

Abstract
The interaction between nuclear receptors and coactivators provides an arena for testing whether protein–protein interactions may be inhibited by small molecule drug candidates. We provide evidence that a short cyclic peptide, containing a copy of the LXXLL nuclear receptor box pentapeptide, binds tightly and selectively to estrogen receptor α. Furthermore, as shown by x-ray analysis, the disulfide-bridged nonapeptide, nonhelical in aqueous solutions, is able to adopt a quasihelical conformer while binding to the groove created by ligand attachment to estrogen receptor α. An i, i+3 linked analog, H-Lys-cyclo(d-Cys-Ile-Leu-Cys)-Arg-Leu-Leu-Gln–NH2 (peptidomimetic estrogen receptor modulator 1), binds with a Ki of 25 nM, significantly better than an i, i+4 bridged cyclic amide, as predicted by molecular modeling design criteria. The induction of helical character, effective binding, and receptor selectivity exhibited by this peptide analog provide strong support for this strategy. The stabilization of minimalist surface motifs may prove useful for the control of other macromolecular assemblies, especially when an amphiphilic helix is crucial for the strong binding interaction between two proteins.
Members of the nuclear receptor (NR) superfamily, which include the steroid receptors, are ligand-activated transcription factors that regulate a wide variety of physiological and developmental processes (1–3). Upon ligand binding, steroid receptors shed their accompanying heat shock proteins to form homodimers, and bind to their cognate DNA elements within the regulatory regions of steroid responsive genes. Steroid receptor agonists are typically hydrophobic molecules and have been demonstrated to bind to a buried hydrophobic pocket within the carboxyterminal ligand-binding domain (LBD) of the receptor. This results in a conformational shift causing repositioning of helix 12, which allows for recognition of coactivator proteins. Many coactivators contain a short pentapeptide motif, known as a NR box (4), that is responsible for recognition of a hydrophobic groove created on the surface of the LBD in response to repositioning of helix 12 upon agonist binding (5). Steroid receptor antagonists, like agonists, are also hydrophobic molecules and bind within the core of the LBD; however, these ligands do not position helix 12 in the correct conformation that would allow the coactivators to recognize the receptor. A large number of proteins characterized as NR coactivators have been identified, and many appear to contain one or, in some cases, multiple copies of the NR box with the consensus sequence LXXLL. McDonnell and coworkers (6, 7) have noted that peptide sequences that mimic this NR interaction motif could function as ER antagonists in cell based models when overexpressed as a component of a fusion protein. Detailed analysis of the interactions between the receptor and coactivators has revealed new possible points of intervention (8). Such targets have recently been proposed as attractive options for new anticancer drugs (9).
In order for this new class of functional NR antagonists to become more “drug-like,” there are several difficulties that must be overcome. We address two of these issues in this study: retention of the α-helical structure of an NR box mimetic in a short peptide and NR specificity. The x-ray work of Shiau et al. (10) served as a template for the design of conformationally constrained analogs that might best mimic the binding face of the NR box domain with the estrogen receptor (ER) α. In particular, analysis of the crystal data revealed that the critical leucine residues are oriented on one face of an α-helix with the XX side chains on the opposite half of the amphiphilic helix. Neighboring residues on the adjacent N- and C-terminal sites of the core LXXLL motif and/or two appropriately spaced dual NR boxes appear to provide selectivity between various members of the NR superfamily (11–15).
In this work we describe our efforts to design a group of linear and cyclic peptides as inhibitors of the coactivator–steroid receptor interaction and correlate their resulting bioactivities with their conformational properties. There are few studies in the literature involving a direct comparison between peptides with side chain cyclic constraints designed to stabilize an α-helical structure (16), although an interesting report on glucagon analogs contrasted i, i+4 amide linkages for helix stabilization with disulfide bridges for inducing turn structures (17). We also provide evidence for an induced fit for our disulfide bridged cyclic peptide in the crystal structure that results in a helical conformation for the inhibitory cyclic peptide and a low nanomolar Ki. This result opens the way for the design of selective antagonists of a broad range of NR targets.
Methods
Peptide Synthesis. The linear and cyclic peptides were synthesized by using Boc-based Merrifield solid phase peptide synthesis (18, 19) using anhydrous hydrogen fluoride for cleavage (20) from the methylbenzhydrylamine resin support (21) to provide the targeted peptide amides. Scheme 1, which is published as supporting information on the PNAS web site, www.pnas.org, summarizes the approach used for the two cyclic variants. The lactam bridge between Glu and Lys was formed on the resin after base-mediated cleavage of the fluorenylmethyl-ester protecting groups. In contrast, disulfide bridge formation was performed off-resin; the DMSO oxidation procedure of Tam et al. (22) provided the best results when using neat sulfoxide reagent.
NMR and CD Studies. Peptides were fully characterized by matrix assisted laser desorption ionization time-of-flight mass spectrometry, amino acid analysis, reversed-phase HPLC, and spectroscopic methods including proton and carbon 13 NMR spectrometry. Two-dimensional NMR methods were used for NMR assignments (Table 3, which is published as supporting information on the PNAS web site), but neither NOESY nor ROESY NMR experiments were able to provide evidence for helical character in 10% D2O, presumably because of rapidly interconverting conformational isomers.
In contrast, CD experiments were supportive of helical character (Figs. 3 and 4, which are published as supporting information on the PNAS web site), but only for the disulfide bridged structure, and only in the presence of 7% trifluoroethanol (23). In view of the partial energy absorption of the disulfide bond in the helical region of interest, no quantitative estimate for helical character was attempted. The linear nonapeptide and the cyclic amide peptides showed either random character or minimal helix tendencies in the two solvents used.
Time-Resolved Fluorescence-Based Coactivator Interaction Assay. The time-resolved fluorescence-based coactivator interaction assay was performed as described (24). Briefly, white 96-well plates were coated overnight with either full-length recombinant baculovirus expressed human ERα or ERβ (PanvVera, Madison, WI). Protein-coated plates were washed five times, then blocked for at least 1 h followed by thorough washing. An NR box peptide–Europium conjugate was prepared by incubating the biotin-labeled NR box peptide with Eu-labeled straeptavidin on ice. The 96-well plate coated with ER was then incubated for at least 1.5 h with a NR box peptide-Eu conjugate in presence of 17β-estradiol (Sigma) and the competitor peptide. The NR box peptide used for ERα was the SRC-1 NR box 2 peptide (LTERHKILHRLLQEGSPSD), whereas the SRC-1 NR box 4 peptide (QAQQKSLLQQLLTE) was used for ERβ experiments. The dissociation constants for both of these peptides for E2-bound ERα or ERβ were previously determined to be 155 ± 21 nM and 261 ± 72 nM, respectively (24). Plates were washed five times followed by incubation with gentle shaking in the presence of enhancement solution for 5 min; this allowed release of the bound Eu label. Plates were read in a Wallac Victor II plate reader using a protocol specific for Europium label (Wallac, Gaithersburg, MD). Assays were performed a minimum of three times, and Ki values were determined by using the Cheng–Prusoff equation with prism software (Graphpad, San Diego).
Crystallography. For crystallographic studies, the human ERα LBD (residues 304–550 with three mutations: C381S, C417S, and C530S) was overexpressed as an N-terminally His-6-tagged protein in BL21(DE3) cells by using expression vector pET19 (Novagen) and purified by PanVera.
Protein solution containing 5 mg/ml ERα LBD, 20 mM Hepes (pH 7.0), 100 mM NaCl, and estradiol and peptidomimetic estrogen receptor modulator (PERM-1) in the amount of 2- to 3-fold molar excess of the protein was used for crystallization. Diffraction-quality crystals of ERα LBD complex were grown by the vapor diffusion technique at 294 K under the reservoir condition 0.1 M Mes, pH 6.8/1 M LiCl/7–15% polyethylene glycol 6000. Crystals belong to orthorhombic space group C2221 with unit cell parameters a = 53.8 Å, b = 102.4 Å, c = 195.3 Å. There are two molecules of the complex per asymmetric unit with a Vm value (25) of 2.28 Å3/Da that corresponds to a solvent content of ≈46%.
The diffraction data (resolution of 2.7 Å; Rmerge = 0.114, and completeness of 95%) were collected by using a MarCCD detector on Industrial Macromolecular Crystallography Association (IMCA) beam line BM-17 at the APS (Advanced Photon Source, Argonne National Laboratories, Argonne, IL). The crystal was cooled at 100 K by using 15–20% glycerol plus the mother liquor as cryoprotectant. The diffraction data were processed by using hkl2000 (26). The crystal structure was determined by the method of molecular replacement with an ERα LBD/estradiol search model (27) by using amore program suite (28). The crystal structure was refined against data between 20 and 2.7 Å by using a maximum likelihood target as incorporated in the program cnx2000 (29) (Rwork = 0.219, Rfree = 0.25, rms = 0.007 Å) (30). The program suite quanta 98 (Molecular Simulation, San Diego) was used for model building between rounds of refinement. An analysis of the geometry showed all parameters were within the values expected for a model at this resolution. All residues were found in the most favorable and additionally allowed regions of a Ramachandran plot.
The peptide PERM-1 was refined with a covalent link between the two sulfur atoms (disulfide bond) by using a weak force constant for this bond. The x-ray coordinates for the ERα-ligand-peptide have been deposited in the Protein Data Bank.
Results and Discussion
Our interest in the design of peptide and protein mimics, together with the suggestion by McDonnell and coworkers (7) that short peptides might serve as antagonists of the estrogen receptor, provided the impetus for the present work. It is clear that the consensus LXXLL pentapeptide motif must assume a helical conformation to provide a productive interaction with the NR. However, there were other elements of the coactivatorderived fragments that caught our attention during the design process. The three leucines provide the bulk of the hydrophobic attraction (Fig. 5, which is published as supporting information on the PNAS web site). However, it seems clear that an additional element, the isoleucine side chain at the –1 position, along with a charge–charge interaction involving the lysine at –3 should also be preserved. Two intermolecular hydrogen bonds, with the Ile-3 amide hydrogen serving as H-bond donor and the Leu-7 carbonyl serving as H-bond acceptor, were also deemed important and constitute the “charge clamp” responsible for specifying the length of the NR box helix (5).
Shorter linear peptides tend to adopt random or β-sheet structures rather than helices. Various strategies have been used to induce helix folding including incorporation of α-alkyl amino acid residues such as aminoisobutyric acid (Aib) (31) or diethylglycine (Deg) (32). This approach may lead to unacceptably high hydrophobic character when matched with an LXXLL sequence. Other options include helix end capping (33) and dipole stabilization, primarily useful for longer sequences.
In view of our synthetic experience with cyclic peptides and pseudopeptides as well as cyclic peptide libraries (34), we focused on published reports describing helix stabilization through side chain to side chain covalent linkages. The cyclization approach was also deemed most suitable in an effort to preserve important protein recognition elements while providing minimal disruption of the delicate balance of hydrophobic and hydrophilic groups required for peptide bioavailability. We reasoned that further manipulations designed to traverse hydrophobic barriers and lead to a viable drug candidate could come later, once the basic tenets of our approach could be validated.
Two primary side chain to side chain tactics have been used to enhance alpha helical character. The first of these involves i to i+4 amide formation, usually through a lysine/glutamic acid side chain bond (35). This linkage was adopted by Felix and coworkers (36) in the design of stabilized analogs of growth hormone-releasing factors. More recently it has been used in the successful stabilization of an antagonist of the thyroid hormone receptor (37). Another cyclization approach involves the formation of a disulfide bridge between cysteine residues located at i and i+3 as described by Schultz and coworkers (38). Both of these general ring types, as well as alterations to the stereo-chemical configurations of each of the amino acid pairs, were considered in our design process.
Molecular dynamics simulations were used to compare the structure of the GRIP1 NR Box II peptide sequence in a helical arrangement against the conformation of our helix-stabilized peptide using the published structure (10). The ability of the peptide analogs to mimic the coactivator fragment was assessed by molecular modeling using macromodel version 7.0 (39), with molecular mechanics and molecular dynamics simulations performed at 300 K using the amber* force field and the GB/SA solvation model (40). It should be noted that explicitly solvated (TIP3P waters) molecular dynamics simulations using the amber suite of programs resulted in a random coil, whereas the implicit solvation simulations maintained helical character. The explicit solvation calculations are consistent with the experimental circular dichroism studies. Nevertheless, our implicit solvation simulations proved appropriate for designing and evaluating the helical nature that would be induced by ERα.
A calculated structure for a linear peptide was superimposed with a linear peptide's crystal structure for validation of future superimpositions. Once agreement was achieved, we replaced the noncritical pairs of amino acids in the coactivator NR Box fragments that would preserve the hydrophobic side chains of Leu and Ile while making the closest fit with the distance between the comparable residues in the linear helical peptide. Table 1 summarizes the distance when varying the replacements for formation of the amide as well as the disulfide linkages. At this stage, both ring structures could adopt a helical like shape, although it appeared that the better fit was retained by the disulfide bridge, and especially when the replacing residues were d-Cys and l-Cys, at positions –2 and X1 (of LX1X2LL). This finding is also consistent with prior modeling and structural reports that support the d,l cysteine motif as the better helical mimetic (41).
Table 1. Calculated interatomic distances for prospective stabilized cyclic analogs of a coactivator fragment in a helical conformation.

Distance given (X, Y) is between two sulfurs in the case of the disulfide compounds and between the carbonyl carbon and amide nitrogen for the lactams, prior to ring closure, assuming that the linear sequence is in an idealized α-helix.
A more detailed fit with other key parameters of the crystal structure also showed the best fit with the d-Cys, l-Cys combination, although the amide bridges were not ruled out as additional synthetic targets.
Thus, our initial synthetic targets included disulfide bridged and amide bridged analogs as well as cyclic peptides with an N-terminal Aib residue for potential enhancement of helicity. N-terminal acetylation 7 proved deleterious to activity but was highly selective for ERα (Table 2); several linear counterparts were also prepared to probe the requirement for a cyclic structure.
Table 2.
Inhibition of estrogen receptor/coactivator recognition as assessed by a time-resolved fluorescence-based coactivator interaction assay (24)
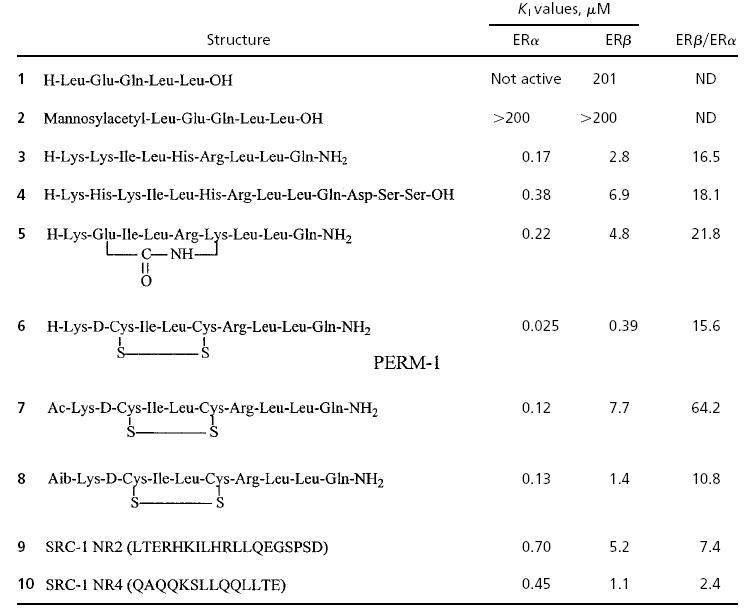
ND, not determined.
As noted in Table 2, most of our compounds were more effective against ERα than against ERβ. The addition of isoleucine at the –1 position was adapted from the solid state x-ray of a linear peptide bound to ERα (10), and this may be primarily responsible for the selectivity differences observed. The linear pentapeptide LXXLL (1) sequence was virtually inactive, as was a mannosylacetyl derivative (3), prepared in an effort to test whether a stabilized C-sugar linkage might enhance binding and/or transport (42). Surprisingly, our linear nonapeptide 3 bound with a Ki of 0.17 μM against ERα, somewhat better than a longer tridecapeptide (4).
Most notably, the analog showing the greatest helical character in the CD study displayed the highest affinity for the ERs. This disulfide-bridged compound 6 (PERM-1) had higher affinity for ERα (Ki = 0.025 μM) than for ERβ (Ki = 0.390 μM). Thus, this compound showed a selectivity ≈15 times higher for ERα over ERβ.
To assess whether the cyclic peptide was indeed binding to the receptor in the predicted fashion, we undertook an x-ray crystal structure determination of the LBD of ERα, together with both the native ligand, estradiol, as well as our most active disulfide bridged peptide, PERM-1. As seen in Fig. 1, the peptide binds to the expected hydrophobic groove formed by helices 3, 4, 5, and 12; the hydrophobic side chains of the three leucines as well as the additional isoleucine residue at position –1 all appear to make close contact with the receptor (Fig. 2). The two XX residues, in this case, Cys–Arg, are oriented away from the groove as expected for an amphiphilic helix. In fact, as suggested previously (41), even the d-Cys, l-Cys bridge does not accommodate a perfect helix. The φ, ψ bond angles are more idealized in the N-terminal portion of the peptide, but tend to divert to larger values at the C-terminal end. Similarly, the disulfide bond distance is somewhat distorted (increased to ≈2.7 Å) from an ideal value of 2.04 Å, although the electron density is fully consistent with the presence of a bond between the sulfurs. Interestingly, the C–S–S–C dihedral angles are also atypical, with values of 69° and 85°, compared with an idealized value of 90°.
Fig. 1.

X-ray structure of the LBD of ERα associated with estradiol and PERM-1, depicting the induced helix formed by the disulfide bridged peptide when bound to the NR dimer LBD. The steroid ligand is shown in red, and the disulfide bridged peptide is shown in green.
Fig. 2.

Interaction of PERM-1 with the LBD of ERα.(A) Superposition of the structure ERα/estradiol/PERM-1 (PDB ID 1PCG) with ERα/DES/GRIP1 (PDB ID 3ERD) is viewed in the coactivator binding site. PERM-1 is colored by elements, whereas GRIP1 NR box II is in orange. The side chains of the two charge clamps in ERα are displayed. (B) The molecular surface of ERα shows a good fit between the hydrophobic residues of PERM-1 (green) and the coactivator binding cleft formed in estradiol bound ERα (yellow). The surface formed by the charge clamps is colored in red and blue to indicate the orientation of the peptide anchored by polar interactions. The N-terminal lysine is positioned at the lower left, and the C-terminal glutamine amide is positioned at the upper right. The image was drawn with pymol (45).
The peptide assumes a tight fit with the receptor, as confirmed by its low Ki. However, in water, the circular dichroism spectrum of PERM-1 shows no evidence of helical character, and it is only with the addition of the structure inducing solvent trifluoroethanol (7%) that the characteristic helical bands appear at 208 and 222 nM. This suggests that the designed NR Box mimic is actually nonhelical in an aqueous environment but is induced to adopt the pseudohelical structure upon receptor binding. It appears that there is also flexibility on the part of the receptor as well, a factor that must be considered in any computational design strategy (43).
The synthesis of both disulfide and amide bridged NR box peptides provided an effective comparison of these two helix-stabilizing strategies. Modeling studies suggested that an i, i+3 disulfide should best mimic an idealized α-helix. The bioassays supported this prediction by providing a highly effective inhibitor of the ERα coactivator fragment peptide binding, with a Ki of 25 nM. In contrast, the i, i+4 amide bridge gave an inhibitor 5 with an order of magnitude lower Ki (0.22 μM for ERα and 4.8 μM for ERβ). X-ray results confirmed that the disulfide bridged analog PERM-1 binds to the ER ligand-binding domain in the predicted fashion. It is anticipated that the d-Cys, l-Cys i, i+3 structural motif will find other applications as a helicomimetic, useful for inhibiting protein–protein interactions when this secondary structure is present.
The observation that the receptor appears to induce the helical structure of the NR box peptide has interesting implications for receptor-induced conformational changes in the coactivator itself and, thus, potential modulation of coactivator activity by the receptor. It was previously demonstrated that PPAR-γ coactivator (PGC)-1's ability to recruit additional coactivators and cointegrators such as SRC-1 (steroid receptor coactivator-1) and CBP (CREB-binding protein) was significantly enhanced when PGC-1 was complexed with a NR, indicating the ability of the receptor to modulate coactivator function (44). Thus, the activated NR may not only recruit the coactivator, but it may play an important role in the modulation of the coactivator activity once bound. Although the mechanism by which the receptor induces the helical structure within the LXXLL peptide is not understood, it may be possible that the “charge clamp” (5) composed of the relatively fixed lysine residue in helix 3 and the glutamic acid residue in the flexible helix 12 that are believed to play a role in specifying the length of the optimal NR box peptide may actually be inducing and stabilizing the helical structure of the peptide as it docks into the hydrophobic surface of the receptor.
These results are supportive of the ability to design constrained fragments of known structural motifs that can affect critical protein–protein binding interactions. The disulfide bridged nonapeptide is considerably more potent than even a comparable linear peptide with 13 residues. These constrained protein mimics should emerge as promising targets for new drug therapies, particularly as new proteins and pairs of controlling recognition sites are identified, such as the interaction between the NRs and the coactivator proteins. Furthermore, because it seems likely that additional receptor selectivity can be introduced through the modification of amino acid residues adjacent to the LXXLL motif, our cyclic construct could lead to new inhibitors of other members of the NR superfamily. With additional replacements of the hydrophobic leucine moieties, the helical constraint should be considered for the preparation of other pharmacophores possessing this common conformational feature.
Additional structural modifications to these cyclic peptides, such as attachment to cell permeable adducts (46) are probably required to confer in vivo activity. Nevertheless, the feasibility of inhibiting protein–protein interactions and the desirability of using selective NR probes to study coactivator function are both evident from this work. Future efforts should focus on improved bioavailability and the design of enhanced selectivity as these helicomimetics are tested against additional NR classes.
Supplementary Material
Acknowledgments
This work was supported by grants from Lilly Research Laboratories (to A.F.S.) and the Phi Beta Psi Research Foundation (to J.L.W.).
Abbreviations: ER, estrogen receptor; LBD, ligand-binding domain; PERM, peptidomimetic estrogen receptor modulator; NR, nuclear receptor.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1PCG).
References
- 1.Mangelsdorf, D. J. (1995) Cell 83, 835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burris, T. P. & McCabe, E. R. B. (2001) Nuclear Receptors and Genetic Disease (Academic, San Diego).
- 3.McKenna, N. J. & O'Malley, B. W. (2002) Cell 108, 465–474. [DOI] [PubMed] [Google Scholar]
- 4.Heery, D. M., Kalkhoven, E., Hoare, S. & Parker, M. G. (1997) Nature 387, 733–736. [DOI] [PubMed] [Google Scholar]
- 5.Nolte, R. T., Wisely, G. B., Westin, S., Cobb, J. E., Lambert, M. H., Kurokawa, R., Rosenfeld, M. G., Willson, T. M., Glass, C. K. & Milburn, M. V. (1998) Nature 395, 137–143. [DOI] [PubMed] [Google Scholar]
- 6.Norris, J. D., Fan, D., Stallcup, M. R. & McDonnel, D. P. (1998) J. Biol. Chem. 273, 6679–6688. [DOI] [PubMed] [Google Scholar]
- 7.Norris, J. D., Paige, L. A., Christensen, D. J., Chang, C.-Y., Huacani, M. R., Fan, D., Hamilton, P. T., Fowlkes, D. M. & McDonnel, D. P. (1999) Science 285, 744–746. [DOI] [PubMed] [Google Scholar]
- 8.Northrop, J. P., Nguyen, D., Piplani, S., Olivan, S. E., Kwan, S., Fei Go, N., Hart, C. P. & Schatz, J. (2000) Mol. Endocrinol. 14, 605–622. [DOI] [PubMed] [Google Scholar]
- 9.Darnell, J. E. (2002) Nat. Rev. Cancer 2, 740–749. [DOI] [PubMed] [Google Scholar]
- 10.Shiau, A. K., Barstad, D., Loria, P. M., Cheng, L., Kushner, P. J., Agard, D. A. & Greene, G. L. (1998) Cell 95, 927–937. [DOI] [PubMed] [Google Scholar]
- 11.McInerney, E. M., Rose, D. W., Flynn, S. E., Westin, S., Mullen, T. M., Krones, A., Inostroza, J., Torchia, J., Notles, R. T., Assa-Munt, N., et al. (1998) Genes Dev. 12, 3357–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darimont, B., Wagner, R., Apriletti, J., Kushner, P., Stallcup, M., Baxter, J., Fletterick, R. & Yamamoto, K. (1998) Genes Dev. 12, 3343–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mak, H. Y., Hoare, S., Henttu, P. M. & Parker, M. G. (1999) Mol. Cell. Biol. 19, 3895–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Voegel, J. J., Heine, M. J., Tini, M., Vivat, V., Chambon, P. & Gronemeyer, H. (1998) EMBO J. 17, 507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ko, L., Cardona, G. R., Iwasaki, T., Bramlett, K. S., Burris, T. P. & Chin, W. W. (2002) Mol. Endocrinol. 16, 128–140. [DOI] [PubMed] [Google Scholar]
- 16.Rizzo, J. & Gierasch, L. M. (1992) Annu. Rev. Biochem. 61, 387–418. [DOI] [PubMed] [Google Scholar]
- 17.Ahn, J.-M., Gitu, P. M., Medeiros, M., Swift, J. R., Trivedi, D. & Hruby, V. J. (2001) J. Med. Chem. 44, 3109–3116. [DOI] [PubMed] [Google Scholar]
- 18.Merrifield, R. B. (1963) J. Am. Chem. Soc. 85, 2149–2154. [Google Scholar]
- 19.Barany, G., Kneib-Cordonier, N. & Mullen, D. G. (1987) Int. J. Peptide Protein Res. 30, 705–739. [DOI] [PubMed] [Google Scholar]
- 20.Sakakibara, S. (1971) in Chemistry and Biochemistry of Amino Acids, Peptides, and Proteins, ed. Weinstein, B. (Dekker, New York), Vol. 1, pp. 51–85. [Google Scholar]
- 21.Gaehde, S. T. & Matsueda, G. R. (1978) Int. J. Peptide Protein Res. 18, 451–458. [DOI] [PubMed] [Google Scholar]
- 22.Tam, J. P., Wu, C.-R., Liu, W. & Zhang, J.-W. (1991) J. Am. Chem. Chem. 113, 6657–6662. [Google Scholar]
- 23.Cammers-Goodwin, A., Allen, T. J., Oslick, S. L., McClure, K. F., Lee, J. H. & Kemp, D. S. (1996) J. Am. Chem. Soc. 118, 3082–3090. [Google Scholar]
- 24.Bramlett, K. S., Wu, Y. & Burris, T. P. (2001) Mol. Endocrinol. 15, 909–922. [DOI] [PubMed] [Google Scholar]
- 25.Matthews, B. W. (1968) J. Mol. Biol. 33, 491–497. [DOI] [PubMed] [Google Scholar]
- 26.Otwinowski, Z. & Minor, W. (1997) Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 27.Brzozowski, A. M., Pike, A. C., Dauter, Z., Hubbard, R. E., Bonn, T., Engström, O., Öhman, L., Greene, G. L., Gustafsson, J.-A. & Carlquist, M. (1997) Nature 389, 753–758. [DOI] [PubMed] [Google Scholar]
- 28.Collaborative Computing Project No. 4 (1994) Acta Crystallogr. D 50, 760–763.15299374 [Google Scholar]
- 29.Badger, J., Kumar, R. A., Yip, P. & Szalma, S. (1999) Proteins Struct. Funct. Genet. 35, 25–33. [PubMed] [Google Scholar]
- 30.Brünger, A. T. (1992) Nature 355, 472–474. [DOI] [PubMed] [Google Scholar]
- 31.Karle, I. L. & Balaram, P. (1990) Biochemistry 29, 6747–6756. [DOI] [PubMed] [Google Scholar]
- 32.Toniolo, C. & Benedetti, E. (1991) Macromolecules 24, 4004–4009. [Google Scholar]
- 33.Scholtz, J. M., Qian, H., York, E. J., Stewart, J. M. & Baldwin, R. L. (1991) Biopolymers 31, 1463–1470. [DOI] [PubMed] [Google Scholar]
- 34.Spatola, A. F., Crozet, Y., Dewit, D. & Yanagisawa, M. (1996) J. Med. Chem. 39, 3842–3846. [DOI] [PubMed] [Google Scholar]
- 35.Bouvier, M. & Taylor, J. W. (1992) J. Med. Chem. 35, 1145–1155. [DOI] [PubMed] [Google Scholar]
- 36.Campbell, R. M., Bongers, J. & Felix, A. M. (1995) Biopolymers 37, 67–88. [DOI] [PubMed] [Google Scholar]
- 37.Geistlinger, T. R. & Guy, R. K. (2001) J. Am. Chem. Soc. 123, 1525–1526. [DOI] [PubMed] [Google Scholar]
- 38.Jackson, D. Y., King, D. S., Chmielewski, J., Singh, S. & Schultz, P. G. (1991) J. Am. Chem. Soc. 113, 9391–9392. [Google Scholar]
- 39.Mohamadi, F., Richards, N. G. J., Guida, W. C., Liskamp, R., Lipton, M., Canfield, C., Chang, G., Hendrickson, T. & Still, W. C. (1990) J. Comput. Chem. 11, 440–467. [Google Scholar]
- 40.Still, W. C., Tempczyk, A., Hawley, R. C. & Hendrickson, T. (1990) J. Am. Chem. Soc. 112, 6127–6129. [Google Scholar]
- 41.Pellegrini, M., Royo, M., Chorev, M. & Mierke, D. F. (1997) J. Pept. Res. 49, 404–414. [DOI] [PubMed] [Google Scholar]
- 42.Brunel, F., Leduc, A.-M., Singh, S., Tang, X., Vogel, D. M., Taylor, K. G. & Spatola, A. F. (2001) Peptides: The Wave of the Future (Am. Peptide Soc., San Diego), pp. 170–171.
- 43.Lin, J., Perryman, A. L., Schames, J. R. & McCammon, J. A. (2002) J. Am. Chem. Soc. 124, 5632–5633. [DOI] [PubMed] [Google Scholar]
- 44.Puigserver, P., Adelmant, G., Wu, Z., Fan, M., Xu, J., O'Malley, B. W. & Spiegelman, B. M. (1999) Science 286, 1368–1371. [DOI] [PubMed] [Google Scholar]
- 45.DeLano, W. L. (2002) The PyMOL Molecular Graphics System (DeLano Scientific, San Carlos, CA).
- 46.Fischer, P. M., Krausz, E. & Lane, D. P. (2001) Bioconjugate Chem. 12, 825–841. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.