

Abstract
We sought to determine whether intramuscular injection of a recombinant adeno-associated virus (rAAV) vector expressing human factor IX (hF.IX) could direct expression of therapeutic levels of the transgene in experimental animals. High titer (1012–1013 vector genomes/ml) rAAV expressing hF.IX was prepared, purified, and injected into hindlimb muscles of C57BL/6 mice and Rag 1 mice. In the immunocompetent C57BL/6 mice, immunofluorescence staining of muscle harvested 3 months after injection demonstrated the presence of hF.IX protein, and PCR analysis of muscle DNA was positive for AAV DNA, but no hF.IX was detected in mouse plasma. Further studies showed that these mice had developed circulating antibodies to hF.IX. In follow-up experiments in Rag 1 mice, which carry a mutation in the recombinase activating gene-1 and thus lack functional B and T cells, similar results were seen on DNA analysis of muscle, but these mice also demonstrated therapeutic levels (200–350 ng/ml) of F.IX in the plasma. The time course of F.IX expression demonstrates that levels gradually increase over a period of several weeks before reaching a plateau that is stable 6 months after injection. In other experiments we demonstrate colocalization of hF.IX and collagen IV in intersitial spaces between muscle fibers. Collagen IV has recently been identified as a F.IX-binding protein; this finding explains the unusual pattern of immunofluorescent staining for F.IX shown in these experiments. Thus rAAV can be used to direct stable expression of therapeutic levels of F.IX after intramuscular injection and is a feasible strategy for treatment of patients with hemophilia B.
Recent reports demonstrate significant progress in the area of gene therapy by in vivo expression of a transgene after direct injection of the vector into skeletal muscle. Adenoviral vectors were used in this manner to express high levels of canine blood coagulation factor IX (F.IX) in immunodeficient mice or in conjunction with immunosuppressive agents to avoid an otherwise strong inflammatory and cytotoxic T lymphocyte response to adenoviral-transduced muscle fibers (1). Other studies suggest that intramuscular injection of replication defective adenovirus can provide long-term expression if the transgene encodes a self-protein so that a strong host immune response is avoided (2, 3). Plasmid DNA injected into mouse muscle directed expression of erythropoietin (4), but has not been shown to be efficient enough for the expression of a gene product such as F.IX, which must be present at much higher levels in the circulation to achieve a therapeutic effect.
Adeno-associated virus (AAV) represents an alternative vehicle for gene delivery into muscle. Recombinant AAV (rAAV) does not contain sequences encoding viral proteins and has the potential to integrate into the chromosomal DNA of the host cell (5, 6). Production and purification procedures are now available that allow the generation of rAAV without significant contamination with wild-type AAV or the helper adenovirus (6–8). While the efficiency of in vivo transduction with rAAV in the absence of helper virus is low for hepatocytes and airway epithelial cells (7), certain postmitotic cells, such as neurons (9) and skeletal muscle fibers (10, 11), can be effectively transduced. Stable expression of lacZ for up to 1½ years has been reported (11). In contrast to adenoviral vectors, intramuscular injection with rAAV in immunocompetent animals does not result in cytotoxic T lymphocyte response against transduced muscle fibers, nor are circulating antibodies against the intracellular lacZ gene product present (10). Based on Southern blot and PCR analysis of genomic DNA from skeletal muscle injected with rAAV, other investigators concluded that rAAV genomes persisted as tandem repeats integrated into chromosomal DNA (10, 11).
There has been a single report of expression of a secreted protein, erythropoietin, after intramuscular injection with rAAV (12), but the levels of protein expression reported (vide infra) were 1–2 orders of magnitude below that needed for a coagulation protein such as F.IX. In this study, we report stable expression (at least 6 months) of therapeutic levels of human F.IX (hF.IX) after intramuscular injection of adult mice with an AAV-F.IX vector. Expression was demonstrated for muscle tissue of immunocompetent animals and reached levels of 200–350 ng/ml in plasma of immunodeficient mice. Therefore, intramuscular injection with rAAV represents a promising approach to provide stable therapeutic amounts of hF.IX in the systemic circulation of hemophilia B patients.
MATERIALS AND METHODS
Production and Purification of rAAV.
rAAV was generated by cotransfection of a F.IX cis plasmid (pAAV-F.IX) and the trans-acting plasmid pAAV/Ad (6) into human embryonic kidney (293) cells infected with an E1-deleted adenovirus as described by Fisher et al. (7). pAAV-F.IX was derived from psub201 (6) and contains the cytomegalovirus (CMV) promoter/enhancer, the hF.IX coding sequence including a 1.4-kb fragment of intron I (13), and the simian virus 40 polyadenylylation signal, flanked by AAV inverted terminal repeat sequences. The size of pAAV-F.IX is 8.34 kb. The AAV rep and cap gene functions were supplied in trans by pAAV/Ad. E1-deleted adenovirus contained a lacZ or alkaline phosphatase reporter gene to trace potential contamination of rAAV stocks with helper virus. Cells were lysed 48 hr after transfection by sonication, and the released viral particles were purified by four rounds of CsCl density gradient centrifugation as described by Fisher et al. (7).
AAV-F.IX particles had a density of 1.37–1.40 g/ml. The titer of the purified AAV-F.IX was determined by slot blot hybridization using a probe specific to either the CMV promoter or intron I sequences and standards of pAAV-F.IX plasmid DNA of known concentration. The ability of AAV-F.IX to transduce cells in vitro was confirmed by transducing growing HeLa cells and measuring the concentration of hF.IX in the culture supernatant 36 hr postinfection with an ELISA specific to hF.IX (14). AAV-F.IX (1012–1013 genomes/ml) was stored at −79°C in Hepes-buffered saline, pH 7.8, including 5% glycerol.
Purified AAV-F.IX routinely lacked detectable amounts of contaminating adenovirus when analyzed by transduction of 293 cells followed by staining for alkaline phosphatase or β-galactosidase as described by Fisher et al. (7). Wild-type AAV was detected at <1 infectious unit per 109 genomes of AAV-F.IX. The assay for wild-type AAV was as described elsewhere (10).
Animal Experiments.
Mouse strains selected for intramuscular injection with rAAV were C57BL/6 (Charles River Breeding Laboratories) and B6, 129, Rag 1 (The Jackson Laboratory). Female mice (4–6 weeks old) were anesthetized with an intraperitoneal injection of ketamine (70 mg/kg) and xylazine (10 mg/kg), and a 1-cm longitudinal incision was made in the lower extremity. AAV-F.IX (2 × 1011 or 1 × 1010 vector genomes per animal in Hepes-buffered saline, pH 7.8) was injected into the tibialis anterior (25 μl) and the quadriceps muscle (50 μl) of each leg using a Hamilton syringe. Incisions were closed with 4–0 Vicryl suture. Blood samples were collected at 7-day intervals from the retro-orbital plexus in microhematocrit capillary tubes and plasma assayed for hF.IX by ELISA (vide infra). For immunofluoresence staining and DNA analysis, animals were sacrificed at selected time points, and injected and noninjected muscles were excised (as well as other tissues). Tissue was placed in OCT embedding compound, snap-frozen in liquid nitrogen-cooled isopentane for 7 sec, and immediately transferred to liquid nitrogen.
Assays for hF.IX.
Human F.IX antigen in mouse plasma was determined by ELISA as described by Walter et al. (14). This ELISA did not crossreact with mouse F.IX. All samples were measured in duplicate. Protein extracts from injected mouse muscle were prepared by maceration of muscle in PBS containing leupeptin (0.5 mg/ml) followed by sonication. Cell debris was removed by microcentrifugation, and 1:10 dilutions of the protein extracts were assayed for hF.IX by ELISA. Extracts from AAV-lacZ-injected muscle were used as negative controls. Protein concentrations were determined with the Bio-Rad assay (Bio-Rad).
Immunofluorescence Staining of Tissue Sections.
Cryosections of muscle tissue (6 μm) were fixed for 15 min in 3% paraformaldehyde in PBS, pH 7.4, rinsed in PBS for 5 min, incubated in methanol for 10 min, washed three times in PBS, and then blocked in PBS/3% BSA for 1 hr. Sections were subsequently incubated overnight with an affinity-purified goat anti-hF.IX antibody (Affinity Biologicals, Hamilton, Ontario, Canada) that was diluted 1:1,000 in PBS/1% BSA. After three washes (10 min each) in PBS/1% BSA, the secondary antibody was applied for 90 min [fluorescein isothiocyanate (FITC)-conjugated rabbit anti-goat IgG (Dako) diluted 1:200 in PBS/1% BSA]. After three additional washes sections were rinsed in distilled water, air-dried, and mounted with Fluoromount G (Fisher Scientific). All incubation steps were at room temperature, except for incubation with the primary antibody (4°C). The same protocol was applied when sections were stained with rabbit anti-human collagen IV as primary antibody (Chemicon) in a 1:500 dilution and FITC-conjugated anti-rabbit IgG (Dako) as secondary antibody. For colocalization studies, a goat anti-hF.IX antibody conjugated to FITC (Affinity Biologicals) was applied simultaneously with the anticollagen IV antibody, and rhodamine-conjugate anti-rabbit IgG (Chemicon) was used to detect collagen IV-antibody complexes. Fluorescence microscopy was performed with a Nikon FXA microscope.
Tests for Circulating Antibody Against hF.IX.
Plasma samples of C57BL/6 mice intramuscularly injected with AAV-F.IX were tested for the presence of antibodies against hF.IX using an ELISA. Microtiter plates were coated with hF.IX (1 μg/ml in 0.1 M NaHCO3, pH 9.2). Dilute plasma samples (1:16) were applied in duplicate, and antibodies against hF.IX detected with horseradish peroxidase-conjugated anti-mouse IgG (Zymed) in a dilution of 1:2,000. Buffer conditions were as described (14). Anti-hF.IX levels were estimated by comparison of absorbance values with monoclonal mouse anti-hF.IX (Boehringer Mannheim) diluted to a final concentration of 1 μg/ml. Negative controls included noninjected and AAV-lacZ-injected mice.
Western blots to demonstrate the presence of anti-hF.IX were performed as outlined by Dai et al. (1), except that a horseradish peroxidase-conjugated goat anti-mouse IgG antibody (Boehringer Mannheim) was used as secondary antibody, thereby allowing the detection of hF.IX-antibody complexes with ECL reagent (Amersham). Samples of mouse plasma were diluted 1:500.
DNA Analyses.
Total genomic DNA was isolated from muscle, liver, spleen, and heart as described for mammalian tissue by Sambrook et al. (15). PCR were carried out as previously described to amplify head-to-tail junctions of rAAV tandem repeats (10). The forward primer 005 (5′-ATAAGCTGCAATAAACAAGT-3′) anneals to the simian virus 40 polyadenylylation signal (bp position 8014–8033), and reverse primers 013 (5′-CATGGTAATAGCGATGACTA-3′) and 017 (5′-GCTCTGCTTATATAGACCTC-3′) bind to the CMV promoter (bp position 4625–4606 and 4828–4809) (see Fig. 5A). PCR products were cloned (for DNA sequence analysis) using the T/A cloning kit (Invitrogen). Southern blot hybridizations were performed using [32P]dCTP random primed labeled probes specific for the CMV promoter (for hybridization to PCR fragments) or for intron I of hF.IX as present in AAV-F.IX (for hybridization to genomic mouse DNA).
Figure 5.
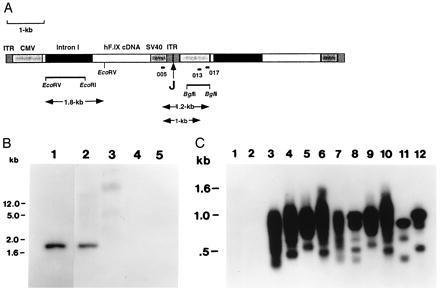
Analyses of DNA isolated from muscle injected with AAV-F.IX. (A) Diagram showing head-to-tail tandem repeats of two vector genomes of AAV-F.IX. Indicated are AAV inverted terminal repeats (ITR), CMV promoter/enhancer (CMV), hF.IX cDNA including coding sequence, and 228 bp of the 3′ untranslated region, a 1.4-kb portion of intron I, simian virus 40 polyadenylylation signal (SV40), and the junction site of the two genomes (J). A 1.2-kb EcoRV–EcoRI fragment from intron I and a 0.7-kb BglII fragment from the CMV promoter fragment were chosen as probes for Southern blot hybridization. Positions of binding sites for primers 005 (forward primer), 013, and 017 (reverse primers) are also shown. (B) Southern blot hybridization of genomic DNA isolated from muscle of Rag 1 mouse 6 weeks postinjection with AAV-F.IX. A radioactively labeled EcoRV–EcoRI fragment from intron I of hF.IX served as a probe. Lane 1, pAAV-FIX plasmid DNA (50 pg). Lanes 2 and 3, DNA isolated from muscle injected with AAV-F.IX. Lanes 4 and 5, DNA isolated from uninjected animal. Lanes 1, 2, and 4, DNA digested with EcoRV. Lanes 3 and 5, undigested DNA. Fifteen micrograms of genomic DNA per lane (lanes 2–5). DNA was separated on a 1% agarose gel before transfer onto a nylon membrane (Schleicher and Schuell). Size markers in the left margin are based on migration of a commercially available set of markers (GIBCO/BRL). (C) Southern blot hybridization of junction fragments of head-to-tail concatemers of AAV-F.IX amplified by PCR. PCR products amplified from genomic DNA using primer pair 005–013 (odd-numbered lanes) or primer pair 005–017 (even-numbered lanes) are shown. Lanes 1 and 2, Uninjected animal. Lanes 3–6, C57BL/6 mice i.m.-injected with AAV-F.IX. Lanes 7–10, Rag 1 mice i.m.-injected with AAV-F.IX. PCR products were from tibialis anterior (lanes 3, 4, 7, and 8) or quadriceps (lanes 5, 6, 9, and 10) muscle DNA. Lanes 11 and 12, PCR products from cell line 10–3.AV 5, which contains at least two monomer copies of integrated AAV-lacZ arranged head-to-tail (10). Note that the sizes of the major PCR products for lanes 11 and 12 are 0.8 and 1 kb, respectively. PCR products were separated on a 2% agarose gel before blotting onto a nylon membrane. A 0.7-kb BglII fragment from the CMV promoter served as a probe. Genomic muscle DNA had been isolated 6 to 8 weeks postinjection.
RESULTS
Expression of hF.IX in Immunocompetent Mice.
The rAAV vector chosen for in vivo experiments contains the hF.IX cDNA including a portion of intron I under transcriptional control of the CMV immediate early gene promoter/enhancer and simian virus 40 polyadenylylation signal. This expression cassette is flanked by AAV inverted terminal repeat sequences and completely lacks AAV protein coding sequences (see Materials and Methods). After intramuscular injection of AAV-F.IX into immunocompetent C57BL/6 mice, hF.IX was detected either transiently or not at all in the plasma of injected animals (Fig. 1). When the same plasma samples were tested for antibodies against hF.IX, a strong antibody response was seen in all injected animals starting at day 11 postinjection (Fig. 2 A and B and data not shown). High levels of circulating antibody persisted for the duration of the experiment. However, protein extracts from injected muscles (tibialis anterior and quadriceps) from animals sacrificed 1 month postinjection revealed the presence of 1.8–2.1 ng of hF.IX per mg of tissue (40–50 ng of hF.IX per mg of protein). The presence of hF.IX in muscle tissues was confirmed by immunofluorescence studies on tissue sections. Fig. 3 B–D shows the expression of hF.IX in muscle fibers of C57BL/6 mice 3 months postinjection. Note that F.IX is present not only in the muscle fibers themselves, but in the interstitial spaces between the fibers as well, where it appears to accumulate. Interestingly, this staining pattern was identical to that seen with a polyclonal antibody against human collagen IV, which also stained the interstitial space (Fig. 4B). Colocalization of the signals for F.IX and collagen IV was confirmed by simultaneous staining with two different fluorescence labels (Fig. 4C). Collagen IV recently has been identified as a binding protein for hF.IX (16). Factor IX was not detected in uninjected muscle (Fig. 3A) or muscle injected with AAV-lacZ (data not shown).
Figure 1.
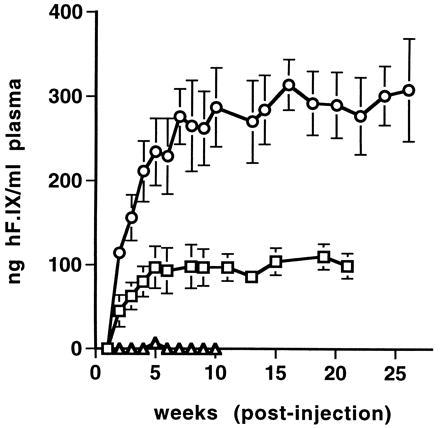
Plasma concentration of hF.IX in experimental mice as a function of time after intramuscular injection with AAV-hF.IX. ▵, C57BL/6 mice after i.m. injection of 2 × 1011 vector genomes/animal. □, Rag 1 mice after i.m. injection of 1 × 1010 vector genomes/animal. ○, Rag 1 mice after i.m. injection of 2 × 1011 vector genomes/animal. Each line represents an average of four animals with error bars denoting SD.
Figure 2.
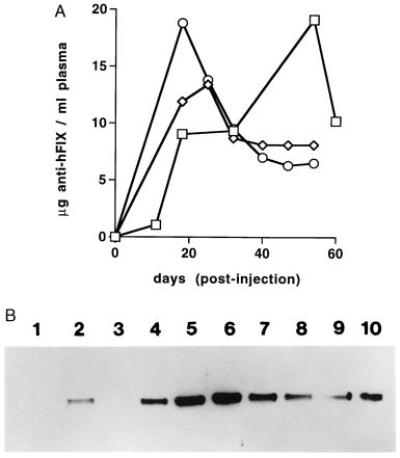
Circulating antibody against hF.IX as a result of i.m. injection of AAV-F.IX in C57BL/6 mice. (A) Time course of anti-hF.IX antibody concentration in plasma after injection with 2 × 1011 vector genomes/animal (n = 3) as determined by ELISA using mouse monoclonal anti-hF.IX (Boehringer Mannheim) as a standard. Each line represents an individual animal. Note that the units on the y axis represent an estimate only, because of probable differences in affinity for hF.IX between the mouse monoclonal used as the standard and the antibodies present in the serum of injected mice. (B) Western blot demonstrating the presence of antibodies against hF.IX in plasma of C57BL/6 mice after i.m. injection. Lane 1, Animal injected i.m. with AAV-lacZ, day 18 postinjection. Lane 2, Animal injected i.m. with rAAV-hF.IX (14), day 20 postinjection. Lanes 3–10, Animals injected i.m. with AAV-F.IX. Lanes 3–7, days 11, 18, 32, 54, and 60 postinjection (same animal). Lanes 8–10, day 18 postinjection (different animals).
Figure 3.
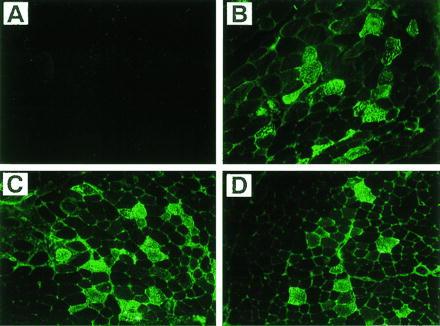
Immunofluorescence staining (with antibody to hF.IX) of tibialis anterior muscle of C57BL/6 mice. (A) Uninjected muscle. (B–D) Muscle 3 months postinjection with AAV-F.IX (3.3 × 1010 vector genomes per injection site). (×200.)
Figure 4.
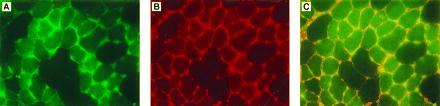
Immunofluorescence staining of muscle sections of tibialia anterior muscle of C57BL/6 mice injected with AAV-F.IX 3 months earlier. Muscle sections were stained simultaneously with FITC-conjugated antibody against hF.IX and a rhodamine-conjugated antibody complex against collagen IV. (A) Fluorescence of FITC (green) showing the presence of hF.IX in muscle fibers and interstitial spaces. (B) Fluorescence of rhodamine (red) showing collagen IV in the extracellular matrix of muscle fibers. (C) Simultaneous excitation of both fluorescence tags. Note the presence of a yellow signal in the interstitial spaces. (×400.)
Inflammation or extensive tissue damage, as described for skeletal muscle injected with recombinant adenovirus (1, 3, 11), was not observed in any of the tissue sections discussed above nor in sections analyzed by hematoxylin/eosin staining (data not shown).
Expression of hF.IX in Immunodeficient Mice.
AAV-F.IX was also delivered to muscles of Rag 1 mice, which are homozygous for a mutation in the recombinase activating gene 1. These animals are therefore functionally equivalent to severe combined immunodeficiency mice and do not produce mature B or T cells. A dose of 2 × 1011 vector genomes per animal resulted in stable expression of hF.IX in mouse plasma (Fig. 1). hF.IX was first detectable by ELISA in the second week after the injection and rose gradually thereafter. Plasma levels reached a plateau 5 to 7 weeks postinjection at 200 to 350 ng of hF.IX per ml of mouse plasma. This level was maintained for the duration of the experiment (6 months postinjection). When a total of 1 × 1010 vector genomes was injected, expression was 3- to 4-fold lower, but still reached therapeutic levels (>100 ng/ml) for some animals (Fig. 1).
Analysis of DNA Introduced into Skeletal Muscle.
Genomic DNA from injected muscle tissue was isolated 6 to 8 weeks postinjection. The plasma level of hF.IX at the time of sacrifice was 225 ng/ml. The presence of introduced vector DNA was demonstrated by digestion with EcoRV, which releases a 1.8-kb fragment from the vector construct including the entire 1.4-kb intron I sequence. A probe specific to intron I hybridized to this fragment (Fig. 5B, lanes 1 and 2) and did not crosshybridize to mouse DNA from an uninjected animal (Fig. 5B lanes 4 and 5). rAAV genome copies per diploid mouse genome can be estimated based on the relative strengths of the signals in lanes 1 and 2, and are ≈0.9 rAAV genomes/diploid mouse genome for injected muscle. Undigested DNA (Fig. 5B, lane 3) showed a hybridization signal in the high molecular mass DNA. Furthermore, PCR primers designed to amplify junction sequences of head-to-tail concatemers of rAAV present in transduced cells (Fig. 5A) successfully amplified such sequences from muscle DNA isolated from AAV-F.IX-transduced tissue (tibialis anterior and quadriceps of immunodeficient and immunocompetent animals). The PCR products were visualized by Southern blot hybridization with a probe specific to the CMV promoter/enhancer (Fig. 5C, lanes 3–10). Primer pair 005–013 produced fragments that were 1.0 kb and smaller; primer pair 005–017 amplified fragments that were 1.2 kb and smaller. As expected, these PCR reactions resulted not in distinct bands of the sizes noted above, but rather in a series of amplification products with a maximum size as predicted, because of imprecise joining of AAV genomes present in these tandem repeats (10, 17). The imprecise joining results from variable deletions of inverted terminal repeat sequences at the junction sites as confirmed by DNA sequencing of cloned PCR products (data not shown). Similar analysis of DNA derived from noninjected muscle, heart, spleen, and liver disclosed no evidence for the presence of AAV-F.IX genomes in these tissues (data not shown).
DISCUSSION
The establishment of an experimental basis for gene therapy of hemophilia B has been hindered by low levels of transgene expression when retroviral vectors are used (18) or by transient expression due to an immune response to the viral vector in the case of adenovirus (1, 14, 19). Recent studies have demonstrated that rAAV vectors can direct persistent expression of reporter genes in mucle fibers of immunocompetent animals (10–12). Why efficient transduction is achieved in these postmitotic cells is unclear. Nevertheless, we sought to exploit this observation to determine whether therapeutic levels of F.IX could be achieved in the circulation of experimental animals after intramuscular injection of a rAAV vector expressing hF.IX. Our studies document that it is possible to achieve therapeutic levels of F.IX using this strategy.
Initial experiments in immunocompetent mice demonstrated that, despite high levels of gene transfer and stable expression of F.IX in injected tissue, it was not possible to detect significant amounts of hF.IX in the circulation, because the animals had developed high-titer antibodies against the circulating foreign protein. [This finding contrasted sharply with our previously reported experience (14), and that of others (20) in which intravenous injection of an adenoviral vector expressing hF.IX did not trigger formation of neutralizing antibodies against hF.IX.] When injection of AAV-F.IX was carried out in immunodeficient (Rag 1) mice, all animals achieved therapeutic levels of hF.IX, in the range of 200–350 ng/ml. These levels, which represent 4–7% of normal circulating levels in plasma, are well within a therapeutic range, and demonstrate that the approach is a feasible one from the standpoint of treating hemophilia. The plasma protein levels achieved here are of particular note because they are crucial to the success of this strategy and because they are considerably higher than those noted in a recent report in which erythropoietin was used as the transgene in a rAAV vector [maximal levels of 750 milliunits/ml, which is equivalent to ≈5 ng/ml of erythropoietin using a specific activity of 119,000 units/mg of protein (12)].
The studies presented here provide some novel insights into the biology of F.IX expression in muscle tissue. Kurachi and colleagues (21) have previously carried out detailed characterization of hF.IX expressed in mouse primary myoblasts and have shown based on analysis of purified hF.IX that the secreted material has full biological activity in a one-stage clotting assay (specific activity 95–125% of plasma-dervied F.IX), is properly processed at the N terminus, and has γ-carboxyglutamic acid content (necessary for biological activity) comparable to that of plasma-derived F.IX. An interesting observation reported earlier by these same investigators was that, after transplantation of transduced myoblasts, the plasma levels of hF.IX were only ≈22% of predicted levels (based on in vitro F.IX synthesis rates in the transduced myoblasts). In our studies, muscle sections from injected tissue showed a characteristic staining pattern, with F.IX localized to some extent intracellularly, but also found extensively in the interstitial spaces between muscle fibers. Although one might expect to see some degree of extracellular staining for a secreted protein such as F.IX, we had not anticipated the finding seen here, specifically what appeared to be a concentration of F.IX in the interstitial spaces. A recent report (16) presents compelling evidence that collagen IV acts as a specific receptor for F.IX. The authors demonstrate that hF.IX binds to collagen IV in the extracellular matrix of endothelial cells in a specific and saturable manner, with a Kd in the nanomolar range. We hypothesized that collagen IV in the basement membrane surrounding muscle fibers might explain the staining pattern seen in the F.IX immunofluorescence studies of muscle tissue. Immunostaining with antibodies to human collagen IV confirmed that the interstitial spaces were indeed rich in this protein. This finding is of interest not only as it relates to the staining pattern seen for F.IX, but also because it may at least partially account for the previously reported finding of low efficiency of transfer of muscle cell-synthesized F.IX into the circulation. Clearly, however, the trapping of F.IX in the interstitial spaces by collagen IV, if it occurs, is not an insuperable obstacle, as the experiments in Rag mice demonstrate.
The time course of expression of hF.IX in the plasma of injected Rag 1 mice is of interest; it allows a quantitative assessment of a trend that is apparent in studies carried out with rAAV-lacZ (7, 22). The absence of expression initially, followed by a slow rise in levels of expression, has been attributed to the necessity for, and the slow rate of, conversion of single-stranded AAV genome to high molecular mass double-stranded DNA. The time course for this conversion in transduced muscle fibers has been documented (10). The point at which single-stranded vector DNA is no longer detectable is reached between day 30 and day 64 postinjection. In the experiments described in this report, circulating levels of F.IX reach a plateau between week 5 and week 7 after injection with rAAV-F.IX. Whether the binding of F.IX to collagen IV in the interstices of muscle fibers further slows the rise cannot be assessed from these data; however, the existence of two F.IX variants with altered affinity for collagen IV (F.IX K5A, no binding to collagen IV; F.IX K5R, Kd 6-fold lower than wild-type F.IX) should allow this hypothesis to be tested (16, 23).
A comparison of the time course of expression of F.IX in the Rag 1 mice and the time course of appearance of antibodies in the C57BL/6 mice helps to explain the data in Fig. 1, the time course of expression of F.IX in the C57BL/6 (immunocompetent) mice. In the first week after injection, no F.IX is detected in plasma even in the Rag 1 mice. Levels of protein expression required to induce antibody formation, however, are quite low, and indeed antibodies are first detected by ELISA (see Fig. 2A) 11 days after injection. The Western blot assay is somewhat less sensitive but documents what appears to be a rising antibody titer beginning 18 days after injection. Thus in the immunocompetent animals, the absence of expression at initial time points is a result of the biology of rAAV expression in muscle, whereas the subsequent absence of detectable F.IX results from the production of antibodies to the foreign protein. The data of Kessler et al. (12) suggest that an immune response to a muscle-expressed transgene is avoided if the expressed protein is autologous; this point will require additional exploration.
The PCR studies presented here (Fig. 5C) demonstrate the presence of head-to-tail concatemers of AAV-F.IX DNA. Whereas head-to-head and tail-to-tail arrangements of AAV genomes can occur during viral replication (24), head-to-tail arrays are more typically associated with AAV that has been integrated into the chromosomal DNA of the transduced cell during latent infection (17, 25, 26). Southern blot data on undigested rAAV-injected muscle cell DNA show that the rAAV DNA derived from host cell genomic DNA 6 weeks after injection is present as a high molecular mass species. The higher signal intensity seen with the restricted DNA (Fig. 5B, compare lanes 2 and 3) likely results from an unmasking effect when the fragments are separated from the bulk of genomic DNA (11). This finding, the presence of a hybridization signal for high molecular mass DNA, is, like the PCR data, consistent with integrative events occurring during transduction. Cloning of junction fragments between chromosomal and vector DNA would be necessary to confirm this interpretation. The data are also consistent with the presence of the DNA as a high molecular mass episomal form; this remains a formal possibility.
Detailed dose–response studies have not yet been carried out, but the data presented in this study may at first suggest a nonlinear response, because mice injected with 2 × 1011 vector genomes had plasma hF.IX levels in the range of 200–350 ng/ml, whereas those receiving a 20-fold lower dose still achieved levels in the range of 50–100 ng/ml. Analysis of the immunofluorescence studies provides some insight into this observation, because the overall appearance of the sections stained for transgene expression is similar, whether a lower (1 × 109 genomes per injection site) or higher (3.3 × 1010 genomes per site) dose is injected (data not shown). Transduction is probably limited by the distance the viral vector can diffuse from the injection site; thus an increase in dose without an increase in the number of injection sites will have only a limited effect on plasma levels of the transgene.
The findings presented here have several implications for development of gene therapy protocols for patients with hemophilia B. First, the levels of F.IX expression obtained here are adequate to achieve a therapeutic effect if efficient scale-up of rAAV production can be achieved. Second, it is clear from these data that multiple intramuscular injection sites will be required; because muscle tissue is abundant this is not an obstacle to treatment with this strategy. The time course of expression documented here is quite different from that seen with adenoviral vectors expressing hF.IX, where therapeutic levels are achieved almost immediately (14, 19). In the case of AAV-F.IX, the gradual rise in plasma F.IX levels over a period of weeks means that patients will need to be covered with F.IX concentrates for the period immediately surrounding the invasive intramuscular injections. The t1/2 of the exogenously administered F.IX in humans is only ≈15 hr; thus plasma F.IX levels will return to baseline before they begin to rise from expression of the transgene, facilitating analysis of results. Finally, data in the literature (12) suggest that expression from muscle tissue transduced with rAAV is persistent for long periods if cross-species boundaries are not transgressed. Thus, administration of rAAV vectors expressing hF.IX to patients with hemophilia B should be a feasible strategy for treatment of the disease.
Acknowledgments
The assistance of the Vector Core and of Ravindra Pathak in the Cell Morphology Core of the Institute for Human Gene Therapy at the University of Pennsylvania is gratefully acknowledged. This work was supported by National Institutes of Health Grants R01 HL53668 and P50 HL54500 to K.A.H.
ABBREVIATIONS
- F.IX
blood coagulation factor IX
- hF.IX
human F.IX
- FITC
fluorescein isothiocyanate
- AAV
adeno-associated virus
- rAAV
recombinant AAV
- CMV
cytomegalovirus
References
- 1.Dai Y, Schwartz E M, Gu D, Zhang W W, Sarvetnick N, Verma I M. Proc Natl Acad Sci USA. 1995;92:1401–1405. doi: 10.1073/pnas.92.5.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tripathy S K, Black H B, Goldwasser E, Leiden J M. Nat Med. 1996;2:545–550. doi: 10.1038/nm0596-545. [DOI] [PubMed] [Google Scholar]
- 3.Yang Y, Haecker S E, Su Q, Wilson J M. Hum Mol Genet. 1996;5:1703–1712. doi: 10.1093/hmg/5.11.1703. [DOI] [PubMed] [Google Scholar]
- 4.Tripathy S K, Svensson E C, Black H G, Goldwasser E, Margalith M, Hobart P M, Leiden J M. Proc Natl Acad Sci USA. 1996;93:10876–10880. doi: 10.1073/pnas.93.20.10876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carter B J. Curr Opin Biotech. 1992;3:533–539. doi: 10.1016/0958-1669(92)90082-t. [DOI] [PubMed] [Google Scholar]
- 6.Skulimowski A W, Samulski R J. Methods Mol Genet. 1995;7:7–12. [Google Scholar]
- 7.Fisher K J, Gao G-P, Weitzman M D, DeMatteo R, Burda J F, Wilson J F. J Virol. 1996;70:520–532. doi: 10.1128/jvi.70.1.520-532.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Samulski R J, Chang L-S, Shenk T. J Virol. 1989;63:3822–3828. doi: 10.1128/jvi.63.9.3822-3828.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaplitt M G, Leone P, Samulski R J, Xiao X, Pfaff D W, O’Malley K L, During M J. Nat Genet. 1994;8:148–154. doi: 10.1038/ng1094-148. [DOI] [PubMed] [Google Scholar]
- 10.Fisher K J, Jooss K, Alston J, Yang Y, Ehlen-Haecker S, High K, Pathak R, Raper S E, Wilson J M. Nat Med. 1997;3:306–312. doi: 10.1038/nm0397-306. [DOI] [PubMed] [Google Scholar]
- 11.Xiao X, Li J, Samulski R J. J Virol. 1996;70:8098–8108. doi: 10.1128/jvi.70.11.8098-8108.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kessler P D, Podsakoff G M, Chen X, McQuston S A, Colosi P C, Matelis L A, Kurtzman G J, Byrne B J. Proc Natl Acad Sci USA. 1996;93:14082–14087. doi: 10.1073/pnas.93.24.14082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurachi S, Hitomi Y, Furukawa M, Kurachi K. J Biol Chem. 1995;270:5276–5281. doi: 10.1074/jbc.270.10.5276. [DOI] [PubMed] [Google Scholar]
- 14.Walter J, You Q, Hagstrom N, Sands M, High K A. Proc Natl Acad Sci USA. 1996;93:3056–3061. doi: 10.1073/pnas.93.7.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. pp. 9-16–9-19. [Google Scholar]
- 16.Cheung W-F, Born J v d, Kühn K, Kjelléen L, Hudson B G, Stafford D W. Proc Natl Acad Sci USA. 1996;93:11068–11073. doi: 10.1073/pnas.93.20.11068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McLaughlin S K, Collins P, Hermonat P L, Muzyczka N. J Virol. 1988;62:1963–1973. doi: 10.1128/jvi.62.6.1963-1973.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kay M A, Rothenberg S, Landen C N, Bellinger D A, Leland F, Toman C, Finegold M, Thompson A R, Read M S, Brinkhous K M, Woo S L C. Science. 1993;262:117–119. doi: 10.1126/science.8211118. [DOI] [PubMed] [Google Scholar]
- 19.Kay M A, Landen C N, Rothenberg S R, Taylor L A, Leland F, Wiehle S, Fang B, Bellinger D, Finegold M, Thompson A R, Read M, Brinkhous K M, Woo S L C. Proc Natl Acad Sci USA. 1994;91:2353–2357. doi: 10.1073/pnas.91.6.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith T A G, Mehaffey M G, Kayda D B, Saunders J M, Yei S, Trapnell B C, McClelland A, Kaleko M. Nat Genet. 1993;5:397–402. doi: 10.1038/ng1293-397. [DOI] [PubMed] [Google Scholar]
- 21.Yao S-N, Smith K J, Kurachi K. Gene Ther. 1994;1:99–107. [PubMed] [Google Scholar]
- 22.Ferrari F K, Samulski T, Shenk T, Samulski R J. J Virol. 1996;70:3227–3234. doi: 10.1128/jvi.70.5.3227-3234.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheung W-F, Hamaguchi N, Smith K J, Stafford D W. J Biol Chem. 1992;267:20529–20531. [PubMed] [Google Scholar]
- 24.Berns K I. Microbiol Rev. 1990;54:316–329. doi: 10.1128/mr.54.3.316-329.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tratschin J D, Miller I L, Smith M G, Carter B J. Mol Cell Biol. 1985;5:3251–3260. doi: 10.1128/mcb.5.11.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muzyczka N. Curr Top Microbiol Immunol. 1992;158:97–129. doi: 10.1007/978-3-642-75608-5_5. [DOI] [PubMed] [Google Scholar]