

Summary
During heart development the second heart field (SHF) provides progenitor cells for most cardiomyocytes and expresses the homeodomain factor Nkx2-5. We now show that feedback repression of Bmp2/Smad1 signaling by Nkx2-5 critically regulates SHF proliferation and outflow tract (OFT) morphology. In the cardiac fields of Nkx2-5 mutants, genes controlling cardiac specification (including Bmp2) and maintenance of the progenitor state were up-regulated, leading initially to progenitor over-specification, but subsequently to failed SHF proliferation and OFT truncation. In Smad1 mutants, SHF proliferation and deployment to the OFT were increased, while Smad1 deletion in Nkx2-5 mutants rescued SHF proliferation and OFT development. In Nkx2-5 hypomorphic mice, which recapitulate human congenital heart disease (CHD), OFT anomalies were also rescued by Smad1 deletion. Our findings demonstrate that Nkx2-5 orchestrates the transition between periods of cardiac induction, progenitor proliferation and OFT morphogenesis via a Smad1-dependent negative feedback loop, which may be a frequent molecular target in CHD.
Introduction
The logic of molecular pathways underpinning heart development is poorly understood and little is known about how such pathways are perturbed in congenital heart disease (CHD). Myocytes of the early heart tube derive from at least two distinct cardiac progenitor cell populations. The cardiac crescent and primary heart tube are derived from cells of the first heart field (FHF) that begin differentiation at the crescent stage. The second heart field (SHF) is a population of undifferentiated multipotent cardiac progenitor cells that proliferate and contribute dynamically to heart tube growth at both inflow and outflow poles, providing the majority of myocytes for the right ventricle (RV) and outflow tract (OFT), as well as contributions to the left ventricle (LV) and atria (Buckingham et al., 2005).
A number of factors have been implicated in the development of SHF progenitors, including transcription factors Tbx1, Islet-1 (Isl1), Foxh1, Mef2c and Hand2, and growth factors Fgf8 and Fgf10 (Buckingham et al., 2005). These genes are expressed in SHF cells and most are down-regulated upon SHF cell differentiation, suggesting progenitor-specific roles (Kelly, 2005). The T-box transcription factor Tbx1 is a positive regulator of SHF proliferation (Xu et al., 2004), potentially via direct control of genes for fibroblast growth factors Fgf8 and Fgf10 (Hu et al., 2004; Vitelli et al., 2002; Xu et al., 2004). Isl1, along with Gata4 and Foxh1, controls expression of Mef2c, a transcription factor essential for specification of both cardiomyocytes and endothelial lineages (Dodou et al., 2004; von Both et al., 2004).
The homeodomain factor Nkx2-5 sits high in the cardiac regulatory hierarchy and is expressed in cells of both the FHF and SHF (Stanley et al., 2002). The human gene, NKX2.5, is to date the most commonly mutated single gene in CHD, accounting for 1–4% of specific malformations including disruption of the inter-atrial wall (atrial septal defect; ASD), mal-positioning of the outflow vessels (double-outlet right ventricle; DORV) and a complex congenital condition arising from stenosis of the pulmonary artery termed tetralogy of Fallot (Benson et al., 1999; Elliott et al., 2003; McElhinney et al., 2003; Schott et al., 1998). Studies in mice show that Nkx2-5 is required for specification and spatial definition of chamber myocardium, and for formation and maintenance of elements of the conduction system (Habets et al., 2002; Jay et al., 2004; Lyons et al., 1995; Pashmforoush et al., 2004; Tanaka et al., 1999). However, expression of Nkx2-5 in cardiac progenitor cells suggests uncharacterized roles for this gene at the earliest stages of cardiogenesis.
Here we use microarray to compare the transcriptomes of Nkx2-5 heterozygous and null mutant embryos and identify an important early role for Nkx2-5 as a negative regulator of genes responsible for cardiac induction and the progenitor state. Furthermore, we provide evidence using Nkx2-5 hypomorphic mice that an Nkx2-5/Bmp2/Smad1 negative feedback pathway is a molecular target in CHD.
Results
To map progenitor cell descendants in Nkx2-5 mutant hearts, we examined expression of region-specific transgenes. The Mlc3f-nlacZ-2E transgene normally marks the LV and right atrium in the looping heart (Fig. 1A and Kelly et al., 1997) and expression in the single ventricle-like chamber of Nkx2-5 null embryos (Fig. 1B) suggests that this chamber is a correlate of the normal LV and mainly derived from the FHF. The narrow, short OFT in mutants was negative for the transgene.
Figure 1.
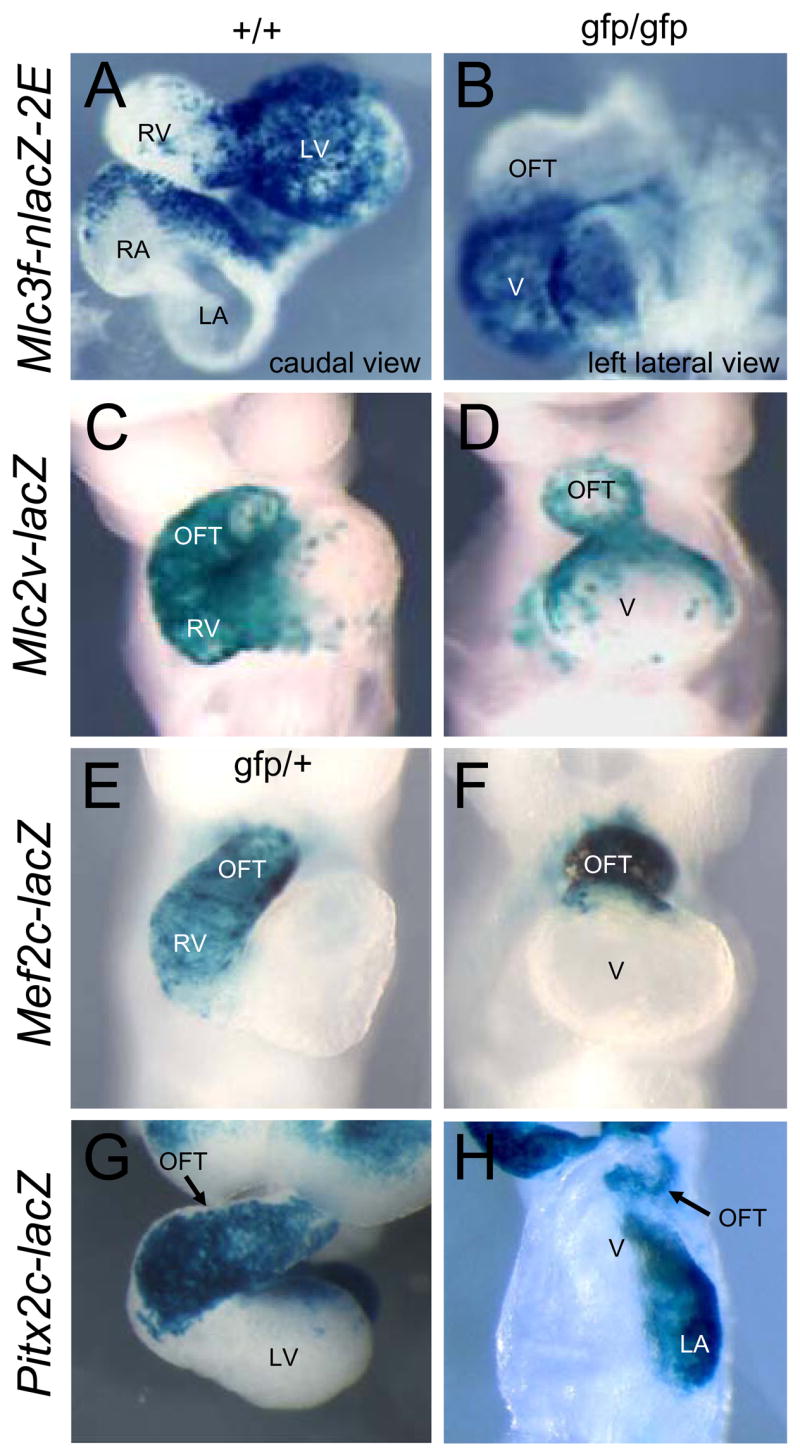
Analysis of FHF and SHF contributions to Nkx2-5 null embryos. A–H. Expression of indicated lacZ transgenes in wildtype (+/+) and Nkx2-5gfp/gfp null (gfp/gfp) embryos at E9.0. Abbreviations: OFT: outflow tract; LV: left ventricle; LA: left atrium; RA: right atrium; RV: right ventricle; V: ventricle-like chamber in null embryos.
To explore the progenitor composition of mutants further, we examined the expression of transgenes Mlc2v-lacZ, Mef2c-lacZ and Pitx2c-lacZ that mark the OFT and RV, derivatives of the anterior SHF (Dodou et al., 2004; Kelly et al., 2001; Ross et al., 1996; Shiratori et al., 2001). All transgenes were expressed in the short outflow region of Nkx2-5 mutant hearts (Fig. 1C–H) showing that this region contains SHF descendants. The Mlc2v-lacZ transgene additionally marks a population of SHF-derived cells at the outflow pole of the LV (Cai et al., 2003; Ross et al., 1996; Verzi et al., 2005) and expression was evident at the outflow pole of the ventricle-like chamber in Nkx2-5 mutants (Fig. 1C,D). This LV SHF contribution was not highlighted by expression of Mef2c-lacZ (Fig. 1E,F) as this transgene is known to be silenced in LV cells (Verzi et al., 2005). Pitx2c-lacZ is expressed in descendants of the left anterior SHF that have been exposed to the left/right morphogen nodal (Shiratori et al., 2001) and reduced expression in the OFT of null embryos (Fig. 1G,H) confirmed that the SHF contribution to the heart tube was compromised. Explants from the SHF region of both wildtype and mutant embryos (Zaffran et al., 2004) expressed Mlc1v-nlacZ-24, marking the SHF (Kelly et al., 2001), but not the LV-specific Mlc3f-nlacZ-2E transgene, and this signature was stable after 48 hrs of in vitro culture (data not shown). These data show that both the FHF and SHF contribute to Nkx2-5 null hearts - a primitive LV is established relatively normally from FHF progenitors (albeit with some delay), but the grossly truncated OFT and indistinct RV highlight a prominent SHF defect.
Microarray profiling of Nkx2-5 null hearts
To identify genes dysregulated in the heart and heart fields of Nkx2-5 mutants, cDNA microarray was performed using cells purified by fluorescence-activated cell sorting (FACS) from embryos carrying an Nkx2-5gfp knock-in allele that expresses enhanced green fluorescent protein (GFP) under Nkx2-5 control (Biben et al., 2000) (Fig. 2A). Sorted cells from single somite-matched embryo pairs (Nkx2-5gfp/+ versus Nkx2-5gfp/gfp) were compared at 9 different stages from E8.0 (5 pairs of somites: ps) to E9.5 (21 ps) (data available from ArrayExpress; accession numbers E-MEXP-533 and -534). Genes with at least a 1.7-fold change in three or more stages were selected (923 of ~16K unique genes). Unsupervised hierarchal clustering (Eisen et al., 1998) correctly ordered all embryonic stages and known Nkx2-5-dependent genes were identified (Fig. 2B). Myofilament protein genes were not selected, confirming that cardiomyocyte differentiation is unaffected in mutants (Lyons et al., 1995).
Figure 2.

Microarray analysis. A. Fluorescence channels 1 (Fl1, 530 nM) and 2 (Fl2, 585 nM) FACS plots of wildtype (+/+), Nkx2-5gfp/+ heterozygous and Nkx2-5gfp/gfp null embryos (both axes log10 scale). Gates define propidium iodide-permeable dead cells (dashed line) and live Nkx2-5-GFP+ cells (solid line). Nkx2-5-GFP+ cells (green) were used for microarray analysis. B. Microarray heat map showing fold-change in mRNA expression between Nkx2-5 null and heterozygous GFP+ cells (see scale). Genes modified at least 1.7-fold at 3 embryonic stages were clustered by standard correlation analysis (see tree diagrams for genes, left, and for embryonic stages, above). Genes known to be down-regulated in Nkx2-5 mutants are arrowed. The prominent cluster of genes up-regulated from 6–21 ps is indicated (*).
Nkx2-5 negatively regulates cardiac progenitor genes
The most numerous and substantial gene expression changes were from 15–21 ps, with functional annotation suggesting a pre-terminal state (see Supp. Fig. 1 legend). We assessed earlier stages and saw up-regulation of a cohort of genes from 6–21 ps in mutants (Fig. 2B). Twenty genes expressed at 8–12 ps were prioritized using a <1/100 false-positive cut-off (Tusher et al., 2001) and their up-regulation was confirmed by quantitative (q) RT-PCR (Supp. Fig. S1A) and in situ hybridization (ISH). Region-specific expression was observed for 9/20 up-regulated genes by ISH. All 9 were expressed in cardiac progenitor cells i.e. precardiac mesoderm and the early crescent, with all bar one (Tbx5) down-regulated in the forming heart tube. Seven genes were previously unrecognized markers for cardiac progenitors. All were expressed, albeit transiently, in the precardiac region and early cardiac crescent, examples being genes for insulin-like growth factor binding protein (Igfbp5), platelet-derived growth factor receptor α (Pdgfra) and transmembrane protein Odz4 (Fig. 3A,B,E,F; Supp. Fig. S2A–C). Later patterns identified novel sub-populations within the SHF e.g. at late crescent stages, genes for Igfbp5, Pdgfra, Odz4, as well as matrix protein tenascin C (Tnc) and homeodomain transcription factor Pbx3, were expressed most strongly in caudal SHF progenitors (Fig. 3A,B,E,F,M,N and Supp. Fig. S2A–D). Tnc was expressed in the lateral reaches of SHF mesoderm (wildtype embryos in Fig. 3M–O), while Pdgfra was expressed more medially in dorsal mesocardium.
Figure 3.

Cardiac progenitor genes/proteins up-regulated in Nkx2-5 null embryos. A–X. Whole mount in situ hybridization and immunohistochemistry with representative sections showing expression of indicated cardiac progenitor cell genes/proteins in wildtype (+/+) and Nkx2-5gfp/gfp embryos at E7.5–E8.5. Regions of up-regulated expression in Nkx2-5 mutants are indicated (arrows, cardiac mesoderm; arrowheads, endoderm).
In Nkx2-5 null embryos, most of these progenitor genes failed to undergo down-regulation in definitive cardiac structures– eg. at E8-8.5 Igfbp5 and Pdgfra expression persisted throughout the entire early heart tube (Fig. 3A–H). We also examined expression of Pdgfra protein. In wildtypes, expression was seen in the cardiac crescent then strongly in caudal progenitors and dorsal mesocardium (Fig. 3I,K), resembling the mRNA pattern (Fig. 3E–G). In null embryos, expression was seen throughout the heart tube (Fig. 3J,L). Expression of Tnc did not persist in the mutant heart tube but was strikingly upregulated in dorsal mesocardium and dorsal pericardial SHF mesoderm (Fig. 3M–P).
Isl1 is a pan-cardiac progenitor marker negatively regulated by Nkx2-5
We examined expression of SHF marker Isl1, not present on our arrays. In wildtype embryos Isl1 transcripts were abundant in the SHF, but undetectable in cardiomyocytes of the crescent and heart tube (Fig. 3Q–S), supporting published data (Cai et al., 2003). However, in Nkx2-5 null embryos, Isl1 expression persisted in cardiomyocytes of the cardiac crescent and heart tube (Fig. 3Q–T). By qRT-PCR, Isl1 mRNA was up-regulated 4-fold in FACS-purified GFP+ cells from Nkx2-5gfp/gfp embryos at 6–12 ps (Supp. Fig. S1A).
In contrast to Isl1 mRNA, Isl1 protein was expressed in E7.5 wildtype embryos throughout the anterior intra-embryonic coelomic walls and proximal head mesenchyme, regions that encompass both the FHF and SHF (Fig. 3U), as well as in ventral and lateral foregut endoderm. Thus, Isl1 is likely to be a pan-cardiac progenitor marker in mouse. By E8.0, Isl1 expression was lost in differentiating cardiomyocytes of the late crescent/forming heart tube (Fig. 3W). In Nkx2-5 null embryos, however, Isl1 levels were increased in anterior coelomic walls and foregut endoderm at E7.5, and Isl1 persisted in cardiomyocytes of the late cardiac crescent and heart tube (Fig. 3V,X). Expression of Mef2c and Tgfβ2, other SHF genes not present on the array, also persisted in the heart tube of Nkx2-5 mutants (Supp. Fig. S2E,F).
Quantitative analysis of cardiac progenitor phenotype in Nkx2-5 mutant embryos
We used the novel cardiac progenitor marker Pdgfra to perform a quantitative FACS analysis of the persistent progenitor phenotype seen in Nkx2-5 null hearts. Nkx2-5 null embryos harbored a ~2-fold increase in the number of Nkx2-5-GFP+/Pdgfra+ progenitor cells from 6–20 ps (Supp. Fig. S3A), attributable to the persistent expression of Pdgfra in the mutant heart tube (Fig. 3E–L). Importantly, mean Pdgfra expression per cell was not altered in Nkx2-5 null embryos (not shown). Other features of this quantitative analysis were noteworthy. In Nkx2-5gfp/+ controls, both cranial and caudal Nkx2-5-GFP+ SHF sub-populations were characterized by ~5-fold lower mean levels of Nkx2-5-GFP compared to differentiating myocytes (inset Supp. Fig. S3B). Furthermore, mean GFP expression per cell was higher in both Nkx2-5-GFP+ and Nkx2-5-GFP+/Pdgfra+ cells isolated from mutants compared to heterozygotes (10–20 ps), suggesting that, as for other progenitor genes, Nkx2-5 is a target of negative feedback regulation (Supp. Fig. 3B–D).
Nkx2-5 limits cardiomyocyte specification
The data above show that Nkx2-5 is essential for repressing early cardiac progenitor genes. We explored whether this repressive activity served to limit cardiac specification. Assessed by FACS, the number of Nkx2-5-GFP+ cells was greater in mutants than in heterozygous controls from 6–15 ps (Fig. 4A). The effect was more pronounced at earlier stages, with ~2-fold more Nkx2-5-GFP+ cells in mutants at 6–8 ps. ISH using a GFP probe suggested expansion of Nkx2-5 expression specifically in SHF cells, i.e. medial to the cardiac crescent at E7.75 and dorso-lateral and anterior to the heart tube at E8.5 (Fig. 4B–D).
Figure 4.
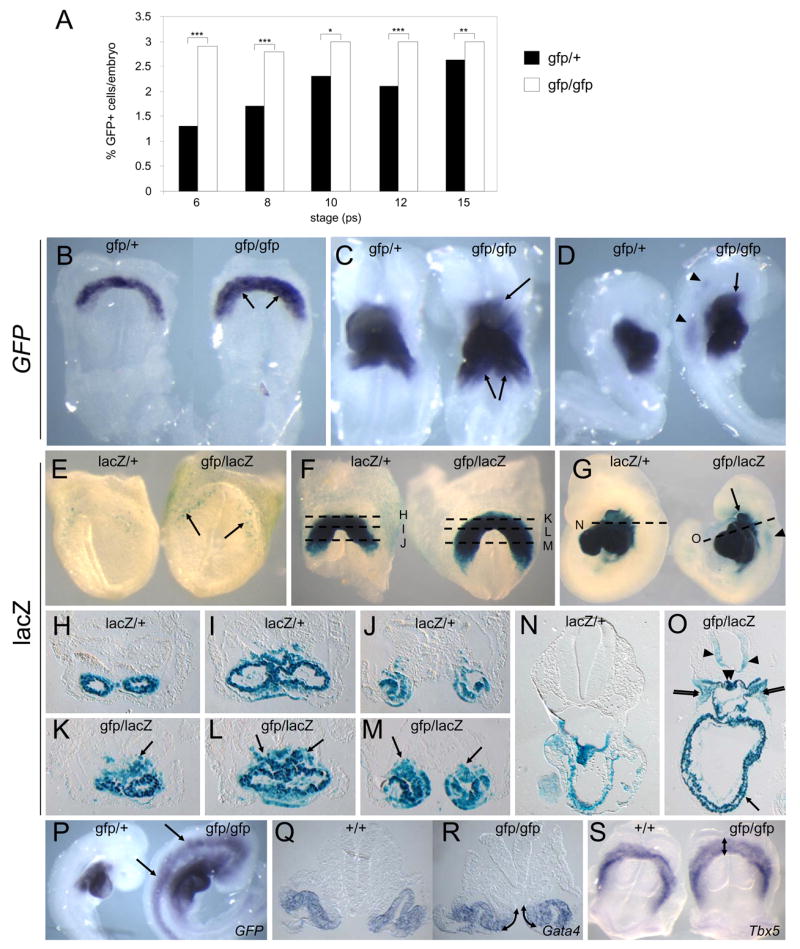
Over-specification of cardiac progenitors in Nkx2-5 null embryos. A. Percentage of Nkx2-5-GFP+ cells/embryo in Nkx2-5gfp/+ control and Nkx2-5gfp/gfp embryos detected by FACS (6–15ps). An average of ~7,200 cells (6–10ps) and 23,000 cells (12–15ps) were counted for each sample. Asterisks show significance by Chi squared analysis (*** p<0.001, ** p<0.01, * p<0.05). B–D. GFP mRNA expression in Nkx2-5gfp/+ and Nkx2-5gfp/gfp null embryos at E7.75 (B) and E8.5 (C,D). Ectopic Nkx2-5-GFP expression is indicated in tissues adjacent to the heart (arrows) and in neural tube (arrowheads). E–O. Xgal staining for Nkx2-5-lacZ expression in Nkx2-5lacZ/+ control and Nkx2-5gfp/lacZ null embryos at E7.5 (E), E8.0 (F and H–M) and E9.5 (G,N,O). Ectopic Nkx2-5-lacZ expression in the cardiac region (arrows), pharyngeal mesenchyme (double arrows), neural tube (arrowheads) and foregut (double arrowheads) is indicated. Dashed lines in F and G indicate planes of sections in H–M and N,O, respectively. P. GFP mRNA expression at E9.0 in Nkx2-5gfp/+ and Nkx2-5gfp/gfp embryos showing induction in neural tube and head mesoderm remote from the cardiac region in mutants. Q,R Expanded expression of Gata4 at E8.0 in the medial wall of the intraembryonic coelom (arrows) in Nkx2-5gfp/gfp embryos. S. Expanded expression of Tbx5 at E7.75 in Nkx2-5gfp/gfp embryos (arrow).
We confirmed an expansion in the number of Nkx2-5-expressing cells using lacZ staining of Nkx2-5lacZ/+ heterozygous and Nkx2-5gfp/lacZ null embryos, matched for dosage of an Nkx2-5-lacZ knock-in allele (Elliott et al., 2006). In null embryos at E8.0 (5 ps), lacZ was more broadly expressed in mesoderm dorsal to the heart tube at all anterior-posterior levels (Fig. 4F,H–M). This finding was not biased by the negative autoregulatory action of Nkx2-5 that became evident only after 10 ps (Supp. Fig. S3B). We examined lacZ expression at the earliest time of its onset in anterior lateral plate mesoderm at E7.5 (Fig. 4E). Even at this stage, expression was evident in more cells in null embryos. Expanded expression of early cardiac transcription factor markers, Gata4 and Tbx5, in null embryos at E7.75–E8.0 (Fig. 4Q–S), confirmed that there was an expansion of the FHF at early stages in mutants. Tbx5 was up-regulated 2-fold in null embryos at E8.0–8.5 by microarray.
Elevated Bmp2 signaling in Nkx2-5 null embryos
Nkx2-5 was also ectopically expressed in the ventral neural tube from ~E8.75, as detected by ISH for GFP and lacZ staining in respective mutant strains, an effect that increased dramatically with further development (Fig. 4D,G,N,O,P and Supp. Fig. S2G). Neural Nkx2-5 expression was not secondary to a pre-terminal state because it was absent in similarly compromised Tbx20lacZ/lacZ null embryos (Supp. Fig. S2H) (Stennard et al., 2005). This suggested the action of a long-range diffusible morphogen accompanying the increased specification of cardiac progenitors in Nkx2-5 null embryos. We therefore examined the expression of known secreted cardiac-inducing factors. Bmp2 and Fgf10 mRNAs were elevated in mutant FACS-purified Nkx2-5-GFP+ cells by microarray and qRT-PCR (3–6 fold and 2–15 fold, respectively; Supp. Fig. S1A,B). By ISH, Bmp2 mRNA was elevated in mutants in the cardiac crescent (E8.0; Fig. 5A–C) and then at the pericardial/myocardial boundaries at the poles of the heart tube (E8.5; Fig. 5D). Bmp4 and Bmp7 were not up-regulated. Bmp signaling through Smad1/5/8 phosphorylation (pSmad1/5/8) was also increased in mutant precardiac mesoderm (E7.75; Fig. 5E,F) then in the fusing heart tube and SHF (E8.0; Fig. 5G–I). Elevated phospho-Smad1/5/8 extended to anterior foregut and visceral endoderm of null mutants (Fig. 5E–I) as well as to neural ectoderm from E8.0–9.5 (Fig. 5H,I). While pSmad was elevated broadly in mutants, Bmp2 up-regulation remained confined to its normal pattern of expression, indicating an enhanced paracrine and indeed long-range effect of mesoderm-expressed Bmp2 on endoderm and ectoderm. Expression of genes encoding Bmp inhibitors, Cer1, Dan and noggin, was not altered in Nkx2-5 mutants (not shown).
Figure 5.
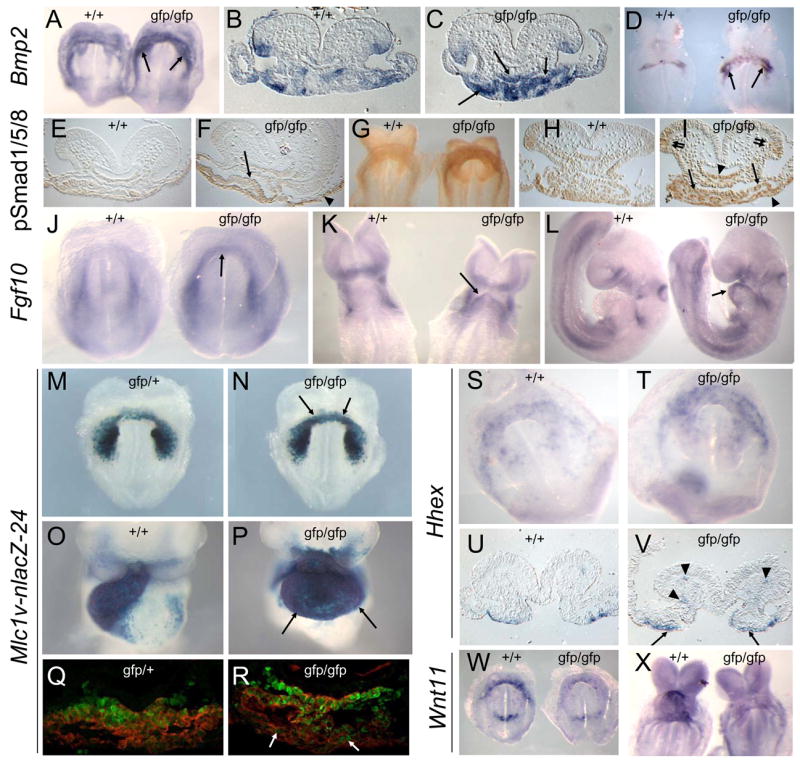
Modified expression of cardiac signaling components in Nkx2-5 null embryos. AD. Bmp2 expression in wildtype (+/+) and Nkx2-5gfp/gfp null embryos at E8.0 (A–C) and E8.5 (D). Elevated expression in mutants arrowed. E–I. Phospho-Smad1/5/8 expression at E7.75 (E,F) and E8.0 (G–I). Elevated expression in mutants indicated in cardiac mesoderm (single arrows), endoderm (arrowheads) and ventral neural tube (double arrows). J–L. Fgf10 expression at E7.5 (J), E8.5 (K) and E9.5 (L), arrows showing ectopic expression. M–P.β-galactosidase activity from the Mlc1v-nlacZ-24 transgene at E8.0 (M,N) and E9.5 (O,P), with arrows showing ectopic expression. Q,R. Co-immuno-detection of β-galactosidase protein from the Mlc1v-nlacZ-24 transgene (green nuclei) and sarcomeric myosin (red) at E7.5 in the cardiac crescent in Nkx2-5gfp/+ control and Nkx2-5gfp/gfp embryos. Arrows indicate expanded expression of β-galactosidase in myosin+ cardiomyocytes in mutants. S–V. Hhex expression at E7.75 showing elevated expression in cardiac endoderm (arrows) and endothelial cells within cranial mesenchyme (arrowheads) of Nkx2-5gfp/gfp embryos. W,X. Wnt11 expression (arrows) is severely reduced in Nkx-2-5 null embryos at E7.75 (Y) and E8.5 (Z).
Fgf10 was also up-regulated and more broadly expressed in the cardiac crescent region of Nkx2-5 mutants at E7.5, as seen by ISH (Fig. 5J), and expression persisted throughout the mutant heart tube (E8.5–9.5; Fig. 5K,L). Fgf8 expression was not elevated (Supp. Fig. S1B). The Mlc1v-nlacZ-24 transgene traps an Fgf10 enhancer expressed in the SHF and in its anterior derivatives, the OFT/RV (Kelly et al., 2001). In mutants, increased β-galactosidase activity was evident in cardiomyocytes of the crescent, then whole heart tube at E9.5 (Fig. 5M–P), similar to findings for Fgf10 mRNA. Double immunostaining of sections from Mlc1v-nlacZ-24 embryos at the late crescent stage showed some overlap between myosin expression, marking the zone of differentiation in FHF and SHF cells, and lacZ expression, which marks the SHF. In Nkx2-5 mutants, there was greater encroachment of β-galactosidase protein expression into the FHF in the cranial region (E7.75; Fig. 5Q,R), indicating a very early effect on Fgf10.
The homeodomain repressor Hhex is expressed in anterior visceral and foregut endoderm, and when over-expressed in frog blastomeres can induce Nkx2-5 expression in adjacent mesoderm (Foley and Mercola, 2005). Hhex was up-regulated in Nkx2-5 mutants in anterior endoderm underlying the cardiac crescent (Fig. 5S–V) as well as in endothelial progenitors within head mesenchyme, again suggestive of the action of a long-range morphogen. Hhex may be induced in mutants by up-regulated Bmp2 via its Smad-responsive enhancer (Zhang et al., 2002). Expression of Dkk1, encoding a canonical Wnt inhibitor that can also induce Hhex (Foley and Mercola, 2005), was unaffected. Cardiogenesis can be stimulated in frog embryos by the non-canonical Wnt pathway ligand, Wnt11 (Pandur et al., 2002). However, Wnt11 was down-regulated in mutants (Figs. 5W,X and 6O,P).
Figure 6.

Nkx2-5 and Smad1 have opposing effects on SHF proliferation and deployment. A,B. Cell proliferation in the SHF of Nkx2-5gfp/gfp mutant versus wildtype control (+/+) embryos (A, E8.0, 6–8ps, C57BL/6 genetic background) and embryos deleted for Smad1 in mesoderm (Mesp1Cre/+/Smad1fl/fl) versus heterozygous deleted controls (Mesp1Cre/+/Smad1fl/+) (B, E8.0, 6–8ps, 75% Quackenbush-Swiss/25% C57BL/6) and for littermate embryos from Nkx2-5+/gfp X Smad1+/− crosses (C, E8.5, 10–12ps, 50% Quackenbush-Swiss/50% C57BL/6). Shown is mean % of phosphohistone H3+ (pHH3) cells +/− standard deviation, n=3 for each genotype. D–F. ISH for Wnt11 mRNA highlighting increased OFT/RV (arrows) size in Mesp1Cre/+/Smad1fl/fl embryos versus Mesp1+/+/Smad1fl/fl and Mesp1Cre/+/Smad1fl/+ controls at E9.5. G–V, Marker analysis of wildtype, Nkx2-5 null, Smad1 null and Nkx2-5/Smad1 double null littermate embryos for Bmp2 (E8.0; G–J), Nkx2-5-GFP (E8.25; sections, K–N), Wnt11 (E8.5; ventral views, O–R; right-hand side views, O′–R′; section through OFT, O″–R″) and Isl1 (E8.25; sections, S–V. Note in Fig. 6L the expanded Nkx2-5-GFP expression in the dorsal pericardium (double arrows), adjacent mesenchyme (arrow), dorsal foregut (arrowhead) and ventral neural tube (double arrowheads) of Nkx2-5/Smad1 double null embryos. In Fig. 6O–R″ Wnt11 expression is indicated in the OFT (arrows) and inflow region (arrowheads) of wildtype (O,O′,O″), Smad1 null (Q,Q′,Q″) and Nkx2-5/Smad1 double null (R,R′,R″) embryos but is absent from the rudimentary OFT of Nkx2-5 null embryos (outlined by dotted lines, P,P′,P″). Persistent Isl1 expression in cardiomyocytes located in the heart tubes of Nkx2-5 null (T) and Nkx2-5/Smad1 double null (V) embryos is indicated (arrows).
Nkx2-5 is required for SHF proliferation through suppression of Bmp2/Smad1 signaling
Impaired proliferation or deployment of SHF cells may underpin the dramatically narrowed and shortened OFT seen in Nkx2-5 mutants. We examined proliferation of SHF progenitors in mutants, scoring for phosphohistone H3. As reported (Tanaka et al., 1999; Tanaka et al., 2001), foregut endoderm proliferation showed a reduction by 44% in mutants (p=0.011; Fig. 6A), but cardiomyocytes were unaffected. However, we also found that proliferation was strikingly reduced in dorsal pericardial SHF cells (80% reduced, p<0.0001) in the absence of effects on cell death (not shown).
The Bmp inhibitor noggin stimulates cell proliferation in chick SHF explants (Waldo et al., 2001). To examine whether Bmp/Smad signaling represses SHF progenitor proliferation in vivo, we deleted loxP-flanked (floxed; fl) Smad1 alleles (Tremblay et al., 2001) specifically in anterior mesoderm, using Mesp1-Cre (Saga et al., 1999). Strikingly, MespCre/+/Smad1fl/fl embryos showed a 2.3-fold increase in proliferation of SHF cells at E8.5 (p<0.0005; Fig. 6B), but no change in myocyte proliferation. Proliferation was unaffected in foregut endoderm (Fig. 6B) that expressed Smad1 (Fig. 5H) but not Mesp1-Cre (Saga et al., 1999) and we conclude, therefore, that the observed effects are not secondary to an altered state of endoderm. Consistent with our hypothesis that SHF proliferation is a driver of OFT/RV morphogenesis, the length (5/7 embryos) or length plus width (2/7 embryos) of the OFT/RV in MespCre/+/Smad1fl/fl hearts was increased at E9.5 (Fig. 6D–F).
We also assessed whether there was a cell autonomous migration defect in Nkx2-5 null SHF cells by examining their ability to be deployed to the OFT/RV in a competitive situation with wildtype cells in embryo chimeras (Supp. Fig. S4). However, we found no evidence for a defect in this assay.
Genetic rescue of OFT formation in Nkx2-5 null embryos
To test genetically whether defective OFT development in Nkx2-5 mutants was due to increased Bmp-Smad signaling, we crossed Nkx2-5 and Smad1 mutant mice. Deletion of one or two Smad1 alleles in Nkx2-5gfp/gfp embryos produced a progressive increase in SHF proliferation (1.5-fold, p=0.059, and 3.1-fold, p=0.004, respectively, compared to Nkx2-5 nulls; Fig. 6C). SHF proliferation in Nkx2-5/Smad1 double null embryos was only 23% less than in wildtype embryos, indicating that most of the anti-proliferative effect of increased Bmp2 was mediated by Smad1. We found no precocious differentiation in the SHF in Nkx2-5 mutants, indicating that the negative effects of increased Bmp2-Smad1 signaling on SHF proliferation were independent of any pro-differentiative activity of Bmps (Supp. Fig. S5). Bmp2 remained up-regulated at the pericardial/myocardial boundary in Nkx2-5gfp/gfp/Smad1−/−embryos (Fig. 6G–J), indicating that Nkx2-5-mediated repression of Bmp2 was independent of Smad1. However, the ectopic expression of Nkx2-5-GFP within the head mesenchyme, dorsal foregut and neural tube of Nkx2-5 mutants was eliminated by Smad1 deletion (Fig. 6K–N). Therefore, upregulated Bmp2-Smad1 signaling can account for elevated cardiac specification and reduced SHF proliferation in Nkx2-5 mutants. We therefore favor the hypothesis that Nkx2-5 acts indirectly to repress progenitor cell specification and proliferation.
In the forming heart, Wnt11 expression marks the inflow and outflow poles (Fig. 6O,O′,O″), which are derivatives of the SHF. At E8.5, Smad1−/− heart tubes were truncated and partially bifid (Fig. 6Q,Q″; M.S, R.P.H., manuscript in preparation). Smad1−/− hearts initially expressed Wnt11 throughout most of the myocardium, suggesting that the primary heart tube in this mutant context is formed predominantly from SHF cells, a possible result of retarded specification of FHF cells and/or increased SHF proliferation. As shown above, Nkx2-5gfp/gfp hearts have a highly compromised SHF-derived component and accordingly showed down-regulated Wnt11 (Fig. 6P,P′,P″). Nkx2-5gfp/gfp/Smad1−/− hearts were also partially bifid, but in striking contrast to Nkx2-5 null hearts had greater morphological development of the outflow region, consistent with improved SHF proliferation and deployment, and prominent Wnt11-positive outflow and inflow poles (Fig. 6R,R′,R″).
We next explored whether the persistent expression of cardiac progenitor genes in differentiated myocytes of Nkx2-5 mutants was also Smad1-dependent. Isl1 was undetectable in the hearts of Smad1−/−embryos, but remained up-regulated in the hearts of Nkx2-5gfp/gfp/Smad1−/− embryos (Fig. 6S–V). Thus, Nkx2-5 represses cardiac progenitor genes through multiple targets.
The Nkx2-5/Bmp2/Smad1 negative feedback loop is a molecular target in CHD
We found that our previously established Nkx2-5-IRES-cre knock-in allele (Stanley et al., 2002) produced ~50% less Nkx2-5 protein than a wildtype allele at E8.0 (data not shown). Nkx2-5 +/IRES-cre and Nkx2-5 IRES-cre/IRES-cre mice were grossly normal, but embryos carrying one Nkx2-5-IRES-cre and one null allele (Nkx2-5gfp/IRES-cre) were hypomorphic and died postnatally, showing a spectrum of cardiac malformations overlapping the more severe defects seen in humans with NKX2-5 mutations (Fig. 7A–D). At mid-late gestation (E14.5 or E17.5; n=29), all hypomorphic hearts showed a ventricular septal defect (VSD), mal-rotation of the OFT vessels and either DORV or a related condition in which the aorta is positioned above a VSD (overriding aorta; OA, Fig. 7A–D,Q). Similar malformations were seen at birth. Other frequent abnormalities included interruption of the inter-atrial septum primum (primum ASD), immature or absent mitral and tricuspid valves, and thin walled ventricles (Fig. 7D,Q–T and Supp. Table S2).
Figure 7.

Genetic rescue of congenital cardiac malformations in Nkx2-5 hypomorphic embryos. A–D. Gross morphology of Nkx2-5 hypomorphic embryos (gfp/IRES-cre) and littermate controls (+/IRES-cre) at E17.5 showing malrotation and rightward displacement of the aorta (dashed lines) and pulmonary artery (solid lines) in hypomorphs (A), with sections from E14.5 embryos showing DORV (C) or OA with large VSD (D). E–H. Morphological analysis of early OFT development showing compromised outflow development (arrows) in hypomorphs. Nkx2-5 expression highlights cardiac structures at E8.5 (E,F). Wnt11 expression marks the RV/OFT at E9.5 (G,H). I–K. Molecular analysis of cardiac progenitor cell defects in Nkx2-5gfp/IRES-cre hypomorphic embryos and Nkx2-5+/IRES-cre controls. Note up-regulated expression of cardiac progenitor cell markers Bmp2 (I, E7.75) and Isl1 (J,K, E7.5) in hypomorphs (arrows, pre-cardiac mesoderm; arrowheads, endoderm). L, Decreased proliferation in the SHF of Nkx2-5gfp/IRES-cre hypomorphic embryos (E8.5, 10–12ps, C57BL/6 genetic background, mean +/− SD, n=3 for each genotype). M–X, Rescue of early OFT defects in Nkx2-5gfp/IRES-cre hypomorphic embryos by deletion of a single allele of Smad1. The immature AV cushions in hypomorphs are indicated by an asterisk (S,T). Abbreviations: Ao: aortic root; ASD: atrial septal defect; DORV: double outlet right ventricle; LV: left ventricle; MV: mitral valve; OA: overriding aorta; PA: pulmonary artery; RV: right ventricle; TV: tricuspid valve; VSD: ventricular septal defect.
Outflow vessel defects seen in Nkx2-5 hypomorphs at mid-gestation were predated by OFT defects in earlier embryos - eg., the OFT was shorter and thinner, and the RV bulge less prominent at E8.5–9.5 compared to Nkx2-5+/IRES-cre controls (Fig. 7E–H). These OFT defects may be a less severe manifestation of those seen in Nkx2-5 nulls. Indeed, at late head fold stages (E7.75), Bmp2 and Isl1 expression were elevated in hypomorphs (Fig. 7I–K), as was Nkx2-5-IRES-cre expression measured by qRT-PCR (1.4-fold increase, p=0.041), the latter indicative of cardiac progenitor over-specification. We also examined proliferation in hypomorphs at E8.5, and found a decrease in the SHF of 51% but no effect on OFT/RV cardiomyocytes (Fig. 7L, p=0.024).
Following logic established for null embryos, we asked whether the OFT/RV defects seen in Nkx2-5 hypomorphs could be rescued by Smad1 deletion. Deletion of one Smad1 allele in the germline, or a floxed Smad1 allele via expression of Cre from the Nkx2-5-IRES-cre allele, was sufficient to increase the size of the OFT and RV swelling at E9.5 (Fig. 7M,N). In conditionally deleted Nkx2-5gfp/IRES-cre/Smad1fl/+ embryos at E14.5, malrotation and rightward displacement of the OFT vessels was largely corrected, and in 11/13 cases the aorta was correctly oriented leftwards and connected directly to the LV (p=0.00024, Supp. Table S2 and Fig. 7O–X). VSD and mitral valve dysmorphogenesis was also less severe in Nkx2-5gfp/IRES-cre hypomorphs with conditionally deleted Smad1 (p=0.0022 and p= 0.018 respectively, Supp. Table S2 and Fig. 7Q–X). RV size was also improved in some embryos (Fig. 7O–X) and in most, maturation of the tricuspid valve was enhanced and an intact atrial septum was restored, although in isolation these results did not reach significance (p=0.053 and p=0.14 respectively, Supp. Table S2). Normal OFT vessel connections, intact atrial septa, and improved RV size, AV valve morphology and VSD severity were also observed in two Nkx2-5gfp/IRES-cre/Smad1fl/fl embryos recovered at E14.5.
Discussion
In Nkx2-5 mutants, the up-regulation of progenitor signature genes in the SHF and the abnormal persistence of expression in differentiating myocytes indicates a major early role for Nkx2-5 in modulating expression of genes associated with cardiac induction and progenitor cell status. The majority of the progenitor genes affected by loss of Nkx2-5 were expressed in both the FHF and SHF. Expression in the FHF was extremely transient, reflecting the early differentiation of these progenitors in the cardiac crescent. For example, Isl1, previously thought to be SHF-specific, was expressed throughout the intra-embryonic coelomic lining at E7.5, which contains components of both progenitor fields, and is therefore likely to be a pan-cardiac progenitor cell marker in mice. Transient FHF Isl1 expression may account for the dearth of marked FHF descendents in a Cre-fate map (Cai et al., 2003). The expression patterns of progenitor markers define new sub-populations and/or their behavioral states. In the SHF, Tnc, Pdgfra and Igfbp5 were expressed most highly in caudal cells, with expression of Pdgfra restricted to its medial portion (dorsal mesocardium) and that of Tnc to the most lateral regions. In contrast to skeletal muscle development where proliferation ceases as differentiation begins and for which there are relatively clear molecular signatures for progenitors (Pax3/7), myoblasts (MyoD, Myf5) and myocytes (myogenin and contractile proteins), cardiac cells continue to proliferate during differentiation. It is possible that some of the identified markers will reflect analogous progressions in the cardiac lineage, although the distinction between progenitor, blast and differentiated states seem less distinct in cardiac muscle.
An Nkx2-5/Bmp2/Smad1 negative regulatory circuit limits cardiac specification
We attribute both over-specification and proliferative failure of cardiac progenitor cells in Nkx2-5 mutants to an over-active Bmp2/Smad1 pathway. Several observations suggested involvement of a long-range diffusible morphogen in the spectrum of defects seen in Nkx2-5 mutants, including up-regulation of Bmp2 in the cardiac fields and increased levels of phospho-Smad1/5/8 in and beyond the fields. Bmp2-related factors are cardiac-inducing molecules in vertebrates and invertebrates, with Nkx2-5 and its Drosophila homologue tinman being direct transcriptional targets of Bmp-Smads (Liberatore et al., 2002; Lien et al., 2002). Therefore, an early and possibly direct function for Nkx2-5 in the cardiac developmental program is to dampen expression of Bmp2, its immediate upstream inducer. Preliminary data show that multiple predicted enhancers of Bmp2 are normally occupied by Nkx2-5 in cardiac cells and these warrant further investigation.
The Bmp repressor noggin can stimulate proliferation of SHF cells in vitro (Waldo et al., 2001) and, as shown here genetically, Smad1 is a critical negative regulator of SHF proliferation in vivo. In contrast to the FHF, SHF cells undergo an extended period of mitotic expansion lasting several days prior to, and overlapping with, their deployment to the OFT/RV and differentiation. The decreased proliferation of the SHF in Nkx2-5 mutants likely accounts for the progressive normalization in numbers of Nkx2-5-GFP+ cells following their initial over-specification. A model for the feedback loop involving Nkx2-5, Bmp2 and Smad1, and its role in cardiac specification, progenitor gene expression and proliferation is presented in Supp. Fig. S6. In the context of developmental time, the repressive action of Nkx2-5 on Bmp2/Smad1 orchestrates the transition between a period of cardiac induction and one of progenitor proliferation, a negative feedback that is likely to be a fundamental property of cardiac developmental circuitry in all vertebrates. Given that Nkx2-5 levels increase from the progenitor to the differentiated state, the negative feedback pathway may be an example of how the number of progenitor cells in a field is controlled by the degree of differentiation – a paradigm that could apply to adult stem cells and organ regeneration. In this paradigm, Nkx2-5 acts as both the sensor of differentiation, and as the arbiter of repression. Because the negative feedback loop balances the number of cardiac progenitors through opposing effects on induction and proliferation, it is self-regulating and thereby shows features of robustness, an essential component of developmental systems.
Co-expression of genes defining cardiac progenitor cells, including Isl1, with cardiomyogenic genes in Nkx2-5 null heart tubes demonstrates that Nkx2-5 also plays a critical role as a molecular switch in ensuring spatial and temporal discrimination of the progenitor and differentiating states. However, persistent expression of Isl1 (and presumably other progenitor genes) in the forming heart tube of Nkx2-5 mutants was not rescued by Smad1 deletion, suggesting that Nkx2-5-dependent negative feedback targets multiple sets of genes with distinct functional consequences for heart development.
A cardiac progenitor cell defect may underlie NKX2-5-related CHD
The decrease in SHF proliferation in Nkx2-5 null embryos has potential ramifications for our understanding of human NKX2-5-related CHD. We describe a hypomorphic Nkx2-5 model in which animals survived until birth with a spectrum of cardiac anomalies overlapping the more severe malformations seen in some humans with NKX2-5 mutations. Of note is the high penetrance of OFT defects accompanied by large VSD in this model. These are not seen in heterozygous Nkx2-5 null mice (Biben et al., 2000), are sporadic findings in human patients (McElhinney et al., 2003), yet are fully penetrant in a heterozygous dominant-negative model of Nkx2-5 deficiency (Elliott et al., 2006). These findings highlight the fine balance between the effective activity of Nkx2-5 and CHD outcome. In humans, the subtle effects of modifier genes on NKX2-5 levels may explain, in some cases, the variable phenotypes seen in patients with NKX2-5 mutations.
Our data provide support for correlative evidence that SHF proliferation is a critical determinant of SHF deployment, and the size and morphology of the OFT/RV. In chick embryos, physical ablation of either the SHF or neural crest, which supports SHF development, leads to a severely decreased contribution of cells to the OFT (Ward et al., 2005; Yelbuz et al., 2002). This produces abnormal looping and rightward displacement of the OFT, manifesting later as an OA (Ward et al., 2005; Yelbuz et al., 2002). Nkx2-5 hypomorphs showed diminished SHF proliferation, and OFT size and morphology were compromised as early as E8.5, leading to abnormal displacement of outflow vessels to the right, OA, and, in more severe cases, DORV. These OFT/RV malformations were partially rescued by deletion of one or both Smad1 alleles. However, not all cardiac defects were rescued in Nkx2-5 hypomorphs by Smad1 deletion. In particular, small VSDs and ventricular chamber defects persisted. These are likely caused by a reduction in chamber differentiation, in which Nkx2-5 is known to have a direct role (Pashmforoush et al., 2004).
OFT defects are amongst the most severe cardiac congenital malformations and often require surgical intervention in the first year of life. Our study suggests that SHF genes and those involved in the Nkx2-5/Bmp2/Smad1 negative feedback loop will be molecular targets and modifiers in CHD. The discovery of this pathway that balances cardiac progenitor specification and proliferation now provides a basis for further mechanistic investigations in this pathological context, as well as in that of the deployment of stem cells that underlies the formation of the heart, and eventually also its repair.
Experimental procedures
Microarray
Amplified RNA from FACS-sorted Nkx2-5gfp/+ and Nkx2-5gfp/gfp cells was labelled with Cy3 or Cy5, and hybridised to cDNA arrays. For the full details (MIAME-compliant) of the clone sets used, array printing, sample preparation, labeling and hybridisation see ArrayExpress database, accession numbers E-MEXP-533 and -534.
Gene/Protein Expression
Controls used in all experiments were somite-matched littermates. ISH was as described (Biben and Harvey, 1997). Probes are described in Suppl. Table S1. For whole-mount immunohistochemistry, PFA-fixed embryos were treated with acetone for 7 mins at −20°C, then blocked for 1 hr in 3% skim milk. For Isl1 and pSmad1, embryos were heated at 95°C for 8 mins in unmasking solution (Vector Laboratories) prior to acetone fixation. Antibodies used were against Pdgfra (APA5, PharMingen), TER-119 (PharMingen), Isl1 (supernatant 39.4D5, raised against chick Isl1 C-terminus, developed by Thomas Jessell and obtained from the Developmental Studies Hybridoma Bank under the auspices of the NICHD and maintained by the University of Iowa, and goat anti-Isl1, raised against full length human Isl1, GT15051, Neuromics), phospho-Smad1/5/8 (9511S, Cell Signaling), β-galactosidase (gift from JF Nicolas, Pasteur Institute), sarcomeric myosin heavy chain (MF20, Developmental Studies Hybridoma Bank) and phosphohistone H3 (06-570, Upstate). Quantitative RT-PCR was as described (Stennard et al., 2005; primers available on request). For statistical methods, see figure legends.
Supplementary Material
Acknowledgments
We thank E Schmied for technical assistance. Work was supported by fellowships from the NHMRC (OWJP), and the Lefoulon-Delalande (FB) and Sasakawa (YW) Foundations, as well as grants (to RPH) from the NHMRC (354400), NGED, NIH NICHHD (HD047858) and Johnson & Johnson Focused Giving Program, and (to MB) from the Pasteur Institute, CNRS and the EU Integrated Project on Heart Repair. No financial conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S, Smalls O, Johnson MC, Watson MS, Seidman JG, et al. Mutations in Nkx2-5, a Cardiac Transcription Factor, Affect Diverse Cardiac Developmental Pathways. J Clin Invest. 1999;104:1567–1573. doi: 10.1172/JCI8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biben C, Harvey RP. Homeodomain factor Nkx2-5 controls left-right asymmetric expression of bHLH eHand during murine heart development. Genes Dev. 1997;11:1357–1369. doi: 10.1101/gad.11.11.1357. [DOI] [PubMed] [Google Scholar]
- Biben C, Weber R, Kesteven S, Stanley E, McDonald L, Elliott DA, Barnett L, Koentgen F, Robb L, Feneley M, Harvey RP. Cardiac septal and valvular dysmorphogenesis in mice heterozygous for mutations in the homeobox gene Nkx2-5. Circ Res. 2000;87:888–895. doi: 10.1161/01.res.87.10.888. [DOI] [PubMed] [Google Scholar]
- Buckingham M, Meilhac S, Zaffran S. Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet. 2005;6:826–835. doi: 10.1038/nrg1710. [DOI] [PubMed] [Google Scholar]
- Cai CL, Liang X, Shi Y, Chu PH, Pfaff SL, Chen J, Evans S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell. 2003;5:877–889. doi: 10.1016/s1534-5807(03)00363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodou E, Verzi MP, Anderson JP, Xu SM, Black BL. Mef2c is a direct transcriptional target of ISL1 and GATA factors in the anterior heart field during mouse embryonic development. Development. 2004;131:3931–3942. doi: 10.1242/dev.01256. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott DA, Kirk EP, Yeoh T, Chandar S, McKenzie F, Taylor P, Grossfeld P, Fatkin D, Jones O, Hayes P, et al. Cardiac homeobox gene NKX2-5 mutations and congenital heart disease: associations with atrial septal defect and hypoplastic left heart syndrome. J Am Coll Cardiol. 2003;41:2072–2076. doi: 10.1016/s0735-1097(03)00420-0. [DOI] [PubMed] [Google Scholar]
- Elliott DA, Solloway MJ, Wise N, Biben C, Costa MW, Furtado MB, Lange M, Dunwoodie S, Harvey RP. A tyrosine-rich domain within homeodomain transcription factor Nkx2-5 is an essential element in the early cardiac transcriptional regulatory machinery. Development. 2006;133:1311–1322. doi: 10.1242/dev.02305. [DOI] [PubMed] [Google Scholar]
- Foley AC, Mercola M. Heart induction by Wnt antagonists depends on the homeodomain transcription factor Hex. Genes Dev. 2005;19:387–396. doi: 10.1101/gad.1279405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habets PE, Moorman AF, Clout DE, van Roon MA, Lingbeek M, van Lohuizen M, Campione M, Christoffels VM. Cooperative action of Tbx2 and Nkx2.5 inhibits ANF expression in the atrioventricular canal: implications for cardiac chamber formation. Genes Dev. 2002;16:1234–1246. doi: 10.1101/gad.222902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu T, Yamagishi H, Maeda J, McAnally J, Yamagishi C, Srivastava D. Tbx1 regulates fibroblast growth factors in the anterior heart field through a reinforcing autoregulatory loop involving forkhead transcription factors. Development. 2004;131:5491–5502. doi: 10.1242/dev.01399. [DOI] [PubMed] [Google Scholar]
- Jay PY, Harris BS, Maguire CT, Buerger A, Wakimoto H, Tanaka M, Kupershmidt S, Roden DM, Schultheiss TM, O’Brien TX, et al. Nkx2-5 mutation causes anatomic hypoplasia of the cardiac conduction system. J Clin Invest. 2004;113:1130–1137. doi: 10.1172/JCI19846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RG. Molecular inroads into the anterior heart field. Trends Cardiovasc Med. 2005;15:51–56. doi: 10.1016/j.tcm.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Kelly RG, Brown NA, Buckingham ME. The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev Cell. 2001;1:435–440. doi: 10.1016/s1534-5807(01)00040-5. [DOI] [PubMed] [Google Scholar]
- Kelly RG, Zammit PS, Schneider A, Alonso S, Biben C, Buckingham ME. Embryonic and fetal myogenic programs act through separate enhancers at the MLC1F/3F locus. Dev Biol. 1997;187:183–199. doi: 10.1006/dbio.1997.8577. [DOI] [PubMed] [Google Scholar]
- Liberatore CM, Searcy-Schrick RD, Vincent EB, Yutzey KE. Nkx-2.5 gene induction in mice is mediated by a Smad consensus regulatory region. Dev Biol. 2002;244:243–256. doi: 10.1006/dbio.2002.0604. [DOI] [PubMed] [Google Scholar]
- Lien CL, McAnally J, Richardson JA, Olson EN. Cardiac-specific activity of an Nkx2-5 enhancer requires an evolutionarily conserved Smad binding site. Dev Biol. 2002;244:257–266. doi: 10.1006/dbio.2002.0603. [DOI] [PubMed] [Google Scholar]
- Lyons I, Parsons LM, Hartley L, Li R, Andrews JE, Robb L, Harvey RP. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeobox gene Nkx2-5. Genes Dev. 1995;9:1654–1666. doi: 10.1101/gad.9.13.1654. [DOI] [PubMed] [Google Scholar]
- McElhinney DB, Geiger E, Blinder J, Benson DW, Goldmuntz E. NKX2.5 mutations in patients with congenital heart disease. J Am Coll Cardiol. 2003;42:1650–1655. doi: 10.1016/j.jacc.2003.05.004. [DOI] [PubMed] [Google Scholar]
- Pandur P, Lasche M, Eisenberg LM, Kuhl M. Wnt-11 activation of a non-canonical Wnt signalling pathway is required for cardiogenesis. Nature. 2002;418:636–641. doi: 10.1038/nature00921. [DOI] [PubMed] [Google Scholar]
- Pashmforoush M, Lu JT, Chen H, Amand TS, Kondo R, Pradervand S, Evans SM, Clark B, Feramisco JR, Giles W, et al. Nkx2-5 pathways and congenital heart disease; loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell. 2004;117:373–386. doi: 10.1016/s0092-8674(04)00405-2. [DOI] [PubMed] [Google Scholar]
- Ross RS, Navankasattusas S, Harvey RP, Chien KR. An HF-1a/HF-1b/MEF-2 Combinatorial Element Confers Cardiac Ventricular Specificity and Establishes an Anterior-Posterior Gradient of Expression via an Nkx2-5 Independent Pathway. Development. 1996;122:1799–1809. doi: 10.1242/dev.122.6.1799. [DOI] [PubMed] [Google Scholar]
- Saga Y, Miyagawa-Tomita S, Takagi A, Kitajima S, Miyazaki J, Inoue T. MesP1 is expressed in the heart precursor cells and required for the formation of a single heart tube. Development. 1999;126:3437–3447. doi: 10.1242/dev.126.15.3437. [DOI] [PubMed] [Google Scholar]
- Schott JJ, Benson DW, Basson CT, Pease W, Silberach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG. Congenital Heart Disease Caused by Mutations in the Transcription Factor NKX2-5. Science. 1998;281:108–111. doi: 10.1126/science.281.5373.108. [DOI] [PubMed] [Google Scholar]
- Shiratori H, Sakuma R, Watanabe M, Hashiguchi H, Mochida K, Sakai Y, Nishino J, Saijoh Y, Whitman M, Hamada H. Two-step regulation of left-right asymmetric expression of Pitx2: initiation by nodal signaling and maintenance by Nkx2. Mol Cell. 2001;7:137–149. doi: 10.1016/s1097-2765(01)00162-9. [DOI] [PubMed] [Google Scholar]
- Stanley EG, Biben C, Elefanty A, Barnett L, Koentgen F, Robb L, Harvey RP. Efficient Cre-mediated deletion in cardiac progenitor cells conferred by a 3′UTR-ires-Cre allele of the homeobox gene Nkx2-5. Int J Dev Biol. 2002;46:431–439. [PubMed] [Google Scholar]
- Stennard FA, Costa MW, Lai D, Biben C, Furtado MB, Solloway MJ, McCulley DJ, Leimena C, Preis JI, Dunwoodie SL, et al. Murine T-box transcription factor Tbx20 acts as a repressor during heart development, and is essential for adult heart integrity, function and adaptation. Development. 2005;132:2451–2462. doi: 10.1242/dev.01799. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Chen Z, Bartunkova S, Yamasaki N, Izumo S. The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development. 1999;126:1269–1280. doi: 10.1242/dev.126.6.1269. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Schinke M, Liao HS, Yamasaki N, Izumo S. Nkx2.5 and Nkx2.6, homologs of Drosophila tinman, are required for development of the pharynx. Mol Cell Biol. 2001;21:4391–4398. doi: 10.1128/MCB.21.13.4391-4398.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay KD, Dunn NR, Robertson EJ. Mouse embryos lacking Smad1 signals display defects in extra-embryonic tissues and germ cell formation. Development. 2001;128:3609–3621. doi: 10.1242/dev.128.18.3609. [DOI] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzi MP, McCulley DJ, De Val S, Dodou E, Black BL. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev Biol. 2005;287:134–145. doi: 10.1016/j.ydbio.2005.08.041. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Taddei I, Morishima M, Meyers EN, Lindsay EA, Baldini A. A genetic link between Tbx1 and fibroblast growth factor signaling. Development. 2002;129:4605–4611. doi: 10.1242/dev.129.19.4605. [DOI] [PubMed] [Google Scholar]
- von Both I, Silvestri C, Erdemir T, Lickert H, Walls JR, Henkelman RM, Rossant J, Harvey RP, Attisano L, Wrana JL. Foxh1 is essential for development of the anterior heart field. Dev Cell. 2004;7:331–345. doi: 10.1016/j.devcel.2004.07.023. [DOI] [PubMed] [Google Scholar]
- Waldo KL, Kumiski DH, Wallis KT, Stadt HA, Hutson MR, Platt DH, Kirby ML. Conotruncal myocardium arises from a secondary heart field. Development. 2001;128:3179–3188. doi: 10.1242/dev.128.16.3179. [DOI] [PubMed] [Google Scholar]
- Ward C, Stadt H, Hutson M, Kirby ML. Ablation of the secondary heart field leads to tetralogy of Fallot and pulmonary atresia. Dev Biol. 2005;284:72–83. doi: 10.1016/j.ydbio.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Xu H, Morishima M, Wylie JN, Schwartz RJ, Bruneau BG, Lindsay EA, Baldini A. Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development. 2004;131:3217–3227. doi: 10.1242/dev.01174. [DOI] [PubMed] [Google Scholar]
- Yelbuz TM, Waldo KL, Kumiski DH, Stadt HA, Wolfe RR, Leatherbury L, Kirby ML. Shortened outflow tract leads to altered cardiac looping after neural crest ablation. Circulation. 2002;106:504–510. doi: 10.1161/01.cir.0000023044.44974.8a. [DOI] [PubMed] [Google Scholar]
- Zaffran S, Kelly RG, Meilhac SM, Buckingham ME, Brown NA. Right ventricular myocardium derives from the anterior heart field. Circ Res. 2004;95:261–268. doi: 10.1161/01.RES.0000136815.73623.BE. [DOI] [PubMed] [Google Scholar]
- Zhang W, Yatskievych TA, Cao X, Antin PB. Regulation of Hex gene expression by a Smads-dependent signaling pathway. J Biol Chem. 2002;277:45435–45441. doi: 10.1074/jbc.M208056200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.