

Abstract
In both human and Old World primates visual information is conveyed by two parallel pathways: the magnocellular (M) and parvocellular (P) streams that project to separate layers of the lateral geniculate nucleus and are involved primarily in motion and color/form discrimination. The present study provides evidence that retinal ganglion cells in the macaque monkey embryo diverge into M and P subtypes soon after their last mitotic division and that optic axons project directly and selectively to either the M or P moieties of the developing lateral geniculate nucleus. Thus, initial M projections from the eyes overlap only in prospective layers 1 and 2, whereas initial P projections overlap within prospective layers 3–6. We suggest that the divergence of the M and P pathways requires developmental mechanisms different from those underlying competition-driven segregation of initially intermixed eye-specific domains in the primate visual system.
Keywords: axon guidance, neuronal specificity, retina, lateral geniculate nucleus, macaque monkey
The segregation of motion and feature vision is a pervasive attribute of primate brain organization at all levels of the neuraxis, from the retina to the frontal lobe (1, 2). In Old World primates motion and color/form discrimination is carried out by separate, magnocellular (M) and parvocellular (P) neuronal pathways that originate in the retina and project to separate M- and P-dedicated layers of the lateral geniculate nucleus (LGN) situated in the thalamus (refs. 3 and 4; Fig. 1). This segregated information then is transmitted from the LGN to M- and P-related sublayers and modules in the visual cortex (5–8). Although the details of the physiology have not been agreed on, it is generally accepted that neurons belonging to these separate streams can be distinguished on the basis of their time of origin (9), morphology (10, 11), connectivity (12, 13), biochemistry (14, 15), and different signaling molecules (16, 17).
Figure 1.
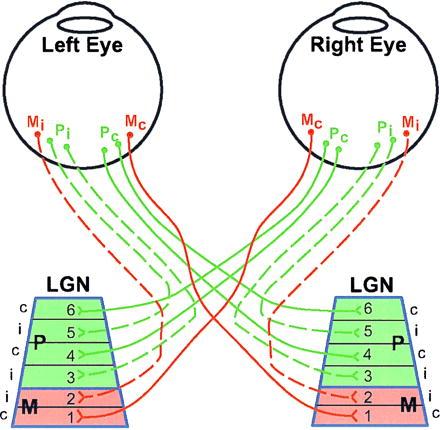
Schematic illustration of neuronal connections between the eyes and the LGN in the macaque monkey. The connections subserving the left and right eyes might be either parvocellular (P, about 80%), magnocellular (M, about 10%) or others (K, I, or S about 10%, not shown). The different types of ganglion cells in the retina are intermixed, although the percentage of P cells is higher in the central retina than at the periphery. In the LGN, layers 1 and 2 are magnocellular (red), and layers 3–6 parvocellular (green). The set of M ganglion cells in a given eye that project to the contralateral LGN terminate in layer 1, whereas those that project ipsilaterally terminate in layer 2. The P ganglion cells project to layers 3 and 5 to the LGN on the same side, and 4 and 6 on the opposite side. For more details see ref. 1–7.
In spite of the significance of the separation of M and P systems for understanding the functional anatomy of the human brain, surprisingly little is known about their development. In contrast, the development of ocular dominance pathways, which transmit information from the eyes via separate synaptic chains to the cerebral cortex, has been extensively studied and used as a premier model system for understanding principles governing the segregation of neuronal connections in the mammalian brain (18–21). A major step in understanding the development of this system was made in the 1970s when the injection of radioactive axonal tracers into the eyes of macaque embryos revealed that visual connections from the eyes initially overlap in the LGN (22, 23). The subsequent discovery that optic axons are more numerous during this early period of overlap than in adults (24), and that prenatal enucleation results in the maintenance of a widespread projection from the remaining eye (25), demonstrated that competitive interactions play an important role in the segregation of initially overlapping retinal projections. These findings, as well as the seminal studies of Hubel, Wiesel, and LeVay (26) on visual deprivation in neonatal primates, led to the concept of activity-dependent regulation of connections subserving binocular vision in a variety of mammalian species (21, 27, 28) and serve as a choice model system for the study of formation of connections in mammals (19, 21, 29, 30). Inexplicably, however, neither prenatal monocular nor binocular enucleation in the monkeys prevented the formation of M and P subsystems in the LGN (25) and the striate cortex (31–35), suggesting that the early divergence of visual projections into M and P functional channels involves activity-independent molecular mechanisms, as has been recently proposed for other systems (36–39).
MATERIALS AND METHODS
Timed-pregnant monkeys were obtained from the New England Regional Primate Research Center (Southborough, MA) and California Primate Research Center (Davis, CA). The gestational ages of the embryos were routinely assessed by ultrasonographic measurements and at the appropriate stage harvested by Caesarean section under general anesthesia in accordance with National Institutes of Health guidelines.
For the immunocytochemical analysis animals were delivered at embryonic (E) days 47, 60, 68, 82, 93, 113, and 120 of the 165-day gestation period in this species. Eyes were rapidly enucleated and placed in chilled PBS. The retina was carefully dissected from other tissue layers, frozen at −70C°, cryosectioned (8–10 μm), and maintained at 20° until the time of processing. Adjacent cryosections were incubated overnight in Brn-3a and Brn-3b antibody (40–42), followed by secondary antibodies conjugated to horseradish peroxidase and processed for visualization of diaminobenzidine reaction product. Immunoreacted retinae were examined on a light microscope coupled to a computer equipped with an image-grabbing board. The pattern of labeled ganglion cells in selected segments of central and peripheral retinae was recorded on a video camera and photographed for illustration.
Study of the neuronal connections was carried out on fetal rhesus monkeys of the following ages: E48, E53, E64, E74, E95, E115, and E135 (gestational period, 165 ± 2 days). The specimens were overdosed with barbiturate and transcardially perfused with heparinized, buffered saline followed by fixative, with the exception of the E48 and E53 embryos, which were immersion fixed. Special care was taken during later dissection not to distort the developing optic nerves, chiasm, and optic tracts when exposing the ventral and dorsal aspect of the brain. Carbocyanine dye crystals (DiI: 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocyanine perchlorate, Molecular Probes) were implanted into the optic nerve to examine the crossed and uncrossed retinal projections. For the older specimens (from E74 onward), due to the increasing distance from the eye to the brain, DiI implants were made into the optic tract. After an appropriate storage time in fixative at 37°C, the brains were embedded in 5% agar and cut coronally at 100- to 200-μm thickness. Brain sections mounted onto gelatinized slides subsequently were counterstained with a solution of 0.0025% bisbenzimide in distilled water and coverslipped before visualization. To increase resolution, labeled retinal axons were viewed using a laser scanning confocal microscope equipped with a filter set matching the excitation-emission characteristics of DiI (MRC 600, Bio-Rad). Sets of optical sections (z-series) were collected through brain sections, and the resulting series of digitally recorded images were reconstructed into a single image. Photomontages were constructed using a computer imaging program (Adobe Photoshop).
RESULTS
Early Divergence of M and P Retinal Ganglion Cells.
Targeted disruptions of the Brn-3 genes in knockout mice resulted in the absence of ganglion cell subpopulations, indicating that these molecules play an essential role in the development of specific ganglion cell types (40, 41). Consistent with these results in rodents, Brn-3a and Bn-3b in the adult macaque monkey are selectively expressed by subsets of retinal ganglion cells that project to the M and P layers of the LGN (42). Thus, in the adult macaque monkey retina, the Brn-3a antibody labels heavily about 10% of P-type ganglion cells and labels lightly approximately 90% of M cells. In contrast, the Brn-3b antibody stains all P-type cells heavily and M-type cells lightly (42). Although these two antibodies cannot be used to determine unequivocally whether an individual ganglion cell belongs to the M or P class, they do provide a reliable assay for assessing the presence of these two separate populations in the retina as a whole. Moreover, the differential expression of the Brn subtypes provides the only available means for assessing diversity among ganglion cells in the embryonic retina. With these considerations in mind, we addressed two questions: (i) when are Brn-positive-staining cells first observed in the embryonic macaque retina; and (ii) at what age does the differential staining pattern correspond to that found in the mature retina?
Brn-3a and Brn-3b positive cells were clearly evident in retinae at all embryonic ages. In the youngest specimen (E47), immunoreactive cells were observed both in the ganglion cell layer and the migratory zone (Fig. 2 a and b). In E68-E82 embryonic retinae, labeling in the central regions was confined to the round-shaped cells of the ganglion cell layer (Fig. 2 c and d). However, in the less mature peripheral regions of these retinae, Brn antisera continued to stain spindle-shaped cells that appeared to be migrating from the proliferative neuroepithelium toward the ganglion cell layer (Fig. 3 b and d).
Figure 2.
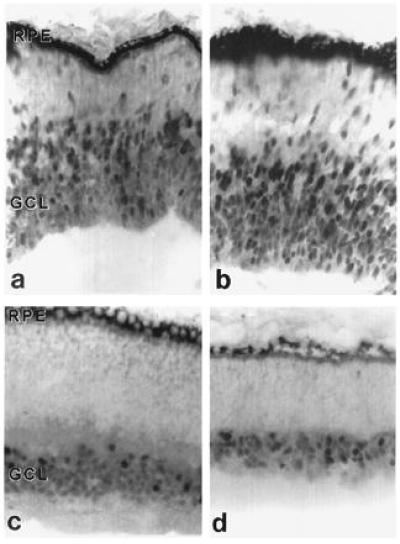
Cross sections of the central retinae of E47 (a and b) and E68 (c and d) macaque embryos immunostained with an antibody generated against Brn-3a (a and c) and Brn-3b (b and d). Most labeled cells in the E47 specimen are situated in the ganglion cell layer (GCL) (a and b), but some appear to be in the process of migrating from their sites of origin near the retinal pigment epithelium toward the GCL. At E68, neurogenesis for the central region of the retina has been completed and all labeled cells are situated within the GCL (c and d).
Figure 3.
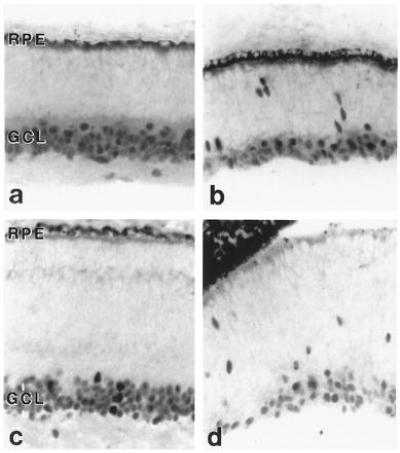
Cross sections of the macaque retinae at E68 (a and b) and E82 (c and d) immunostained with an antibody for Brn-3b. In the more mature central regions, labeled cells are situated exclusively in the ganglion cell layers (GCL) (a and c). However, in the immature, peripheral regions of the retina (b and d), cohorts of migrating immunopositive cells appear to be streaming from the neuroepithelium, near the retinal pigment epithelium to the GCL.
Adult-like patterns of light and heavy labeled cells were observed with either Brn-3a or Brn-3b antibodies throughout the embryonic period, suggesting that POU (the gene complex that comprises the family of Brn transcription factors) domain proteins are expressed differentially in separate ganglion cell classes from the onset of retinal development. An alternative explanation for these results may be that differences in labeling intensity signify a cell’s transition from an immature to mature state. However, our observations do not support this possibility. First, the same differential pattern of labeling was found both before and after the arrival of cells at the ganglion cell layer. Even migrating cells situated close to their origin in the proliferative zone could be classified into heavily and lightly labeled categories, indicating that the two populations are separated soon after their last cell division (Fig. 2). Furthermore, if light cells transform into dark cells during the course of development, we should observe an increasing percentage of darkly labeled cells at later embryonic ages. However, we did not observe an overall increase in the number of darkly labeled cells. Rather, as described below, the change in labeling pattern is entirely consistent with the emergence of regional density differences that characterize M and P ganglion cells in the mature primate retina.
Based on the distribution of heavily and lightly labeled subsets of Brn-3b positive cells, the ratio of M and P ganglion cells changes systematically during development at various retinal eccentricities. Between E47 and E82, the density of M ganglion cells in central retinal regions was equal to or slightly higher than P cell density (Fig. 4 b–d). By E113-E120, however, the very same regions were dominated by P cells (Fig. 4a). It may be significant that the period of decrease in the proportion of central M cells corresponds to the onset of the rapid loss of retinal ganglion cells through apoptosis (43). The initial ratio of M and P cells in central retina may be altered in favor of P cells by the selective elimination of some M ganglion cells, possibly in response to a change in the number of M-dedicated neurons in the LGN by E80 (44). This hypothesis is supported by the fact that a proportionally larger number of M than P cells is eliminated from the monkey LGN during the course of normal development (44).
Figure 4.
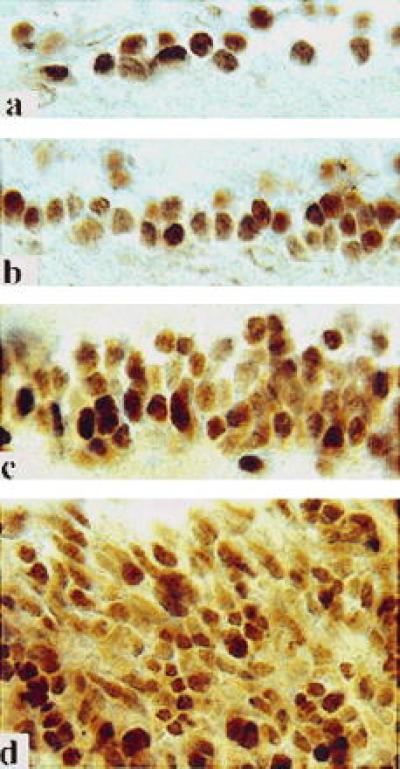
Representative sections of the central retinae from the E113 (a), E82 (b), E68 (c), and E47 (d) embryo immunostained with Brn-3b antibodies to illustrate proportions of heavily and lightly reacted cells at various embryonic ages.
Differentiation of embryonic cells to a generic ganglion cell phenotype minutes after their final mitotic division has been observed in the avian and rodent retina using a variety of markers (45–47). Our detection of the early and selective expression of Brn-3a and Brn-3b in the embryonic monkey retina extends these findings by showing that, in primates, ganglion cells have the capacity to differentiate into M and P neuronal subtypes shortly after being generated. Members of the POU family of homeodomain-containing proteins are good candidates for establishing specific cellular phenotypes because they act as transcription factors and are capable of inducing changes in basic cellular programs (40, 41). As indicated below, such early divergence of ganglion cell subtypes may be an essential step in development of M and P pathways, enabling subtype-specific axons to identify appropriate M and P domains in the LGN.
Specificity of Retinogeniculate Projections in Embryo.
Previous studies on the development of retinogeniculate pathways in macaque embryos using intraocular transport of radioactive tracers demonstrated a complete overlap of ipsilateral and contralateral projections between E64 and E78 (22, 23). In the present study, we made implants of DiI crystals into the optic nerve of fixed embryos at an earlier stage, beginning at E48, which is only a few days after retinal axons begin to navigate across the optic chiasm (48). At this age, crossed ganglion cell axons already have reached the anlage of the LGN (Fig. 5A) and all geniculate neurons have been generated and have completed their migration (49). However, despite the availability of thalamic target neurons, the initial contingent of retinal fibers bypasses the dorsal thalamus to innervate the retinorecipient zones of the midbrain. Several days later, retinal fibers coursing through the LGN anlage begin to sprout short branches that terminate selectively in the medial segment of the nucleus (Fig. 5B). With continued development, there is robust innervation of the medial segment of the geniculate, with very few fibers terminating in its lateral aspect (Fig. 5C). This highly selective innervation of the LGN is displayed by both crossed and uncrossed retinal axons, although the ingrowth of ipsilateral fibers lags at least 5 days behind. At still later stages of gestation, the LGN anlage rotates so that the medial and lateral segments achieve a dorsal-ventral orientation (49) (Fig. 5 D-E). During the course of this rotation, significant retinal innervation of the ventral region of the LGN, formerly the lateral segment, was first observed at E74 (Fig. 5D), nearly a month after fibers colonized the medial segment. Because lamination of the geniculate does not occur until about E91 (49), a precise boundary between M and P laminae is not directly visible at that age. However, based on the outside-to-inside temporal generation sequence of LGN neurons (49), it can be inferred that the early-innervated medial segment corresponds to the region that will form the P layers, whereas the later-innervated lateral segment differentiates into the M laminae.
Figure 5.
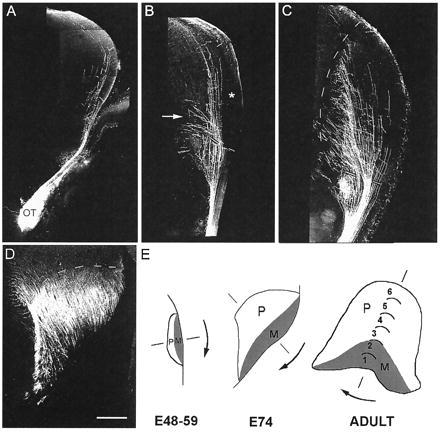
Photomontages of confocal images through representative coronal sections of the diencephalon showing the distribution of DiI-labeled retinal axons in the embryonic thalamus. (A–D) Dashed lines indicate the border of the dorsal lateral geniculate (dLGN) with a dorsal orientation to the top and a lateral orientation to the right. (A–C) The contralateral side to the DiI optic nerve implants. (A) At E48, retinal axons navigate the contralateral optic tract before being deflected away from the pial surface as they approach the geniculate. A few axons course dorsally past the dLGN toward the midbrain. (B) At E53, there is greater ingrowth of retinal fibers, some of them elaborating medially directed branches. Note that the lateral aspect of the dLGN remains totally devoid of retinal axons (∗). Branches derived from the axon trunks are concentrated at the bottom third of the dLGN, and in some cases these extend past its medial border in the external medullary lamina (arrow). (C) At E64, an increasing number of axonal branches invade the medial region of the nucleus. Note that, at this age, the lateral segment of the dLGN is still virtually free of retinal afferents. The section shown is from the rostral part of the dLGN, but essentially the same pattern is observed throughout the rostro-caudal extent of the nucleus. (D) At E74, DiI crystals were implanted into the optic tract to reduce the distance of diffusion. Virtually the entire extent of the nucleus now receives a retinal innervation. The coronal section is from the caudal aspect of the dLGN. (E) Schematic representation of the dLGN rotation (arrows) from E48 to adulthood. The presumptive M layers (shaded area) rotate from a lateral to a ventral position whereas the presumptive P layers rotate from a medial to a dorsal position (46). (Scale bar: A–C, and E, 500 μm and D, 400 μm.)
In adult primates, M and P retinogeniculate arbors are characterized by distinct morphologies (8). Remarkably, clear differences were evident in the present study between retinogeniculate axons innervating the M and P moieties from the time that terminal arbors first became elaborated. For instance, the first retinal terminal arbors were formed in the P region of the LGN by E95, and these were characterized by multiple ramified processes (Fig. 6A), resembling the cluster-like P arbors of mature animals (8). By contrast, retinal arbors in the M layers were much simpler at this age, consisting of only a few short terminal processes (Fig. 6B). Moreover, assessment of the total number of branch points of individual retinogeniculate arborizations revealed that the degree of terminal branching was significantly greater in the early-innervated P segment than in the M segment (Fig. 7). Taken together, the selective innervation of the M and P moieties of the LGN by retinal axons, and the distinctive morphological features of their terminal arbors, demonstrate a highly precise specification of these functional visual subsystems in the primate embryo. Although we cannot rule out that some M and P ganglion cells may initially innervate inappropriate segments of the LGN, these data suggest a highly specific molecular-based recognition by M and P ganglion cells of their respective LGN target neurons.
Figure 6.
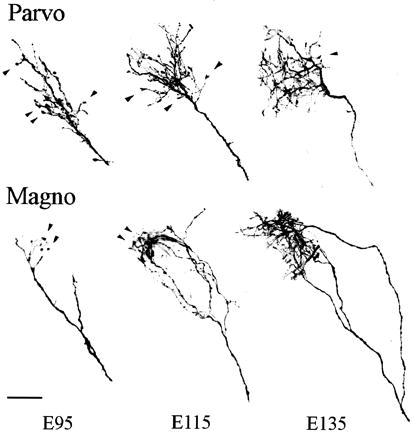
Confocal images of representative terminal retinogeniculate arbors viewed from the coronal plane. Selected arborizations centered on a 200-μm thick section counterstained with bisbenzimide were reconstructed and localized in relation to the border between the M and P layers, viewed under UV illumination. Upper shows retinal axons arborizing in the P compartment, whereas lower illustrates axonal terminals in the M compartment. Arrowheads point to growth cones at the distal end of terminal branches. (Upper) A dense clump of terminal processes is already apparent at E95 in the P region of the dLGN. Increasingly complex arbors, still bearing growth cones, are seen at E115 and E135. (Lower) Long bifurcated axonal branches are apparent in the M region at E95, with higher order processes just beginning to form. From E115 onwards axonal branches emerge gradually from the main trunk and ramify typically in two overlapping clumps of terminals, resembling the morphology of adult M arborizations. (Scale bar = 50 μm.)
Figure 7.
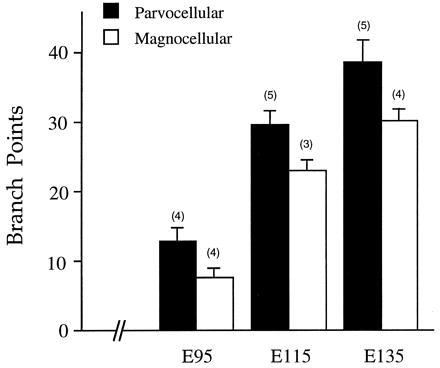
Analysis of terminal arborization complexity of single retinogeniculate axons in M (open bar) and P (solid bar) layers at three fetal ages. The number of arbors analyzed is indicated in parenthesis above each bar, and the vertical bracket denotes the SEM. Note that, at all fetal ages, examined P arbors had a significantly greater number of branch points than M arbors (P < .05 on a t test). During this developmental period there was also a trend for the overall size of P arbors to be larger than M arbors (not shown), but this was not statistically significant.
DISCUSSION
The early and precise specification of the M and P retinogeniculate system that we have demonstrated here stands in contrast to the lack of specificity in initial target selection by the very same projections originating from the left and right eyes, which are intermixed before segregating into appropriate ipsilateral and contralateral layers of the LGN in primates (22) and in other species (50, 51). The selective elimination of initially supernumerary ganglion cells (43) and LGN neurons (25) may contribute to binocular segregation (22, 25, 52). Furthermore, the refinement of retinogeniculate terminal arbors by activity-dependent events also plays a significant role in this process (21, 53).
Although neuronal activity may adjust the ratio of M and P cells in the retina and in the LGN (41), it does not appear to induce neuronal assignments into these two basic functional subsystems. Moreover, at very early stages of development retinal ganglion cells are incapable of discharging action potentials due to their low density of sodium-voltage gated channels (54). Activity-mediated segregation of ganglion cell axons into ipsilateral and contralateral layers of the LGN occurs at much later stages of development and only within the appropriate moiety, so that M retinogeniculate terminals, originating from the left and right eyes, eventually compete only for M layers 1 and 2, whereas P ganglion cell axons compete only for P layers 3 to 6. Thus, it seems reasonable to infer that the selective innervation patterns of M and P layers that we have documented here reflect the expression of laminar-specific molecular markers. In this respect, factors underlying the formation of the M and P channels may be fundamentally similar to those proposed for guiding the selective innervation of cortical layer 4 by thalamic afferents (55–58). Our results also indicate that the development of the functionally distinct visual connections may follow similarly precise molecular markers as observed in the olfactory system (36).
The developmental mechanisms underlying the early formation of separate M and P projections versus the late segregation of initially intermixed binocular projections would seem to reflect the functional differences between these two systems. While the M and P streams are destined to carry different aspects of visual perception, the binocular system is concerned with the allocation of identical input transmitting from each eye to the two sides of the brain. The distinct M and P systems emerge separated from the start. In contrast, the initially intermixed inputs from the two eyes become segregated by competition for available synaptic targets. This may explain why manipulation of retinal input in embryos has different effects on the development of these two visual subsystems. Monocular enucleation in monkey embryos results in dramatically enlarged input and territory of the brain subserving the remaining eye (2, 22). In contrast, neither prenatal monocular nor binocular enucleation prevents the emergence of basic cytoarchitectonic and chemoarchitectonic features of the M and P systems (31–35). These findings in previously enucleated monkeys, coupled with the present results, raise the question of whether the segregation of binocular connections that occurs late in development through competitive interaction provides a general model for the formation of distinct functional connections.
The projections of M and P retinogeniculate projections into separate territories in the LGN are unique to primates, although parallel visual channels exist in other mammalian species (59–61). In particular, studies in the cat have called attention to the role of competitive interactions among different classes of retinogeniculate terminals in refining immature X and Y pathways (62, 63). Such a mechanism may be operative in this species because different cell types are largely intermingled within the A laminae of the geniculate (64, 65). In this respect, the organization of these functional subsystems in primate visual system is different from nonprimate species, even at early stages of embryonic development. The early differentiation and segregation of M and P pathways in an Old World primate demonstrated here is likely to occur during formation of the human visual system.
Acknowledgments
We are grateful to Dr. Jeremy Nathans for the antibodies against Brn transcription factors, Dr. Maria Donoghue for help with affinity purification of the antibodies, and Dr. Oguz Algan for help with illustrations. This work was supported by U.S. Public Health Service Grants EY02593 (P.R.), EY09917 (K.W.), EY03391 (L.M.C.), and Human Frontier Program (C.M.).
ABBREVIATIONS
- M
magnocellular
- P
parvocellular
- LGN
lateral geniculate nucleus: E, embryonic, postconceptual day
- DiI
1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocyanine perchlorate
- dLGN
dorsal lateral geniculate
References
- 1.Fellman D J, Van Essen D C. Cereb Cortex. 1991;1:1–47. doi: 10.1093/cercor/1.1.1-a. [DOI] [PubMed] [Google Scholar]
- 2.Wilson F A W, O’Scalaidhe S P, Goldman-Rakic P S. Science. 1993;260:1955–1958. doi: 10.1126/science.8316836. [DOI] [PubMed] [Google Scholar]
- 3.Livingstone M, Hubel D. Science. 1988;240:740–749. doi: 10.1126/science.3283936. [DOI] [PubMed] [Google Scholar]
- 4.Maunsell J H R. Curr Opin Neurobiol. 1992;2:502–510. doi: 10.1016/0959-4388(92)90188-q. [DOI] [PubMed] [Google Scholar]
- 5.Leventhal A G, Rodieck R W, Dreher B. Science. 1981;213:1139–1142. doi: 10.1126/science.7268423. [DOI] [PubMed] [Google Scholar]
- 6.Merigan W H, Katz L M, Maunsell J H R. J Neurosci. 1991;11:946–952. doi: 10.1523/JNEUROSCI.11-04-00994.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shapley R. Annu Rev Psychol. 1990;41:635–658. doi: 10.1146/annurev.ps.41.020190.003223. [DOI] [PubMed] [Google Scholar]
- 8.Conley M, Fitzpatrick D. Visual Neurosci. 1989;2:287–296. doi: 10.1017/s0952523800001206. [DOI] [PubMed] [Google Scholar]
- 9.Rapaport D H, Fletcher J T, LaVail M M, Rakic P. J Comp Neurol. 1992;322:577–588. doi: 10.1002/cne.903220411. [DOI] [PubMed] [Google Scholar]
- 10.Hockfield S, McKay R D, Hendry S H C, Jones E G. Cold Spring Harbor Symp Quant Biol. 1993;48:877–886. doi: 10.1101/sqb.1983.048.01.090. [DOI] [PubMed] [Google Scholar]
- 11.Rodieck R W. J Comp Neurol. 1985;233:115–132. doi: 10.1002/cne.902330107. [DOI] [PubMed] [Google Scholar]
- 12.Lachica E A, Mavity-Hudson J A, Casagrande V A. Brain Res. 1991;564:1–11. doi: 10.1016/0006-8993(91)91344-z. [DOI] [PubMed] [Google Scholar]
- 13.Fitzpatrick D, Lund J S, Blasdel G G. J Neurosci. 1985;5:3329–3349. doi: 10.1523/JNEUROSCI.05-12-03329.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carrol E W, Wong-Riley M T T. Neuroscience. 1985;15:959–976. doi: 10.1016/0306-4522(85)90246-5. [DOI] [PubMed] [Google Scholar]
- 15.Horton J D, Hocking D R. J Neurosci. 1996;16:1791–1807. doi: 10.1523/JNEUROSCI.16-05-01791.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuljis R O, Rakic P. Visual Neurosci. 1989;2:57–62. doi: 10.1017/s0952523800004326. [DOI] [PubMed] [Google Scholar]
- 17.Mrzljak L, Levey A I, Rakic P. Proc Natl Acad Sci USA. 1996;93:7337–7340. doi: 10.1073/pnas.93.14.7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiesel T N. Nature (London) 1982;299:583–591. doi: 10.1038/299583a0. [DOI] [PubMed] [Google Scholar]
- 19.Rakic P. Trends Neurosci. 1986;9:11–15. [Google Scholar]
- 20.Hubel D. Eye, Brain, and Vision. New York: Freeman; 1988. [Google Scholar]
- 21.Shatz C J. Proc Natl Acad Sci USA. 1996;93:602–608. doi: 10.1073/pnas.93.2.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rakic P. Nature (London) 1976;261:467–471. doi: 10.1038/261467a0. [DOI] [PubMed] [Google Scholar]
- 23.Rakic P. Philos Trans R Soc London. 1977;278:245–260. doi: 10.1098/rstb.1977.0040. [DOI] [PubMed] [Google Scholar]
- 24.Rakic P, Riley K P. Science. 1983;209:1441–1444. doi: 10.1126/science.6828871. [DOI] [PubMed] [Google Scholar]
- 25.Rakic P. Science. 1981;2l4:928–931. doi: 10.1126/science.7302569. [DOI] [PubMed] [Google Scholar]
- 26.Hubel D H, Wiesel T N, LeVay S. Philos Trans R Soc London B. 1977;278:377–409. doi: 10.1098/rstb.1977.0050. [DOI] [PubMed] [Google Scholar]
- 27.Stryker M P, Harris W A. J Neurosci. 1986;6:2117–2133. doi: 10.1523/JNEUROSCI.06-08-02117.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sretavan D W, Shatz C J, Stryker M P. Nature (London) 1988;336:468–471. doi: 10.1038/336468a0. [DOI] [PubMed] [Google Scholar]
- 29.Fregnac Y, Imbert M. Physiol Rev. 1984;64:324–434. doi: 10.1152/physrev.1984.64.1.325. [DOI] [PubMed] [Google Scholar]
- 30.Miller K D, Keller J B, Stryker M P. Science. 1989;245:605–615. doi: 10.1126/science.2762813. [DOI] [PubMed] [Google Scholar]
- 31.Rakic P. Science. 1988;241:170–176. doi: 10.1126/science.3291116. [DOI] [PubMed] [Google Scholar]
- 32.Dehay C, Horsburgh V, Berland M, Killackey H, Kennedy H. Nature (London) 1991;337:265–267. doi: 10.1038/337265a0. [DOI] [PubMed] [Google Scholar]
- 33.Kuljis R O, Rakic P. Proc Natl Acad Sci USA. 1990;87:5303–5306. doi: 10.1073/pnas.87.14.5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kennedy H, Dehay G. Cereb Cortex. 1993;3:171–186. doi: 10.1093/cercor/3.3.171. [DOI] [PubMed] [Google Scholar]
- 35.Rakic P, Lidow M S. J Neurosci. 1995;15:2561–2574. doi: 10.1523/JNEUROSCI.15-03-02561.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Momberts P, Wang F, Dulac C, Chao S K, Nemes A, Mendelsohn M, Edmondson J, Axel R. Cell. 1996;15:675–686. doi: 10.1016/s0092-8674(00)81387-2. [DOI] [PubMed] [Google Scholar]
- 37.Agmon A, Yang L T, Jones E G, O’Dowd D K. J Neurosci. 1995;15:549–561. doi: 10.1523/JNEUROSCI.15-01-00549.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamagata M, Herman J-P, Sanes J R. J Neurosci. 1995;15:4556–4571. doi: 10.1523/JNEUROSCI.15-06-04556.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tessier-Lavigne M, Goodman C S. Science. 1996;274:1123–1133. doi: 10.1126/science.274.5290.1123. [DOI] [PubMed] [Google Scholar]
- 40.Erkman L, McEvilly R J, Lou L, Ryan K A, Hoeshmand F, O’Connel S M, Keithly E M, Rapaport D H, Ryan A F, Rosenfeld M G. Nature (London) 1996;381:6903–606. doi: 10.1038/381603a0. [DOI] [PubMed] [Google Scholar]
- 41.Gan L, Xiang M, Zhou L, Wagner D S, Klein W H, Nathans J. Proc Natl Acad Sci USA. 1996;93:3920–3925. doi: 10.1073/pnas.93.9.3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiang M, Zhou L, Macke J, Yoshioka T, Hendry S H C, Eddy R, Shows T B, Nathans J. J Neurosci. 1996;15:4762–4785. doi: 10.1523/JNEUROSCI.15-07-04762.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rakic P, Riley K P. Nature (London) 1993;305:135–137. doi: 10.1038/305135a0. [DOI] [PubMed] [Google Scholar]
- 44.Williams R W, Rakic P. J Comp Neurol. 1988;272:424–436. doi: 10.1002/cne.902720310. [DOI] [PubMed] [Google Scholar]
- 45.McLoon S C, Barnes R B. J Neurosci. 1989;9:1424–1432. doi: 10.1523/JNEUROSCI.09-04-01424.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trisler D, Rutin J, Pessac B. Proc Natl Acad Sci USA. 1996;93:6269–6274. doi: 10.1073/pnas.93.13.6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wald D K, McLoon S C. Neuron. 1995;14:117–124. doi: 10.1016/0896-6273(95)90245-7. [DOI] [PubMed] [Google Scholar]
- 48.Meissirel C, Chalupa L M. Proc Natl Acad Sci USA. 1994;91:3906–3910. doi: 10.1073/pnas.91.9.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rakic P. J Comp Neurol. 1977;176:23–52. doi: 10.1002/cne.901760103. [DOI] [PubMed] [Google Scholar]
- 50.Linden D C, Guillery R W, Cucchiaro J. J Comp Neurol. 1981;203:189–211. doi: 10.1002/cne.902030204. [DOI] [PubMed] [Google Scholar]
- 51.Sretavan D W, Shatz C J. J Neurosci. 1986;6:234–251. doi: 10.1523/JNEUROSCI.06-01-00234.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chalupa L M, Williams R W. Hum Neurol. 1984;3:103–107. [PubMed] [Google Scholar]
- 53.Hahm J O, Langdon R B, Sur M. Nature (London) 1991;351:568–570. doi: 10.1038/351568a0. [DOI] [PubMed] [Google Scholar]
- 54.Skaliora I, Scobey R P, Chalupa L M. J Neurosci. 1993;13:313–323. doi: 10.1523/JNEUROSCI.13-01-00313.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Molnar A, Blakemore C. Nature (London) 1991;351:475–477. doi: 10.1038/351475a0. [DOI] [PubMed] [Google Scholar]
- 56.Bolz J, Novak N, Staiger V. J Neurosci. 1992;12:3054–3070. doi: 10.1523/JNEUROSCI.12-08-03054.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gotz M, Novak N, Bastmeyer M, Bolz J. Development. 1992;116:507–519. [Google Scholar]
- 58.Antonini A, Stryker M P. J Neurosci. 1993;13:3549–3573. doi: 10.1523/JNEUROSCI.13-08-03549.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rodieck R W. Annu Rev Neurosci. 1979;2:193–225. doi: 10.1146/annurev.ne.02.030179.001205. [DOI] [PubMed] [Google Scholar]
- 60.Stone J. Parallel Processing in the Visual System. New York: Plenum; 1983. [Google Scholar]
- 61.Sherman S M. Progr Psychobiol Physiol Psychol. 1985;11:233–314. [Google Scholar]
- 62.Sur M, Humphrey A L, Sherman S M. Nature (London) 1982;300:183–185. doi: 10.1038/300183a0. [DOI] [PubMed] [Google Scholar]
- 63.Garraghty P E, Sur M. Physiol Rev. 1993;73:529–545. doi: 10.1152/physrev.1993.73.3.529. [DOI] [PubMed] [Google Scholar]
- 64.Cleland B G, Dubin M W, Levick W R. J Physiol. 1971;217:473–496. doi: 10.1113/jphysiol.1971.sp009581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fukuda Y, Stone J. J Neurophysiol. 1974;37:749–772. doi: 10.1152/jn.1974.37.4.749. [DOI] [PubMed] [Google Scholar]