

Abstract
Phototransduction in Drosophila occurs through inositol lipid signaling that results in Ca2+ mobilization. In this system, we investigate the hitherto unknown physiological roles of calmodulin (CaM) in light adaptation and in regulation of the inward current that is brought about by depletion of cellular Ca2+ stores. To see the effects of a decreased Ca–CaM content in photoreceptor cells, we used several methods. Transgenic Drosophila P[ninaCΔB] flies, which have CaM-deficient photoreceptors, were studied. The peptide inhibitor M5, which binds to Ca–CaM and prevents its action, was applied. A Ca2+-free medium, which prevents Ca2+ influx and thereby diminishes the generation of Ca–CaM, was used. The decrease in the Ca–CaM level caused the following effects. (i) Fluorescence of Ca2+ indicator revealed an enhanced light-induced Ca2+ release from internal stores. (ii) Measurements of the light-induced current in P[ninaCΔB] cells showed a reduced light adaptation. (iii) Internal dialysis of M5 initially enhanced excitation and subsequently disrupted the light-induced current. (iv) An inward dark current appeared after depletion of the Ca2+ stores with ryanodine and caffeine. Importantly, application of Ca–CaM into the photoreceptor cells prevented all of the above effects. We propose that negative feedback of Ca–CaM on Ca2+ release from ryanodine-sensitive stores mediates light adaptation, is essential for light excitation, and keeps the store-operated inward current under a tight control.
Keywords: phototransduction, phosphoinositide signaling, ryanodine-sensitive stores
Drosophila phototransduction is a phosphoinositide-mediated and Ca2+-regulated signaling system, readily accessible to a combination of genetic and physiological dissection in vivo (for reviews, see ref. 1–5). A major unresolved issue is the role and regulation of the internal Ca2+ stores in Drosophila phototransduction. It is generally agreed that light adaptation in Drosophila is mediated by an increase in cellular Ca2+ (6–8); however, both the site and the mechanism of action of Ca2+ in light adaptation are still unknown.
Genetic studies in Drosophila photoreceptors have unequivocally demonstrated that phospholipase C is required for light excitation (9, 10). However, an excitatory action of Ca2+ could not be demonstrated in isolated Drosophila ommatidia subjected to photolysis of caged Ca2+ (11). Likewise, fluorimetric measurements using Ca2+ indicators failed to detect light-induced release of Ca2+ from intracellular stores (12–14). Several investigators have, therefore, argued that it is unlikely that Ca2+ release from intracellular stores is involved in visual excitation of Drosophila (refs. 13 and 14, but see also ref. 15).
In muscle, neurons, and many types of cells, including insect photoreceptors (16, 17), the endoplasmic reticulum has both receptors for d-myo-inositol 1,4,5-trisphosphate and another Ca2+ release channel, the ryanodine receptor (17–20). The ryanodine receptor responds to an increase in cellular Ca2+ by Ca2+-induced Ca2+ release, a mechanism that appears to be tightly regulated by Ca–calmodulin (Ca–CaM; ref. 21 and 22). The Drosophila ryanodine receptor shows 45–47% homology with the amino acid sequence of the mammalian ryanodine receptors, including the preserved CaM binding sites (16). The visual mutant designated neither inactivation nor afterpotential C (ninaC; ref. 23) encodes two photoreceptor cell-specific unconventional myosins, p132 and p174, consisting of fused protein kinase and myosin head domains (24). The NINAC proteins have been found to be the major CaM binding proteins in Drosophila photoreceptors (25). In the transgenic flies, P[ninaCΔB], the CaM binding domains of NINAC have been deleted, resulting in a dramatic reduction of the CaM content in the photoreceptor cells (25, 26). CaM binding domains have also been demonstrated in the putative channel proteins of Drosophila, TRPL (27) and TRP (28), but their physiological function is unknown (for review, see ref. 5).
In the present study, the effects of Ca–CaM on light adaptation, light- and chemically induced Ca2+ release from internal stores, and the resulting inward current were examined using whole-cell patch-clamp and fluorimetric recordings from Drosophila photoreceptor cells. We find that negative regulation of Ca–CaM on Ca2+ release from ryanodine-sensitive stores is essential for light excitation and light adaptation, and for keeping the store-operated current (ISOC) under a tight control.
MATERIALS AND METHODS
Fly Stocks.
White-eyed Drosophila melanogaster of the Oregon-R-strain (wild type; WT), white-eyed P[ninaCΔB] transgenic flies, and a white-eyed null allele of the rhodopsin Rh1 mutant ninaEoraJK84 were used for the experiments as indicated.
Preparation.
Dissociated ommatidia were prepared from newly emerged flies (<4 h after eclosion) and whole-cell, patch-clamp recordings were performed in isolated ommatidia as described (6, 12). Recordings were made at 21°C with use of patch pipettes with a resistance of 5–10 MΩ. Series resistance was carefully compensated (≈80%) during all the experiments (6). Signals were amplified using an Axopatch-1D (Axon Instruments, Foster City, CA) patch-clamp amplifier, sampled at 5 kHz, and filtered below 2 kHz through a 4-pole Bessel filter. For fluorescence measurements, data were sampled at either 300 Hz (see Fig. 2) or 30 Hz (see Fig. 5) and analyzed using a Codas waveform scrolling card (DATAQ, Akron, OH).
Figure 2.
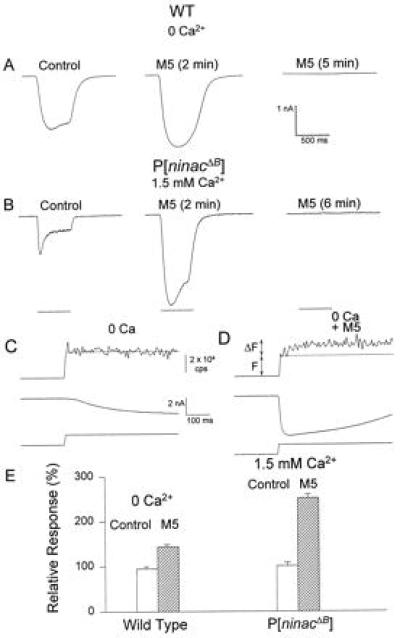
Reduction of cellular Ca–CaM by M5 enhances, temporarily, both the LIC and Ca2+ release from internal stores and subsequently suppresses the photoresponse. M5 facilitated and then blocked the LIC of WT flies (A) bathed in Ca2+-free medium or P[ninaCΔB] cells bathed in normal Ringer’s solution (B). The control LIC was measured in Ca2+-free Ringer solution (no Ca2+ buffers added) ≈10 min before M5 was applied to another cell of the same retina (first trace). Application of 40 μM M5 into the pipette enhanced and slowed the LIC. The LIC was abolished 5 min after M5 application (third trace, same cell). The orange light pulses were attenuated by 1 log unit. (C and D) A reduction in cellular Ca–CaM by M5 enabled detection of light-induced release of Ca2+ from internal stores. Simultaneous recordings of LIC (second traces) and the fluorescence of the Ca2+ indicator fluo-3 (100 μM, upper traces) measured in Ca2+-free medium (0.2 mM EGTA) in P[ninaCΔB] cells. The fluorescence data were sampled at 300 samples per second, and the background fluorescence was subtracted. The onset of maximal intensity Xenon light (indicated by a photocell current, bottom trace) elicited a LIC and excited the fluorescent dye. Because the LIC lagged behind the indicator fluorescence, the first two to four data points represent the resting [Ca2+]i (F, thin line). The fluorescence increment above F reflects the rise in [Ca2+]i (ΔF). ΔF/F is used to compare the rise in [Ca2+]i between cells. (C) Control. (D) M5 (100 μM) was present in the recording pipette (same retina). (E) A histogram summarizing the results of A and B. The relative peak amplitudes of the LIC of WT and the P[ninaCΔB] cells, before and after the effect of M5 are compared. The peak LIC 2 min after application of M5 was divided by the peak of the LIC of the same cell 30 s after the formation of whole-cell recordings, before M5 had any significant effect. The geometrical average of the ratios is presented in the bar graphs. Control histograms are presented in a similar manner. The error bars are the SEM (n = 4–8). The averaged ratios are statistically different from each other (P < 0.001).
Figure 5.
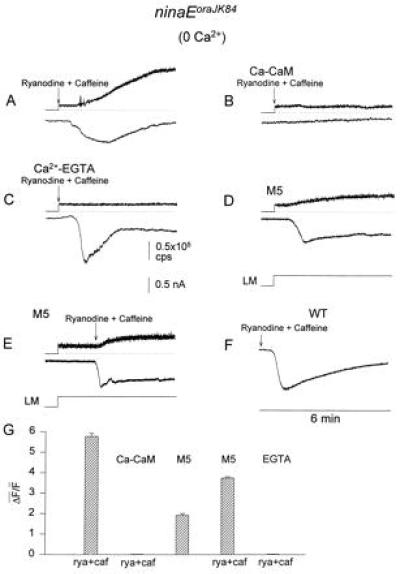
Ryanodine plus caffeine, either alone or with M5, induces an inward current (ISOC, lower traces) and Ca2+ release from internal stores in ninaEoraJK84 flies (upper traces). Ca2+-free medium was used (0.2 mM EGTA). The fluorescence data were sampled at 30 samples per second and are presented in counts per second. M5, Ca–CaM and EGTA were included in the recording pipette. A large variability in the waveform and peak amplitude of ISOC was found in the various experiments. (A) Application of 4 μM ryanodine plus 10 mM caffeine. (B) Inclusion of 5 μM CaM plus 10 μM Ca2+ in the pipette prevented the ryanodine plus caffeine-induced current and the increase in cytosolic Ca2+. Experimental conditions as Fig. A. Only the first 6 min of the 12-min recordings are shown. (C) Internal dialysis with Ca-EGTA buffer (5 mM Ca2+ and 10 mM EGTA), which buffered the cell to an ≈200 nM Ca2+ (14), followed by bath application of ryanodine plus caffeine prevented the increase in cellular Ca2+ but not the induction of ISOC. (D) Application of M5 produced an inward current in the dark which was accompanied by an increase in fluo-3 fluorescence. Experimental paradigm similar to A except that 100 μM M5 was applied through the pipette. Formation of whole-cell recordings started 4 min before the beginning of the trace in D. The inward current was accompanied by an increase in fluo-3 fluorescence in four cells. In one cell, the inward current was approximately two times slower and smaller and no increase in fluorescence was observed. (E) M5 accelerates the ryanodine plus caffeine-induced current. Experimental paradigm similar to that of A except that 100 μM M5 was applied through the recording pipette for 2 min and then ryanodine plus caffeine were applied to the bath. The inward current was induced within 20 s. (F) Application of 4 μM ryanodine plus 10 μM caffeine to WT cells in the dark induced ISOC (n = 6). Experimental paradigm similar to A except that fluo-3 was not included in the recording pipette and cells were maintained in darkness during the entire experiment. (G) Quantitative summary of the fluorescent measurements of ninaEoraJK84 cells (traces in A–E above). To estimate the release of Ca2+ from intracellular stores by ryanodine (rya) plus caffeine (caf), the geometrical average ΔF̄/F̄ was calculated. The bar graphs show the geometrical average of the ΔF/F ratios calculated from different cells (4–6 cells). Control experiments in which the Ca2+-insensitive dye, fluorescein, was used instead of fluo-3 indicated that the increase in fluorescence was not due to volume changes.
Solution.
The bath solution contained 120 mM NaCl, 5 mM KCl, 10 mM TES buffer [N-Tris-(hydroxymethyl)-methyl-2-amino-ethanesulfonic acid, pH 7.15], 4 mM MgSO4, and 1.5 mM CaCl2 (except when Ca2+-free medium was used as indicated). The whole-cell recording pipette contained 100 mM potassium gluconate, 10 mM TES (pH 7.15), 2 mM MgSO4, 4 mM MgATP, and 0.4 mM Na2GTP. For measurements of I–V curves, an internal solution, which blocked K+ channels, containing 100 mM CsCl, 15 mM tetraethylammonium chloride, 2 mM MgSO4, 10 mM TES buffer (pH 7.15), 4 mM MgATP, 0.4 mM Na2GTP, and 30 mM sucrose, was added to the external solution. The chemicals caffeine, heparin, and CaM were obtained from Sigma and ryanodine from Alamone Laboratories (Jerusalem). For bath application of the chemicals, the external solution was replaced during 1 min through a perfusion system at a rate of eight chambers per minute.
Fluorescent Measurements.
The Ca2+ indicator used in this study was fluo-3 (100 μM, pentapotassium salt, Molecular Probes). The optical setup and photon counting were only slightly modified from the previously described setup (12). A Zeiss Axiovert 35 inverted microscope with a Fluar ×40 objective (n.a. = 1.3) was used. Two light sources illuminated the preparation. A high-intensity Xenon source (XBO, 150 W) was used to provide epi-illumination for measuring Ca-indicator fluorescence and provided intense stimulation of the photoreceptors. A tungsten (12-V, 100-W halogen lamp, in conjunction with Schott RG 630 red edge filter or OG 590 orange edge filters and 2 KG3 heat filters) transillumination light provided illumination for viewing the preparation (the red filter) and for stimulation of the photoreceptors (the orange filter) in all the experiments (except where otherwise indicated). The effective intensity of the orange light stimulus was 2 log units below the saturating intensity of the light-induced current (LIC). The maximal light intensity of the transillumination light was attenuated by 1 log unit (except where otherwise indicated).
RESULTS
Ca–CaM Restores the Photoresponse Following Disruption by Ryanodine or Caffeine.
Caffeine is known to bring about the release of Ca2+ from internal stores (17), whereas treatment with ryanodine either blocks (17) or leads to a release of Ca2+ from ryanodine-sensitive stores, depending on the experimental conditions (17, 18, 29–31).
To test the possible existence of ryanodine-sensitive stores in Drosophila, isolated ommatidia were perfused with caffeine (10 mM) or ryanodine (4 μM) 1 min after formation of whole-cell recordings. Throughout the experiments, a light pulse was applied every 1 min. The first response to light was recorded before drug application (Fig. 1 A and B, Control). Fig. 1 A and B (second traces) shows that bath application of caffeine or ryanodine initially eliminated the sustained response to prolonged illumination. This effect was followed by total collapse of the photoresponse upon additional light stimuli (Fig. 1 A and B, third traces). Similar disruption of the photoresponse was obtained by inclusion of ryanodine or caffeine in the recording pipette (n = 4). The photoresponse was also disrupted when photoreceptors were internally dialyzed with heparin (Fig. 1C), a known blocker of the d-myo-inositol 1,4,5-trisphosphate receptor in photoreceptors (32). To demonstrate the essential role of Ca–CaM in light excitation, the LIC was first disrupted by prolonged (12–40 min) bath application of ryanodine or caffeine. Subsequently, the LIC was completely restored (in other cells of the same retina) by application of Ca–CaM through the recording pipette (5 μM CaM plus 10 μM Ca2+, Fig. 1 B and C, fourth traces). Application of free CaM (40 μM, n = 5), free Ca2+ (10 μM, n = 5), or Ca2+ buffers [0.4 mM ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) or 40 μM 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA), n = 5] did not rescue the LIC. In some experiments, the time course of the Ca–CaM-induced recovery from the effect of ryanodine or caffeine was measured and found to be <1 min (n = 5). Ca–CaM did not rescue the LIC after heparin treatment (Fig. 1C, fourth trace). These results clearly show that illumination combined with ryanodine, caffeine, or heparin abolishes the LIC and that Ca–CaM restored the LIC of ryanodine- and caffeine-treated (but not of heparin-treated) cells.
Figure 1.
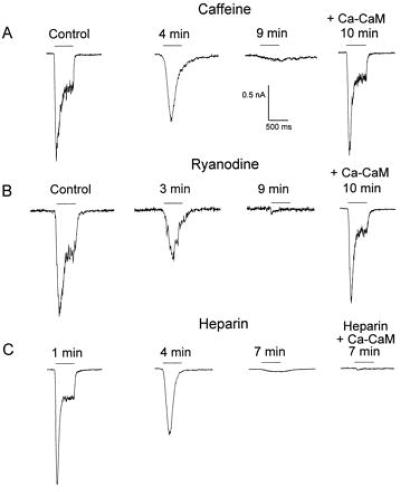
Chemically induced disruption of the photoresponse in WT cells and its rescue by Ca–CaM. Inward currents are shown in response to orange (OG-590 Schott) light pulses [horizontal bars above the traces (log I = −1, A and C; log I = −2, B] at holding potentials of −60 mV. All the experiments were carried out at normal Ringer’s solution. The time from formation of the whole-cell recordings is indicated. Cells were incubated in a ryanodine- or caffeine-containing solution for 12–40 min. Control photoresponses were recorded before drugs application. Following the disruption of the LIC, internal dialysis with Ca–CaM (5 μM CaM plus 10 μM Ca2+, in other cells of the same retina) rescued the LIC in the ryanodine- or caffeine-treated cells (A and B, fourth trace). Disruption of the LIC was defined as a reduction, to less then 1%, of the geometrical average of the ratios between the peak LIC 10–12 min after drug application and the peak LIC before drug application. A restored LIC was obtained when this ratio was not statistically different from control LIC of 100% (Student’s t test, P > 0.05). The control result was obtain by the same experimental procedure but without application of the chemicals. Throughout the experiments n = 4–7 in each experiment. (A and B) Perfusion of cells with Ringer’s solution containing caffeine (A, 10 mM in 0.1% dimethyl sulfoxide) or ryanodine (B, 4 μM in 0.1% dimethyl sulfoxide). Application of 0.1% dimethyl sulfoxide to the bath for 1 h had virtually no effect on the LIC (n = 4). (C) Heparin sulfate (10 mg/ml) was applied through the whole-cell recording pipette. The first trace shows a normal LIC recorded 1 min after formation of whole-cell recordings, before heparin had any significant effect. Internal dialysis with Ca–CaM (5 μM CaM plus 10 μM Ca2+) together with heparin did not rescue the LIC (fourth trace, recorded from another cell of the same retina).
Light Excitation Depends on Regulation by Ca–CaM.
Because we assume that the light-induced Ca2+ influx increases the Ca–CaM levels of Drosophila photoreceptors, we sought to prevent this increase by removing external Ca2+. In the presence of extracellular Ca2+, the LIC rapidly decreases to a plateau level during light (Fig. 1, first traces), whereas in the absence of extracellular Ca2+, this decrease does not occur (Fig. 2A, first trace). An alternative and more specific protocol to decrease the Ca–CaM level in the cell is to apply M5, a known specific inhibitor of Ca–CaM (33, 34). Fig. 2A (second trace) shows that inclusion of M5 (40 μM) in the whole-cell recording pipette during perfusion with Ca2+-free medium enhanced the amplitude (Fig. 2E) and slowed down the light response. The facilitation of the LIC by M5 in Ca2+-free medium, however, was transient, and long (>3 min) applications of M5 in Ca2+-free medium suppressed and then completely eliminated the LIC (Fig. 2A, third trace). The disruption of the LIC was accompanied by production of an inward current in the dark (see below). At normal (1.5 mM) external Ca2+, M5 had virtually no effect on the LIC (n = 5), apparently due to the presence of excessive amounts of endogenous CaM (25) and production of Ca–CaM upon Ca2+ influx. Thus, under these conditions generation of endogenous Ca–CaM was probably in excess of the maximal amount of M5 (100 μM) that we used. A more direct evaluation of the effect of M5 on the waveform of the LIC was made possible using transgenic Drosophila flies, P[ninaCΔB], in which the endogenous level of CaM in the photoreceptors is markedly reduced (25). Application of M5 to P[ninaCΔB] cells bathed in Ca2+-containing solution, had effects similar to application of M5 into WT photoreceptors bathed in Ca2+-free solution (Fig. 2 A and B, second traces). In P[ninaCΔB] flies, dialysis of the cells with M5 initially enhanced the LIC, producing a “square-like” large photoresponse (Fig. 2 B, second trace, and E) but subsequently caused a total elimination of the photoresponse (Fig. 2B, third trace). This enhancement and subsequent suppression of the LIC by M5 was eliminated by dialysis of the cells with M5 together with Ca–CaM (Fig. 3A, third trace; see below). These results clearly demonstrate that endogenous Ca–CaM is essential for light excitation.
Figure 3.
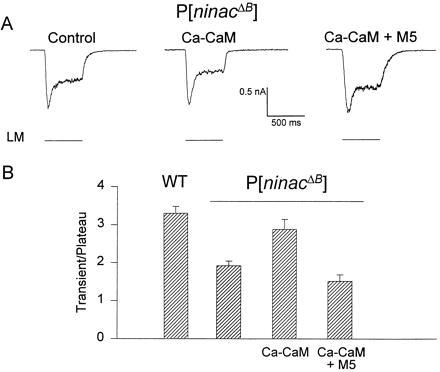
Inhibition of Ca–CaM by M5 in P[ninaCΔB] cells reduces the transient to plateau ratio of the LIC. (A) Ca–CaM (40 μM CaM and 80 μM Ca2+) protected the LIC from disruption by M5 (40 μM M5) in P[ninaCΔB] photoreceptors in normal Ca2+ medium (third trace). Ca–CaM alone lowered the plateau phase (second trace) relative to control (first trace). LM, light monitor. (B) The transient to plateau ratios of the LIC in P[ninaCΔB] cells depends on regulation by Ca–CaM. The peak transients of the LICs were divided by the corresponding plateau values measured 500 ms after light onset (orange light, attenuated by 1 log unit). Geometrical averages of the ratios are presented for each experimental condition as indicated. The recording pipette contained 40 μM CaM and 80 μM Ca2+ (Ca–CaM), or 40 μM M5, 40 μM CaM, and 80 μM Ca2+ (M5 plus Ca–CaM). The averaged ratios are statistically different from each other (P < 0.001). Four to eight cells were used.
Ca–CaM Inhibits Ca2+ Release from Internal Stores and Regulates Light Adaptation.
To examine if M5 causes an excessive release of Ca2+ from internal stores, simultaneous measurements of the LIC and fluorescence of Ca2+ indicator (fluo-3) were conducted in mutant photoreceptors, P[ninaCΔB], bathed in Ca2+-free medium (including 0.2 mM EGTA). To estimate the release of Ca2+ from intracellular stores, the ratio ΔF̄/F̄ was calculated (F̄ is the average fluorescence level reflecting the resting intracellular concentration of Ca2+, [Ca2+]i, and ΔF̄ is the average peak increase in fluorescence reflecting the increase in [Ca2+]i). In agreement with previous studies on WT flies (12–14), the photoreceptors of P[ninaCΔB] flies, bathed in Ca2+-free medium, failed to show light-induced increase in [Ca2+]i above the resting [Ca2+]i (ΔF̄/F̄ = 0.00, n = 4, Fig. 2C). However, internal dialysis for 2 min with M5 (100 μM, same retina) caused a significant increase of [Ca2+]i in response to light (ΔF̄/F̄ = 0.61 ± 0.02, n = 4, Fig. 2D). The LIC, which accompanied the fluo-3 fluorescence during dialysis with M5, was larger and faster than the LIC that was recorded without M5. Similar results were described for WT cells, bathed in Ca2+-free medium during dialysis with M5 (15). The results of Fig. 2 thus suggest that Ca–CaM regulates light-induced Ca2+ release from internal stores.
The decrease of the LIC from transient to plateau is a typical manifestation of light adaptation (35). Fig. 3 shows that the LIC in P[ninaCΔB] flies is characterized by a smaller ratio between the peak transient and plateau phases than WT flies (Figs. 1 and 3) as previously found for ninaC flies (36). However, the basis for this phenotype has not been studied. We found that application of Ca–CaM without M5 decreased the plateau level of the P[ninaCΔB] response and increased the peak transient to plateau ratio, thus, making the LIC of the transgenic flies more similar to that of WT (Fig. 3). Furthermore, dialysis of the cells with M5 together with Ca–CaM had the opposite effect to that of Ca–CaM alone because it made the response more “square-like” and reduced the transient to plateau ratio (Fig. 3). Accordingly, the difference in the waveforms of the LIC between WT (Fig. 1, first traces) and P[ninaCΔB] (Figs. 2 and 3) appears to result from a reduced CaM level in the transgenic flies. This low CaM level may reduce light adaptation by decreasing the inhibitory effect of Ca–CaM on Ca2+ release from internal stores.
To support the above notion, the intensity-response function of P[ninaCΔB] cells, which were internally dialyzed with Ca–CaM, was compared with that curve of untreated cells (Fig. 4). A significant shift of the curve to a range of more intense lights was observed in photoreceptors dialyzed with Ca–CaM, indicating that addition of Ca–CaM confers light adaptation properties to the mutant photoreceptors. In WT photoreceptors, internal dialysis with Ca–CaM had no effect (n = 5), probably because of the larger amount of endogenous Ca–CaM (25, 37). The above results thus demonstrate that Ca–CaM regulates light adaptation.
Figure 4.
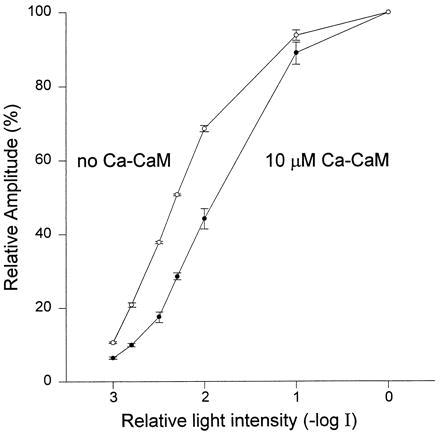
Application of Ca–CaM into P[ninaCΔB] cells shifts the intensity-response function to stronger lights. Normalized peak LIC of cells bathed in normal medium is plotted against increasing intensities of 500-ms white flashes with 1-min interval between test pulses (halogen lamp, which provided transillumination, log I = −2.5 was equivalent to log I = −1.0 of the orange lights used in the other figures). The effect of internal dialysis of Ca–CaM (10 μM CaM and 20 μM Ca2+) was measured 2 min after formation of whole-cell recordings (•, n = 6) and compared with the intensity-response curve measured before application of Ca–CaM (○, n = 6). All the current traces were normalized to the averaged peak amplitude of the untreated cells in response to maximal intensity white light (the averaged maximal amplitude was 6.9 nA for untreated cells and 6.8 nA for treated cells).
Ca–CaM Prevents Ca2+ Store Depletion and Thereby Inhibits Production of an Inward Current.
To measure changes in cellular Ca2+ solely resulting from store depletion by chemical agents and not by light activation of the phototransduction cascade, we used the ninaEoraJK84 mutant. The major fraction of photoreceptors of this mutant do not respond to light because of the absence of a major opsin, Rh1 (38). Therefore, the mutant photoreceptors that were used did not show light-induced Ca2+ release during excitation of the fluorescent Ca2+ indicator. Fig. 5A shows that bath application of ryanodine plus caffeine to ninaEoraJK84 cells bathed in Ca2+-free solution produced a slow inward current, referred to as store-operated current (ISOC) and an increase in fluo-3 fluorescence (Fig. 5G). The increase in fluo-3 fluorescence was due to release of Ca2+ from internal stores because Ca2+ influx was prevented by the Ca2+-free external medium that included EGTA (0.2 mM, 12). The presence of Ca–CaM in the recording pipette prevented both the release of Ca2+ and the induction of ISOC, following bath application of ryanodine plus caffeine (Fig. 5B). Treatment with ryanodine plus caffeine also caused production of ISOC when cellular Ca2+ was maintained at ≈200 nM (14) either by 10 mM EGTA and 5 mM Ca2+ (Fig. 5 C and G), or by 10 mM BAPTA and 6 mM Ca2+ applied through the pipette (n = 4). This procedure prevented an increase in the averaged cellular Ca2+ (Fig. 5 C and G), suggesting that ISOC is not caused by an increase of cellular Ca2+ per se but rather by depletion of the Ca2+ stores. Control experiments in cells perfused with the same Ca2+ buffers, but without application of ryanodine plus caffeine, did not produce ISOC or an increase in fluo-3 fluorescence within a 15-min time interval (n = 4). Fig. 5D shows that application of M5 to ninaEoraJK84 cells bathed in Ca2+-free medium also resulted in an inward current similar to ISOC which was accompanied by a small release of Ca2+ from internal stores (Fig. 5G). The slow onset of ISOC from the time of drug application (Fig. 5A) could be due to a strong negative effect of endogenous Ca–CaM on Ca2+ release from internal stores. This notion was supported by bath application of ryanodine plus caffeine during dialysis with M5, which shortened the onset time of ISOC and Ca2+ release from >1 min (Fig. 5A) to <20 s (Fig. 5E) measured from the time of drugs application. The relatively small increase in cellular Ca2+ observed in Fig. 5 E and G could be due to a slow, undetectable release of Ca2+ caused by M5 before ryanodine plus caffeine application. ISOC was also produced in WT cells, either by bath application of ryanodine plus caffeine (Fig. 5F) or by their application through the recording pipette. The onset of this current was also relatively slow (n = 5). To estimate the release of Ca2+ from intracellular stores by ryanodine plus caffeine or M5 under the various conditions in the ninaEoraJK84 mutant, the geometrical average of the ratios ΔF/F (ΔF̄/F̄) was calculated (Fig. 5G). The results of Fig. 5 suggest that ryanodine plus caffeine, which release Ca2+ and presumably deplete internal Ca2+ stores also induced an inward current (ISOC) in the dark, whereas application of Ca–CaM prevented both the release of Ca2+ from the internal stores and the induction of ISOC.
Characteristics of the Store-Operated Current.
The light-sensitive channels of Drosophila frequently undergo spontaneous uncontrolled activation during prolonged whole-cell recordings, producing inward current in the dark (39). Depletion of the internal Ca2+ stores by ionomycin mimics this uncontrolled activation of the light-sensitive channels in the dark (40). This dark current is characterized by a close to zero reversal potential, inward and outward rectification, a maximal amplitude in a divalent free solution, a dose-dependent inactivation by Ca2+ or Mg2+, and a total block by lanthanum (39, 41, 42).
To test the hypothesis that ISOC is caused by activation of the light-sensitive channels, we compared the main properties of ISOC to those of the above current. The current–voltage relationship (I–V curve) was measured from a family of current traces elicited by voltage steps between −80 mV and plus 100 mV in intervals of 20 mV. The I–V curves were obtained in WT photoreceptors 2–8 min after induction of ISOC by ryanodine plus caffeine (Fig. 6) when the LIC was already disrupted. The I–V curve of Fig. 6 (external [Ca2+] = 0) shows an inward and outward rectification that is typical for the I–V curve of the light-activated channels in cells bathed in Ca2+-free medium during spontaneous activation (41, 43). Fig. 6 also shows that at 1.5 mM external [Ca2+], the inward current was inactivated (41). The reversal potential (Erev), which was measured from the I–V curve, was ≈3 mV (Erev of the LIC in WT is ≈17 mV). Inactivation of the chemically induced current in Ca2+-free medium was also obtained by increasing external Mg2+ from 4 to 11 mM (n = 4; ref. 42). Application of La3+ blocked ISOC in a manner similar to the La3+ block of the spontaneously activated current (n = 4; ref. 39). Bath application of either ryanodine or caffeine may induce inward current in the dark similar to ISOC, but because of their slow action, the resulting current could not be reliably separated from the spontaneous uncontrolled openings of the light-sensitive channels. Our results, therefore, suggest that the current produced in the dark by application of ryanodine plus caffeine (ISOC) is similar to the current produced by the spontaneous uncontrolled activation of the light-sensitive channels, suggesting that ISOC reflects activation of the light-sensitive channels following Ca2+ store depletion with ryanodine plus caffeine or M5.
Figure 6.
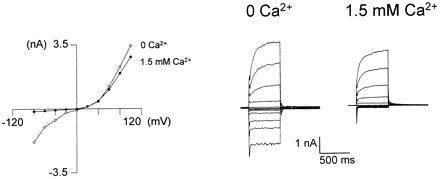
ISOC arises from activation of the light-sensitive channels. Increasing external [Ca2+] inactivated ISOC in WT photoreceptors. The I–V curves were measured 2–8 min after ISOC was induced by ryanodine plus caffeine (4 μM ryanodine and 10 mM caffeine), and examples of curves from a single cell are presented. The pipette solution included ions that blocked K+ channels. Increasing external Ca2+ inactivated the inward current. Current traces were elicited by 500-ms voltage steps (−80 to +100 mV in intervals of 20 mV) in a cell bathed in Ca2+-free medium (at a holding potential of −20 mV; 0 Ca2+, ○) and 2 min after 1.5 mM Ca2+ was added to the bathing solution (1.5 Ca2+, •, recorded from the same cell). The graphs are the current–voltage relationships (I–V curves) measured from the peak amplitudes of the presented currents (right traces, n = 4).
DISCUSSION
Calmodulin Regulation of Ca2+ Release from Ryanodine-Sensitive Stores Is Essential for Light Excitation.
The LIC was rescued by Ca–CaM after ryanodine- or caffeine-induced (but not heparin-induced) disruption of the light response. This rescue was prevented by M5 (15), suggesting that functional ryanodine-sensitive stores are essential for light excitation and that Ca–CaM specifically antagonized the disruption by ryanodine and caffeine (Figs. 1 and 2).
Recent studies in Drosophila have failed to detect light-induced Ca2+ release from internal stores, suggesting that release of Ca2+ from internal stores is not required for visual excitation (13, 14). Our results suggest that this failure is most likely due to a problem in signal detection. The small size of these stores in Drosophila (44, 45) and the negative feedback that is exerted by Ca–CaM on the release mechanism (Fig. 2C) appear to produce only a small and local Ca2+ release. This release is difficult to detect by the integral of the Ca2+ indicator fluorescence that is measured over the entire cell (12–14). The suppression of the photoresponse by ryanodine, caffeine, and M5 is most easily explained by assuming that release of Ca2+ from internal stores is required for light excitation. This release is normally controlled by a negative feedback through Ca–CaM. Accordingly, excessive release by the above agents can lead to a complete depletion of the stores and disruption of the light response. Increasing the negative control by application of exogenous Ca–CaM may curb the excessive release and restore the photoresponse because it prevents exhaustion of the Ca2+ stores.
Ca–CaM Regulates Light Adaptation.
It is well established that light adaptation of invertebrate photoreceptors is mediated by Ca2+ (6, 7, 35). In the present study, we have demonstrated that Ca2+ exerts its action through calmodulin. The action of Ca–CaM on light adaptation can be best illustrated in P[ninaCΔB] photoreceptors in which Ca–CaM is a limiting factor. Accordingly, modulation of the transient to plateau ratio of the LIC (Figs. 2 and 3) and the shift of the intensity-response function to intense lights following application of Ca–CaM (Fig. 4) indicate that Ca–CaM enhances adaptation, whereas M5 antagonizes this effect. We, therefore, suggest that the role of Ca2+ influx in light adaptation of Drosophila is to produce Ca–CaM, which then limits Ca2+ release from internal stores through a negative-feedback mechanism. Such a process has been previously described for skeletal and cardiac muscles (21, 22, 46, 47). Release of Ca2+ from internal stores also creates Ca–CaM complexes that inhibit Ca2+ release, thereby further contributing to the negative-feedback mechanism.
Ca–CaM Prevents Store Depletion That Activates the Light-Sensitive Channels in the Dark.
We found that ryanodine plus caffeine caused release of Ca2+ from internal stores and induced ISOC after a long delay. M5 accelerated the action of ryanodine plus caffeine, whereas Ca–CaM prevented the induction of ISOC (Fig. 5). Production of ISOC, by ryanodine plus caffeine, also occurred when cellular Ca2+ was maintained at 200 nM by Ca2+-EGTA or Ca2+-BAPTA buffers, which prevented the rise in cellular Ca2+ during ISOC. This suggests that the store-depletion process, and not the average rise in cellular Ca2+, is the mechanism that activates the surface membrane channels. The similarity between ISOC and the current resulting from a spontaneous uncontrolled activation of the light-sensitive channels may be due to an accelerated induction of this current by the chemical agents. The effect of thapsigargin, the microsomal Ca-ATPase inhibitor (48), on Drosophila photoreceptors has been previously studied, but no excitatory effect has been found (ref. 13 but see also refs. 40 and 49). However, coexpression of Drosophila TRP and TRPL (but not of each one individually) in Xenopus oocytes resulted in activation of store-operated inward current following Ca2+ store depletion with thapsigargin (49). It is possible that thapsigargin fails to deplete the Ca2+ stores in Drosophila photoreceptors due to an insufficient leak of Ca2+ from the internal stores (13). It should be noted that the physiological function of the store-operated inward current as well as the role of Ca–CaM binding to TRP, TRPL, and possibly other proteins in Drosophila phototransduction require further studies.
CONCLUSIONS
In the present study, the physiological role of Ca–CaM was mainly examined by measuring the LIC—the last step of the phototransduction cascade. Ca–CaM is known to regulate multiple cellular processes; therefore, several stages of phototransduction are probably affected by Ca–CaM. Nevertheless, the simplest hypothesis that explains the results of the present study is that light releases only a small amount of Ca2+ from the d-myo-inositol 1,4,5-trisphosphate-sensitive stores. The resulting modest rise in intracellular Ca2+ triggers Ca2+-induced Ca2+ release (17) from ryanodine-sensitive stores and this amplifies the light response. Hence, amplification through Ca2+ release from internal stores is essential for light excitation. The failure of photolyzed, caged Ca2+ to excite the cells (11) further suggests that a rise in cellular Ca2+ by itself is not sufficient to excite the cells and that store depletion may also be required for excitation. Ca2+-induced Ca2+ release is a powerful mechanism for amplification, but it can easily lead to exhaustion of Ca2+ in the stores, as occurs artificially upon treatment with ryanodine and caffeine. The strong negative effect of Ca–CaM on Ca2+ release is, therefore, an essential safety mechanism to prevent exhaustion of the Ca2+ stores and disruption of the photoresponse. Importantly, Ca–CaM also regulates light adaptation by negative feedback on Ca2+ release. The present study thus strongly suggests a central role for Ca–CaM in negative regulation of Ca2+ release from the ryanodine-sensitive stores; Ca–CaM is essential for light excitation, regulates light adaptation, and keeps the store-operated inward current under a tight control.
Acknowledgments
We thank Drs. Simon Levy and Ze’ev Paroush for invaluable comments on the manuscript and Dr. Y. Salomon for the generous gift of M5. We also thank Dr. A. Peretz and Ms. H. Cohen for help with the experiments. This work was supported by Grant EY03529 from the National Eye Institute and the Minerva Foundation (B.M. and Z.S.) and Grant EY10852 (C.M.) from the the US–Israel Binational Science Foundation and the German Israeli Foundation (B.M.).
ABBREVIATIONS
- CaM
calmodulin, [Ca2+]i, intracellular Ca2+ concentration
- NINAC
neither inactivation nor afterpotential protein
- ISOC
store-operated current
- LIC
light-induced current
- EGTA
ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- BAPTA
1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- WT
wild type
References
- 1.Selinger Z, Doza Y N, Minke B. Biochim Biophys Acta. 1993;1179:283–299. doi: 10.1016/0167-4889(93)90084-3. [DOI] [PubMed] [Google Scholar]
- 2.Pak W L. Invest Ophthalmol Visual Sci. 1995;36:2340–2357. [PubMed] [Google Scholar]
- 3.Ranganathan R, Malicki D M, Zuker C S. Annu Rev Neurosci. 1995;18:283–317. doi: 10.1146/annurev.ne.18.030195.001435. [DOI] [PubMed] [Google Scholar]
- 4.Hardie R C, Minke B. Cell Calcium. 1995;18:256–274. doi: 10.1016/0143-4160(95)90023-3. [DOI] [PubMed] [Google Scholar]
- 5.Minke B, Selinger Z. Curr Opin Neurobiol. 1996;6:459–466. doi: 10.1016/s0959-4388(96)80050-x. [DOI] [PubMed] [Google Scholar]
- 6.Hardie R C. Proc R Soc London Ser B. 1991;245:203–210. [Google Scholar]
- 7.Ranganathan R, Harris G L, Stevens C F, Zuker C S. Nature (London) 1991;354:230–232. doi: 10.1038/354230a0. [DOI] [PubMed] [Google Scholar]
- 8.Hardie R C, Minke B. Neuron. 1992;8:643–651. doi: 10.1016/0896-6273(92)90086-s. [DOI] [PubMed] [Google Scholar]
- 9.Bloomquist B T, Shortridge R D, Schneuwly S, Perdew M, Montell C, Steller H, Rubin G, Pak W L. Cell. 1988;54:723–733. doi: 10.1016/s0092-8674(88)80017-5. [DOI] [PubMed] [Google Scholar]
- 10.Pearn M T, Randall L L, Shortridge R D, Burg M G, Pak W L. J Biol Chem. 1996;271:4937–4945. doi: 10.1074/jbc.271.9.4937. [DOI] [PubMed] [Google Scholar]
- 11.Hardie R C. J Neurosci. 1995;15:889–902. doi: 10.1523/JNEUROSCI.15-01-00889.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peretz A, Suss-Toby E, Rom-Glas A, Arnon A, Payne R, Minke B. Neuron. 1994;12:1257–1267. doi: 10.1016/0896-6273(94)90442-1. [DOI] [PubMed] [Google Scholar]
- 13.Ranganathan R, Bacskai B J, Tsien R Y, Zuker C S. Neuron. 1994;13:837–848. doi: 10.1016/0896-6273(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 14.Hardie R C. J Neurosci. 1996;16:2924–2933. doi: 10.1523/JNEUROSCI.16-09-02924.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arnon A, Cook B, Montell C, Selinger Z, Minke B. Science. 1997;275:1119–1122. doi: 10.1126/science.275.5303.1119. [DOI] [PubMed] [Google Scholar]
- 16.Takeshima H, Nishi M, Iwabe N, Miyata T, Hosoya T, Masai I, Hotta Y. FEBS Lett. 1994;337:81–87. doi: 10.1016/0014-5793(94)80634-9. [DOI] [PubMed] [Google Scholar]
- 17.Walz B, Baumann O, Zimmermann B, Ciriacy Wantrup E V. J Gen Physiol. 1995;105:537–567. doi: 10.1085/jgp.105.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berridge M J. Nature (London) 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 19.McPherson P S, Campbell K P. J Biol Chem. 1993;268:19785–19790. [PubMed] [Google Scholar]
- 20.Ehrlich B. Curr Opin Neurobiol. 1995;5:304–309. doi: 10.1016/0959-4388(95)80042-5. [DOI] [PubMed] [Google Scholar]
- 21.Sitsapesan R, McGarry S J, Williams A J. Trends Pharmacol Sci. 1995;16:386–391. doi: 10.1016/s0165-6147(00)89080-x. [DOI] [PubMed] [Google Scholar]
- 22.Tripathy A, Xu L, Mann G, Meissner G. Biophys J. 1995;69:106–119. doi: 10.1016/S0006-3495(95)79880-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsumoto H, Isono K, Pye Q, Pak W L. Proc Natl Acad Sci USA. 1987;84:985–989. doi: 10.1073/pnas.84.4.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montell C, Rubin G M. Cell. 1988;52:722–757. doi: 10.1016/0092-8674(88)90413-8. [DOI] [PubMed] [Google Scholar]
- 25.Porter J A, Yu M, Doberstein S K, Pollard T D, Montell C. Science. 1993;262:1038–1042. doi: 10.1126/science.8235618. [DOI] [PubMed] [Google Scholar]
- 26.Porter J A, Minke B, Montell C. EMBO J. 1995;14:4450–4459. doi: 10.1002/j.1460-2075.1995.tb00124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phillips A M, Bull A, Kelly L E. Neuron. 1992;8:631–642. doi: 10.1016/0896-6273(92)90085-r. [DOI] [PubMed] [Google Scholar]
- 28.Chevesich J, Kreuz A J, Montell C. Neuron. 1997;18:95–105. doi: 10.1016/s0896-6273(01)80049-0. [DOI] [PubMed] [Google Scholar]
- 29.Klee C B, Vanaman T C. Adv Protein Chem. 1982;35:213–321. doi: 10.1016/s0065-3233(08)60470-2. [DOI] [PubMed] [Google Scholar]
- 30.Meissner G. J Biol Chem. 1986;261:6300–6306. [PubMed] [Google Scholar]
- 31.Furuichi T, Kohda K, Miyawaki A, Mikoshiba K. Curr Opin Neurobiol. 1994;4:294–303. doi: 10.1016/0959-4388(94)90089-2. [DOI] [PubMed] [Google Scholar]
- 32.Frank T M, Fein A. J Gen Physiol. 1991;97:697–723. doi: 10.1085/jgp.97.4.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kennelly P J, Edelman A M, Blumenthal D K, Krebs E G. J Biol Chem. 1987;262:11958–11963. [PubMed] [Google Scholar]
- 34.Blumenthal D K, Charbonneau H, Edelman A M, Hinds T R, Rosenberg G B, Storm D R, Vincenzi F F, Beavo J A, Krebs E G. Biochem Biophys Res Commun. 1988;156:860–865. doi: 10.1016/s0006-291x(88)80923-9. [DOI] [PubMed] [Google Scholar]
- 35.Lisman J E, Brown J E. J Gen Physiol. 1972;59:701–719. doi: 10.1085/jgp.59.6.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hofstee C A, Henderson S, Hardie R C, Stavenga D G. Visual Neurosci. 1996;13:897–906. doi: 10.1017/s0952523800009147. [DOI] [PubMed] [Google Scholar]
- 37.de Couet H G, Jablonski P P, Perkin J L. Cell Tissue Res. 1986;244:315–319. [Google Scholar]
- 38.O’Tousa J E, Leonard D S, Pak W L. J Neurogenet. 1989;6:41–52. doi: 10.3109/01677068909107099. [DOI] [PubMed] [Google Scholar]
- 39.Hardie R C, Minke B. J Gen Physiol. 1994;103:389–407. doi: 10.1085/jgp.103.3.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hardie R C. Cell Calcium. 1996;20:315–327. doi: 10.1016/s0143-4160(96)90037-8. [DOI] [PubMed] [Google Scholar]
- 41.Hardie R C, Minke B. J Gen Physiol. 1994;103:409–427. doi: 10.1085/jgp.103.3.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hardie R C, Mojet M H. J Neurophysiol. 1995;74:2590–2599. doi: 10.1152/jn.1995.74.6.2590. [DOI] [PubMed] [Google Scholar]
- 43.Pollock J A, Assaf A, Peretz A, Nichols C D, Mojet M H, Hardie R C, Minke B. J Neurosci. 1995;15:3747–3760. doi: 10.1523/JNEUROSCI.15-05-03747.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsumoto Suzuki E, Hirosawa K, Hotta Y. J Neurocytol. 1989;18:87–93. doi: 10.1007/BF01188427. [DOI] [PubMed] [Google Scholar]
- 45.Hardie R C, Minke B. Trends Neurosci. 1993;16:371–376. doi: 10.1016/0166-2236(93)90095-4. [DOI] [PubMed] [Google Scholar]
- 46.Meissner G, Henderson J S. J Biol Chem. 1987;262:3065–3073. [PubMed] [Google Scholar]
- 47.Fuentes O, Valdivia C, Vaughan D, Coronado R, Valdivia H H. Cell Calcium. 1994;15:305–316. doi: 10.1016/0143-4160(94)90070-1. [DOI] [PubMed] [Google Scholar]
- 48.Jackson T R, Patterson S I, Thastrup O, Hanley M R. Biochem J. 1988;253:81–86. doi: 10.1042/bj2530081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gillo B, Chorna I, Cohen H, Cook B, Manitersky I, Chorev M, Arnon A, Pollock J A, Selinger Z, Minke B. Proc Natl Acad Sci USA. 1996;93:14146–14151. doi: 10.1073/pnas.93.24.14146. [DOI] [PMC free article] [PubMed] [Google Scholar]