

Abstract
Autosomal Dominant Polycystic Kidney Disease (ADPKD) is estimated to affect 1/600–1/1000 individuals worldwide. The disease is characterized by age dependent renal cyst formation that results in kidney failure during adulthood. Although ultrasound imaging may be an adequate diagnostic tool in at risk individuals older than 30, this modality may not be sufficiently sensitive in younger individuals or for those from PKD2 families who have milder disease. DNA based assays may be indicated in certain clinical situations where imaging cannot provide a definitive clinical diagnosis. The goal of this study was to evaluate the utility of direct DNA analysis in a test sample of 82 individuals who were judged to have polycystic kidney disease by standard clinical criteria. The samples were analyzed using a commercially available assay that employs sequencing of both genes responsible for the disorder. Definite disease causing mutations were identified in 34 (~42%) study participants. An additional 30 (~37%) subjects had either in frame insertions/deletions, non-canonical splice site alterations or a combination of missense changes that were also judged likely to be pathogenic. We noted striking sequence variability in the PKD1 gene, with a mean of 13.1 variants per participant (range 0–60). Our results and analysis highlight the complexity of assessing the pathogenicity of missense variants particularly when individuals have multiple amino acid substitutions. We conclude that a significant fraction of ADPKD mutations are caused by amino acid substitutions that need to be interpreted carefully when utilized in clinical decision-making.
Keywords: Autosomal dominant polycystic kidney disease, ADPKD, mutation analysis, PKDx®, PKD1, PKD2, DNA testing
Introduction
Autosomal dominant polycystic kidney disease (ADPKD [MIM 173900]) is one of the most common inherited disorders in humans, affecting ~1/600–1/1000 individuals [1]. The disease is characterized by age dependent growth of renal cysts such that end stage renal disease (ESRD) typically ensues during mid adulthood. Approximately 85% of ADPKD cases are caused by mutations in the PKD1 gene [MIM 601313], which is located on chromosome 16, while the remaining cases are due to mutations in PKD2 [MIM 173910] located on chromosome 4 [2–7].
Renal ultrasound is a sensitive method of diagnosing ADPKD in those individuals older than 30 years of age who are at risk for inheriting PKD1 mutations. Since cyst formation is an age dependent process, the false negative rate of ultrasound is higher in younger individuals or in those with PKD2 mutations, which are associated with later onset disease [8, 9]. There are a number of clinical scenarios in which DNA based testing for ADPKD might be indicated. Ultrasound imaging might not provide a sufficiently certain diagnosis in at risk individuals younger than 30 years of age who wish to donate a kidney to a relative with renal failure. In addition, clinicians may encounter patients with atypical cystic disease in whom the diagnosis may not be obvious. Finally, new therapies for ADPKD are on the horizon and there is evidence to suggest that these may be most effective if initiated early in the disease course, perhaps even before cystic disease is apparent [10–12].
Linkage analysis for ADPKD has been commercially available for some time, but requires the participation of at least two and preferably more affected family members. In cases where family members are not available, direct DNA analysis offers the possibility of establishing a molecular diagnosis. Direct DNA testing for ADPKD has posed a unique set of challenges. PKD1 analysis in particular has been complicated because the 5′ portion of the gene (exons 1–34) is replicated in at least 5 highly homologous copies (<2% divergence) elsewhere on chromosome 16 [6]. Several groups have now been able to analyze the bona fide PKD1 sequence by using gene-specific primers to amplify large products that can then be screened via nested PCR using direct sequencing [13–24] or other techniques such as denaturing high-performance liquid chromatography (DHPLC) [25–28].
In the current study, we report an analysis of direct sequencing results for both PKD1 and PKD2 in a cohort of 82 individuals with the clinical features of polycystic renal disease (PKDx®, Athena Diagnostics, Inc). Since a major indication for direct DNA testing is the absence of family members for linkage analysis, we chose to study one individual from each family. We identified truncating mutations in ~42% (N=34) of the participants. An additional 30 (~37%) study subjects had either in frame insertions/deletions, non canonical splice site alterations or a combination of missense changes that were judged likely to be pathogenic. Direct sequencing is a sensitive method of detecting DNA variability in PKD1 and PKD2, but given the significant proportion of non-truncating mutations, the need for strict interpretation is essential if the results are to be used in clinical decision-making. To our knowledge, this is the first report of direct sequencing of the entire coding regions of both PKD1 and PKD2.
Materials and Methods
Patient Recruitment and Clinical Evaluation
Eighty-two unrelated ADPKD patients were recruited from outpatient nephrology clinics at the Johns Hopkins Hospital and the University of Toronto. Patients were seen at various stages of the disease process. Institutional review boards at the Johns Hopkins School of Medicine and the University of Toronto approved the study. Informed consent was obtained from each participant. We based the diagnosis of ADPKD on the Ravine ultrasound criteria [8]. A detailed medical history and a coded blood sample were obtained at the time of entry into the study. Blood samples were sent to Athena Diagnostics, Inc. for mutation analysis using the same methodology as for commercially obtained samples (http://www.athenadiagnostics.com/content/test-catalog/find-test/service-detail/q/id/249). In most cases, routine laboratory data was obtained as part of the standard medical evaluation.
Mutation Analysis
Sequence analysis was performed using previously described methods optimized at Athena [13–15, 19, 21, 29, 30]. Briefly, genomic DNA was derived from whole blood using Puregene® DNA extraction kit (Gentra Systems, Inc.) and used as template for specific long-range PCR amplification of 8 segments encompassing the entire PKD1 duplicated region. The long-range PCR products served as template for 43 nested PCR reactions while the unique region of the PKD1 gene and the entire PKD2 gene were amplified from genomic DNA in 28 additional gene segments. All 71 PCR products were bi-directionally sequenced including the coding regions and exon-intron splice junctions of both PKD1 and PKD2.
Analysis of Normal Samples
The normal population was selected from anonymized samples, older than 65, submitted to Athena for ataxia testing. PCR products from a minimum of 171 individuals were sequenced to determine the frequency of certain common variants in either PKD1 or PKD2. Complete DNA analysis was not performed for these samples.
Cleavage Assay
Missense variants were generated using the QuickChange Site–Directed Mutagenesis Kit (Stratagene). The full-length, wild type PKD1 cDNA, four additional constructs (Q3016R, F3064L, F2853S, E2771K) and transfection methods have been previously described [31, 32]. After transfection, the cells were lysed and lysates were immunoprecipitated using ANTI-FLAG® M2 beads (SIGMA) and resolved on NuPAGE® 3–8% Tris-Acetate Gels (Invitrogen). The products were electro-blotted onto an Immobilon™ transfer membrane (MILLIPORE) and probed with α-Leucine-rich-repeat (LRR) and α-C-terminus (CT) antibodies for polycystin-1 (PC1) [32, 33].
Web Resources
The following GenBank sequences (http://www.ncbi.nlm.nih.gov/Web/Genbank/) served as reference files: L39891 for PKD1 genomic nucleotide position, L33233 for PKD1 cDNA position, AAC37576 for PKD1 amino acid position, V50928 for PKD2 genomic nucleotide position, NM000297 for PKD2 cDNA position and NP00288 for PKD2 amino acid position. Other Web Resources: Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/; Splice Site Prediction by Neural Network (SSPNN; http://fruitfly.org/seqtools/splice.html [34]; Automated Splice Site Analyses (ASSA), https://splice.cmh.edu/[35, 36]; Simple Modular Architecture research tool (SMART), http://smart.embl.de/; Pfam, http://www.sanger.ac.uk/Software/Pfam/. MDRD equation: http://nephron.com/cgi-bin/MDRDSI.cgi.
Results
Patient Characteristics
We selected an outpatient cohort that was judged to have polycystic kidney disease based on standard ultrasound criteria (Table 1) [8].
Table 1.
Study Cohort Characteristics.
| % Femalea | 50% |
| Average Age at time of Testa | 46.5 (range 1–73y) |
| % ESRDa,b | 20.7% |
| Average GFR (ml/min)c | 68.7 (range 14–126) |
| % Liver cystsa | 74.3% |
| % Vascular complicationsa | 9.8% |
| % Unknown or no Family Historya | 28% |
Data available for N=82 subjects.
ESRD defined as transplant, dialysis or MDRD (Modification of Diet in Renal Disease) GFR <10.
Data available for 80 patients.
Classification of Sequence Alterations
DNA sequencing results fell into three classes (Table 2). Class I tests were defined as those that had definitive pathogenic sequence variants, such as stop codons, frameshifts or canonical splice site alterations. Class II tests consisted of those demonstrating in frame deletion/insertions, non-canonical splice site mutations, or amino acid substitutions that were judged likely to be pathogenic based on the application of various algorithms. Class III tests included those where no pathogenic changes could be confirmed. The location of Class I and Class II variants are summarized in Figure 1.
Table 2.
Distribution Of Class I and Class II Tests
| Class I
|
Class II
|
Total (%) | |||||
|---|---|---|---|---|---|---|---|
| Stop codon | Frameshift | Splice | In frame | Splice | Missense | ||
| PKD1 | 8 (9.85%) | 14 (17.1%) | 2 (2.4%) | 6 (7.3%) | 4 (4.9%) | 17 (20.7%) | 51 (62.2%) |
| PKD2 | 6 (7.3%) | 3 (3.65%) | 1 (1.2%) | 2 2.4%) | 1 (1.2%) | 0 (0%) | 13 (15.8%) |
|
|
|
||||||
| Total (%) | 34 (41.5%) | 30 (36.6%) | 64 (78%) | ||||
Class I tests are those with a definitive pathogenic mutation predicted to result in premature truncation, including stop codons, frameshifts or canonical splice site alterations. Class II tests contained either in-frame deletion/insertions, unique intronic variants predicted to disrupt splicing or at least one amino acid substitution deemed likely to be pathogenic. See text for details.
Figure 1. Schematic Representation of Polycysin-1 and 2 (PC-1 and PC-2).
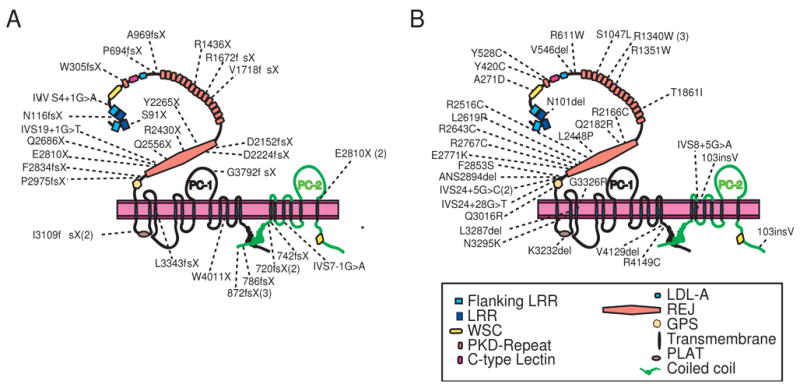
The location of Class I and Class II mutations are indicated in Panels A and B, respectively. Class I tests contain variants predicted to result in premature truncation of either protein. Class II tests consist of either in-frame deletion/insertions, unique intronic variants predicted to disrupt splicing or at least one amino acid substitution deemed likely to be pathogenic. See text for details.
Class I Tests
Thirty-four study participants (~42%) were found to have Class I variants (Table 2 and Supp Table 1 and 2). Twenty-four mutations occurred in PKD1 (~29% of total sample) and ten in PKD2 (~12% of total sample).
Class II Tests
Thirty study participants had Class II variants (Table 2 and Supp Tables 2–4). Participants with class II tests had a positive family history in 77% of cases, similar to 79% for Class I.
We detected a total of 7 unique in-frame deletions (6 PKD1 and 1 PKD2) and 1 unique in-frame insertion (PKD2) in the study population (Supp Table 3). In each instance the in-frame change affected one or more residues that were fully or highly conserved between the Fugu rubripes and Mus musculus polycystin proteins. It seems likely that most if not all of these changes are pathogenic but we cannot exclude the possibility that a functional protein might be synthesized from these alleles.
There were 10 individuals with no other truncating PKD mutations who had unique intronic variants. Two of these sequence alterations (PKD1: IVS24+5 G>C in JHU573 and JHU595; PKD2: IVS8+5 G>A in JHU105) occurred at the 5th base pair from a splice-donor site, which is highly conserved as a guanine in 84% of cases [37]. These variants as well as two others were predicted to result in improper splicing by both the ASSA and SSPNN programs (Supp Table 2). In addition, IVS37-10C>A (JHU 604), was previously reported to segregate with ADPKD in a European family [38]. Although these intronic variants are likely to represent splicing mutations, aberrant splicing could not be confirmed at the RNA level using this DNA based assay.
Most of the remaining participants (N=29) had a combination of amino acid substitutions, primarily in PKD1 (Supp Table 4). We used three major criteria to judge the pathogenicity of each missense variant. We examined the conservation of the altered residue between human polycystin-1 and both the Fugu and mouse proteins. In addition, we assigned a pathogenicity score for each missense variant using the matrix of Miller and Kumar [39], which defines the relative likelihood that a missense change represents a pathogenic alteration versus a polymorphism. This algorithm was developed by using interspecies sequence comparisons coupled with Grantham’s chemical difference matrix to determine the common attributes of amino acid replacement mutations across 7 other disease genes. Other investigators have also used this strategy to assist in characterizing amino acid substitutions [40]. Finally, we reviewed the literature to determine whether any of the variants had been reported by others to occur in normals. Our analysis of individual amino acid substitutions is summarized in Supp Table 4. We found that 17 participants had at least one variant that was likely to be pathogenic.
Several missense changes were predicted to disrupt key structural determinants of PKD1 (Figures 1 and 2). Three participants had variants that had been previously shown to disrupt polycystin-1 cleavage, a property that is critical for the protein’s function [32]. We suspected that other amino acid substitutions near the polycystin-1 GPS site might also disrupt cleavage. In order to test this, we generated full-length mutant constructs and expressed them in HEK293 cells. We confirmed that E2771K, Q3016R and F2853S disrupt cleavage, as do three of these additional missense changes, R2643C, R2767C and L2619P (Figure 3).
Figure 2. Missense Changes Affecting PKD Repeats and C-Lectin Domain.
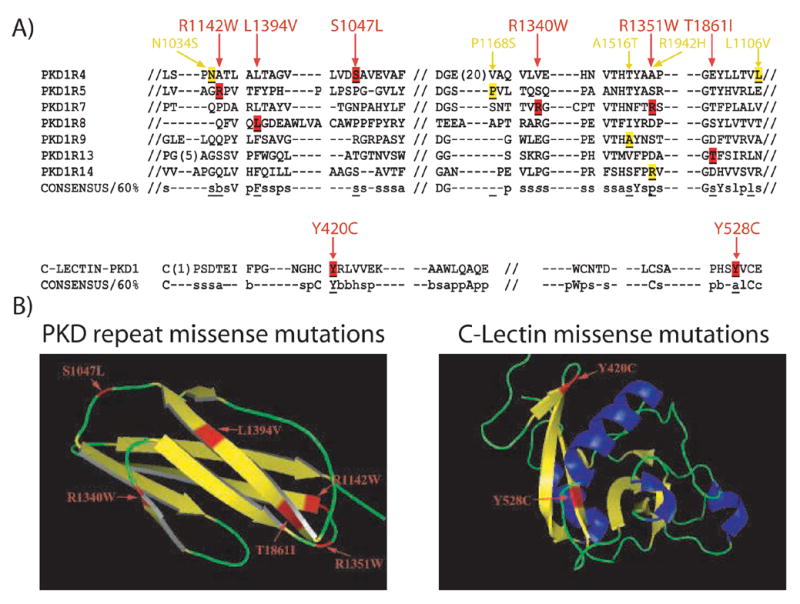
A) Alignment of the consensus sequences for the PKD repeats and C-Lectin domain. The location of missense changes that disrupt the consensus sequence are shown in red, and those that do not are in yellow. Consensus sequence code: l (Aliphatic), a (aromatic), c (Charged), s (small residue), p (polar residue), b (big residue), h (hydrophobic) and capital letter (corresponding amino acid code). B) Ribbon diagrams of PKD repeat and C-Lectin domain. Potentially pathogenic missense changes are demonstrated in red. Helix (blue)-Sheet (yellow)-Loop (green). The secondary structure was determined by PyMol, DeLano Scientific LLC.
Figure 3. Missense Substitutions Affect PKD1 Cleavage.
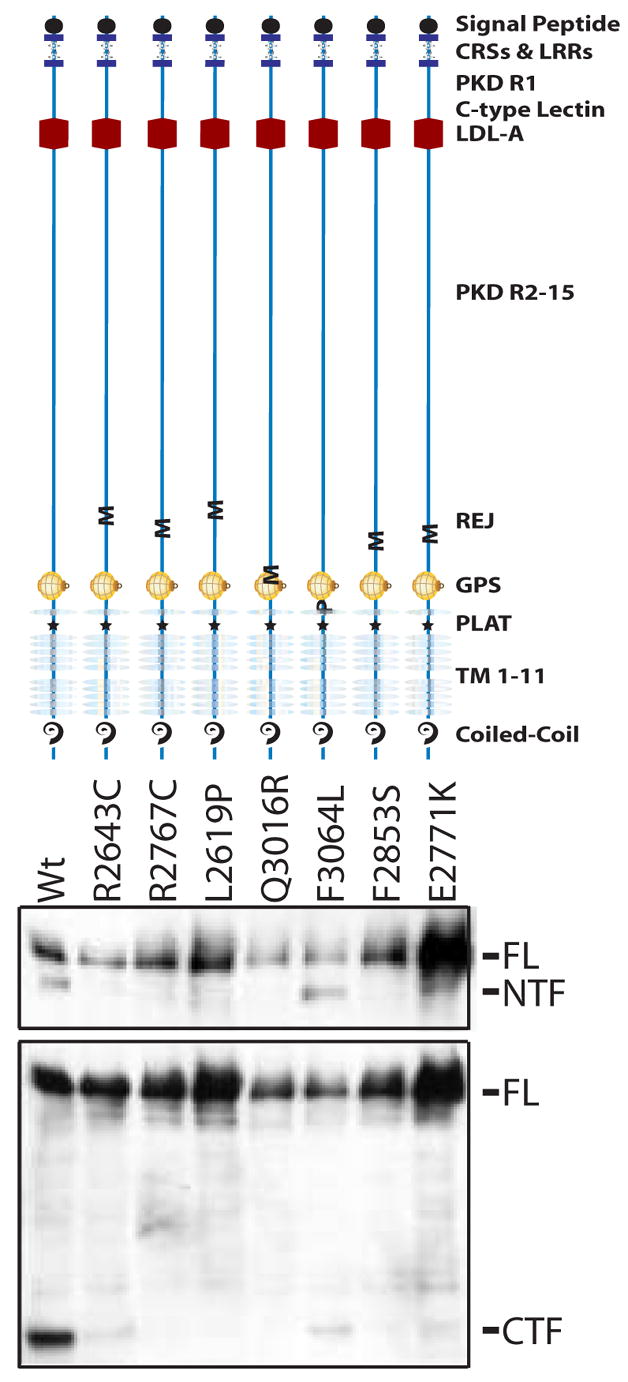
Top Panel shows a schematic of PKD1 mutant polypeptides with the location of each amino acid substitution indicated by M (missense) or P (polymorphism). HEK cells were transfected with Flag-tagged, full length PKD1 constructs for each mutant protein. Western blots were prepared and loaded in the corresponding lanes below each schematic (middle and bottom Panels). In the middle panel, cell lysates were immunoprecipitated with M2 beads and Western blots were probed with an antibody to the LRR (leucine rich repeat). In the bottom panel, Western blots were probed with an antibody to the C-terminus of PKD1. Absence of the NTF fragment is indicative of aberrant cleavage. Six of the missense changes (E2771K, Q3016R, F2853S, R2643C, R2767C, L2619P) disrupt cleavage. In contrast a PC-1 construct with a previously described polymorphism (F3064L) cleaved with the wild type pattern. FL: Full-Length, PKD1 NTF: (N-terminal cleavage fragment). Wt: Wild-type PC-1.
There were several recurrent PKD1 variants (R2200C, Q739R, G2814R, Q2182R) that had a higher pathogenic potential by the Miller/Kumar matrix [39] but which were also present in individuals with chain terminating mutations (Supp Tables 4 and 5). For example, R2200C was present in four participants and one (JHU111) also had a PKD1 frame shift. This association suggested that R2200C might represent a polymorphism. We sequenced 342 normal chromosomes to examine a subset of these missense changes and identified R2200C in a small (1.4%) fraction but greater than the polymorphism threshold of 1%. Likewise Q739R (this study, 6.4%) and G2814R (Rossetti, et al ~0.9%) have also been reported in a small percentage of the unaffected population and are or may be polymorphisms, respectively [16, 26].
Five participants, JHU602 (N=2), JHU100 (N=3), JHU588 (N=2), JHU411 (N=2) and JHU114 (N=2), had more than one PKD1 amino acid variant that met the criteria for pathogenicity (Supp Table 4). These observations raise the possibility that a combination of missense changes in cis might cooperatively result in a diminished level of functional protein [41].
In contrast with PKD1, there were only two PKD2 amino acid substitutions and both were predicted to be polymorphisms (Supp Table 4). M800L (JHU559) did not segregate with disease in a PKD2 family [42]. A second PKD2 substitution, A190T, was identified in 3.2% of normal chromosomes.
Class III Tests
After conducting the analysis described above, there remained 18 subjects who lacked a definitive pathogenic sequence alteration (Individuals without shaded variants not underlined in Supp Table 4). Of these, 9 had a clear family history of ADPKD. All of the remainder (N=9) had enlarged cystic kidneys.
There are several reasons why we might have failed to identify a pathogenic change in this subset of individuals. Mutational events in individuals with class III tests could involve introns or other regulatory regions that were not assayed by the methodology that was used. Direct sequencing might also miss deletions or duplications, which would appear as an area of homozygous normal sequence. Alternatively, our stringent criteria may have identified some missense changes as benign when they may in fact be pathogenic. For example JHU617, with an extensive family history of ADPKD, was found to have a unique leucine to valine change in PKD repeat 4 that was judged more likely to be a polymorphism by the matrix of Miller/Kumar [39]. Nevertheless, this change does disrupt the structure of PKD repeat 4 and could be pathogenic (Figure 2). In addition, as reported by Reynolds, missense variants may unexpectedly activate cryptic splice sites, thereby reducing the level of normal transcript [43].
Polymorphisms, Private Normal Variants And Variability In PKD Genes
There was an impressive degree of sequence variability, particularly in the PKD1 gene. In addition to the sequence alterations described in Supp Tables 1–5, we detected a large number of polymorphisms and private, non-pathogenic variants (see Supp Table 6). The density of variants was noted to be dramatically higher in PKD1 as compared with PKD2. On average, the 82 study participants had 13.1 PKD1 sequence variants per patient (range 0 to 60, Supp Tables 3–5). In contrast, there was an average of only 1.1 PKD2 variants per patient (range of 0–3).
Discussion
We anticipate that there will continue to be a need for molecular diagnosis among individuals from ADPKD families. DNA testing may be relevant in a variety of clinical circumstances including the evaluation of living donors from ADPKD families, early detection for treatment with new therapeutic agents and in individuals with atypical clinical presentations where a diagnosis of ADPKD is in question. In situations where ultrasound is not sufficiently sensitive and where family members are unavailable for linkage studies, direct DNA testing has the potential to provide genetic information to an isolated proband. Direct sequencing of PKD1 and PKD2 forms of the basis for the analysis in this study. We have evaluated the utility of this test in 82 individuals with a clinical diagnosis of ADPKD.
We found that 42% of the study participants (N=34) had a definitive pathogenic mutation predicted to result in premature truncation of either PKD protein (Class I). The remaining study subjects had in frame deletions/insertions, amino acid substitutions or intronic changes requiring further analysis to assess pathogenicity (Class II). We chose not to study the segregation of these variants since the lack of available family members is an important indication for the use of direct sequencing. Instead we utilized several in silico approaches to determine which variants were more likely to be disease-related. This extended analysis allowed us to conclude that 30 individuals with Class II test results had at least one variant that was deemed likely to be pathogenic. Further evidence of disease relatedness for a subset of variants was provided by in vitro functional studies demonstrating that six of the missense amino acid substitutions resulted in loss of polycystin-1 cleavage [32]. If one combines Class I and Class II tests, mutations were detected in approximately 78% (N=64) of the study population.
Only one other group has published a study similar to ours. Rossetti et al used DHPLC to screen PKD1 and PKD2 in 45 genetically uncharacterized individuals with clinical ADPKD [26]. In their research cohort, 34 (76%) subjects had class I or class II tests. This is not statistically different from our results (78%, p≤1.0, Chi-square test). The differences in the incidence of truncating mutations between our cohort and Rossetti’s may be related to characteristics of the study population. Our cohort was not identified on the basis of ESRD so we may have selected for a group with a milder phenotype and hence a lower prevalence of truncating mutations.
There are several reports that have employed a combination of screening methods including protein truncation, DHPLC and selective sequencing to identify mutations in portions of PKD1 or PKD2 [13–19, 21–27, 29]. We note that several of the missense and splicing variants classified as pathogenic in those studies were found in association with chain terminating changes in our cohort (for example, R2200C and IVS39-25del72bp in JHU111, N116fsX and JHU104, 3792fsX, respectively) [22, 27, 44]. R2200C was subsequently found in a small fraction of normal chromosomes and observed in homozygosity (personal communication, JGJ), suggesting that it can occur on a normal haplotype. These reports demonstrate the importance of complete sequencing of both genes in order to identify the bona fide gene mutation.
The DNA sequence analysis reported here reveals a remarkable degree of sequence variability particularly in PKD1 and suggests that a substantial fraction of PKD mutations will be amino acid substitutions. These results also highlight the complexity of assessing the pathogenicity of missense variants particularly in individuals who may harbor multiple such amino acid substitutions. In order to develop the most robust algorithm for classifying missense substitutions, complete sequence data from a reference population of unaffected, ethnically diverse individuals would be invaluable in assessing the pathogenicity of missense changes. These data also would provide an estimate of the frequency of missense changes in normal controls and expand the spectrum of known, non-pathogenic sequence variants. This information would be particularly helpful in analyzing cases where the a priori probability of having ADPKD is less than 50%.
In summary, we estimate that in individuals from ADPKD families, approximately 40–60% of DNA tests will yield definitive results. The major limitation of DNA testing in ADPKD is the large number of missense changes that may be challenging to interpret even with the best in silico approaches. Since clinical decision-making demands an added level of certainty beyond that required in research situations, Class II results may often be non-diagnostic. Therefore, in some situations, such as the evaluation of prospective renal donors from small ADPKD families, we recommend initial testing of the affected recipient to identify the disease-causing mutation. If a diagnostic mutation is identified, we then proceed with directed testing of the prospective donor. In other situations the possibility of an indeterminate result versus the impact of a positive diagnosis on clinical management must be factored into the decision to pursue DNA testing.
Despite the high frequency of missense mutations, direct DNA testing for ADPKD can provide valuable information in certain clinical circumstances. In the future, sequence analyses of PKD1 and PKD2 in normal populations coupled with data from functional research studies should assist in the interpretation of missense variants, thereby expanding the diagnostic utility of molecular genetic testing for ADPKD.
Supplementary Material
Acknowledgments
This work was supported by Becas FPI de investigacion (MCYT, Spain) to MGG, R01DK70617 to TW, PKD Center of Excellence DK57325, R37DK48006 to GGG. GGG is the Irving Blum Scholar of the Johns Hopkins University School of Medicine. We thank Ms. Elizabeth Lyden, University of Nebraska Medical Center for assistance with the statistical analysis.
Footnotes
Conflict of Interest Disclosure:
Under a licensing agreement between Athena Laboratories and the Johns Hopkins University, Dr. Watnick and Dr. Germino are entitled to a share of royalty received by the University on sales of products described in this article. They have elected to donate their share of the royalty to the Polycystic Kidney Disease Research Foundation. The terms of this arrangement are being managed by the Johns Hopkins University in accordance with its conflict of interest policies.
References
- 1.Gabow PA. Autosomal Dominant Polycystic Kidney Disease. 1993;329:332–42. doi: 10.1056/NEJM199307293290508. [DOI] [PubMed] [Google Scholar]
- 2.Peters DJ, Sandkuijl LA. Genetic heterogeneity of polycystic kidney disease in Europe. Contrib Nephrol. 1992;97:128–39. doi: 10.1159/000421651. [DOI] [PubMed] [Google Scholar]
- 3.The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. The European Polycystic Kidney Disease Consortium. Cell. 1994;77(6):881–94. doi: 10.1016/0092-8674(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 4.Burn TC, Connors TD, Dackowski WR, Petry LR, Van Raay TJ, Millholland JM, Venet M, Miller G, Hakim RM, Landes GM, et al. Analysis of the genomic sequence for the autosomal dominant polycystic kidney disease (PKD1) gene predicts the presence of a leucine-rich repeat. The American PKD1 Consortium (APKD1 Consortium) Hum Mol Genet. 1995;4(4):575–82. doi: 10.1093/hmg/4.4.575. [DOI] [PubMed] [Google Scholar]
- 5.Polycystic kidney disease: the complete structure of the PKD1 gene and its protein. The International Polycystic Kidney Disease Consortium. Cell. 1995;81(2):289–98. doi: 10.1016/0092-8674(95)90339-9. [DOI] [PubMed] [Google Scholar]
- 6.Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millan JL, Gamble V, Harris PC. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet. 1995;10(2):151–60. doi: 10.1038/ng0695-151. [DOI] [PubMed] [Google Scholar]
- 7.Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, Kimberling WJ, Breuning MH, Deltas CC, Peters DJ, Somlo S. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272(5266):1339–42. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- 8.Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet. 1994;343(8901):824–7. doi: 10.1016/s0140-6736(94)92026-5. [DOI] [PubMed] [Google Scholar]
- 9.Hateboer N, v Dijk MA, Bogdanova N, Coto E, Saggar-Malik AK, San Millan JL, Torra R, Breuning M, Ravine D. Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet. 1999;353(9147):103–7. doi: 10.1016/s0140-6736(98)03495-3. [DOI] [PubMed] [Google Scholar]
- 10.Torres VE. Therapies to slow polycystic kidney disease. Nephron Exp Nephrol. 2004;98(1):e1–7. doi: 10.1159/000079926. [DOI] [PubMed] [Google Scholar]
- 11.Grantham JJ, Torres VE, Chapman AB, Guay-Woodford LM, Bae KT, King BF, Jr, Wetzel LH, Baumgarten DA, Kenney PJ, Harris PC, Klahr S, Bennett WM, Hirschman GN, Meyers CM, Zhang X, Zhu F, Miller JP. Volume progression in polycystic kidney disease. N Engl J Med. 2006;354(20):2122–30. doi: 10.1056/NEJMoa054341. [DOI] [PubMed] [Google Scholar]
- 12.Walz G. Therapeutic approaches in autosomal dominant polycystic kidney disease (ADPKD): is there light at the end of the tunnel? Nephrol Dial Transplant. 2006;21(7):1752–7. doi: 10.1093/ndt/gfl246. [DOI] [PubMed] [Google Scholar]
- 13.Watnick TJ, Piontek KB, Cordal TM, Weber H, Gandolph MA, Qian F, Lens XM, Neumann HP, Germino GG. An unusual pattern of mutation in the duplicated portion of PKD1 is revealed by use of a novel strategy for mutation detection. Hum Mol Genet. 1997;6(9):1473–81. doi: 10.1093/hmg/6.9.1473. [DOI] [PubMed] [Google Scholar]
- 14.Watnick TJ, Gandolph MA, Weber H, Neumann HP, Germino GG. Gene conversion is a likely cause of mutation in PKD1. Hum Mol Genet. 1998;7(8):1239–43. doi: 10.1093/hmg/7.8.1239. [DOI] [PubMed] [Google Scholar]
- 15.Watnick T, Phakdeekitcharoen B, Johnson A, Gandolph M, Wang M, Briefel G, Klinger KW, Kimberling W, Gabow P, Germino GG. Mutation detection of PKD1 identifies a novel mutation common to three families with aneurysms and/or very-early-onset disease. Am J Hum Genet. 1999;65(6):1561–71. doi: 10.1086/302657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomas R, McConnell R, Whittacker J, Kirkpatrick P, Bradley J, Sandford R. Identification of mutations in the repeated part of the autosomal dominant polycystic kidney disease type 1 gene, PKD1, by long-range PCR. Am J Hum Genet. 1999;65(1):39–49. doi: 10.1086/302460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perrichot RA, Mercier B, Simon PM, Whebe B, Cledes J, Ferec C. DGGE screening of PKD1 gene reveals novel mutations in a large cohort of 146 unrelated patients. Hum Genet. 1999;105(3):231–9. doi: 10.1007/s004390051094. [DOI] [PubMed] [Google Scholar]
- 18.Afzal AR, Florencio RN, Taylor R, Patton MA, Saggar-Malik A, Jeffery S. Novel mutations in the duplicated region of the polycystic kidney disease 1 (PKD1) gene provides supporting evidence for gene conversion. Genet Test. 2000;4(4):365–70. doi: 10.1089/109065700750065108. [DOI] [PubMed] [Google Scholar]
- 19.Phakdeekitcharoen B, Watnick TJ, Ahn C, Whang DY, Burkhart B, Germino GG. Thirteen novel mutations of the replicated region of PKD1 in an Asian population. Kidney Int. 2000;58(4):1400–12. doi: 10.1046/j.1523-1755.2000.00302.x. [DOI] [PubMed] [Google Scholar]
- 20.Perrichot R, Mercier B, Quere I, Carre A, Simon P, Whebe B, Cledes J, Ferec C. Novel mutations in the duplicated region of PKD1 gene. Eur J Hum Genet. 2000;8(5):353–9. doi: 10.1038/sj.ejhg.5200459. [DOI] [PubMed] [Google Scholar]
- 21.Phakdeekitcharoen B, Watnick TJ, Germino GG. Mutation analysis of the entire replicated portion of PKD1 using genomic DNA samples. J Am Soc Nephrol. 2001;12(5):955–63. doi: 10.1681/ASN.V125955. [DOI] [PubMed] [Google Scholar]
- 22.Rossetti S, Strmecki L, Gamble V, Burton S, Sneddon V, Peral B, Roy S, Bakkaloglu A, Komel R, Winearls CG, Harris PC. Mutation analysis of the entire PKD1 gene: genetic and diagnostic implications. Am J Hum Genet. 2001;68(1):46–63. doi: 10.1086/316939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inoue S, Inoue K, Utsunomiya M, Nozaki J, Yamada Y, Iwasa T, Mori E, Yoshinaga T, Koizumi A. Mutation analysis in PKD1 of Japanese autosomal dominant polycystic kidney disease patients. Hum Mutat. 2002;19(6):622–8. doi: 10.1002/humu.10080. [DOI] [PubMed] [Google Scholar]
- 24.Burtey S, Lossi AM, Bayle J, Berland Y, Fontes M. Mutation screening of the PKD1 transcript by RT-PCR. J Med Genet. 2002;39(6):422–9. doi: 10.1136/jmg.39.6.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizoguchi M, Tamura T, Yamaki A, Higashihara E, Shimizu Y. Mutations of the PKD1 gene among Japanese autosomal dominant polycystic kidney disease patients, including one heterozygous mutation identified in members of the same family. J Hum Genet. 2001;46(9):511–7. doi: 10.1007/s100380170032. [DOI] [PubMed] [Google Scholar]
- 26.Rossetti S, Chauveau D, Walker D, Saggar-Malik A, Winearls CG, Torres VE, Harris PC. A complete mutation screen of the ADPKD genes by DHPLC. Kidney Int. 2002;61(5):1588–99. doi: 10.1046/j.1523-1755.2002.00326.x. [DOI] [PubMed] [Google Scholar]
- 27.Rossetti S, Chauveau D, Kubly V, Slezak JM, Saggar-Malik AK, Pei Y, Ong AC, Stewart F, Watson ML, Bergstralh EJ, Winearls CG, Torres VE, Harris PC. Association of mutation position in polycystic kidney disease 1 (PKD1) gene and development of a vascular phenotype. Lancet. 2003;361(9376):2196–201. doi: 10.1016/S0140-6736(03)13773-7. [DOI] [PubMed] [Google Scholar]
- 28.Zhang DY, Sun TM, Zhang SZ, Tang B, Dai B, Zhang WL, Mei CL. [Mutation detection of PKD2 gene in Chinese by denaturing high-performance liquid chromatograph] Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2004;21(3):211–4. [PubMed] [Google Scholar]
- 29.Watnick TJ, Torres VE, Gandolph MA, Qian F, Onuchic LF, Klinger KW, Landes G, Germino GG. Somatic mutation in individual liver cysts supports a two-hit model of cystogenesis in autosomal dominant polycystic kidney disease. Mol Cell. 1998;2(2):247–51. doi: 10.1016/s1097-2765(00)80135-5. [DOI] [PubMed] [Google Scholar]
- 30.Watnick T, Germino GG. Molecular basis of autosomal dominant polycystic kidney disease. Semin Nephrol. 1999;19(4):327–43. [PubMed] [Google Scholar]
- 31.Hanaoka K, Qian F, Boletta A, Bhunia AK, Piontek K, Tsiokas L, Sukhatme VP, Guggino WB, Germino GG. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature. 2000;408(6815):990–4. doi: 10.1038/35050128. [DOI] [PubMed] [Google Scholar]
- 32.Qian F, Boletta A, Bhunia AK, Xu H, Liu L, Ahrabi AK, Watnick TJ, Zhou F, Germino GG. Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1-associated mutations. Proc Natl Acad Sci U S A. 2002;99(26):16981–6. doi: 10.1073/pnas.252484899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boletta A, Qian F, Onuchic LF, Bhunia AK, Phakdeekitcharoen B, Hanaoka K, Guggino W, Monaco L, Germino GG. Polycystin-1, the gene product of PKD1, induces resistance to apoptosis and spontaneous tubulogenesis in MDCK cells. Mol Cell. 2000;6(5):1267–73. doi: 10.1016/s1097-2765(00)00123-4. [DOI] [PubMed] [Google Scholar]
- 34.Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4(3):311–23. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- 35.Nalla VK, Rogan PK. Automated splicing mutation analysis by information theory. Hum Mutat. 2005;25(4):334–42. doi: 10.1002/humu.20151. [DOI] [PubMed] [Google Scholar]
- 36.Rogan PK, Faux BM, Schneider TD. Information analysis of human splice site mutations. Hum Mutat. 1998;12(3):153–71. doi: 10.1002/(SICI)1098-1004(1998)12:3<153::AID-HUMU3>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 37.Lewin . Genes. Oxford University Press, Inc; 1997. p. 888. [Google Scholar]
- 38.Bogdanova N, McCluskey M, Sikmann K, Markoff A, Todorov V, Dimitrakov D, Schiavello T, Thomas M, Kalaydjieva L, Dworniczak B, Horst J. Screening the 3′ region of the polycystic kidney disease 1 (PKD1) gene in 41 Bulgarian and Australian kindreds reveals a prevalence of protein truncating mutations. Hum Mutat. 2000;16(2):166–74. doi: 10.1002/1098-1004(200008)16:2<166::AID-HUMU9>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 39.Miller MP, Kumar S. Understanding human disease mutations through the use of interspecific genetic variation. Hum Mol Genet. 2001;10(21):2319–28. doi: 10.1093/hmg/10.21.2319. [DOI] [PubMed] [Google Scholar]
- 40.Sharp AM, Messiaen LM, Page G, Antignac C, Gubler MC, Onuchic LF, Somlo S, Germino GG, Guay-Woodford LM. Comprehensive genomic analysis of PKHD1 mutations in ARPKD cohorts. J Med Genet. 2005;42(4):336–49. doi: 10.1136/jmg.2004.024489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bouba I, Koptides M, Mean R, Costi CE, Demetriou K, Georgiou I, Pierides A, Siamopoulos K, Deltas CC. Novel PKD1 deletions and missense variants in a cohort of Hellenic polycystic kidney disease families. Eur J Hum Genet. 2001;9(9):677–84. doi: 10.1038/sj.ejhg.5200696. [DOI] [PubMed] [Google Scholar]
- 42.Reiterova J, Stekrova J, Peters DJ, Kapras J, Kohoutova M, Merta M, Zidovska J. Four novel mutations of the PKD2 gene in Czech families with autosomal dominant polycystic kidney disease. Hum Mutat. 2002;19(5):573. doi: 10.1002/humu.9035. [DOI] [PubMed] [Google Scholar]
- 43.Reynolds DM, Hayashi T, Cai Y, Veldhuisen B, Watnick TJ, Lens XM, Mochizuki T, Qian F, Maeda Y, Li L, Fossdal R, Coto E, Wu G, Breuning MH, Germino GG, Peters DJ, Somlo S. Aberrant splicing in the PKD2 gene as a cause of polycystic kidney disease. J Am Soc Nephrol. 1999;10(11):2342–51. doi: 10.1681/ASN.V10112342. [DOI] [PubMed] [Google Scholar]
- 44.Peral B, San Millan JL, Ong AC, Gamble V, Ward CJ, Strong C, Harris PC. Screening the 3′ region of the polycystic kidney disease 1 (PKD1) gene reveals six novel mutations. Am J Hum Genet. 1996;58(1):86–96. [PMC free article] [PubMed] [Google Scholar]
- 45.Nauli SM, Rossetti S, Kolb RJ, Alenghat FJ, Consugar MB, Harris PC, Ingber DE, Loghman-Adham M, Zhou J. Loss of polycystin-1 in human cyst-lining epithelia leads to ciliary dysfunction. J Am Soc Nephrol. 2006;17(4):1015–25. doi: 10.1681/ASN.2005080830. [DOI] [PubMed] [Google Scholar]
- 46.Veldhuisen B, Saris JJ, de Haij S, Hayashi T, Reynolds DM, Mochizuki T, Elles R, Fossdal R, Bogdanova N, van Dijk MA, Coto E, Ravine D, Norby S, Verellen-Dumoulin C, Breuning MH, Somlo S, Peters DJ. A spectrum of mutations in the second gene for autosomal dominant polycystic kidney disease (PKD2) Am J Hum Genet. 1997;61(3):547–55. doi: 10.1086/515497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rossetti S, Bresin E, Restagno G, Carbonara A, Corra S, De Prisco O, Pignatti PF, Turco AE. Autosomal dominant polycystic kidney disease (ADPKD) in an Italian family carrying a novel nonsense mutation and two missense changes in exons 44 and 45 of the PKD1 Gene. Am J Med Genet. 1996;65(2):155–9. doi: 10.1002/(SICI)1096-8628(19961016)65:2<155::AID-AJMG15>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 48.Peral B, Gamble V, Strong C, Ong AC, Sloane-Stanley J, Zerres K, Winearls CG, Harris PC. Identification of mutations in the duplicated region of the polycystic kidney disease 1 gene (PKD1) by a novel approach. Am J Hum Genet. 1997;60(6):1399–410. doi: 10.1086/515467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCluskey M, Schiavello T, Hunter M, Hantke J, Angelicheva D, Bogdanova N, Markoff A, Thomas M, Dworniczak B, Horst J, Kalaydjieva L. Mutation detection in the duplicated region of the polycystic kidney disease 1 (PKD1) gene in PKD1-linked Australian families. Hum Mutat. 2002;19(3):240–50. doi: 10.1002/humu.10045. [DOI] [PubMed] [Google Scholar]
- 50.Aguiari G, Savelli S, Garbo M, Bozza A, Augello G, Penolazzi L, De Paoli Vitali E, La Torre C, Cappelli G, Piva R, del Senno L. Novel splicing and missense mutations in autosomal dominant polycystic kidney disease 1 (PKD1) gene: expression of mutated genes. Hum Mutat. 2000;16(5):444–5. doi: 10.1002/1098-1004(200011)16:5<444::AID-HUMU11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 51.Torra R, Viribay M, Telleria D, Badenas C, Watson M, Harris P, Darnell A, San Millan JL. Seven novel mutations of the PKD2 gene in families with autosomal dominant polycystic kidney disease. Kidney Int. 1999;56(1):28–33. doi: 10.1046/j.1523-1755.1999.00534.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.