

Abstract
In view of evidence that growth hormone (GH) and insulin-like growth factors (IGF) may play a role in the development of renal cell carcinoma (RCC), we investigated the effects of growth hormone-releasing hormone (GH-RH) antagonist MZ-4-71 on the proliferation of the human renal adenocarcinoma cell line Caki-I in vitro and in vivo. Male nude mice bearing xenografts of human Caki-I RCC were treated for 4 weeks with MZ-4-71 injected s.c. twice daily at a dose of 20 μg per animal. Tumor growth, serum, liver, and tumor IGF levels and IGF-I receptor concentrations in Caki-I cell membranes were measured. After 4 weeks of therapy, the final volume of Caki-I tumors in nude mice treated with MZ-4-71 was significantly (P < 0.01) decreased to 52.6 ± 12.3 mm3 as compared with controls that measured 504.2 ± 104.1 mm3. Treatment with GH-RH antagonist also significantly reduced tumor weight, serum levels of GH and IGF-I, liver concentrations of IGF-I, and tumor levels of IGF-I and IGF-II. High-affinity binding sites for IGF-I were detected in the cell membranes of Caki-I tumors. IGF-I and IGF-II stimulated the proliferation of Caki-I cells in tissue cultures. Antagonist MZ-4-71 could inhibit in vitro growth of Caki-I cells, but only at high concentrations. Our findings demonstrate that GH-RH antagonist MZ-4-71 can significantly inhibit the growth of Caki-I RCC. MZ-4-71 may exert its suppressive effect on tumor growth through a reduction in GH release from the pituitary and the subsequent decrease in the production of IGF-I in the liver and IGF-I and II by the tumors. The efficacy of MZ-4-71 suggests that this compound could be considered for the therapy of recurrent or metastatic RCC.
Keywords: kidney neoplasms, renal cell carcinoma, insulin-like growth factor
Renal cell carcinoma (RCC) is the most common renal tumor, and in 1996 approximately 30,000 patients in the United States are expected to be diagnosed with this neoplasm, with an estimated 12,000 deaths (1). About 30% of patients with RCC have evidence of metastatic disease at initial presentation, and the 2-year survival rate in this group is poor, ranging from 10% to 20% (2). For the advanced stages of RCC, no effective therapy is available. Cytotoxic chemotherapy shows marginal efficacy at best and fails to show statistically significant survival advantages for responding patients (3). Radiotherapy has proven ineffective for the treatment of local recurrent or metastatic disease (4). Immunotherapy, with the administration of interferons and interleukins, have shown some promising results, but is still in experimental phases (4).
The mechanisms responsible for tumor growth in RCC are still unclear. However, there is good evidence that hormones like growth hormone (GH) and somatostatin, and growth factors such as insulin-like growth factor (IGF)-I, epidermal growth factor, transforming growth factor-α and -β, angiogenin, and tumor necrosis factor-α might have direct or indirect regulatory effects on the function and growth of this carcinoma (4–7). Kellerer et al. (8) have shown that the number of IGF-I binding sites is doubled in kidney tumor tissue, with a significantly increased IGF-I receptor autophosphorylation by an augmented tyrosine-kinase activity.
The Caki-I cell line was established in 1971 and derived from a human renal adenocarcinoma metastatic to the skin (9). It has been shown that IgG1 mAbs against this cell line crossreact with antigens present in most human RCCs, but not with any normal adult kidney tissue (10). Caki-I cell line can be xenografted into nude mice, forming poorly differentiated G3 tumors (11). Caki-I cells express c-myc oncogene, which is known to stimulate epidermal growth factor receptor expression and the multidrug-resistant phenotype (gp170) (12). This cell line is susceptible to cytokines and interferons and shows many characteristics of human RCC.
Various potent GH-releasing hormone (GH-RH) antagonists have been synthesized in our laboratory, including [isobutyryl0, d-arginyl2, para-chlorophenylalanyl6, α-aminobutyryl15, norleucyl27]hGH-RH(1–28)Agm (MZ-4-71) (13). This antagonist has been previously tested for its effects on human osteosarcomas and human small and nonsmall cell lung cancers in athymic nude mice (14, 15). In the present study we evaluated the effect of the GH-RH antagonist MZ-4-71 on the growth of Caki-I RCC cell line xenografted into nude mice. The effects of IGF-I and GH-RH antagonist MZ-4-71 on the proliferation of Caki-I cells in vitro were also investigated.
MATERIALS AND METHODS
Peptide and Reagents.
GH-RH antagonist MZ-4-71, [isobutyryl0, d-arginyl2, para-chlorophenylalanyl6, α-aminobutyryl15, norleucyl27]hGH-RH(1–28)Agm, was synthesized by solid phase methods and purified in our laboratory (13). For daily injections, MZ-4-71 was dissolved in 0.1% dimethyl sulfoxide in 10% propylene glycol/saline solution (Sigma).
Animals.
Male athymic (NCr nu/nu) nude mice, approximately 5 weeks old on arrival, were obtained from the National Cancer Institute (Bethesda, MD) and housed in a laminar airflow cabinet under pathogen-free conditions with a 12-hr light/12-hr dark schedule and fed autoclaved standard chow and water ad libitum throughout the experiment. Their care was in accord with institutional guidelines.
Cell Culture.
The human renal adenocarcinoma cell line Caki-I was obtained from the American Type Culture Collection and cultured in McCoy’s 5A medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 100 units/ml sodium penicillin-G, 100 μg/ml streptomycin sulfate, and 0.25 μg/ml amphotericin B (all from GIBCO), at 37°C in a humidified 95% air/5% CO2 atmosphere. Tumor cells growing exponentially were harvested by a brief incubation with 0.25% trypsin-EDTA solution (GIBCO). Xenografts were initiated by s.c. injection of 1 × 107 cells into the left flanks of five male nude mice.
Measurement of Cell Growth in Vitro by the [3H]Thymidine-Incorporation Assay.
Incorporation of [methyl-3H]thymidine into DNA (14) was used to determine the effect of MZ-4-71, hGH(1–43) (Bachem), IGF-I (GIBCO/BRL), and IGF-II (Bachem) on the Caki-1 cell line in vitro. Cells were seeded into 96-well microplates (Falcon) in culture medium cited above. After 3–4 days, the confluent cultures were washed with McCoy’s 5A medium and maintained in McCoy’s 5A medium containing 2% fetal bovine serum for 24–30 hr. Several concentrations of each compound diluted with media were then added to the quiescent cultures. Control cultures received the media without addition (14). After a 20-hr incubation, all cultures were pulsed with 0.25 μCi per well of [methyl-3H]thymidine (specific activity 25 Ci/mmol; Amersham) in a total volume of 175 μl per well for 4–6 hr. The cells were fixed with ice-cold 10% trichloroacetic acid (TCA), washed twice with TCA at 4°C, and solubilized overnight in 0.2M NaOH at 37°C. Radioactivity was determined by liquid scintillation counting (Searle Analytic Model 6880). Inhibition or stimulation of growth as compared with controls was expressed using the formula
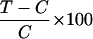 |
where T = average dpm of test cultures and C = average dpm of control cultures.
Experimental Protocol.
Caki-I tumors resulting after 4 weeks were aseptically dissected and mechanically minced; 3-mm3 pieces of tumor tissue were transplanted s.c. by trocar needle into 25 male animals. The tumor take rate was 95%. Two weeks after transplantation, tumor had grown to a volume between 20 and 35 mm3. The tumor-bearing mice were then divided into two experimental groups of 10 animals each, which received the following treatments: group 1 (control), s.c. injection of saline containing 0.1% dimethyl sulfoxide; group 2, MZ-4-71 at a dose of 20 μg/twice daily s.c. The treatment was continued for 4 weeks. The tumors were measured once a week with microcalipers, and the tumor volume was calculated as length × width × height × 0.5236 (14). Tumor volume doubling time was calculated between the start and the end of the experiment. At the end of the treatment period, mice were anesthetized with methoxyflurane (Metofane; Pitman–Moore, Mundelein, IL), killed by decapitation, and trunk blood was collected. The serum was separated for hormone analyses. Body weights were recorded, and various organs were removed and weighed. Tumors were cleaned and weighed, and samples were taken for IGF measurements and receptor studies.
Method of Tissue Extraction.
Tumor and liver tissue concentrations of IGF-I and IGF-II were determined by an adaptation of the methods described previously (14, 16). The tissue was homogenized and centrifuged at 2,000 × g for 20 min at 4°C. The supernatants were combined, lyophilized, and reconstituted in 0.1 M phosphate buffer, pH 7.6. The Bio-Rad protein assay kit was used for protein determination.
Radioimmunoassays of GH, IGF-I, and IGF-II.
GH was determined by using materials provided by A. F. Parlow (Pituitary Hormones and Antisera Center, Torrance, CA; mouse GH reference preparation AFP10783B, mouse GH antigen AFP10783B, and anti-rat GH-RIA-5/AFP-411S). All serum and reconstituted tissue samples for IGF-I and IGF-II determination were extracted by a modified acid-ethanol method described earlier (17, 18). The extracted IGF-I was measured by RIA using IGF-I (88-G4;, Genentech) as a standard in the range of 2–500 pg/tube and also for iodination by the chloramine-T method. Antibodies UB3–189 and UB2–495 (a gift from L. E. Underwood and J. van Wyk, University of North Carolina, Chapel Hill) obtained from the National Institute of Diabetes and Digestive and Kidney Diseases were used at the final dilution of 1:10,000 and 1:14,000, respectively in the RIA.
IGF-II was measured using human recombinant IGF-II (Bachem) in the range of 2–500 pg/tube. IGF-II was iodinated by lactoperoxidase method and purified by reverse-phase HPLC using Vydac C18 column. For the assay, Amano mAb generated against rat IGF-II (10 μg/ml) was used at the final dilution of 1:14,285 (Amano International Enzymes, Troy, VA). This antibody crossreacts 100% with human IGF-II and rat IGF-II.
Receptor Assay.
Measurement of receptors for IGF-I in the membranes of Caki-I tumors was performed as previously described (14). The ligand PC computerized curve-fitting program of Munson and Rodbard (19) was used to determine the types of receptor binding, dissociation constant (Kd) values, and the maximal binding capacity (Bmax) of receptors.
Statistical Methods.
All data are expressed as the mean ± SEM, and statistical analyses of the tumor data were performed using Duncan’s new multiple range test (20). All P values are based on two-sided hypothesis testing.
RESULTS
The effects of treatment with GH-RH antagonist MZ-4-71 on final tumor volume, body, and tumor weights, and tumor doubling time are shown in Table 1. After 4 weeks of therapy, when the experiment was terminated, there were no significant differences in body weights between untreated and treated animals (Table 1). Fig. 1 shows the tumor volume as measured at weekly intervals. Mean tumor volume and weight were significantly (P < 0.01) reduced in animals receiving GH-RH antagonist MZ-4-71 to 52.6 ± 12.3 mm3 and 52 ± 18 mg, as compared with the control group (504.2 ± 104.1 mm3 and 357 ± 118 mg), corresponding to a 89% and 85% decrease in tumor volume and weight, respectively. Tumor doubling time in mice receiving MZ-4-71 was extended to 59.9 ± 29.6 days from 7.7 ± 0.6 days calculated for control animals (Table 1).
Table 1.
Effect of treatment with GH-RH antagonist MZ-4-71 on body and tumor weight, tumor volume, and tumor doubling time in nude mice bearing xenografts of the human kidney adenocarcinoma cell line Caki-I
| Treatment group | Tumor volume, mm3
|
Body weight, g | Tumor weight, mg | Tumor doubling time, days | |
|---|---|---|---|---|---|
| Initial | Final | ||||
| Control | 26.5 ± 3.1 | 504.2 ± 104.1 | 29.1 ± 0.7 | 357 ± 118 | 7.7 ± 0.6 |
| MZ-4-71 | 24.8 ± 0.9 | 52.6 ± 12.3** | 30.1 ± 0.8 | 52 ± 18* | 59.9 ± 29.6* |
Values are mean ± SEM; ∗, P < 0.05 vs. control; ∗∗, P < 0.01 vs. control.
Figure 1.
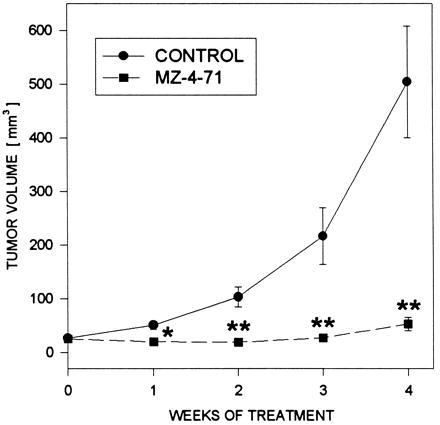
Tumor volumes in athymic nude mice bearing subcutaneously xenografted Caki-I carcinomas during treatment with the GH-RH antagonist MZ-4-71 administered at a dose of 20 μg B.I.D. per animal, s.c. Treatment was started when the tumors measured approximately 24–27 mm3 and lasted for 4 weeks. Vertical bars represent standard error, ∗, P < 0.5 versus control, ∗∗, P < 0.01 versus control.
Effect of MZ-4-71 on Serum and Tissue IGF-I, IGF-II, and GH Levels.
The serum levels of GH, IGF-I, and IGF-II in controls and in nude mice treated with the peptide analog are shown in Table 2. In Caki-I tumor bearing mice, serum GH levels were significantly reduced in the group treated with MZ-4-71 to 1.29 ± 0.06 ng/ml, as compared with control animals (1.9 ± 0.15 ng/ml). The GH-RH antagonist significantly (P < 0.05) reduced serum IGF-I levels to 94.8 ± 7.9 ng/ml as compared with 127.7 ± 10.7 ng/ml for controls, whereas serum IGF-II levels were similar in both groups (Table 2). Control tumor tissue showed detectable amounts of IGF-I and IGF-II (726 ± 126 pg/100 μg protein and 4,580 ± 628.1 pg/100 μg protein, respectively). In tumor tissues from animals treated with MZ-4-71, levels of IGF-I and IGF-II were significantly (P < 0.05) decreased to 362.5 ± 4.5 and 2,438.5 ± 103.5 pg/100 μg protein, respectively (Table 3). After therapy with MZ-4-71, levels of IGF-I in liver tissue of nude mice bearing Caki-I tumors were significantly reduced from 135.2 ± 10.4 to 97.0 ± 6.6 pg/100 μg protein (P < 0.01) (Table 3).
Table 2.
Serum GH, IGF-I, and IGF-II in nude mice bearing Caki-I human kidney cancer cell line xenografts after treatment with GH-RH antagonist MZ-4-71
| Treatment group | GH, ng/ml | IGF-I, ng/ml | IGF-II, ng/ml |
|---|---|---|---|
| Control | 1.90 ± 0.15 | 127.7 ± 10.7 | 25.1 ± 2.0 |
| MZ-4-71 | 1.29 ± 0.06* | 94.8 ± 7.9* | 26.2 ± 3.4 |
Values are mean ± SEM; ∗, P < 0.05 vs. control
Table 3.
Tumor and liver IGF-I and IGF-II levels in athymic nude mice bearing Caki-I xenografts after treatment with the GH-RH antagonist MZ-4-71
| Treatment group | Tumor
|
Liver
|
|
|---|---|---|---|
| IGF-I | IGF-II, pg/100 μg protein | IGF-I, pg/100 μg protein | |
| Control | 726 ± 126 | 4580 ± 628.1 | 135.2 ± 10.4 |
| MZ-4-71 | 362.5 ± 45* | 2,438.5 ± 103.5* | 97.0 ± 6.6* |
All values are means ± SEM. ∗, P < 0.05 versus control.
Effect of Treatment with GH-RH Antagonist MZ-4-71 on IGF-I Receptors.
The binding characteristics of receptors for IGF-I in the membranes of Caki-I tumors were analyzed after treatment with GH-RH antagonist MZ-4-71, and the results are presented in Table 4. High-affinity binding sites for IGF-I were present in the membranes of Caki-I tumors (Kd = 0.89 ± 0.08 nm). A significant (P < 0.05) decrease in binding capacity of IGF-I receptors was observed after treatment with MZ-4–71 (Bmax = 71.5 ± 8.85 fmol/mg protein vs. Bmax = 126.3 ± 5.9 fmol/mg protein for control) (Table 4).
Table 4.
Binding characteristics of receptors for IGF-I in membranes of Caki-I renal adenocarcinomas after in vivo treatment with GH-RH antagonist MZ-4-71
| Treatment group | Kd, nM | Bmax, fmol/ mg protein |
|---|---|---|
| Control | 0.89 ± 0.08 | 126.3 ± 5.9 |
| MZ-4-71 | 0.49 ± 0.14 | 71.5 ± 8.85* |
Binding characteristics were obtained from 10-point displacement experiments. Significance was calculated with Duncan’s new multiple range test. All values represent mean ± SEM of 2 or 3 experiments, each done in triplicate. ∗, P < 0.05 vs. control.
Effects of IGF-I, IGF-II, GH, and GH-RH Antagonist MZ-4-71 on Cell Proliferation in Vitro.
To evaluate the effects of IGF-I, IGF-II, GH, and GH-RH antagonist MZ-4-71 on the Caki-I cell line in vitro, the [3H]thymidine uptake assay was used. IGF-I at concentrations of 10 and 20 ng/ml stimulated significantly the proliferation of this cell line by 66% and 101%, respectively. IGF-II at concentrations of 50 and 90 ng/ml increased the [3H]thymidine uptake by 209% and 302%, respectively. The addition of GH to the culture medium (1 to 25 ng/ml) had no stimulatory effect on Caki-I cell proliferation. At a concentration of 42.3 μg/ml (10−5 M), GH-RH antagonist MZ-4-71 decreased the proliferation of Caki-I cells by 59%, as compared with the controls. However, lower concentrations of the antagonist did not affect the growth of Caki-I cells in vitro (data not shown).
DISCUSSION
Caki-I is a valuable human renal adenocarcinoma cell line that can be xenografted into nude mice. Our work shows that GH-RH antagonist MZ-4-71 effectively inhibits growth of Caki-I RCC in nude mice and prolongs tumor doubling time by more than 700%. Previously, MZ-4-71 was shown to inhibit tumor growth of human osteosarcomas (14) and human small-cell and non-small-cell lung carcinomas (15).
GH receptors have been identified in a large number of tissues, among them kidney and RCC (21). Treatment with MZ-4-71 significantly reduced serum GH levels in nude mice bearing Caki-I carcinomas. These findings suggest that some of the inhibitory effect of the GH-RH antagonist could be attributed to the reduced serum GH concentration. GH directly regulates the IGF-I synthesis in the liver, the kidney, and other organs (22, 23). A significant fall in GH levels, induced by the GH-RH antagonists, could, through mechanisms involving suppression of IGFs, be of major importance for the inhibition of tumor growth. In nude mice treated with the GH-RH antagonist, serum IGF-I levels were significantly lower than in control animals.
In our study, IGF-I also was measured in liver tissue of treated and untreated Caki-I tumor-bearing animals. Liver IGF-I levels were significantly reduced in animals after chronic administration of GH-RH antagonist MZ-4-71. This observation is in accord with many previous studies that indicated that IGF-I synthesis depends on GH stimulation (14, 22, 23). We also recorded for the first time, to our knowledge, high tissue concentrations of IGF-I and IGF-II in Caki-I tumors of untreated animals. This new observation suggests that an autocrine or paracrine production of IGF-I and IGF-II might be involved in the stimulation of cell proliferation of Caki-I tumor cells. These high tissue concentrations of IGF-I and IGF-II were significantly decreased after treatment with the GH-RH antagonist. Thus, the mechanism of action of MZ-4–71 on Caki-I RCC might be based on the suppression of the endocrine, autocrine, or paracrine production of IGF-I and II. Inhibition of a direct stimulatory effect of endogenous GH on autocrine secretion of IGF-I and IGF-II by Caki-I cells also might be involved. Irrespective of the exact mechanism, our findings are in agreement with previous studies on osteosarcoma and lung cancers in nude mice (14, 15).
The presence of high-affinity binding sites for IGF-I in membranes of Caki-I RCC was detected. Chronic treatment of nude mice with MZ-4-71 significantly decreased the concentration of IGF-I receptors of Caki-I tumor membranes. IGF-I and IGF-II bind with different affinities to IGF receptors, which are thought to mediate the biological effects of both ligands through tyrosine kinase-type activity (24). However, it is more likely that the inhibition of tumor growth in MZ-4–71-treated animals was due to the reduction of IGF-I and IGF-II synthesis rather than to the decrease of IGF-I receptors.
Our in vitro experiments show for the first time, to our knowledge, that addition of IGF-I or IGF-II, but not GH, to the culture medium resulted in a stimulation of proliferation of the Caki-I cell line. This reinforces the view that the growth of this RCC model depends on IGFs. A statistically significant inhibition of proliferation of Caki-I cells was obtained with 10−5 M MZ-4-71, but the proliferation of this cell line was not decreased at lower concentrations of the antagonist. This result points to a non-GH-RH specific phenomenon. GH-RH is a member of the glucagon-secretin family of peptides, which demonstrate considerable animo acid sequence homology (25, 26). These peptides bind to common receptor proteins, such as that for vasoactive intestinal peptide, and produce similar biological responses in most of their target organs (27, 28). Thus, GH-RH antagonists might bind with reduced affinity to one of these receptors.
Our study shows that GH-RH antagonist MZ-4-71 effectively inhibits growth of Caki-I RCC for at least 4 weeks. This effect was linked to a reduction in serum GH and IGF-I levels, IGF-I concentration in liver tissue and IGF-I and IGF-II levels in tumors. Examination of tumor growth curve suggests the possibility that the growth inhibitory effects of GH-RH antagonists could diminish in time and the tumors might start growing faster with a decreased tumor doubling time. These results should be extended using other RCC cell lines and surgical specimens of RCC. Our findings support the merit of further investigations aimed at evaluating the possible use of GH-RH antagonists for the palliative therapy of advanced RCC.
Acknowledgments
We thank Dora Rigo, Elena Glotser, and Harold Valerio for their technical assistance and Prof. Julian Frick for his support. The gifts of materials used in RIA from the National Hormone and Pituitary Program are greatly appreciated. This work was supported by a grant from Pierre Fabre Medicament to Tulane University School of Medicine and by the Medical Research Service of the Veterans Affairs Department (A.V.S.). A.J. is a recipient of a fellowship from the Fond zur Förderung der wissenschaftlichen Forschung, Austria.
ABBREVIATIONS
- GH
growth hormone
- GH-RH
GH-releasing hormone
- RCC
renal cell carcinoma
- IGF
insulin-like growth factor
References
- 1.Parker S L, Tong T, Bolden S, Wingo P A. Ca Cancer J Clin. 1996;44:5–27. doi: 10.3322/canjclin.46.1.5. [DOI] [PubMed] [Google Scholar]
- 2.Libertino J A. J Urol. 1996;155:446–447. doi: 10.1016/s0022-5347(01)66419-x. [DOI] [PubMed] [Google Scholar]
- 3.Yagoda A, Petrylak D, Thompson S. Urol Clin North Am. 1993;20:303–321. [PubMed] [Google Scholar]
- 4.deKernion J B, Belldegrun A. In: Campbell’s Urology. Walsh P C, Retik A B, Stamey T A, Vaughan E D Jr, editors. Vol. 2. Philadelphia: Saunders; 1992. pp. 1053–1093. [Google Scholar]
- 5.Reubi J C, Kvols L. Cancer Res. 1992;52:6074–6078. [PubMed] [Google Scholar]
- 6.Pekonen F, Partanen S, Rutanen E M. Int J Cancer. 1989;43:1029–1033. doi: 10.1002/ijc.2910430612. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi A, Sasaki H, Kim S J, Tobisu K-I, Kakizoe T, Tsukamoto T, Kumamoto Y, Sugimura T, Terada M. Cancer Res. 1994;54:4233–4237. [PubMed] [Google Scholar]
- 8.Kellerer M, von Eye Corleta H, Muehlhoefer A, Capp E, Mosthaf L, Bock S, Petrides P E, Haering H U. Int J Cancer. 1995;62:501–507. doi: 10.1002/ijc.2910620502. [DOI] [PubMed] [Google Scholar]
- 9.Fogh J, Wright W C, Loveless J D. J Natl Cancer Inst. 1977;58:209–214. doi: 10.1093/jnci/58.2.209. [DOI] [PubMed] [Google Scholar]
- 10.Luner S J, Ghose T, Chatterjee S, Cruz H N, Belitsky P. Cancer Res. 1986;46:5816–5820. [PubMed] [Google Scholar]
- 11.Korhonen M, Sariola H, Gould V E, Kangas L, Virtanen I. Cancer Res. 1994;54:4532–4538. [PubMed] [Google Scholar]
- 12.Mizutani Y, Bonavida B, Koishihara Y, Akamatsu K-I, Ohsugi Y, Yoshida O. Cancer Res. 1995;55:590–596. [PubMed] [Google Scholar]
- 13.Zarandi M, Horvath J E, Halmos G, Pinski J, Nagy A, Groot K, Schally A V. Proc Natl Acad Sci USA. 1994;91:12298–12302. doi: 10.1073/pnas.91.25.12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pinski J, Schally A V, Groot K, Halmos G, Szepeshazi K, Zarandi M, Armatis P. J Natl Cancer Inst. 1995;87:1787–1794. doi: 10.1093/jnci/87.23.1787. [DOI] [PubMed] [Google Scholar]
- 15.Pinski J, Schally A V, Jungwirth A, Groot K, Halmos G, Armatis P, Zarandi M, Vadillo-Buenfil M. Int J Oncol. 1996;9:1099–1105. doi: 10.3892/ijo.9.6.1099. [DOI] [PubMed] [Google Scholar]
- 16.D’Ercole A J, Stiles A D, Underwood L E. Proc Natl Acad Sci USA. 1984;81:935–939. doi: 10.1073/pnas.81.3.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Breier B H, Gallaher B W, Gluckman P D. J Endocrinol. 1991;128:347–357. doi: 10.1677/joe.0.1280347. [DOI] [PubMed] [Google Scholar]
- 18.Daughaday W H, Mariz I K, Blethen S L. J Clin Endocrinol Metab. 1980;51:781–788. doi: 10.1210/jcem-51-4-781. [DOI] [PubMed] [Google Scholar]
- 19.Munson J P, Rodbard D. Anal Biochem. 1980;107:220–239. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- 20.Steel R G D, Torrie J. Principles and Procedures of Statistics. New York: McGraw-Hill; 1976. p. 114. [Google Scholar]
- 21.Reiter E, Kecha O, Hennuy B, Lardinois S, Klug M, Bruyninx M, Closset J, Hennen G. Endocrinology. 1995;136:3338–3345. doi: 10.1210/endo.136.8.7628369. [DOI] [PubMed] [Google Scholar]
- 22.Macaulay V M. Br J Cancer. 1992;65:311–320. doi: 10.1038/bjc.1992.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Froesch E R, Schmid C, Schwander J, Zapf J. Annu Rev Physiol. 1985;47:443–467. doi: 10.1146/annurev.ph.47.030185.002303. [DOI] [PubMed] [Google Scholar]
- 24.Cohen P, Peehl D M, Rosenfeld R G. Horm Metab Res. 1994;26:81–84. doi: 10.1055/s-2007-1000777. [DOI] [PubMed] [Google Scholar]
- 25.Ciampani T, Fabbri A, Isidori A, Dufau M L. Endocrinology. 1992;131:2785–2792. doi: 10.1210/endo.131.6.1332849. [DOI] [PubMed] [Google Scholar]
- 26.Christophe J, Svoboda M, Dehaye J P, Winand J, Vandermeers-Pire M C, Vandermeers A, Cauvin A, Gouerlet P, Robberecht P. In: Peptide Hormones as Prohormones: Processing, Biological Activity, Pharmacology. Martinez J, editor. New York: Halsted; 1989. pp. 211–242. [Google Scholar]
- 27.Robberecht P, Waelbroeck M, Coy D, DeNeef P, Camus J C, Christophe J. Peptides. 1986;7:53–59. doi: 10.1016/0196-9781(86)90164-6. [DOI] [PubMed] [Google Scholar]
- 28.Laburthe M, Couvineau A. Ann NY Acad Sci. 1988;527:296–313. doi: 10.1111/j.1749-6632.1988.tb26988.x. [DOI] [PubMed] [Google Scholar]