

Abstract
PI3K pathway exerts its function through its downstream molecule AKT in regulating various cell functions including cell proliferation, cell transformation, cell apoptosis, tumor growth and angiogenesis. PTEN is an inhibitor of PI3K, and its loss or mutation is common in human prostate cancer. But the direct role and mechanism of PI3K/PTEN signaling in regulating angiogenesis and tumor growth in vivo remain to be elucidated. In this study, by using chicken chorioallantoic membrane (CAM) and in nude mice models, we demonstrated that inhibition of PI3K activity by LY294002 decreased PC-3 cells-induced angiogenesis. Reconstitution of PTEN, the molecular inhibitor of PI3K in PC-3 cells inhibited angiogenesis and tumor growth. Immunohistochemical staining indicated that PTEN expression suppressed HIF-1α, VEGF and PCNA expression in the tumor xenographs. Similarly, expression of AKT dominant negative mutant also inhibited angiogenesis and tumor growth, and decreased the expression of HIF-1α and VEGF in the tumor xenographs. These results suggest that Inhibition of PI3K signaling pathway by PTEN inhibits tumor angiogenesis and tumor growth. In addition, we found that AKT is the downstream target of PI3K in controlling angiogenesis and tumor growth, and PTEN could inhibit angiogenesis by regulating the expression of HIF-1 and VEGF expression through AKT activation in PC-3 cells.
Keywords: PTEN, AKT, Hypoxia inducible factor 1, Vascular endothelial growth factor
1. Introduction
The phosphatidylinotidol 3-kinase (PI3K) is a heterodimeric enzyme composed of a 110-kDa catalytic subunit and an 85-kDa regulatory subunit [1]. PI3K catalyzes the production of PtdIns-3,4-P2 and PtdIns-3,4,5-P3 helping to recruit AKT (protein kinase B) and PDK1 to the membrane [1-5]. When activated, AKT transmits survival signals from growth factors and inactivates the apoptotic pathways [6,7].
PTEN (phosphatase and tensin homolog deleted on chromosome ten)/MMAC (mutated in multiple advanced cancers) is a tumor suppressor gene [3,8]. The PTEN tumor suppressor gene is lost or mutated in a large number of human cancers including prostate cancer [8-10]. Deregulation of PI3K pathway is commonly observed in many human cancers. This deregulation can be caused either by loss of PTEN, or by constitutive activation of PI3K or its target AKT. Activation of PI3K/AKT may potentiate cell survival, cell migration, proliferation, and cytoskeletal rearrangement. In addition, PI3K pathway is implicated in upregulating vascular endothelial growth factor (VEGF). Studies show that overexpression of PI3K or AKT correlates with increased VEGF levels [11-14], and overexpression of PTEN in chicken embryo inhibited embryonic angiogenesis [15].
Angiogenesis is formation of new blood vessels from preexisted ones. It is critical for tumor growth. The tumor cannot grow more than 1-2 mm in diameter without angiogenesis [16]. Tumor angiogenesis is stimulated by a few angiogenic factors, among which VEGF is a fundamental regulator [17]. Hypoxia inducible factor 1 (HIF-1) is a transcription factor that activates the transcription of many genes, including VEGF [18]. HIF-1 is composed of HIF-1α and β subunits [19,20]. HIF-1α expression can be stimulated by a low concentration of oxygen or loss of function of VHL and PTEN [21,22]. HIF-1α expression can be induced by growth factors through activation of PI3K/AKT signaling [23,24]. PI3K/AKT signaling regulates tumor angiogenesis via HIF-1α/VEGF.
Prostate cancer is one of the most commonly diagnosed male malignancy in the United States, affecting 1 in 9 men over 65 years of age [25]. Several lines of evidence implicate that loss of PTEN is a major factor causing human prostate carcinogenesis. PTEN was found to be mutated or deleted in many prostate cancer cell lines including LNCaP, PC-3, NCI H660 [8,10]. It was shown that PTEN is frequently lost in prostate cancer cells [26], that it undergoes homozygous deletions in ∼10% of primary prostate tumors [27], and that alterations are frequently occurred in metastatic prostate cancer [28].
Elevated levels of VEGF are found in the serum and urine of prostate cancer patients, suggesting that VEGF plays an important role in prostate cancer. PI3K/PTEN pathway regulated HIF-1α and VEGF expression in prostate cancer cell lines [11,14,28], indicating that this pathway may play an important role in regulating prostate tumor angiogenesis. However, there is a lack of direct evidence to show that PI3K/PTEN pathway regulates prostate tumor angiogenesis in vivo. We have successfully established tumor cells-induced angiogenesis and tumor growth models on chicken chorioallantoic membrane (CAM) and in nude mice [12,15,29]. The goal of this study is to investigate: 1) whether PI3K activity is required for angiogenesis of prostate tumor; 2) whether restoration of PTEN expression in prostate cancer cells inhibits tumor angiogenesis and growth on CAM in vivo; 3) whether the level and activity of PI3K and PTEN affect HIF-1 and VEGF expression in tumors; and 4) whether AKT is downstream target of PI3K for regulating angiogenesis.
2. Materials and methods
2.1. Reagents and antibodies
The antibodies against phospho-AKT, AKT, and PTEN were purchased from Cell Signaling (Beverly, MA). Antibodies against VEGF and PCNA were products of Santa Cruz (Santa Cruz, CA). The antibodies against β-actin and CD31 were from Sigma (St. Louis, MO) and BD Bioscience (Bedford, MA), respectively. Trizol reagent was from Invitrogen. AMV Reverse Transcriptase and oligo(dT) primer were from Promega (Madison, WI).
2.2. Cell culture
The PC-3 cells were cultured in RPMI 1640 medium, and human embryonic kidney 293 cells were cultured in MEM medium, supplemented with 10% fetal bovine serum, 100 unit/ml penicillin, and 100 μg/ml streptomycin. The cells were incubated under 5% CO2 at 37 °C as described [30].
2.3. Infection of cells with adenovirus
The PTEN, PTEN mutant with cysteine 124 to serenine (PTEN-C124S), and AKT dominant negative mutant (AKT-DN) were previously described [15]. These constructs were inserted into adenoviral vector using Ad-Easy System (Stratagene, San Diego, CA). The PC-3 cells were infected with adenovirus carrying GFP (Ad-GFP), PTEN (Ad-PTEN), PTEN-C124S (Ad-PTEN-C124S), or AKT-DN (Ad-AKT-DN) at 20 MOI. The infection rate of the cells was approximately 80%.
2.4. Angiogenesis and tumor growth assay on the CAM
Fertilized White Leghorn chicken eggs were incubated at 37°C under conditions of constant humidity. The developing CAM was separated from the shell by opening a window at the broad end of the egg above the air sac on Day 9. The opening was sealed and the eggs were returned to the incubator. To investigate tumor angiogenesis, the cells were suspended in medium containing 50% Matrigel (BD Biosciences, Bedford, MA). Aliquots (30 μl) of the mixture were then applied onto the CAM of 9-day-old embryos. The area around the implanted Matrigel was photographed 4 days after the implantation, and the blood vessels were counted by two observers in a double-blind manner. Assays for each treatment were carried out using 8–10 chicken embryos. For determining tumor growth, the cells were implanted onto the CAM to grow tumors for 9 days.
2.5. Matrigel plug angiogenesis assay and tumor growth experiments in nude mice
Male BALB/cA-nu nude mice (4 weeks old) were purchased from Shanghai Experimental Animal Center (Chinese Academy of Sciences, China) and maintained in pathogen-free conditions. PC-3 cells were infected with adenovirus carrying GFP, PTEN, PTEN -C124S or AKT-DN. The cells were harvested 24 h after infection, and re-suspended in serum-free medium. The cells (0.2 ml, 4 million cells) were mixed with 0.4 ml of growth factor-reduced Matrigel, and the mixture was immediately injected into the flanks of nude mice. The mice were sacrificed 11 days after the injection, and the Matrigel plugs were taken out of the mice. The hemoglobin content of the Matrigel was determined using Drabkin reagent. Each treatment had 8 mice and the experiment was repeated twice. For tumor growth assay, the infected PC-3 cells were re-suspended in culture medium and 4 million of the cells (in 0.1 ml) were injected into the nude mice at the flank sites. Tumor growth was determined by measuring the size of the tumors, starting at Day 11 and ending at Day 21. Tumor volumes were calculated according to the formula (width2 × length)/2.
2.6. Immunoblotting and immunohistochemistry
Immunoblotting and immunohistochemistry were performed as described previously [29]. Tumor tissues were fixed in 10% buffered formalin for 24 h and processed by conventional method. For CD31 staining, tumor tissues were fixed in zinc fixative (BD Biosciences) and embedded in paraffin. The relative angiogenesis was calculated as micro-vessel density (MVD) as described [31]. Briefly, slides were first scanned under low power (×40) in order to determine three ‘hot-spots’ or areas with the maximum number of micro-vessels, which were then evaluated at magnification of 200. The number of micro-vessels in each field was determined and expressed as MVD per field.
2.7. RT-PCR
Tumor tissues prepared from the cells infected by Ad-GFP and Ad-PTEN groups were ground in liquid nitrogen to extract total RNAs. Total RNAs were used for cDNA synthesis by reverse transcription, and cDNAs of VEGF and GAPDH were amplified by PCR. The primers used for the PCR are as follows: VEGF upstream primer 5'-TCGGGCCTCCGAAACCATGA-3', VEGF downstream primer 5'-CCTGGTGAGAGATCTGGTTC-3'; GAPDH upstream primer 5'-CCACCCATGGCAAATTCCATGGCA-3', GAPDH downstream primer 5'- CCTGGTGAGAGATCTGGTTC-3'.
2.8. Tunnel assay and Statistical analysis
In Situ Cell Death Detection kit was purchased from Roche Co, Switzerland. Tunnel staining of the tissue sections was performed per the manufacture's instruction. The data represent mean ± SD from three independent experiments except where indicated. Statistical analysis was performed by student t test at a significant level (p< 0.05).
3. Results
3.1. LY294002 inhibited PC-3 xenograft-induced angiogenesis on the CAM
We used the CAM to test whether PI3K in PC-3 cells plays a role in tumor-induced angiogenesis. PC-3 cells greatly induced angiogenesis (Fig. 1A). PI3K inhibitor LY294002 decreased PC-3 xenograft-induced angiogenesis in a concentration dependent manner (Fig. 1). This result showed that PI3K activation in PC-3 cells is required for tumor-induced angiogenesis.
Fig. 1.
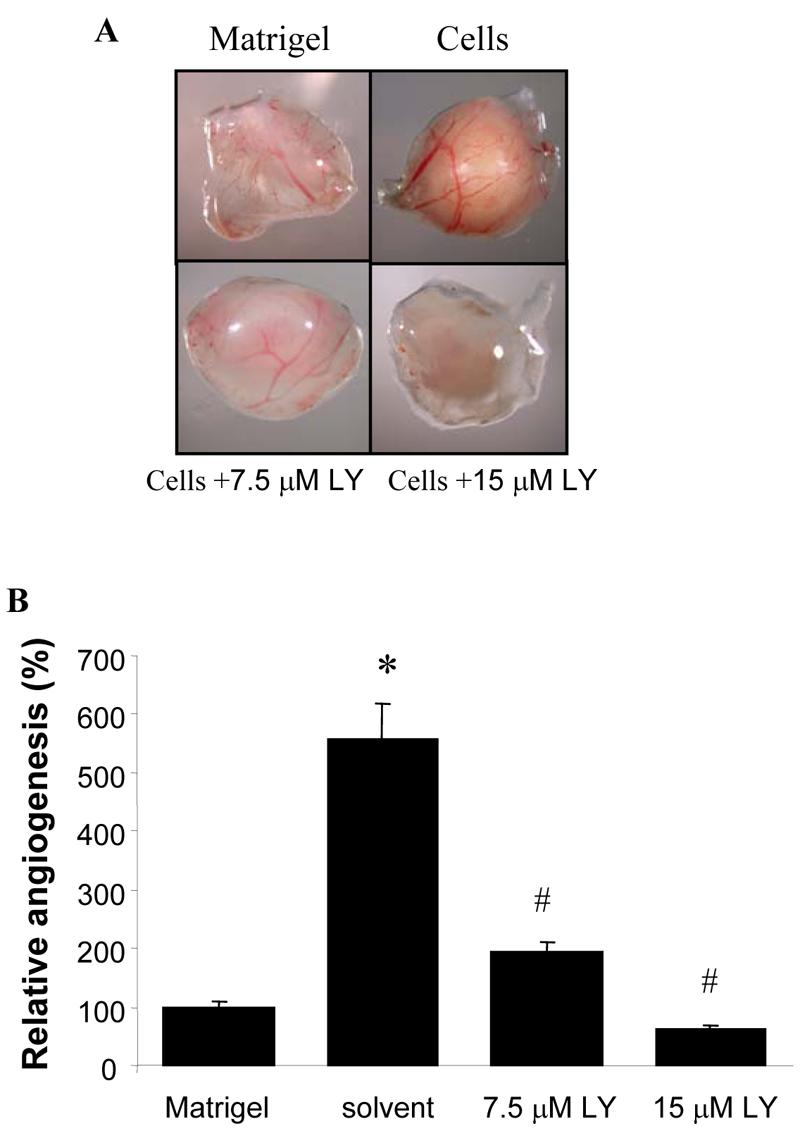
LY294002 inhibited tumor-induced angiogenesis. PC-3 cells (15 μl) were mixed with 15 μl Matrixgel in the presence or absence of LY294002. The cell mixture was implanted onto the CAM of a 9-day old chicken embryo. After 4 days of implantation, the CAM was cut off, and the amount of blood vessels on the CAM induced by the PC-3 xenografts was determined from eight different replicated experiments. (A) Representative tumors on the CAM from Matrixgel alone, PC-3 cells alone, PC-3 cells with 7.5 μM LY294002, or 15 μM LY294002. (B) The relative angiogenesis in the CAM. The number of blood vessels were counted from replicate experiments, and normalized to that of the Matrigel alone as relative angiogenesis. The experiment was performed twice, and the data are mean and SD from 8 replicates. * indicates the significant difference when compared to the Matrigel alone, # indicates the significant difference when compared to the PC-3 cells alone (p< 0.05).
3.2. Over-expression of PTEN inhibited angiogenesis and tumor growth
Since PC-3 cells contain a mutated PTEN gene, we transected a wild type PTEN gene to restore PTEN expression in PC-3 cells. The cells were infected by adenovirus carrying GFP or wild-type PTEN. The PTEN expression in PC-3 cells was confirmed by immunoblotting, which inhibited AKT phosphorylation and activation (Fig. 2A). PTEN expression decreased cell proliferation when compared to the cells expressing GFP and the parental cells (Fig. 2B). To analyze the effect of PTEN restoration in PC-3 cells in affecting angiogenesis, the cells alone or expressing GFP or wild-type PTEN were used for the angiogenesis assay in vivo. PC-3 xenografts induced angiogenesis on the CAM, and overexpression of PTEN significantly inhibited tumor-induced angiogenesis while the tumor size was similar 4 days after the implantation (Fig. 2C). This result suggested that PTEN expression initially inhibited angiogenesis before affecting tumor growth. To test the effect of PTEN on tumor growth, the tumor sizes were analyzed 9 days after the implantation of PC-3 cells on the CAM cells. Over-expression of PTEN greatly inhibited the tumor growth when it was analyzed 9 days after the implantation (Fig. 2D). Immunohistochemical staining of the tumor sections showed that PTEN restoration in PC-3 cells decreased the expression of HIF-1 and VEGF in tumor sections (Fig. 2E). The levels of HIF-1α protein and VEGF mRNA from the tumor tissues infected by adenovirus carrying PTEN showed 70% decrease when compared to those from the Ad-GFP control (Fig. 2F), further confirming the crucial role of PTEN in regulating the expression of HIF-1 and VEGF during the tumor angiogenesis.
Fig. 2.
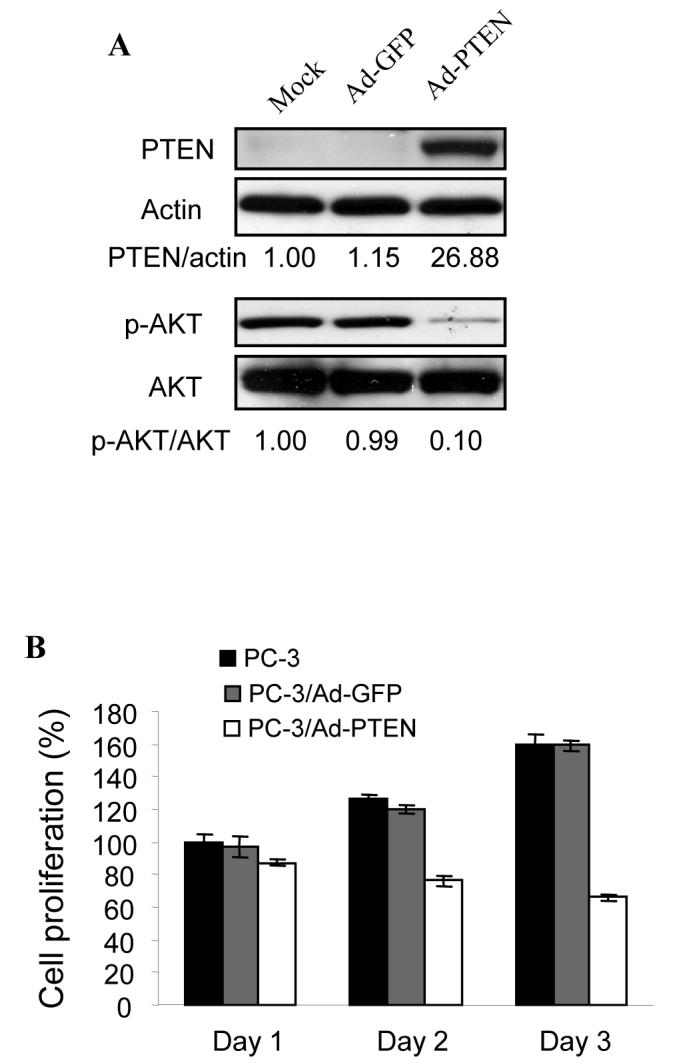

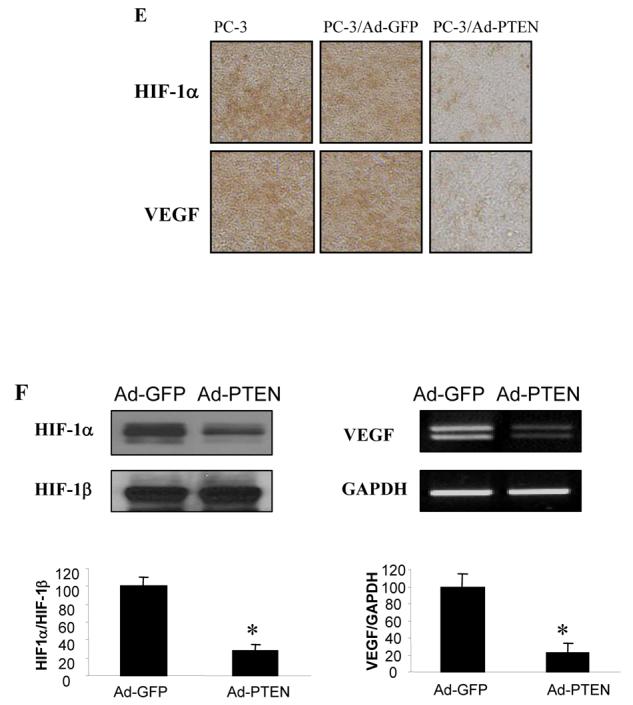
Over-expression of PTEN inhibited angiogenesis and growth of tumor. The PC-3 cells were infected with adenovirus vector or adenovirus carrying PTEN. Relative density signals were quantified, and normalized to those of actin levels and to those of the mock control. (A) The infected cells were harvested 48 h after the infection, and used for immunoblotting analysis. (B) The infected cells were split into 96-well plates at 1× 104cells/well. Cell proliferation was assayed 24 h post-infection (Day 1) using MTT assay [30]. The value of MTT reduction at Day 1 was defined as 100%. (C) PC-3 cells were infected with adenovirus or Ad-PTEN. The cells were harvested 24 h after the infection, and re-suspended in the serum-free medium. The cells were mixed with Matrigel at 1:1 ratio, and 30 μl of the mixture (3× 106 cells) was implanted onto the CAM of a 9-day old chicken embryo. The CAM was cut off 4 days after the implantation, and the number of blood vessels on the CAM was counted under microscope. At least five unit areas of each CAM were counted under the tumor. The results are mean and SD from two experiments (n = 8). * indicates the significant difference when compared to the GFP control (p< 0.05). (D) PC-3 cells were implanted onto the CAM as above. The tumors were harvested and weighed 9 days after the implantation. The data indicate the mean ± SD (n = 8) of three independent experiments. * indicates the significant difference when compared to the GFP control (p< 0.05). (E) Immunohistochemical staining of tissues using antibodies against HIF-1α and VEGF. (F) PC-3 cells were infected with Ad-GFP and Ad-PTEN for 24 h. The cells were mixed and implanted onto the CAM of a 9-day old chicken embryo as above. After 9 days of incubation, tumor tissues were grounded by liquid nitrogen. Tissue lysates were analyzed by immunoblotting for HIF-1α and HIF-1β expression (left, up panel). Relative HIF-1α protein signals were quantified and normalized to those of HIF-1β and to the control from three replicate experiments. Total RNAs were extracted from the tumor tissues, and VEGF mRNA levels were determined by RT-PCR (right, up panel). Relative VEGF mRNA levels were from three replicate experiments. * indicates the significant difference when compared to the vector control (p< 0.05).
To test whether PTEN restoration in PC-3 cells expression has similar effect in regulating angiogenesis and tumor growth in a different animal model, we performed similar experiments using nude mice. Tumor angiogenesis was determined using Matrigel plug assay. PC-3 cells dramatically induced angiogenesis, and PTEN expression decreased the angiogenesis (Fig. 3A). The relative angiogenesis was analyzed by the levels of hemoglobin in the Matrigel plugs. PTEN expression in the cells leads to the hemoglobin level 6-fold less than that of the control in the Matrigel plugs (Fig. 3A). We also studied tumor growth by the subcutaneous injection of the cells in the nude mice.
Fig. 3.
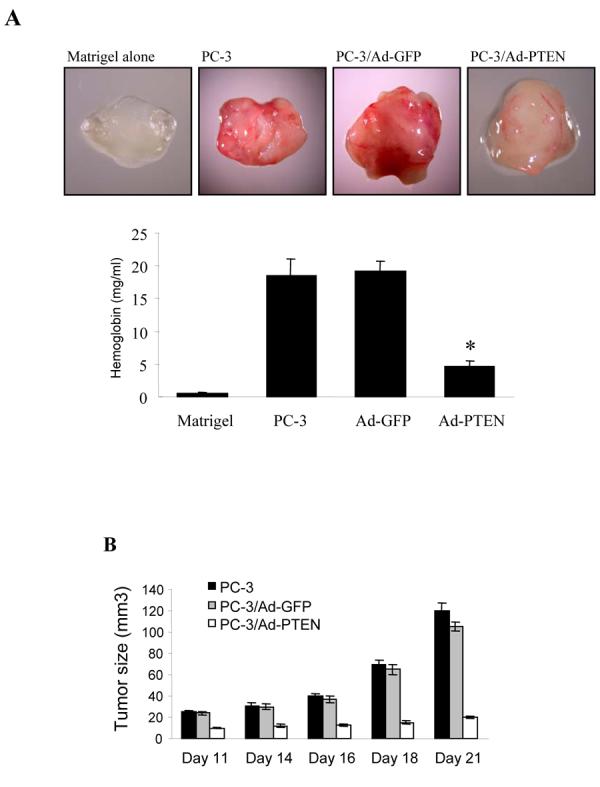
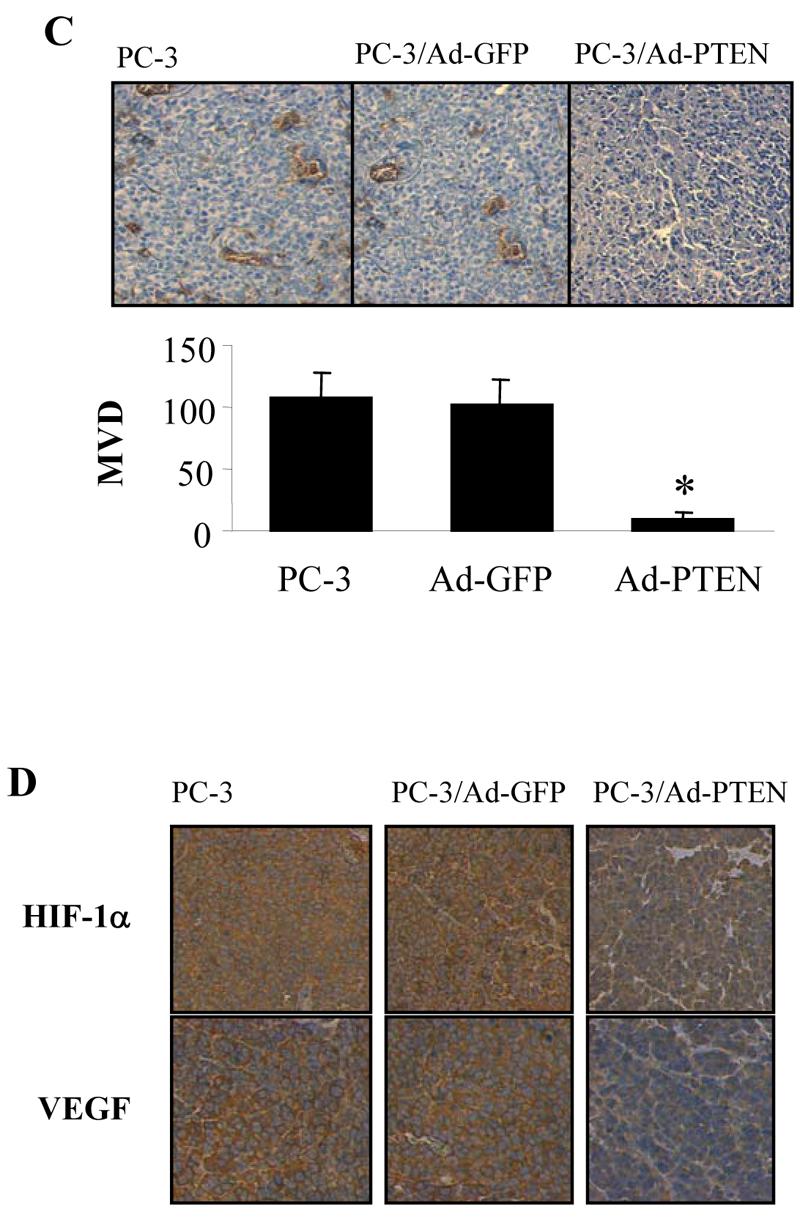
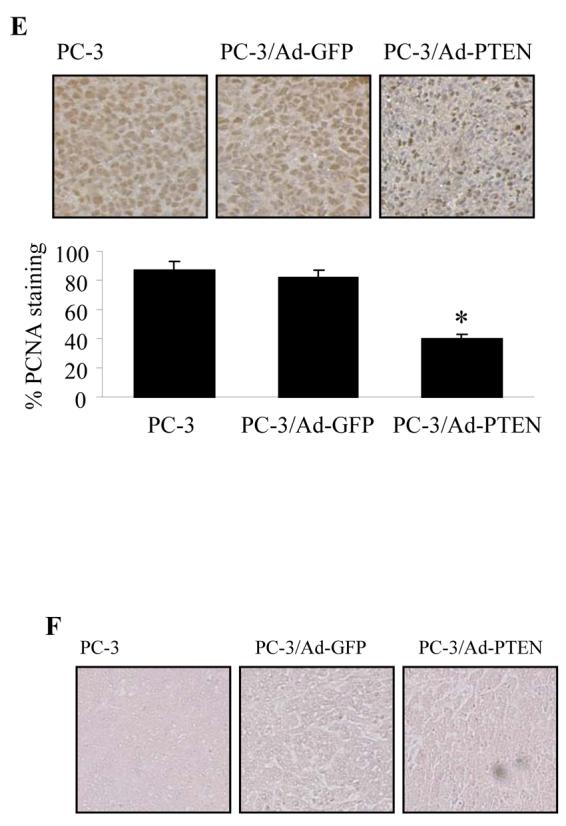
Over-expression of PTEN inhibited tumor-induced angiogenesis and tumor growth. (A) PC-3 cells were infected as described above. The cells were harvested 24 h after infection, and re-suspended in serum-free medium. The cells (0.2 ml, 4 million cells) were mixed with 0.4 ml of growth factor-reduced Matrigel, and the mixture was immediately injected into the flanks of nude mice. The mice were sacrificed 11 days after the injection, and the Matrigel plugs were taken out of the mice. The hemoglobin content of the Matrigel was determined using Drabkin reagent. Each treatment had 8 mice and the experiment was repeated twice. * indicates the significant difference when compared to the GFP control (p< 0.05). (B) The infected PC-3 cells were re-suspended in culture medium and 4 million of the cells (in 0.1 ml) were injected into the nude mice at the flank sites. Tumor growth was determined by measuring the size of the tumors, starting at Day 11 and ending at Day 21. The data indicate mean ± SD from two experiments (n = 8). (C) CD31 immunohistochemical staining. Tumor sections were stained using antibodies against CD31, and microvessel density of the tumors was analyzed as described under Materials and Methods. * indicates the significant difference when compared to the GFP control (p< 0.05). (D) Immunohistochemical staining for HIF-1α and VEGF expression in tumor sections. (E) The PCNA staining of the tumor sections. Proliferating cells were quantified by counting the PCNA-positive (brown) cells and the total number of cells at 5 arbitrarily selected fields at ×200. The data are mean ± SD (n = 5). * indicates the significant difference when compared to the GFP control (p< 0.05). (F) Tunnel assay of the tumor sections.
The tumor growth assay showed that PTEN expression in PC-3 cells significantly inhibited the tumor growth when compared to the control (Fig. 3B). Micro-vessel density (MVD) of tumor sections was determined as an index of tumor angiogenesis. Expression of PTEN in PC-3 cells significantly decreased the tumor MVD (Fig. 3C). The expression of HIF-1α and VEGF was decreased in tumor tissues from the cells infected with Ad-PTEN, indicating that PTEN may inhibit angiogenesis through the expression of HIF-1 and VEGF (Fig. 3D). We stained the tumor sections using antibodies against PCNA, and found that more than 80% of the cells were PCNA-positive in the control tumors versus only about 40% of the cells that were PCNA-positive in the PTEN-expressing tumors (Fig. 3E). Tunnel assay indicated that there was no apparent difference of apoptotic cells between the control and PTEN-treated tumors (Fig. 3F). Taken together, these results suggest that the inhibition of tumor growth by PTEN is partially due to the cell proliferation decrease.
3.3 .PTEN lipid phosphotase activity is required for inhibiting tumor-induced angiogenesis and tumor growth
To study whether PTEN lipid phosphotase activity is required for PTEN-inhibiting angiogenesis, we expressed PTEN-C124S, a point mutant of PTEN that abolishes the lipid phosphatase activity. The PC-3 cells were infected with adenovirus carrying GFP or PTEN-C124S. Over-expression of PTEN-C124S was confirmed by immunoblotting, which did not affect the level of phospho-AKT (Fig. 4A) and cell proliferation (Fig. 4B), suggesting that PTEN phosphatase activity is required for inhibiting AKT activation and cell proliferation. The tumor angiogenesis assay was performed using the CAM assay. The forced expression of PTEN-C124S in PC-3 cells did not inhibit tumor-induced angiogenesis using the CAM (Fig. 4C), suggesting that PTEN phosphatase activity is required for inhibiting PC-3 tumor-induced angiogenesis. We used Matrigel plug assay, and also showed that PTEN-C124S expression did not inhibit PC-3 cell-induced angiogenesis in nude mice (Fig. 4D). To determine the effect of PTEN-C124S expression in PC-3 cells in tumor growth, the cells were implanted onto the CAM to grow tumor for 9 days. Expression of PTEN-C124S did not inhibit tumor growth and the levels of HIF-1α and VEGF in the tumor (Fig. 5B).
Fig. 4.
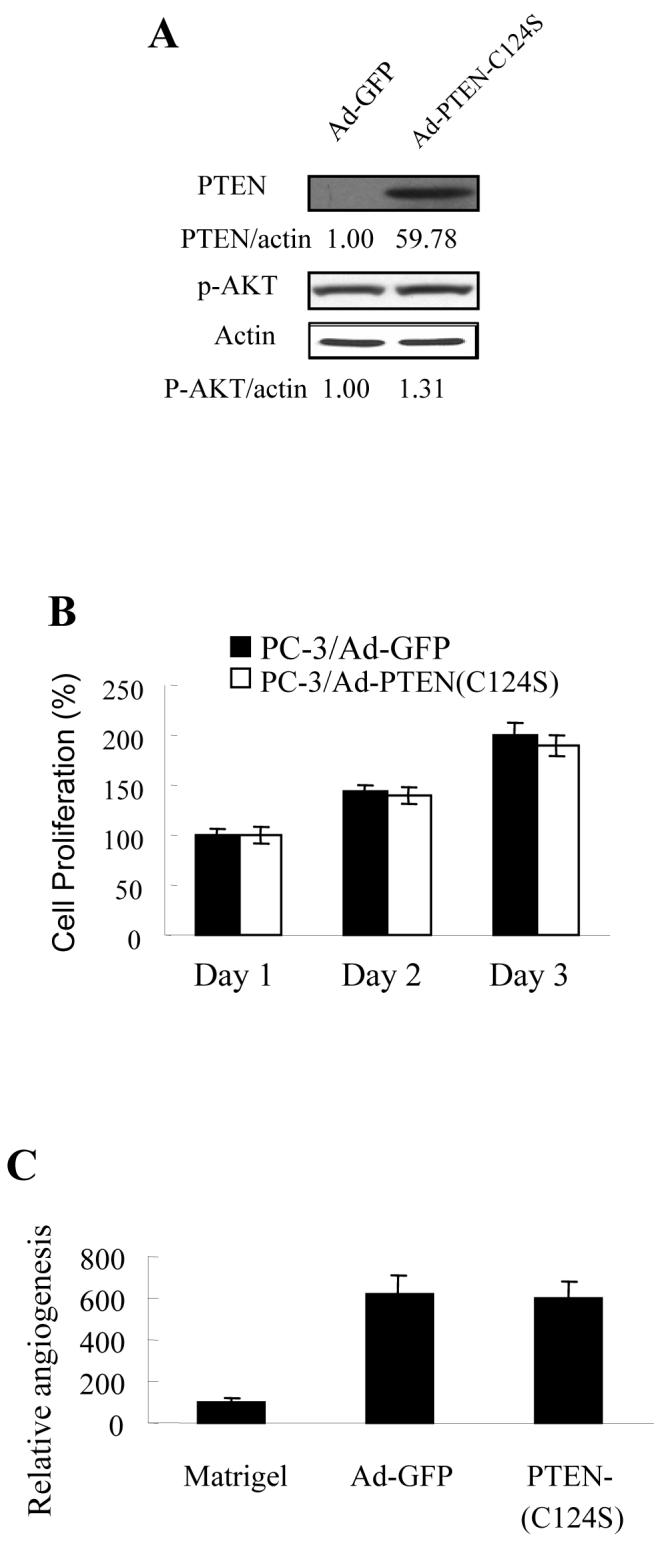
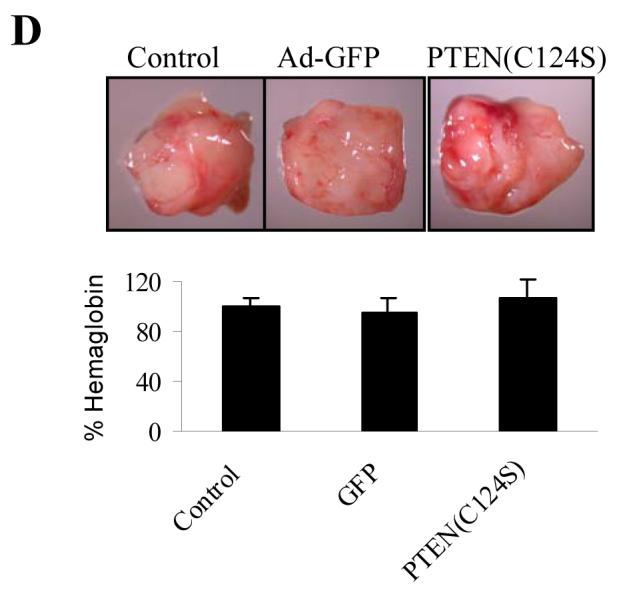
Over-expression of PTEN-C124S did not inhibit angiogenesis. The PC-3 cells were infected by adenovirus carrying GFP or PTEN-C124S. (A) The infected cells were harvested 48 h after infection, and used for immunoblotting analysis. Relative values of density were quantified as Fig. 1A. (B) The infected cells were split into 96-well plates (1× 104 cells/well) 24 h post-infection. The cell proliferation was assayed as in Fig. 2B. (C) PC-3 cells were infected with adenovirus carrying GFP or PTEN-C124S. The infected cells were harvested 24 h after infection, mixed with Matrigel, and implanted onto the CAM of 9-day old embryos as described in Fig. 2C. The CAM was excised 4 days after implantation, and blood vessels were counted as described in Fig. 2C. * indicates the significant difference when compared to the GFP control (p< 0.05). (D) PC-3 cells were infected with adenovirus carrying GFP or PTEN-C124S. The infected cells were harvested 24 h after infection, mixed with Matrigel, and injected into the nude mice. The mice were sacrificed 11 days after implantation, and the Matrigel plugs were harvested for hemoglobin assay as described above. * indicates the significant difference when compared to the GFP control (p< 0.05).
Fig. 5.
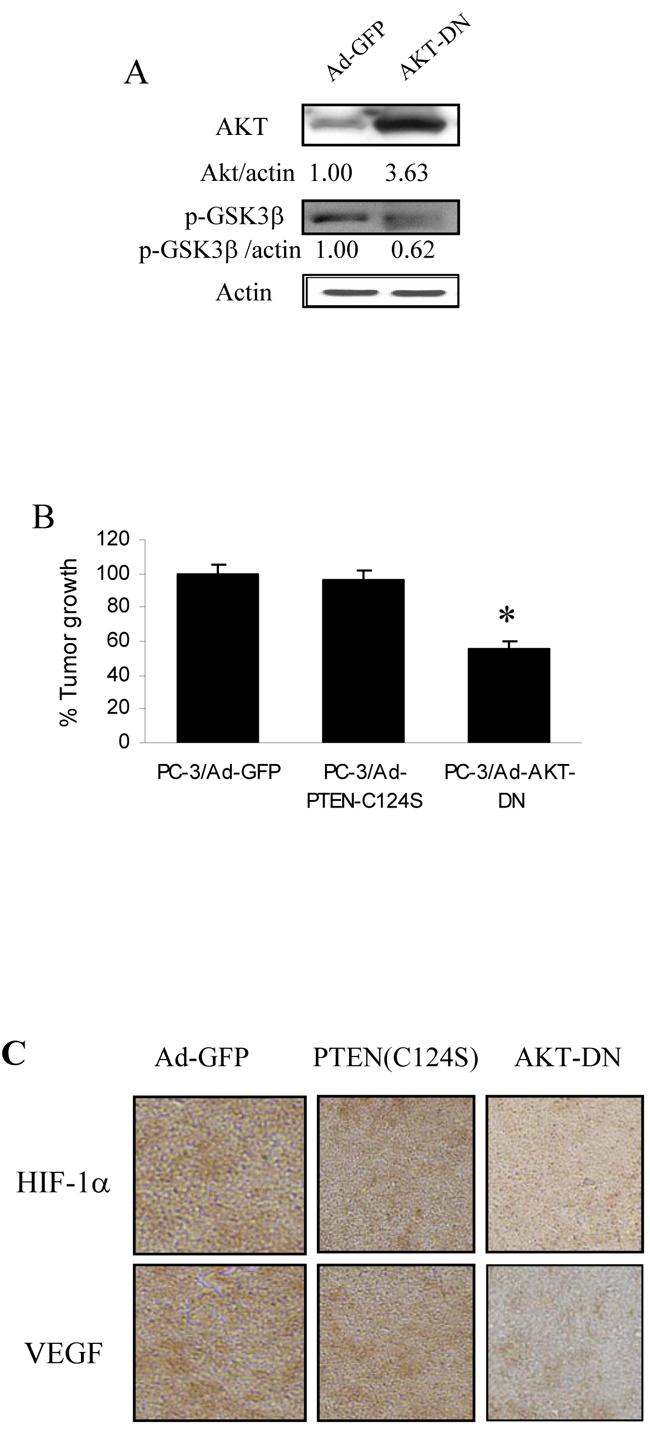
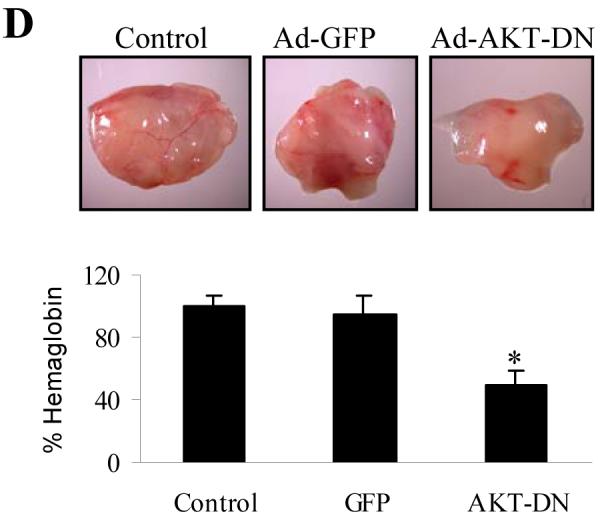
Expression of AKT-DN inhibited PC-3-induced angiogenesis and tumor growth. PC-3 cells were infected with adenovirus carrying GFP, PTEN-C124S or AKT-DN. The cells were harvested 24 h after infection. (A) The cells were used for immunoblotting for total AKT and phospho-GSK3 levels. Relative values of density were quantified as previously described. (B) The cells were mixed with mixed with Matrigel, and implanted onto the CAM of 9-day old embryos as described above. Total cells implanted on each embryo were 3 million. The tumors were harvested and weighed 9 days after implantation. The data are the mean ± SD of 8 tumor weight. * indicates the significant difference when compared to the GFP control (p< 0.05). (C) Immunohistochemical staining for HIF-1α and VEGF expression in tumor sections from representative tumors from the above experiment. (D) The cells infected with adenovirus carrying GFP, PTEN-C124S or AKT-DN were mixed with Matrigel, and injected into the nude mice. The mice were sacrificed 11 days after implantation, and the Matrigel plugs were harvested for hemoglobin assay as described above. * indicates the significant difference when compared to the GFP control (p< 0.05).
3.4 AKT is required for tumor-induced angiogenesis and tumor growth
We next determined the effect of AKT dominant negative mutant (AKT-DN) in PC-3 cells in regulating angiogenesis. PC-3 cells were infected with adenovirus carrying GFP or AKT-DN. The AKT-DN expression in the cells was confirmed by immunoblotting, and its inhibitory effect was showed through decreasing the level of phospho-GSK-3β, a downstream target of AKT in the cells (Fig. 5A). Forced expression of AKT-DN inhibited tumor growth (Fig. 5B). The immunohistochemical staining of the tumor sections showed that expression of AKT-DN in PC-3 cells inhibited the expression of HIF-1α and VEGF (Fig. 5C). Expression of AKT-DN in PC-3 cells greatly inhibited PC-3 cell-induced angiogenesis (Fig. 5D), showing that AKT is a PTEN-inhibited downstream target that is required for PC-3 cell-induced angiogenesis in vivo.
4. Discussion
The PI3K/AKT signaling has been shown to play an important role in survival and proliferation of human prostate cancer cells [32-35]. The tumor suppressor PTEN is frequently mutated in human prostate cancer. It has been shown that inhibition of PI3K by LY294002 abrogated the expression of HIF-1α and VEGF in prostate cancer cell lines [11,14]. However, the role and mechanism of PI3K and AKT in prostate tumor angiogenesis in vivo remain to be elucidated. To determine the role and mechanism of PTEN in tumor growth and angiogenesis, in this study we used PC-3 human prostate cancer cell line to perform angiogenesis assay in vivo using the CAM and nude mouse models. PC-3 contains PTEN mutation, which is good to test the functions of the reconstituted PTEN construct in the tumor growth without the interference of endogenous PTEN.
The PC-3 xenograft greatly induced angiogenesis on the CAM, and inhibition of PI3K by LY294002 significantly suppressed angiogenesis, suggesting that PI3K is required for prostate tumor angiogenesis. PTEN is a molecular inhibitor of PI3K, and is one of the most commonly mutated tumor suppressor genes, which is ranked the Number 2 after p53 mutations [3]. PTEN mutation is frequently observed in prostate cancer [8,10,26,27]. Inactivation of PTEN leads to the activation of PI3K/AKT signaling. The mutation of PTEN in PC-3 cells leads to the high levels of HIF-1α and VEGF expression. In order to determine the role and mechanism of PTEN in PI3K-regulated angiogenesis and tumor growth, we found that introduction of PTEN into PC-3 cells inhibited AKT activation and cell proliferation. Over-expression of PTEN inhibited angiogenesis and tumor growth in CAM model. Immunohistochemical staining of the tumor xenographs showed that the expression of PTEN decreased HIF-1α and VEGF protein levels, suggesting that the inhibition of angiogenesis could due to the decreased expression of HIF-1α and VEGF. To further confirm our results obtained by CAM model, we used similar method to test anigogenesis and tumor growth in nude mice model. CD31, which is a specific marker to determine vascular endothelial cells, was used to determine tumor angiogenesis. We found that the number of CD31-positive microvessels and the expression of HIF-1α and VEGF decreased significantly in tumor xenographs from the Ad-PTEN-treated group, confirming our previous results that over-expression of PTEN inhibited angiogenesis through HIF-1α and VEGF expression. Previous studies have showed that angiogenesis is required for tumor growth [16,36]. In addition, inhibition of PI3K resulted in inactivation of AKT and decreased cell proliferation. PCNA staining showed that the cell proliferation in PTEN-expressing cells was inhibited while there was no apparent difference of apoptotic cells between the control and PTEN-treated tumors, indicating that the inhibition of tumor growth was also caused by the inhibition of cell proliferation by PTEN. Our results suggest that PI3K is required and plays an important role in prostate tumor growth and angiogenesis.
Next, in order to determine whether the function of PTEN is required in inhibiting angiogenesis and tumor growth, we introduced mutant PTEN-C124S to PC-3 cells. Expression of mutant PTEN had no effects on phospho-AKT and cell proliferation of PC-3 cells. Mutant PTEN expression in PC-3 cells had no inhibitory effect on angiogenesis and tumor growth induced by PC-3 xenografts. Immunohistochemical staining of the tumor xenographs indicated that expression of PTEN mutant could not inhibit HIF-1 and VEGF expression, either. These results suggest that as a tumor suppressor, PTEN plays an important role in regulating angiogenesis and tumor growth.
AKT is one of the important downstream targets of PI3K. Finally, we determined whether AKT activation is necessary during tumor angiogenesis and tumor growth. As shown in Fig. 4A, expression of AKT dominant-negative inhibited AKT activity, and decreased cell proliferation. Inhibition of AKT by Ad-AKT-DN infection significantly suppressed PC-3 cells-induced angiogenesis and tumor growth with decreased expression of HIF-1α and VEGF by immunohistochemical staining. AKT lies downstream of PTEN, and does not affect PTEN expression in the PC-3 cells. These results suggest that the inhibition of AKT resulted in abrogation of HIF-1α and VEGF expression, which could lead to the inhibition of angiogenesis and tumor growth, suggesting that AKT plays an important role in angiogenesis. In summary, this study shows that PI3K/PTEN/AKT signaling plays an important role in regulating tumor angiogenesis and growth through HIF-1α and VEGF expression.
Acknowledgment
This work was supported by NIH Grants CA109460, RR016440, and CA123675; and by American Cancer Society Research Scholar Grant 04-076-01-TBE.
Abbreviation
- CAM
chicken chorioallantoic membrane
- PI3K
phosphatidylinsitol 3-kinase
- MVD
microvessel density
- MOI
multiplicity of infection
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carpenter CL, Duckworth BC, Auger KR, Cohen B, Schaffhausen BS, Cantley LC. J. Biol. Chem. 1990;265:19704. [PubMed] [Google Scholar]
- 2.Auger KR, Serunian LA, Soltoff SP, Libby P, Cantley LC. Cell. 1989;57:167. doi: 10.1016/0092-8674(89)90182-7. [DOI] [PubMed] [Google Scholar]
- 3.Cantley LC, Neel BG. Proc. Natl. Acad. Sci. U. S. A. 1999;96:4240. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Franke TF, Kaplan DR, Cantley LC. Cell. 1997;88:435. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 5.Whitman M, Downes CP, Keeler M, Keller T, Cantley L. Nature. 1988;332:644. doi: 10.1038/332644a0. [DOI] [PubMed] [Google Scholar]
- 6.Chan TO, Rittenhouse SE, Tsichlis PN. Ann. Rev. Biochem. 1999;68:965. doi: 10.1146/annurev.biochem.68.1.965. [DOI] [PubMed] [Google Scholar]
- 7.Duronio V, Scheid MP, Ettinger S. Cell. Signal. 1998;10:233. doi: 10.1016/s0898-6568(97)00129-0. [DOI] [PubMed] [Google Scholar]
- 8.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R. Science. 1997;275:1943. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 9.Di Cristofano A, Pandolfi PP. Cell. 2000;100:387. doi: 10.1016/s0092-8674(00)80674-1. [DOI] [PubMed] [Google Scholar]
- 10.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV. Nat. Genet. 1997;15:356. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 11.Jiang BH, Jiang G, Zheng JZ, Lu Z, Hunter T, Vogt PK. Cell Growth Differ. 2001;12:363. [PubMed] [Google Scholar]
- 12.Xia C, Meng Q, Cao Z, Shi X, Jiang BH. J. Cell. Physiol. 2006;209:56. doi: 10.1002/jcp.20707. [DOI] [PubMed] [Google Scholar]
- 13.Zhang L, Yang N, Katsaros D, Huang W, Park JW, Fracchioli S, Vezzani C, Rigault de la Longrais IA, Yao W, Rubin SC, Coukos G. Cancer Res. 2003;63:4225. [PubMed] [Google Scholar]
- 14.Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, Simons JW, Semenza GL. Cancer Res. 2000;60:1541. [PubMed] [Google Scholar]
- 15.Jiang BH, Zheng JZ, Aoki M, Vogt PK. Proc. Natl. Acad. Sci. U. S. A. 2000;97:1749. doi: 10.1073/pnas.040560897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Folkman J. Semin. Oncol. 2002;29:15. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- 17.Plate KH, Breier G, Weich HA, Risau W. Nature. 1992;359:845. doi: 10.1038/359845a0. [DOI] [PubMed] [Google Scholar]
- 18.Semenza GL. Nat. Rev., Cancer. 2003;3:721. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 19.Jiang BH, Rue E, Wang GL, Roe R, Semenza GL. J. Biol. Chem. 1996;271:17771. doi: 10.1074/jbc.271.30.17771. [DOI] [PubMed] [Google Scholar]
- 20.Wang GL, Jiang BH, Rue EA, Semenza GL. Proc. Natl. Acad. Sci. U. S. A. 1995;92:5510. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. Nature. 1999;399:271. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 22.Zundel W, Schindler C, Haas-Kogan D, Koong A, Kaper F, Chen E, Gottschalk AR, Ryan HE, Johnson RS, Jefferson AB, Stokoe D, Giaccia AJ. Genes Dev. 2000;14:391. [PMC free article] [PubMed] [Google Scholar]
- 23.Blancher C, Moore JW, Robertson N, Harris AL. Cancer Res. 2001;61:7349. [PubMed] [Google Scholar]
- 24.Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. Mol. Cell. Biol. 2001;21:3995. doi: 10.1128/MCB.21.12.3995-4004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abate-Shen C, Shen MM. Genes Dev. 2000;14:2410. doi: 10.1101/gad.819500. [DOI] [PubMed] [Google Scholar]
- 26.Vlietstra RJ, van Alewijk DC, Hermans KG, van Steenbrugge GJ, Trapman J. Cancer Res. 1998;58:2720. [PubMed] [Google Scholar]
- 27.Wang SI, Parsons R, Ittmann M. Clin. Cancer Res. 1998;4:811. [PubMed] [Google Scholar]
- 28.Koul D, Shen R, Garyali A, Ke LD, Liu TJ, Yung WK. Int. J. Oncol. 2002;21:469. [PubMed] [Google Scholar]
- 29.Liu LZ, Fang J, Zhou Q, Hu X, Shi X, Jiang BH. Mol. Pharmacol. 2005;68:635. doi: 10.1124/mol.105.011254. [DOI] [PubMed] [Google Scholar]
- 30.Fang J, Xia C, Cao Z, Zheng JZ, Reed E, Jiang BH. FASEB J. 2005;19:342. doi: 10.1096/fj.04-2175com. [DOI] [PubMed] [Google Scholar]
- 31.Weidner N, Semple JP, Welch WR, Folkman J. N. Engl. J. Med. 1991;324:1. doi: 10.1056/NEJM199101033240101. [DOI] [PubMed] [Google Scholar]
- 32.Gao N, Zhang Z, Jiang BH, Shi X. Biochem. Biophys. Res. Commun. 2003;310:1124. doi: 10.1016/j.bbrc.2003.09.132. [DOI] [PubMed] [Google Scholar]
- 33.Murillo H, Huang H, Schmidt LJ, Smith DI, Tindall DJ. Endocrinology. 2001;142:4795. doi: 10.1210/endo.142.11.8467. [DOI] [PubMed] [Google Scholar]
- 34.Priulla M, Calastretti A, Bruno P, Amalia A, Paradiso A, Canti G, Nicolin A. Prostate. 2007;67:782. doi: 10.1002/pros.20566. [DOI] [PubMed] [Google Scholar]
- 35.Zi X, Singh RP, Agarwal R. Carcinogenesis. 2000;21:2225. doi: 10.1093/carcin/21.12.2225. [DOI] [PubMed] [Google Scholar]
- 36.Blagosklonny MV. Cancer Cell. 2004;5:13. doi: 10.1016/s1535-6108(03)00336-2. [DOI] [PubMed] [Google Scholar]