 ± 0.064 | abcde | 0.77 |
| coxIII | 556 | 0.091 ± 0.013 | abc | 165 | 0.620 ± 0.100 | abcde | 0.58 |
| atp6 | 499 | 0.084 ± 0.014 | abc | 137 | 0.627 ± 0.094 | abcde | 0.99 |
| nad4 | 993 | 0.077 ± 0.009 | abcd | 280 | 0.488 ± 0.054 | cdef | 1.15 |
| cob | 821 | 0.077 ± 0.010 | abcd | 229 | 0.546 ± 0.064 | bcdef | 1.07 |
| nad5 | 1408 | 0.076 ± 0.008 | abcd | 386 | 0.589 ± 0.056 | abcdef | 0.83 |
| nad1 | 220 | 0.075 ± 0.019 | abcd | 68 | 0.475 ± 0.110 | ef | 0.75 |
| nad3 | 238 | 0.068 ± 0.019 | bcd | 59 | 0.313 ± 0.089 | f | 2.05 |
| coxII | 517 | 0.053 ± 0.011 | cd | 134 | 0.502 ± 0.090 | def | 0.77 |
| coxI | 1131 | 0.036 ± 0.006 | cd | 330 | 0.676 ± 0.067 | abc | 0.90 |
| atp9 | 148 | 0.015 ± 0.010 | d | 47 | 0.851 ± 0.294 | ab | 0.99 |
| Total | 9234 | 2603 | |||||
| Weighted average‡ | 0.108 ± 0.013 | 0.601 ± 0.081 |
M, Marchantia polymorphia; Z, Zea mays; T, Triticum aestiyum; Or, Oryza sativa; Sb, Sorghum bicolor; V, Vicia faba; Pi, Pisum sativum; G, Glycine max; Ph, Phaseolus vulgaris; Oe, Oenothera berteriana; R, Raphanus sativus; Be, Beta vulgaris; C, Citrullus lanatus; H, Helianthus annuus; N, Nicotiana tabaccum; Pe, Petunia hybrida; D, Daucus carota; La, Lactuca sativa; Ly, Lycopersicon esculentum; Pa, Panax ginseng; St, Solanum tuberosum; A, Arabidopsis thaliana; Br, Brassica campestris; La, number of nonsynonymous sites; LS, number of synonymous sites.
Taxa used for each gene; coxI: M, Z, T, Or, Sb, Pi, G, Oe, R, Be; coxII: M, Z, T, Or, Pi, G, Oe, Be, Pe, D; coxIII: M, Z, T, Or, V, G, Oe, H; atp1: M, Z, T, Or, Pi, G, Ph, Oe, R, Be, H, N, Br; atp6: M, Z, T, Or, Sb, V, G, Oe, R, N: atp9: M, Z, T, Or, Sb, V, Pi, G, OE, Be, H, N, Pe; rps3: M, Z, Or, Oe, Pe, Ly; rps12: M, Z, T, Or, Pe, Pa, Br; rps13: M, Z, T, Oe, N, D; orf25: M, Z, T, Or, N, A; cob: M, Z, T, Or, V, Oe, St, A; nad1: M, T, Oe, C, Pe, A; nad3: M, Z, T, Or, Oe, Pe, Pa, Br; nad4 (exons 1, 2, 3, and 4), M, Z, T, La, Br; and nad5: M, T, Oe, A.
Kruskal–Wallis multiple test of mean comparison: genes with the same letter are not significantly different (level P < 0.05 corrected for multiple test).
SE were calculated according to described methods (1).
Table 3.
Evolutionary rates of mitochondrial and nuclear paralog sequences for coxII and rps12
| Taxon | Nucleotide differences, n | ts | tv | ts/tv | Ka | Ks |
|---|---|---|---|---|---|---|
| coxII* | ||||||
| Zea mays | 78 | 33 | 45 | 0.73 | 0.057 ± 0.011‡ | 0.506 ± 0.093‡,§ |
| Triticum aestivum | 76 | 32 | 44 | 0.73 | 0.057 ± 0.011‡ | 0.464 ± 0.084‡,§ |
| Oryza sativa | 79 | 34 | 45 | 0.76 | 0.057 ± 0.011‡ | 0.499 ± 0.089‡,§ |
| Oenothera berteriana | 93 | 41 | 52 | 0.79 | 0.069 ± 0.012 | 0.593 ± 0.107 |
| Pisum sativum | 78 | 35 | 43 | 0.81 | 0.053 ± 0.010‡,§ | 0.457 ± 0.081‡,§ |
| Daucus carota | 78 | 34 | 42 | 0.86 | 0.047 ± 0.010‡,§ | 0.509 ± 0.089‡,§ |
| Petunia hybrida | 85 | 36 | 49 | 0.74 | 0.062 ± 0.011‡ | 0.504 ± 0.089‡,§ |
| Beta vulgaris | 73 | 35 | 38 | 0.92 | 0.041 ± 0.009‡,§ | 0.468 ± 0.081‡,§ |
| Glycine max (ps) | 77 | 35 | 42 | 0.83 | 0.050 ± 0.010‡,§ | 0.480 ± 0.086‡,§ |
| Vignia radiata (nc) | 120 | 62 | 58 | 1.07 | 0.090 ± 0.014 | 0.878 ± 0.137 |
| Glycine max (nc) | 110 | 54 | 56 | 0.96 | 0.080 ± 0.013 | 0.774 ± 0.127 |
| rps12† | ||||||
| Zea mays | 72 | 39 | 33 | 1.18 | 0.113 ± 0.022 | 0.741 ± 0.145 |
| Triticum aestivum | 72 | 39 | 33 | 1.22 | 0.113 ± 0.022 | 0.696 ± 0.135 |
| Oryza sativa | 71 | 38 | 33 | 1.15 | 0.118 ± 0.023 | 0.689 ± 0.131 |
| Petunia hybrida | 66 | 37 | 29 | 1.28 | 0.106 ± 0.022 | 0.584 ± 0.116¶ |
| Panax ginseng | 67 | 35 | 32 | 1.09 | 0.106 ± 0.022 | 0.627 ± 0.119¶ |
| Brassica campestris | 70 | 31 | 39 | 0.80 | 0.112 ± 0.022 | 0.708 ± 0.137 |
| Oenothera hookeri (nc) | 87 | 45 | 42 | 1.07 | 0.124 ± 0.024 | 1.143 ± 0.231 |
Comparisons are between angiosperm taxa and the outgroup Marchantia polymorpha. Results from nuclear sequences are shown in bold type. ts, transitions; tv, transversions; Ka, numbers of nonsynonymous substitutions per site ± SE; Ks, numbers of synonymous substitutions per site ± SE; ps, pseudogene; nc, nuclear paralog.
coxII results compiled from 651 nucleotide sites for all comparisons.
rps12 results compiled from 345 nucleotide sites for all comparisons.
Significantly different than the nuclear paralog of Vigna radiata using a relative rate test with Marchantia as outgroup (P < 0.05).
Significantly different than the nuclear paralog of Glycine max using a relative rate test with Marchantia as outgroup (P < 0.05).
Significantly different than the nuclear paralog of Oenothera hookeri using a relative rate test with Marchantia as outgroup (P < 0.05).
Table 4.
Overall numbers (±SE) of substitutions per site (Ko) for six plant mitochondrial exons and introns
| Gene* | Sites for introns, n† | Numbers of substitutions per site
|
|||||
|---|---|---|---|---|---|---|---|
| Within monocots
|
Within dicots
|
Between monocots and dicots
|
|||||
| Exons | Intron | Exons | Intron | Exons | Intron | ||
| coxII | 771 | 0.006 ± 0.003 | 0.010 ± 0.003 | 0.042 ± 0.008 | 0.042 ± 0.008 | 0.073 ± 0.011 | 0.086 ± 0.011 |
| nad1 | 721 | NA | NA | 0.007 ± 0.005 | 0.051 ± 0.009 | 0.009 ± 0.005 | 0.069 ± 0.010 |
| nad4 | 972 | 0.005 ± 0.003 | 0.018 ± 0.004 | 0.017 ± 0.005 | 0.051 ± 0.007 | 0.044 ± 0.008 | 0.067 ± 0.009 |
| nad5-1, 2 | 796 | NA | NA | 0.016 ± 0.003 | 0.054 ± 0.008 | 0.031 ± 0.005 | 0.098 ± 0.012 |
| nad5-4, 5 | 862 | 0.006 ± 0.004 | 0.009 ± 0.003 | 0.011 ± 0.005 | 0.060 ± 0.009 | 0.031 ± 0.008 | 0.120 ± 0.013 |
| rps3 | 1352 | 0.011 ± 0.003 | 0.019 ± 0.004 | 0.040 ± 0.007 | 0.091 ± 0.009 | 0.066 ± 0.009 | 0.142 ± 0.011 |
| Weighted average§ | 0.007 ± 0.003 | 0.015 ± 0.004 | 0.024 ± 0.005 | 0.060 ± 0.008 | 0.046 ± 0.007 | 0.101 ± 0.011 | |
Taxa used for each gene (see Table 1 for taxon abbreviations): cox2: Z, T, Or, Be, Pe, D; nad1: T, Oe, C, Pe, A; nad4: Z, T, Or, La, Br; nad5-1, 2: T, Oe, Be, A; nad5-4, 5: Z, T, Oe, A; and rps3: Z, Or, Oe, Pe.
See Table 1 for number of sites for exons except nad4 for which only exons 1 and 2 were used here for a total of 648 sites.
SE were calculated according to described methods (1).
NA, not available.
DNA Amplification and Sequencing of the coxI Gene.
The sampling scheme was completed by determining the nucleotide sequence of the subunit 1 of cytochrome oxydase (coxI) for five woody perennial angiosperm taxa: Chamaerops humilis “Elegans argentea” (monocot-Arecaceae), Sabal palmetto (monocot-Arecaceae), Magnolia stellata “Rubra” (dicot-Magnoliaceae), Populus tremuloides (dicot-Salicaceae), and Betula papyrifera (dicot-Betulaceae). DNA was extracted from young leaves of one individual of each species using a cetyltrimethylammonium bromide procedure (24). The gene was amplified as overlapping fragments using eight primers internal to the ORF and two primers, one upstream (−46+) and one downstream (1640−) from the coding region, which were designed from the alignment of previously published coxI sequences of annual herbaceous dicots and monocots. The six sets of primers used for symmetrical PCR amplification were: −46+ (5′-A/TGGGGCCCCTCTCTG/CATAAGG-3′) and 550− (5′-ATCTATGCATAGTCATTCCAGG-3′); 25+ (5′-TTCTCCACTAACCACAAGGATAT-3′) and 550−; 280+ (5′-GACATGGCATTTCCACGATTAA-3′) and 947− (5′-ACAGCTATGATCATGGTAGCTGC-3′); 529+ (5′-CCTGGAATGACTATGCATAGAT-3′) and 1344− (5′-AGCATCTGGATAATCTGGAATG-3′); 1129+ (5′-GCACATTTCCATTATGTACTTTC-3′) and 1550− (5′-GGAAGTTCTCCAAAAGTATGA-3′) and 1129+; and 1640− (5′-ATTGAAATGTTCTGTTAGGTTCTT-3′).
The PCR products were purified with a QIAquick PCR purification kit (Qiagen, Chatsworth, CA), and the two DNA strands were separated using Dynabeads Template Preparation Kit according to recommendations of the manufacturer (Dynal, Oslo). To do so, one primer per set (550−, 280+, 1129+, and 1344−) was biotinylated to ensure separation of the two DNA strands. Direct sequencing of the two DNA strands was conducted with the dideoxynucleotide chain termination procedure using the appropriate amplification primers and a Sequenase version 2.0 kit (United States Biochemical). DNA amplification and sequencing followed described procedures (25) with slight modifications.
Alignment of DNA Sequences.
Each protein-coding gene was aligned from predicted amino acid sequences using the procedure PILEUP in the GCG package (26), and final adjustments were made by eye using the procedure LINEUP. Codons affected by potential or confirmed edited sites were removed from the data sets. Edited sites were found by comparing cDNA sequences with their corresponding genomic sequences for each gene. In addition, when no specific cDNA were available, sites also were considered as potentially edited when there was a C to T nonsynonymous change between angiosperm and liverwort sequences. This procedure excluded from 2.2% (gene atp1) to 14.3% (gene nad3) of the codons, depending on the gene. To render numbers of substitutions per site comparable among various levels of taxonomical subdivision for a given gene, regions in the alignment for which the homology between sequences were uncertain and codons corresponding to gap positions present in one or more sequences were removed from all sequences.
Numerical Analysis and Relative Rate Tests.
Overall numbers of substitutions per site (Ko) were calculated according to the two-parameter model of Kimura (27), and numbers of synonymous (Ks) and nonsynonymous (Ka) substitutions per site were calculated according to the method of Li (28). Calculations were conducted with the programs lwl (28) and mega (29). Homogeneity of substitution rates between taxa and between lineages was assessed by a relative rate test procedure that takes into account the covariances between sequences and that reduces the number of tests to be performed and type II errors (30). The statistic of this test follows approximately the standardized normal distribution.
RESULTS AND DISCUSSION
Numbers of Substitutions per Site for Protein-Coding Sequences.
The 15 mitochondrial protein-coding genes analyzed were found to have evolved at strikingly different rates (Table 1). The weighted averages obtained indicated that synonymous changes occurred six times more often than nonsynonymous changes, but gene-to-gene variation was less for the numbers of synonymous substitution (Ks) than for the numbers of nonsynonymous substitutions (Ka) per site; gene-to-gene differences were 4-fold for Ks and up to 30-fold for Ka. The highest values (either Ka or Ks) were observed for the gene orf25. The lowest value for Ks was observed for the gene nad3, and the lowest value for Ka was observed for the gene atp9. The genes coxI and atp9, which had low Ka, showed two of the highest Ks. The opposite picture was observed for the gene rps13, which showed a high value of Ka but a low value of Ks. A multiple comparison test based on ranks revealed that genes clustered in four overlapping groups for Ka and in six groups for Ks (Table 1). Transversions occurred more frequently than transitions for 11 genes of 15 (Table 1). These results also were consistent with those of Wolfe et al. (1), who observed that transitions make up <50% of the substitutions in the plant mitochondrial genome.
The average weighted values of numbers of synonymous and nonsynonymous substitutions per site between monocots and dicots (data not shown) were slightly lower than published estimates (1). This may be because codons with edited sites were removed from the present analysis but not in the previous study, which was published before the discovery of plant mitochondrial mRNA editing (22, 23). The addition of new genes has resulted in an increase in gene-to-gene variation of Ks and Ka. For some genes, high values of numbers of substitutions per site were obtained when considering either within angiosperm or liverwort–angiosperm comparisons. The numbers of substitutions per site dropped drastically when taxa of the same family were compared (Poaceae, Papilionoideae, Solanaceae: down to 0 for Ka of genes coxII, atp9, and nad3 and for Ks of genes atp9 and nad3). In general, Ks dropped by an order of magnitude for comparisons within a family. Such results confirm the small numbers of potentially informative sites in mitochondrial exon sequences when intrafamilial taxa are to be compared. Consequently, their use as molecular markers in phylogenetic studies should be limited to the order level and higher (3). Although these new estimates confirm the rule that mitochondrial genes display lower rates of synonymous substitution per site compared with genes of the other two genomes (1), some mitochondrial genes showed high values for monocot–dicot comparisons; atp9 had 0.4434 synonymous substitution per site and orf25 had 0.1135 nonsynonymous substitution per site, both in the range of some chloroplast genes (1).
Rate Heterogeneity Between Woody Perennials and Annuals for coxI.
The coxI genes of woody perennial and herbaceous annual taxa were found to have evolved at different rates. The woody perennial taxa considered, either monocots or dicots, showed a slower rate of substitution when compared with herbaceous annual taxa (Table 2). In fact, a 2-fold difference was observed between these two groups for transitions, transversions, and Ks.
Table 2.
Comparisons of the evolutionary rates of coxI gene sequences in angiosperms between woody perennial and annual taxa
| Comparison* | Nucleotide differences, n | ts | tv | ts/tv | Ka | Ks |
|---|---|---|---|---|---|---|
| Woody monocots vs. woody dicots | 37 ± 7 | 17 ± 2 | 20 ± 5 | 0.84 ± 0.16 | 0.006 ± 0.002 | 0.090 ± 0.018 |
| Woody monocots vs. annual dicots | 53 ± 8 | 23 ± 6 | 30 ± 5 | 0.81 ± 0.26 | 0.009 ± 0.003 | 0.134 ± 0.022 |
| Woody dicots vs. annual monocots | 63 ± 8 | 28 ± 3 | 35 ± 6 | 0.81 ± 0.13 | 0.008 ± 0.003 | 0.182 ± 0.027 |
| Annual dicots vs. annual monocots | 74 ± 9 | 35 ± 5 | 39 ± 4 | 0.92 ± 0.10 | 0.009 ± 0.003 | 0.211 ± 0.029 |
Average numbers are given for each comparison. The bold type indicates the small number of substitutions for woody perennial taxa. ts, transitions; tv, transversions, Ka, numbers of nonsynonymous substitutions per site ± SE; Ks, numbers of synonymous substitutions per site ± SE.
Taxa used: woody monocots: Chamaerops humilis, Sabal palmetto; annual monocots: Zea mays, Triticum aestivum, Oryza sativa, Sorghum bicolor; woody dicots: Magnolia stellata, Betula papyrifera, Populus tremuloides; annual dicots: Pisum sativum, Glycine max, Oenothera berteriana, Solanum tuberosum, Raphanus sativus, and Beta vulgaris.
In the groupwise relative rate test procedure used to test the hypothesis of homogeneity of substitution rates for coxI, C. humilis (a woody perennial monocot) and T. aestivum (an annual monocot) were used as reference taxa in the tests involving the dicots. For the tests involving the monocots, M. stellata (a woody perennial dicot) and O. berteriana (an annual dicot) were used as reference taxa. Significant heterogeneity in Ks was detected between woody perennial and annual monocots (P < 0.01), the former having evolved more slowly. In the dicots, the difference between perennials and annuals was significant at P < 0.01 when C. humilis was used as reference taxon and at P < 0.05 when T. aestivum was used as reference taxon. Again, the woody perennials were shown to have evolved more slowly. In the tests based on Ka, a significant difference (P < 0.01) was observed only between woody perennial and annual dicots when C. humilis was used as the reference taxon. The rate heterogeneity observed for the mitochondrial gene coxI between woody perennial and annual taxa correlated with that observed for the chloroplastic gene rbcL (8–10).
Several hypotheses have been suggested to explain this rate heterogeneity among plant taxa that clearly follows a trend related to life history. Factors, such as generation time, which affects mutation rate, but also efficiency of DNA replication and population size have been proposed to account for such rate heterogeneity (8, 10, 31–33). Generation time (the time to reach sexual maturity) could be a factor here because the woody perennials analyzed, which have longer generation times compared with annual taxa, showed lower numbers of nucleotide substitutions per site. This trend was more obvious at synonymous than at nonsynonymous sites. Such a more visible trend at synonymous sites would be expected from a generation time effect because selection is likely less stringent for synonymous positions.
The effects of population size and speciation rate could also be invoked and are likely confounding factors with generation time; the woody perennial taxa analyzed here generally show larger effective population sizes and more archaic and slow developing reproductive isolation mechanisms compared with annual forms, hence leading to a lower frequency of bottlenecks and adaptation events (8). Thus, the resulting slower rate of speciation in perennials would translate in lower probabilities of fixation of mutations.
Although more rate heterogeneity was found at nonsynonymous sites for rbcL (8), the heterogeneity of substitution rates for coxI between annuals and woody perennials was more evident at synonymous sites. The lack of significant rate heterogeneity at nonsynonymous sites is apparently due to the low number of substitutions observed at these sites for coxI because codons including confirmed and potential edited sites were excluded from the analysis (4.4% of the codons for coxI). Indeed, a posteriori analysis including edited or potentially edited codons showed significant differences (at P < 0.05 and at P < 0.01 depending on the test) at nonsynonymous sites between woody perennial and annual taxa in both monocots and dicots when using Magnolia, Chamaerops, or Triticum as reference taxa (data not shown).
Heterogeneity Between Mitochondrial and Nuclear Paralogs.
The nuclear genes coxII and rps12, which have been translocated from the mitochondria to the nucleus (17, 18), showed significant differences in their evolutionary rates when compared with their mitochondrial counterparts. The neighbor-joining trees based on overall numbers of substitutions per site (Ko) for the genes coxII and rps12 show long branches leading to the nuclear paralogs (Fig. 1). The three nuclear paralogs analyzed, two for coxII and one of rps12, clearly have accumulated more substitutions per site in their nucleotide sequence than their mitochondrial counterparts, and this difference is greatest at synonymous sites (Table 3).
Figure 1.
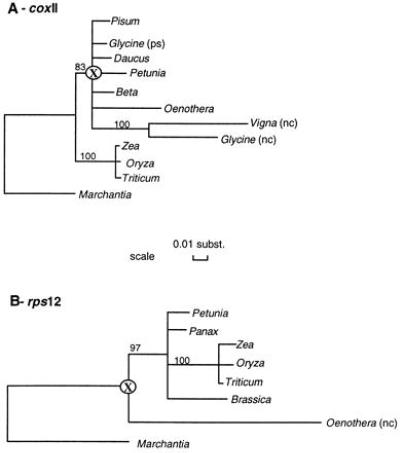
Eighty percent boot strap concensus trees constructed with the neighbor-joining method using overall numbers of substitutions per site (Ko). Branch lengths are proportional to numbers of substitutions per site. Numbers at nodes indicate bootstrap values based on 500 replicates. X, the most recent common ancestor between nuclear and mitochondrial paralogs; nc: nuclear paralog; ps: pseudogene.
In the pairwise relative rate tests (using Marchantia as a reference taxon), all mitochondrial coxII gene sequences (except the one of Oenothera) were found to have evolved at a significantly slower rate at synonymous sites than the two nuclear coxII sequences (P < 0.05). These rate differences were less obvious at nonsynonymous sites; the nuclear coxII gene of Vigna radiata was found to have evolved significantly faster than eight of nine mitochondrial paralogs (P < 0.05), and the other nuclear coxII gene belonging to Glycine max was found to have evolved at a significantly faster rate at nonsynonymous sites in four of nine comparisons with mitochondrial paralogs. The coxII pseudogene sequence of Glycine (17, 34) has not evolved at a significantly different rate, for either synonymous and nonsynonymous substitutions, when compared with the other mitochondrial coxII genes, except for Oenothera at nonsynonymous sites, which suggests a relatively recent lost of function for this pseudogene. For the gene rps12, pairwise relative rate tests (using M. polymorpha as a reference taxon) showed significant differences (at P < 0.05) only for synonymous substitutions between the nuclear gene of Oenothera and the mitochondrial paralogs of Petunia and Panax. Although fewer significant differences in numbers of nonsynonymous substitutions per site were found for both genes, an acceleration of evolutionary rate also could be observed for the nuclear paralogs at nonsynonymous sites. Here also, the small number of observed nonsynonymous differences between nuclear and mitochondrial gene sequences renders the relative rate tests less sensitive in declaring significant differences between nuclear and mitochondrial copies.
Hence, mitochondrial gene sequences transferred and integrated into the nuclear genome show significant modification of their substitution rates, particularly at synonymous sites, when compared with their mitochondrial counterparts. Since the divergence from their common ancestors (nodes X; Fig. 1), there has been, on average, seven times more synonymous substitutions per site in nuclear than in mitochondrial paralogs, which is similar to the ratio of 5 between nuclear and mitochondrial genes reported by Wolfe et al. (1). This suggests (i) relatively ancient transfers, which is likely (see below), and/or (ii) little time needed before the nucleotide sequence of a transferred gene reflects completely its new subcellular compartment. Moreover, because the nuclear paralogs have been shown to be functional (17, 18, 34), differences in numbers of substitutions between the nuclear and the mitochondrial paralogs are more likely the result of different mutation rate rather than differential selection, even if observed differences at nonsynonymous sites were not all statistically significant.
How many transfers have occurred? For the gene coxII, and according to the data analyzed here, only one transfer would have occurred early in the evolution of dicots. It would have been after the split between monocots and dicots and before the diversification of the Papilionoideae (Fig. 1A) (results confirmed with a parsimony analysis not shown). Thus, it is likely that other dicot families also have a nuclear paralog of the gene coxII. For the gene rps12, one transfer seems to have occurred early during the angiosperm evolution, before the split between monocots and dicots (Fig. 1B) (results confirmed with a parsimony analysis not shown). Hence, it is likely from the phylogenetic evidence that most extant angiosperm taxa, both in monocots and dicots, also have a nuclear paralog of the gene rps12.
Rates of Evolution of Mitochondrial Introns.
Homologous sequences were available from at least five angiosperm taxa for each of six introns analyzed (Table 4). However, no introns were found to interrupt the genes nad1 and rps3 in the mitochondrial genome of Marchantia (5). Although the genes nad4, nad5, and coxII were interrupted by introns in liverwort, it was not possible to align them adequately with corresponding ones in angiosperms (average sequence identity of 30%; data not shown). In contrast, exon sequences for the same genes were strongly conserved from liverwort to flowering plants. These introns in liverwort are presumably nonhomologous with those in angiosperms because they were not inserted between the same sites.
Overall numbers of substitutions per site in exon and intron sequences were estimated with complete deletion of gap sites to render estimates comparable among various levels of taxonomical subdivision. For each gene, the overall numbers of substitutions per site (Ko) always were found to be higher in introns than in exons, except in one case: comparisons within dicots for the gene coxII, for which the numbers of substitutions per site were similar between exons and the intron (Table 4). This confirms the highly conserved nature of this class II intron (35). The weighted averages of overall numbers of substitutions per site were found to be at least twice higher for introns than for exons (Table 4). However, when only synonymous substitutions were taken into account for exons, the numbers of substitutions per site were similar between mitochondrial introns and exons [corresponding weighted averages for Ks(within monocots) = 0.0247, Ks(within dicots) = 0.054, Ks(monocots–dicots) = 0.113]. These differences in numbers of substitutions per site between mitochondrial introns and exons are in the range of those observed between introns and exons of the nuclear and the chloroplast genomes (36, 37), indicating, despite the different mutation rates among genomes, that intron sequences are evolutionarily less constrained than exons in the same fashion for the three genomes.
Insertions–deletions (indels) observed in introns (37, 38) and intergenic spacers (39) of the chloroplastic genome may fall within different classes according to the mechanisms involved in their formation (39, 40). It can be safely argued that similar mechanisms can account for the indels observed in mitochondrial introns. Indeed, short indels (1–10 bp) that represented >50% of these events were observed for all introns and are likely due to slipped-strand DNA mispairing (40). Other indels were longer and are likely promoted by the presence of long stem–loop structures in introns (41, 42). Such indels (of more than 100 bp) and stem–loop structures were observed in plant mitochondrial introns (data not shown).
For each intron, the numbers of indels per site were estimated by summing up all indels in each pairwise taxa comparison and dividing by the number of sites. Each indel was considered as a single event irrespective of its length. To correlate the numbers of indels per site with corresponding most precise numbers of nucleotide substitutions per site for each pairwise comparison between taxa, an estimate of the numbers of nucleotide substitutions was obtained for each comparison with gaps deleted only in the sequences concerned and not across all taxa analyzed, thus maximizing the number of nucleotide sites sampled. Few or no indels were necessary to align the exon sequences compared with intron sequences, so no such correlations were estimated for the exon sequences.
The numbers of indels per site estimated for the six mitochondrial introns were always lower than the numbers of substitutions per site (Table 5). Similarly as for the numbers of substitutions, the numbers of indels per site varied by a factor of two from intron to intron. On average, the ratio of substitutions over indels in introns indicated that substitutions occurred twice as frequently as indels in monocot comparisons, five times more frequently in dicot comparisons, and six times more frequently in monocot–dicot comparisons (Table 5). Because of the increase of this ratio with taxonomical divergence, relatively more substitutions than indels could be detected when distantly related taxa were considered. This could be due to multiple indel events at the same sites that could not be properly identified between more distantly related taxa, such as in the monocot–dicot comparisons (39) or to indels that were tolerated only at a limited number of sites.
Table 5.
Average numbers (±SE) of substitutions (Ko) and numbers of indels (I) per site for six mitochondrial introns and correlations expressed as R2 values between the two rates
| Intron* | Within monocots
|
Within dicots
|
Between monocots and dicots
|
R2‡ | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ko | I† | Ko/I | Ko | I† | Ko/I | Ko | I† | Ko/I | ||
| coxII | 0.012 ± 0.004 | 0.005 ± 0.002 | 2.6 | 0.063 ± 0.007 | 0.011 ± 0.003 | 6.0 | 0.113 ± 0.011 | 0.017 ± 0.003 | 6.5 | 0.91§ (15) |
| nadI | NA | NA | NA | 0.057 ± 0.008 | 0.009 ± 0.003 | 6.2 | 0.109 ± 0.010 | 0.016 ± 0.003 | 6.7 | 0.88§ (10) |
| nad4 | 0.019 ± 0.004 | 0.011 ± 0.003 | 1.8 | 0.069 ± 0.007 | 0.022 ± 0.004 | 3.1 | 0.084 ± 0.090 | 0.023 ± 0.004 | 3.7 | 0.90§ (10) |
| nad5-1,2 | NA | NA | NA | 0.055 ± 0.008 | 0.010 ± 0.003 | 5.8 | 0.100 ± 0.012 | 0.013 ± 0.004 | 7.8 | 0.54 (10) |
| nad5-4,5 | 0.009 ± 0.003 | 0.003 ± 0.002 | 2.8 | 0.071 ± 0.009 | 0.012 ± 0.003 | 6.1 | 0.133 ± 0.013 | 0.014 ± 0.004 | 9.4 | 0.58 (6) |
| rps3 | 0.019 ± 0.004 | 0.007 ± 0.002 | 2.7 | 0.093 ± 0.008 | 0.022 ± 0.004 | 4.3 | 0.144 ± 0.011 | 0.029 ± 0.004 | 4.9 | 0.99§ (6) |
| Weighted average¶ | 0.015 ± 0.004 | 0.006 ± 0.002 | 2.3 | 0.068 ± 0.008 | 0.014 ± 0.003 | 4.8 | 0.114 ± 0.011 | 0.019 ± 0.004 | 6.1 | |
See bottom of Table 4 for taxa used.
SE were calculated according to the binomial law.
In parentheses, number of pairwise comparisons (data points) available to calculate correlations.
Correlations significant at P < 0.001; other correlations were unsignificant (P > 0.05).
SE were calculated according to described methods (1).
NA, not available.
However, for a given intron, the correlations between the numbers of substitutions and the numbers of indels per site were positive in all cases and were high for four of six introns (Table 5). The two nad5 introns, which showed the lowest R2 values, were also those for which many fewer indels were necessary to correctly align the sequences. But overall, the indels appeared to be fixed in the introns investigated as regularly as substitutions were although at a slower pace (see above). These results are well in line with those observed for chloroplastic intergenic sequences of monocots (39) and for mitochondrial and nuclear noncoding sequences of primates (43). Thus, in introns, indel events appear to be of potential complementary value to substitutions to estimate phylogenies within angiosperms.
This study has underlined several parameters of the modes and tempos of evolution of plant mitochondrial gene sequences. Gene-to-gene differences in rate of evolution are much larger than previously thought, which indicates that constraints of different power exist on mitochondrial proteins. More important, intra-gene heterogeneity in rates of evolution related to life history of taxa or to gene transfer toward the nuclear genome has been described, indicating that great care must be taken when proceeding to sequence sampling and estimation of phylogenetic trees with mitochondrial genes. It has been shown that mitochondrial introns could represent valuable sources of polymorphic markers because they seem to have fixed nucleotide and indel mutations more readily than coding sequences. However, the evolutionary constraints imposed by intron secondary structures should be investigated. Finally, because introns in liverwort were found to be so different than those in angiosperms, their homology remains to be verified.
Acknowledgments
We thank D. Perry, Centre de Recherche en Biologie Forestière, Université Laval, and two anonymous reviewers for comments on previous drafts of this manuscript. This work was supported by grants from Fonds pour les Chercheurs et l’Aide à la Recherche of Québec and Natural Sciences and Engineering Research Council of Canada to J.B.
ABBREVIATIONS
- Ka
number of nonsynonymous substitutions per site
- Ks
number of synonymous substitutions per site
- Ko overall number of substitutions per site
indels, insertions–deletions
Footnotes
References
- 1.Wolfe K H, Li W-H, Sharp P M. Proc Natl Acad Sci USA. 1987;84:9054–9058. doi: 10.1073/pnas.84.24.9054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palmer J D. In: Molecular Evolutionary Genetics. MacIntyre R J, editor. New York: Plenum; 1985. pp. 131–240. [Google Scholar]
- 3.Hiesel R, von Haesler A, Brennicke A. Proc Natl Acad Sci USA. 1994;91:634–638. doi: 10.1073/pnas.91.2.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laroche J, Li P, Bousquet J. Mol Biol Evol. 1995;12:1151–1156. [Google Scholar]
- 5.Oda K, Yamato K, Ohta E, Nakamura Y, Takemura M, Nozato N, Akashi K, Kanegae T, Ogura M, Kohchi K, Ohyama K. J Mol Biol. 1992;223:1–7. doi: 10.1016/0022-2836(92)90708-r. [DOI] [PubMed] [Google Scholar]
- 6.Ritland K, Clegg M T. Am Nat. 1987;130:S74–S100. [Google Scholar]
- 7.Doebley J, Durbin M, Golenberg E M, Clegg M T, Ma D P. Evolution. 1990;44:1097–1108. doi: 10.1111/j.1558-5646.1990.tb03828.x. [DOI] [PubMed] [Google Scholar]
- 8.Bousquet J, Strauss S H, Doerksen A H, Price R A. Proc Natl Acad Sci USA. 1992;89:7844–7848. doi: 10.1073/pnas.89.16.7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson M A, Gaut B, Clegg M T. Mol Biol Evol. 1990;7:303–314. doi: 10.1093/oxfordjournals.molbev.a040605. [DOI] [PubMed] [Google Scholar]
- 10.Gaut B S, Muse S V, Clark W D, Clegg M T. J Mol Evol. 1992;35:292–303. doi: 10.1007/BF00161167. [DOI] [PubMed] [Google Scholar]
- 11.Yokoyama S, Harry D E. Mol Biol Evol. 1993;10:1215–1226. doi: 10.1093/oxfordjournals.molbev.a040073. [DOI] [PubMed] [Google Scholar]
- 12.Mackay J J, Liu W, Whetten R, Sederoff R, O’Malley D M. Mol Gen Genet. 1995;247:537–545. doi: 10.1007/BF00290344. [DOI] [PubMed] [Google Scholar]
- 13.Hillis D M. Nature (London) 1996;383:130–131. doi: 10.1038/383130a0. [DOI] [PubMed] [Google Scholar]
- 14.Savard L, Li P, Strauss S H, Chase M W, Michaud M, Bousquet J. Proc Natl Acad Sci USA. 1994;91:5163–5167. doi: 10.1073/pnas.91.11.5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moon E, Kao T-H, Wu R. Mol Gen Genet. 1988;213:247–253. doi: 10.1007/BF00339588. [DOI] [PubMed] [Google Scholar]
- 16.Baldauf S L, Palmer J D. Nature (London) 1990;344:262–265. doi: 10.1038/344262a0. [DOI] [PubMed] [Google Scholar]
- 17.Nugent J M, Palmer J D. Cell. 1991;66:473–481. doi: 10.1016/0092-8674(81)90011-8. [DOI] [PubMed] [Google Scholar]
- 18.Grohmann L, Brennicke A, Schuster W. Nucleic Acids Res. 1992;20:5641–5646. doi: 10.1093/nar/20.21.5641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakazono M, Hirai A. Mol Gen Genet. 1993;236:341–346. doi: 10.1007/BF00277131. [DOI] [PubMed] [Google Scholar]
- 20.Gojobori T, Li W-H, Graur D. J Mol Evol. 1982;18:360–369. doi: 10.1007/BF01733904. [DOI] [PubMed] [Google Scholar]
- 21.Li W-H, Wu C-I, Luo C-C. J Mol Evol. 1984;21:58–71. doi: 10.1007/BF02100628. [DOI] [PubMed] [Google Scholar]
- 22.Covello P S, Gray M W. Nature (London) 1989;341:662–666. doi: 10.1038/341662a0. [DOI] [PubMed] [Google Scholar]
- 23.Gualberto J M, Lamattina L, Bonnard G, Weil J-H, Grienenberger J-M. Nature (London) 1989;341:660–662. doi: 10.1038/341660a0. [DOI] [PubMed] [Google Scholar]
- 24.Bousquet J, Simon L, Lalonde M. Can J For Res. 1990;20:254–257. [Google Scholar]
- 25.Bousquet J, Strauss S H, Li P. Mol Biol Evol. 1992;9:1076–1088. doi: 10.1093/oxfordjournals.molbev.a040779. [DOI] [PubMed] [Google Scholar]
- 26.Devereux J, Habeli P, Smities O. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kimura M. J Mol Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 28.Li W-H. J Mol Evol. 1993;36:96–99. doi: 10.1007/BF02407308. [DOI] [PubMed] [Google Scholar]
- 29.Kumar S, Tamura K, Nei M. mega. University Park, PA: Pennsylvania State Univ.; 1993. [Google Scholar]
- 30.Li P, Bousquet J. Mol Biol Evol. 1992;9:1185–1189. doi: 10.1093/oxfordjournals.molbev.a040779. [DOI] [PubMed] [Google Scholar]
- 31.Wu C-I, Li W-H. Proc Natl Acad Sci USA. 1985;82:1741–1745. doi: 10.1073/pnas.82.6.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Britten R J. Science. 1986;231:1393–1398. doi: 10.1126/science.3082006. [DOI] [PubMed] [Google Scholar]
- 33.Stephan W, Langley C H. Genetics. 1992;132:567–574. doi: 10.1093/genetics/132.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Covello P S, Gray M W. EMBO J. 1992;11:3815–3820. doi: 10.1002/j.1460-2075.1992.tb05473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Albrizio M, De Gara L, De Benedetto C, Arrigoni O, Gallerani R. Plant Sci. 1994;100:179–186. [Google Scholar]
- 36.Gaut B S, Clegg M T. Proc Natl Acad Sci USA. 1991;88:2060–2064. doi: 10.1073/pnas.88.6.2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gielly L, Taberlet P. Mol Biol Evol. 1994;11:769–777. doi: 10.1093/oxfordjournals.molbev.a040157. [DOI] [PubMed] [Google Scholar]
- 38.Downie S R, Katz-Downie D S, Cho K-J. Mol Phylogenet Evol. 1996;6:1–18. doi: 10.1006/mpev.1996.0053. [DOI] [PubMed] [Google Scholar]
- 39.Golenberg E M, Clegg M T, Durbin M L, Doebley J, Ma D P. Mol Phylogenet Evol. 1993;2:52–64. doi: 10.1006/mpev.1993.1006. [DOI] [PubMed] [Google Scholar]
- 40.Levinson G, Gutman G A. Mol Biol Evol. 1987;4:203–221. doi: 10.1093/oxfordjournals.molbev.a040442. [DOI] [PubMed] [Google Scholar]
- 41.Buroker N E, Brown J R, Gilbert T A, O’Hara P J, Beckenback A T, Thomas W K, Smith M J. Genetics. 1990;124:157–163. doi: 10.1093/genetics/124.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Learn G H, Shore J S, Furnier G R, Zurawski G, Clegg M T. Mol Biol Evol. 1992;9:856–871. doi: 10.1093/oxfordjournals.molbev.a040765. [DOI] [PubMed] [Google Scholar]
- 43.Saitou N, Ueda S. Mol Biol Evol. 1993;11:504–512. doi: 10.1093/oxfordjournals.molbev.a040130. [DOI] [PubMed] [Google Scholar]