

Summary
The human synuclein family includes the protein α-synuclein, which has been linked to both familial and sporadic Parkinson’s disease, as well as the highly homologous proteins β- and γ-synuclein. Mutations in α-synuclein cause autosomal dominant early onset Parkinson’s, and the protein is found deposited in a fibrillar form in both hereditary and idiopathic forms of the disease. No genetic links between β- and γ-synuclein and any neurodegenerative diseases have been established, and it is generally considered that these two family members are not highly pathogenic. In addition, both β- and γ-synuclein are reported to aggregate less readily than α-synuclein in vitro. Indeed, β-synuclein has been reported to protect against α-synuclein aggregation in vitro, as well as α-synuclein mediated toxicity in vivo. We have previously compared the structural properties of the highly helical states adopted by all three synucleins in association with detergent micelles in an attempt to delineate the basis for functional differences between the three proteins. Here we report a comparison of the structural and dynamic properties of the free states of all three proteins in order to shed light on differences that may help to explain their different propensities to aggregate, which in turn may underlie their differing contributions to the etiology of Parkinson’s disease. We find that γ-synuclein closely resembles α-synuclein in its free state residual secondary structure, consistent with the more similar propensities of the two proteins to aggregate in vitro. β-synuclein, however, differs significantly from α-synuclein, exhibiting a lower predisposition towards helical structure in the second half of its lipid-binding domain, and a higher preference for extended structures in its C-terminal tail. Both β- and γ-synuclein also show less extensive transient long-range structure than that observed in α-synuclein. These results raise questions regarding the role of secondary structure propensities and transient long-range contacts in directing synuclein aggregation reactions.
Keywords: alpha-synuclein, beta-synuclein, gamma-synuclein, Parkinson’s disease, Intrinsically unstructured proteins
Introduction
Synucleins are small highly conserved vertebrate proteins that are highly expressed in neurons 1; 2; 3. In humans, three proteins, α-synuclein (aS), β-synuclein (bS), and γ-synuclein (gS), are included in the synuclein family 4; 5; 6. aS and bS are predominantly localized at presynaptic nerve terminals and are largely absent from peripheral tissues 7; 8. In contrast, gS is abundant in the peripheral nervous system, but is also expressed in other tissues, including the brain, as well as breast and ovarian cancers 3; 6; 9; 10. In addition to their overlapping expression patterns, synuclein family members share a high level of sequence homology. The N-terminal domains of all three proteins, defined by their lipid-interactions, are especially highly conserved and include several imperfect 11 residues repeats, while the C-terminal domains are highly acidic and are more diverse between synucleins (Figure 1). Within the N-terminal domain, the most notable sequence difference between the proteins is the deletion of 11 residues in the bS sequence that correspond to parts of repeats 6 and 7 of aS and gS and are located in the most hydrophobic region of the proteins, which is referred to as the NAC region in aS 11. Within the C-terminal domain, both aS and bS contain two 16-residue imperfect repeats 12, while gS has a relatively shorter C-terminal tail that does not contain these repeats. All three synucleins are intrinsically unstructured when isolated under physiological conditions 13; 14; 15; 16 and adopt highly helical structures in their N-terminal domains upon binding to lipid vesicles or detergent micelles in vitro 17; 18; 19; 20.
Figure 1.
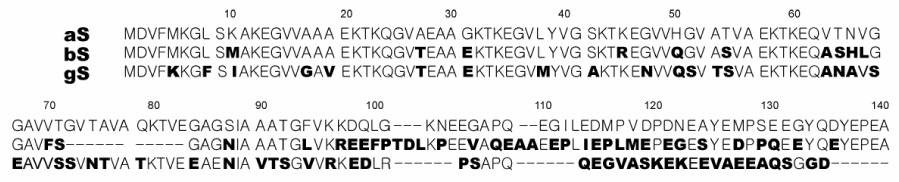
Sequence alignment of human aS, bS and gS produced by the program ClustalW 103. The imperfect 11-residue repeats are delineated by spaces. Boldface characters indicate a difference from the aS sequence.
Among the three synucleins, aS has been intensely studied because it is linked with the etiology of Parkinson’s disease (PD). aS is a major component of Lewy bodies and Lewy neurites 21, proteinaceous deposits associated with functionally damaged brain regions in PD, where it is found aggregated in the form of amyloid fibrils 22; 23. Furthermore, three missense mutations, A53T 24, A30P 25, and E46K 26 in the α-synuclein gene are associated with familial Parkinsonism, and gene duplication 27 or triplication 28 can also cause early onset familial PD. Despite extensive study, however, both the normal functions of aS and their relation to the mechanism by which aS contributes to PD remain unclear. Consistent with its localization to synaptic terminals, aS appears to be involved in synaptic plasticity 29. Additional and mounting evidence suggests a role for the protein in neurotransmitter release pathways. For example, aS has bee shown to regulate the production of phosphatidic acid by phospholipase D 30, an activity that is thought to be important in the formation and fusion of synaptic vesicles 31. aS can also act to rescue neurodegeneration caused by the absence of cysteine-string protein α, a protein responsible for assisting in fusion-mediating SNARE complex assembly 32. Accumulation of aS causes defects in ER-Golgi traffic in yeast, and increased neuronal expression of Rabs, proteins involved in vesicle trafficking, rescues aS-induced cell death 33. Finally, aS has been show to modulate neurotransmitter release in a number of systems 34; 35; 36; 37; 38.
The role of aS aggregation in PD also remains unclear. All three PD-linked aS mutations increase the rate at which the protein forms oligomeric species in vitro 39; 40; 41; 42; 43, suggesting that this effect may underlie the ability of these mutations to cause disease. A number of models for how aggregation may lead to toxicity exist, but conclusive evidence for any one model has proven difficult to obtain. Nevertheless, extensive efforts have been made to understand the process by which aS converts into amyloid fibrils, including detailed structural studies of the free protein isolated in vitro 15; 44; 45; 46, representing the initiation point of the aggregation reaction, as well as of the aggregated fibrillar form of the protein, representing the end point of the reaction 47; 48.
Unlike aS, relatively little is known about bS and gS. In contrast to aS, no mutations in the bS and gS genes in familial PD have been identified 49; 50; 51 and they are not found in Lewy bodies or Lewy neurites 23, excluding a primary role for either protein in the pathogenesis of PD. Nevertheless, some evidence exists for bS and gS deposition in PD or dementia with Lewy Bodies (DLB) 52, and for a genetic linkage between bS and DLB 53. In vitro, bS and gS are less prone to aggregation than aS. gS can form amyloid fibrils under typical conditions that lead to aS fibril formation, but does so less readily 14; 54. bS was initially reported to be incapable of forming fibrils 14; 54, but more recently was shown to do so in the presence of metals 55. Interestingly, bS 14; 56; 57 and gS 14 were shown to inhibit aS fibril formation when incubated with aS in vitro. Additionally, double-transgenic mice, expressing human aS and bS show an ameliorated PD-like phenotype when compared with singly transgenic mice expressing human aS alone 56.
Here we report a detailed comparison of the structural properties of bS and gS with those of aS in the free state of each protein isolated in solution. Because bS and gS are not clearly linked to PD despite their high sequence homology to aS, and are also less prone to aggregate in vitro, structural differences between the three proteins may be informative regarding those properties of aS which are crucial for facilitating its aggregation, and for determining its role in the etiology of PD. We further investigate potential interactions of bS and gS with aS in an attempt to understand the inhibitory effects of the two proteins on aS aggregation and toxicity.
Results
Circular dichroism
To assess the gross secondary structure content of all three synucleins under the conditions used in our NMR experiments, we collected far-UV CD spectra of all three proteins (Supplementary Data Figure 1). The spectra confirm the highly unstructured nature of the polypeptides, as indicated by the intense negative band near 195 nm and the absence of a strong signal at either the β-sheet characteristic band around 218 nm or the helix indicative bands at 208 and 222 nm. Nevertheless, the deviation of the strong negative band away from 195 nm towards longer wavelengths, combined with the weak signal for all three proteins at 222 nm are suggestive of the presence of a small degree of residual helical structure.
NMR chemical shifts
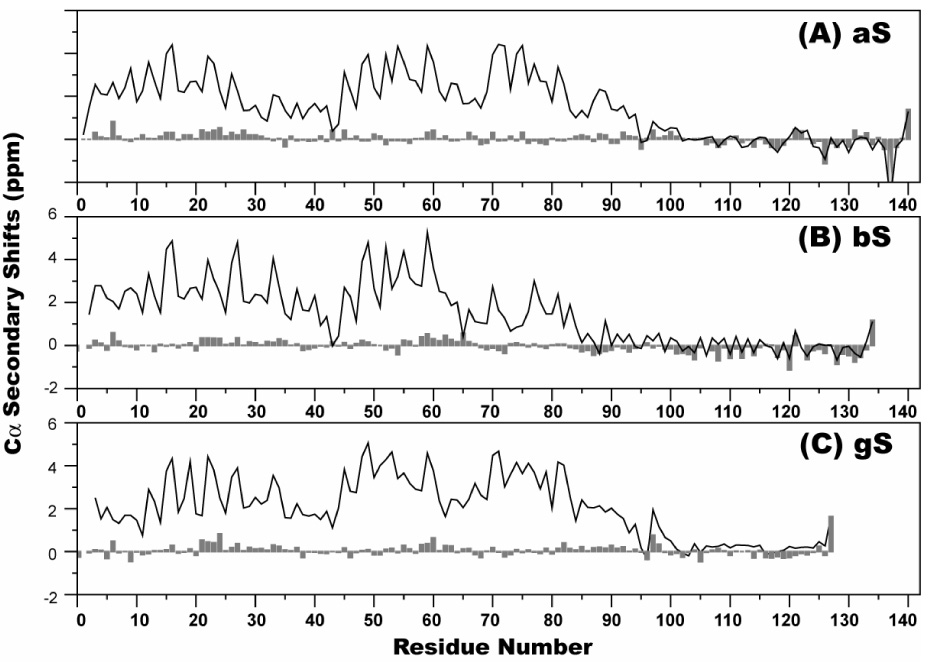
To investigate sequence specific residual structure in bS and gS, the backbone NMR resonances of the free states of both proteins were assigned. Consistent with the lack of significant secondary structure observed in the CD spectra, the ¹H-15N heteronuclear single quantum coherence (HSQC) spectra of bS and gS show a degree of resonance dispersion that is much lower than that observed for well-structured proteins (Figure 2). To investigate the potential for residual structure in both proteins, the deviations of the chemical shifts of the Cα nuclei from those expected for a fully random coil ensemble of conformations 58 were calculated for both proteins. These so-called secondary chemical shifts can be used as indicators of secondary structure preferences in protein 59; 60. Residues with positive Cα secondary chemical shifts have preferences for helical regions of ϕ-ψ space, while residues that populate more extended conformations show the opposite pattern. Figure 3 shows the Cα secondary shifts of bS and gS as a function of residue number as well as the values previously determined for aS 15; 46 for comparison. Although the secondary shifts are small, distinct preferences towards positive or negative values can be seen in the N-terminal and C-terminal domains, respectively, of all three proteins. bS exhibits generally positive Cα secondary shifts from its very N-terminus until approximately residue 65 (mean value of 0.09 PPM). Between residues 66 and 83, the bS secondary shifts are slightly negative or close to zero (mean value of −0.04 PPM). Between residues 84 and the C-terminus of the protein, the secondary shifts are predominantly negative (mean value of −0.25 PPM). For gS, a preference for positive secondary shifts is maintained from the N-terminus through approximately residue 98 (mean value of 0.09 PPM), after which a reversal to negative secondary shifts is again observed (mean value of −0.04 PPM). The data for gS closely resemble those previously obtained for aS, whereas for bS the change from positive to negative shifts occurs much earlier within the sequence.
Figure 2.
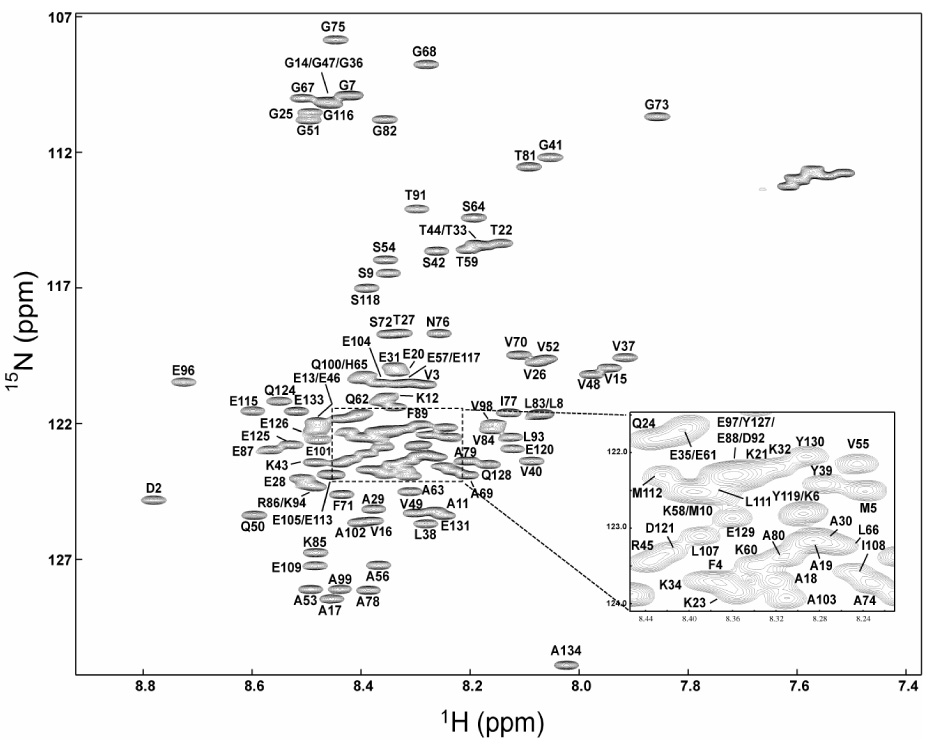
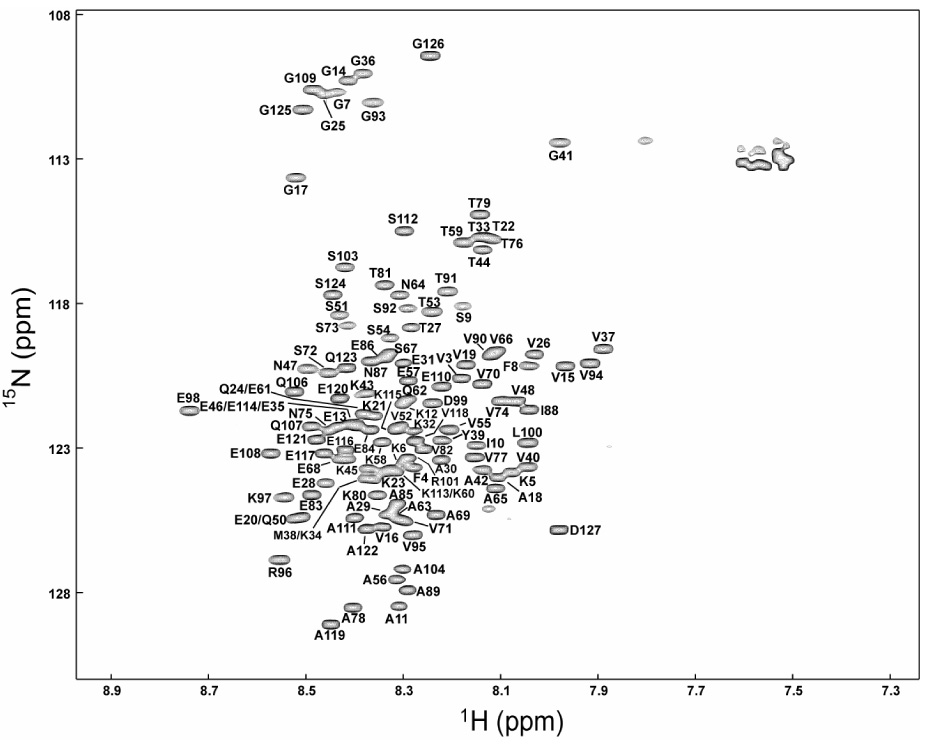
¹H-15N heteronuclear single quantum coherence (HSQC) spectra of bS (A) and gS (B) at pH 7.4, at 10 ° C. Sequence-specific resonance assignments are indicated. Unlabeled peaks correspond to side-chain resonances.
Figure 3.
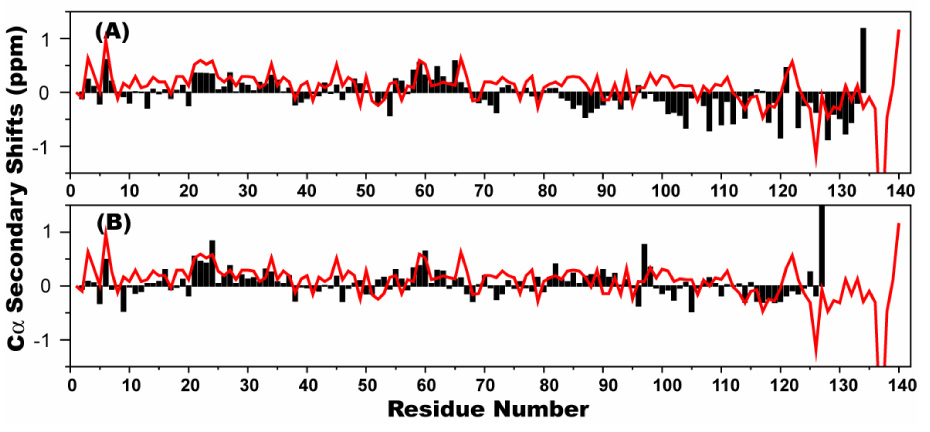
The deviation of Cα chemical shifts from random coil values (so called secondary shifts) for bS (bars, top), gS (bars, bottom), and for aS (line, top and bottom) as previously published 46.
Previously, we have shown that the highly conserved N-terminal regions of all three synucleins adopt a highly helical structure in the presence of SDS micelles, while the C-terminal regions remain largely unstructured 18; 20. We also noted in our studies of aS that the structural preferences observed for the free state of the protein reflected the well formed structure observed in the micelle-bound state, particularly in the lipid-binding N-terminal domain. To investigate whether a similar correspondence exists between the free and micelle-bound states of bS and gS, the Cα secondary chemical shifts in the two states were compared (Figure 4). For gS, the end of the helical structure in the micelle-bound state occurs at residue 95. Slightly positive Cα secondary shifts continue past the end of the helical structure, however, as do significant sequential HN-HN NOEs, indicating that a number of residues past the end of the helical structure adopt a somewhat compact ordered conformation in the micelle-bound state 20. In the free state, there is a dip in the secondary shifts at positions 95–96 corresponding to the end of the micelle-bound helical structure, but a return to positive values for the subsequent two residues, reflecting the non-helical extension. For bS, the micelle-bound helical structure ends at residue 84. Correspondingly, a shift to negative secondary shifts is observed in the free state data at position 83. A unique additional feature of micelle-bound bS is that the C-terminal half of the second of the two micelle-bound helices is strongly destabilized, resulting in significantly decreased Cα secondary shifts beyond position 65. This transition also appears to be reflected in the free state data, as the secondary shifts decrease past position 65 and remain near zero until position 83, where the further shift to negative values takes place.
Figure 4.
Comparison of the deviation of Cα chemical shifts from random coil values in the presence (line) and in the absence (bars) of SDS micelles for aS (A), bS (B) and gS (C). Values for the SDS-bound states are taken from previous reports 18; 20.
Interestingly, the Cα secondary shifts in the C-terminal tails of all three synucleins are very similar in the free and micelle-bound states. Indeed, they are nearly identical for aS and bS, and although for gS the micelle-bound shifts are slightly more positive in the C-terminal tail, the effect is systematic suggesting that there may be a slight offset in the referencing of the data for micelle-bound gS. This close correspondence in the Cα secondary shifts indicates that the secondary structure propensities of the C-terminal tail are not affected by micelle-binding. This does not mean that the C-terminal tail is entirely insensitive to micelle or lipid binding, as the amide proton chemical shifts of the C-terminal tail of aS do exhibit small changes upon micelle binding, indicating some degree of change in the environment of the acidic tail region 61.
Backbone dynamics
To investigate the backbone dynamics of free bS and gS, 15N R1, R2 and steady state heteronuclear ¹H-15N NOE (hNOE) values were measured (Figure 5). The R1 relaxation rates are quite uniform away from the polypeptide termini and are very similar for all three synucleins. The R2 relaxation rates are less uniform reflecting both the greater sensitivity of this rate constant to protein motions and the lower signal-to-noise that is typically achievable in these measurements. A consistent feature in the data for all three proteins is a somewhat decreased R2 value in the region of residues 70 – 80, suggesting a greater degree of mobility in this region, an observation which was previously noted for aS 46. Although other variations occur throughout the R2 data for all three proteins, there is no clear evidence for slow time scale conformational exchange or of other easily interpretable variations in fast time scale motions. The steady state NOE data are also relatively uniform. For aS, these data also exhibit slightly decreased values in the NAC region, as previously reported, but this trend is not as clear cut for either bS or gS. Overall the relaxation data clearly indicate that all three proteins are highly dynamic, as the hNOE data fall well below values indicative rigid structure, and the R2 values are only slightly greater than the R1 values, indicating a high degree of flexibility with individual sites approaching the extreme narrowing limit.
Figure 5.
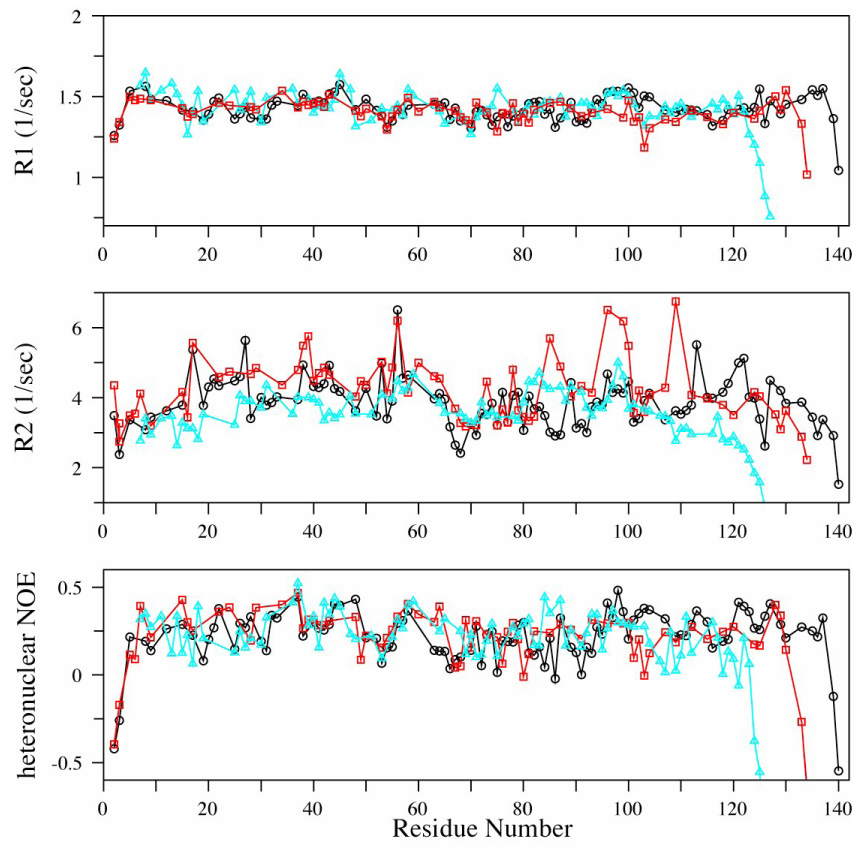
R1, R2 and steady state ¹H-15N NOE relaxation parameters at 800 MHz for backbone 15N nuclei in aS (black ○), bS (red ◻), and gS (cyan △) at pH 7.4, at 10 ° C.
Paramagnetic relaxation enhancement
By attaching spin labels to specific sites in a protein, it is possible to establish the physical proximity of the spin-labeled site to other locations within the protein through the degree of broadening that is observed in the NMR signals originating from such locations 62; 63. Recent studies have utilized this approach to document long-range intramolecular interactions within aS 44; 45. We applied this approach to compare long-range contacts in the three synuclein proteins (Figure 6). For aS, consistent with previous reports, spin labels at N-terminal locations lead to broadening in the C-terminal tail, and spin labels at C-terminal locations lead to broadening in the NAC region as well as near the N-terminus of the protein. Specifically, introduction of a spin label at residue 20 caused broadening of the resonances originating from the vicinity of residue 128, introduction of a spin label at residue 120 caused broadening in the vicinity of residue 90 as well as near residue 10, and introduction of a spin label at position 85 leads to broadening through much of the C-terminal domain. In addition, spin labels at both position 20 and 85 caused local broadening that extended significantly beyond what would be expected for an idealized random coil conformation, indicating that these regions of the protein are significantly more compact than an ideal random coil.
Figure 6.

Paramagnetic Relaxation Enhancement for aS, bS and gS. The histograms represent the intensity ratios for each resolved cross-peak in the ¹H–15N HSQC spectra of nitroxide spin-labeled and reduced protein samples. Solid red lines indicated the broadening predicted using an idealized random coil model of the polypeptide chain.
In contrast to aS, introduction of spin labels at similar positions within bS and gS did not lead to broadening at corresponding distant regions within these proteins. Specifically, spin labels at position 20 in both bS and gS lead to little or no broadening in the C-terminal regions of either protein, spin labels at position 114 in both bS and gS lead to no significant broadening either in the N-terminal regions of either proteins or in central regions corresponding to the NAC region of aS, and spin labels at position 74 for bS and 85 for gS did not cause appreciable broadening in the C-terminal domain of either protein. Thus, the long-range contacts inferred to exist in the free state of aS appear to be largely absent in both bS and gS.
Notably, as for aS, spin labels within the N-terminal domain of both bS and gS also lead to broadening that extends significantly beyond the region that would be effected for an idealized random coil. For the spin label at position 20, the effect is particularly strong in gS, leading to significant broadening from the very N-terminus to around residue 50, similar to that observed for aS. In bS, broadening is observed in a similarly extensive region, but the degree of broadening is significantly diminished. For gS, labeling at position 85 leads to significant broadening through residue 105 on the C-terminal side of the label, whereas on the N-terminal side, the broadening profile is similar to that expected for a model random coil, extending only to around position 75. For bS, labeling at position 74 leads to broadening ranging from positions 50 through 100, covering a range similar to that observed in aS upon labeling position 85. Interestingly, for both bS and gS labeling at position 74 or 85, respectively, leads to some broadening of first 10 to 20 N-terminal residues. This is a feature not observed in the aS data, suggesting the potential for contacts within the bS and gS lipid-binding domains that are absent in aS.
For both bS and gS, labeling within the C-terminal tail at position 114 leads to a broadening profile that closely resembles that predicted for a model random coil, indicating that the C-terminal tails of both proteins are more highly disordered than the N-terminal domains, and also validating the model used to predict the extent of broadening expected for an idealized random coil. For gS, a slight degree of additional broadening is observed N-terminal to the calculated profile, possibly reflecting the proximity of the spin label to the compact region inferred from the data obtained with the label at position 85.
Residual dipolar couplings
Recently residual dipolar couplings (RDCs) have been measured from unfolded proteins to provide additional structural information 64, although the interpretation of such data remains challenging in this context. In the case of the random flight or Gaussian modes for polypeptide chains lacking local structural preferences, RDCs are expected to be essentially uniform throughout the sequence except near the termini, where values fall off due to increased motions and a closer approximation to spherical symmetry 65. Deviations from such a uniform distribution may report on residual structure, which may in principle reflect either local or long-range structure. A study of RDCs in acid-denatured apomyoglobin demonstrated a high degree of correspondence between RDCs and local residual secondary structure 66. For the free state of aS, measurements of RDCs have been reported 45 and have been analyzed in the context of calculations based on ensembles of simulated coil-like structures 67 or on local sequence properties such as side chain bulkiness 68. Both of these studies reported a disparity between the calculated and measured RDCs, which was interpreted to reflect the presence of long-range structure with free aS. Here we have measured RDCs from all three synucleins in stretched polyacrylamide gels (Figure 7). The RDC profiles for all three proteins deviate from the uniform distribution expected for a guassian coil, consistent with the presence of residual structure. A consistent feature among all three synucleins is the presence of higher amplitude RDCs in the C-terminal tail regions. Given the previously observed correlation between residual secondary structure and RDCs for acid-denatured apomyoglobin, we decided to compare the Cα secondary shifts observed for the three synucleins with the measured RDCs. We observe a qualitative correlation between the RDC and secondary shift data in the C-terminal regions of all three proteins. Negative secondary shifts are consistently associated with larger amplitude RDCs, and those few regions with positive secondary shifts within the C-terminal domains consistently exhibit decreased RDC values. These observations are commensurate with a local preference for extended local structure in the C-terminal domain of all three proteins, which leads to the preferential alignment of the polypeptide chain along the direction of the alignment medium, and the consequent alignment of the NH bond vectors perpendicular to the external magnetic field, resulting in negative RDC values. Within the N-terminal lipid-binding domains of all three proteins, some degree of correlation between the secondary shift and RDC data is also observed. In particular, the region with the strongest chemical shift indications for helical structure in aS, encompassing residues 18–31, shows the lowest measured RDC values, and this trend is largely maintained in all three proteins. Although well populated helical structure would be expected to lead to sign-reversal of the RDC values, the observed decrease in the RDCs is consistent with either more highly transient helical structure in equilibrium with extended structures, or with pre-helical compact conformations that randomize the alignment of the NH bond vectors. Interestingly, our results do not show the previously reported high amplitude RDCs for residues at the very N-terminus of aS 45. This may reflect differences in the experimental conditions used.
Figure 7.
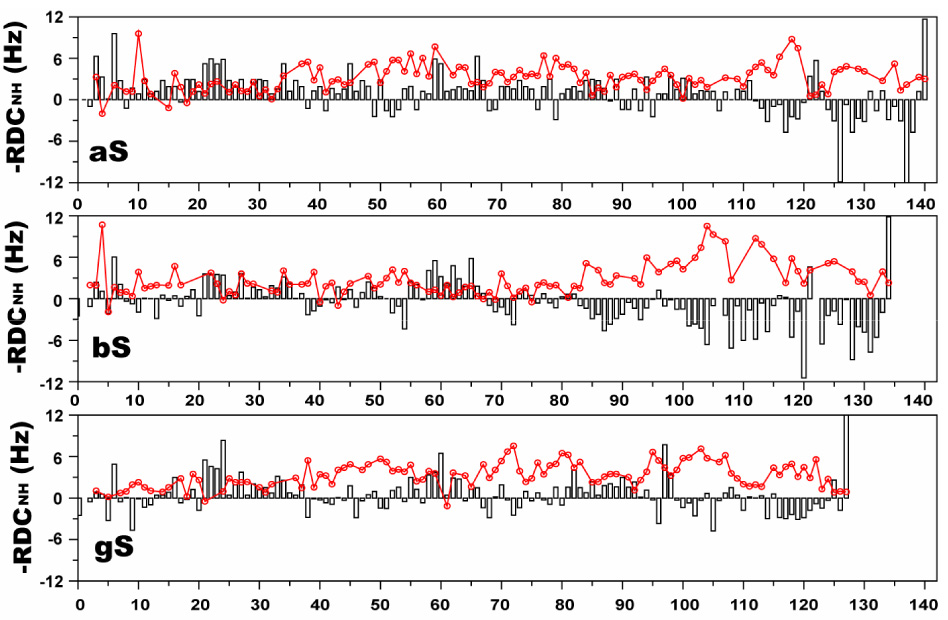
15N-¹H residual dipolar couplings for aS (bars, top), bS (bars, middle) and gS (bars, bottom) in stretched polyacrylamide gels. Cα secondary shift values for each protein are overlayed onto the RDC data as solid lines. Note that the RDCs are plotted on an inverted axis (i.e. negative values are plotted above the zero line), in accord with convention.
Chemical shift mapping
The perturbations of chemical shifts of a protein upon interacting with another protein or ligand are a sensitive tool for probing such interactions and and for mapping binding sites. Because both bS and gS have been reported to inhibit the aggregation of aS, we looked for evidence of an interaction between the free state of aS with bS or gS by monitoring ¹H-15N HSQC spectra of 15N-labeled aS titrated with increasing amounts of unlabeled bS or gS. Notable spectral changes were initially observed throughout the C-terminal tail of aS, as well as for the few residues surrounding His 50. Because we have previously observed that the resonance of His 50 is extremely sensitive to even small perturbations in pH, and since our experiments are performed in phosphate-buffered saline at pH 7.4, well away from the pKa of phosphate, we suspected some of the observed changes might reflect changes in sample pH upon titration. We therefore examined the effects of decreased pH on HSQC spectra of aS. We found that spectra at pH 6.0 led to chemical shift changes that reproduced those observed near His 50 in the titration experiments (Figure 9). Direct post-titration pH measurements of samples used in equivalent titration experiments confirmed variable drops in pH from the starting value of 7.4, and microdialysis of such titrated samples against pH 7.4 buffer strongly attenuated spectral changes around His 50. Surprisingly, restoring the titrated samples to pH 7.4 also eliminated the chemical shift changes observed in the C-terminal tail of aS, and the spectra collected at pH 6.0 reproduced these changes, which are most pronounced for residues 109–137. Spectra of bS and gS collected at pH 6.0 revealed similar chemical shift changes in the C-terminal tails of both proteins (most pronounced for residues 90–134 of bS and 106–127 of gS) as well as in the vicinity of bS His 65 (the sole histidine in the bS sequence). Thus, lowering the pH from 7.4 to 6.0 leads to significant chemical shift changes in the C-terminal tails of all three synucleins. In contrast, the addition of a molar excess of bS or gS to aS, and of aS to bS or gS, did not lead to comparable chemical shift changes once accompanying alterations in pH were eliminated.
Figure 9.
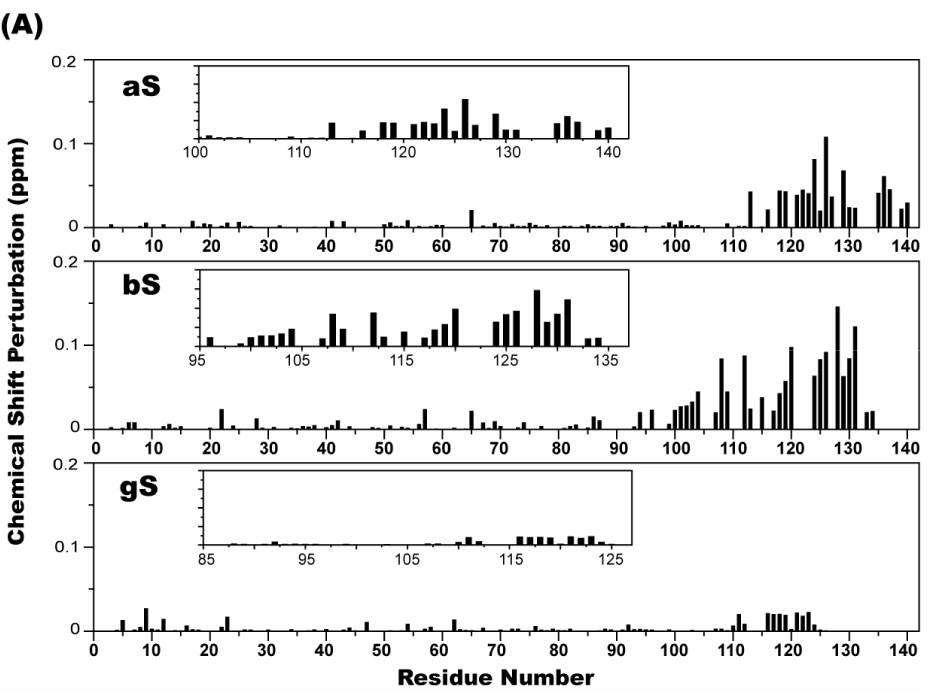
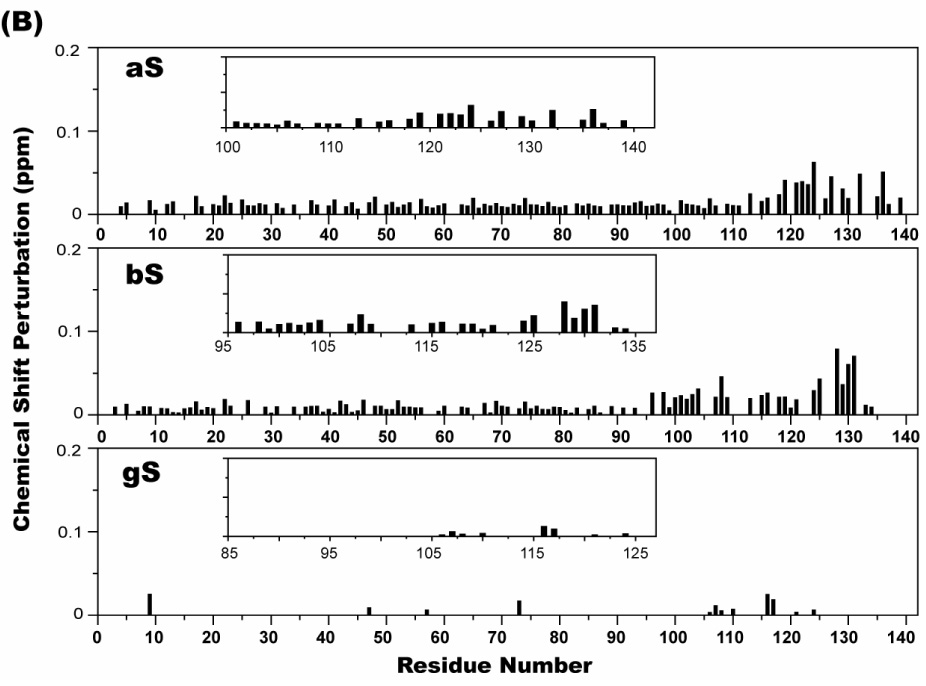
Chemical shift perturbations in ¹H- 15N HSQC spectra of 70 µM 15N-labeled aS, bS, and gS in the presence of 100 µg/ml poly-L-lysine (A), and 50 µg/ml poly-L-argine (B). The weighted average of the ¹H and 15N chemical shifts of a given residue was calculated as [(Δδ²NH + Δδ²NN/25)/2]1/2. The insets provide a blow up of the chemical shift perturbations in the C-terminal tail regions.
aS has also been shown to bind to polycations, including poly-L-lysine and poly-L-arginine, spermine, and spermidine; an interaction that accelerates the rate of aS fibril formation 69; 70. We used NMR to investigate whether bS and gS also bind to polycations and if so, where such binding occurs. Figure 9 shows the weighted average perturbations of chemical shifts in HSQC spectra of all three synucleins upon addition of poly-L-lysine or poly-L-arginine. Clear effects were observed for residues in the C-terminal regions of both aS and bS while more marginal perturbations were observed for gS. In addition to the larger perturbations in the C-terminal regions, smaller perturbations were induced throughout the N-terminal regions of aS and bS by poly-L-Arg, but not to the same extent by Poly-L-Lys, prossibly reflecting the higher molecular weight of the former (55 kD vs. 29 kD). Interestingly, we did not observe any alterations in the intensities of resonances originating from the N-terminal lipid-binding domain of either aS, bS or gS upon polycation binding, in contrast to previously reported increases in aS NAC region resonance intensities upon polycation binding 71, which were concluded to imply a polycation-induced release of interactions between the C-terminal tail of aS and the NAC region.
Discussion
The role of synuclein function and aggregation in disease
Despite the high degree of sequence homology between the members of the human synuclein family, only aS is clearly linked to the pathogenesis of PD. Within the N-terminal lipid binding domain, extending through aS residue 94, bS contains 14 single residue substitutions and an 11 residue deletion when compared with aS, and gS contains 30 single residue substitutions. Within their C-terminal tails, a much greater degree of sequence diversity is evident for the synucleins, with 30 substitutions for bS and 24 substitutions as well as 18 missing residues for gS. These primary sequence differences must necessarily underlie the functional and pathogenic differences between aS, bS and gS, but the mechanism by which the sequence alterations lead to biological consequences remains unclear. Because protein sequence determines protein structure, we previously investigated structural differences between aS, bS and gS in their highly helical micelle-bound conformations 18; 20. We found that the lipid-binding domains of aS and gS were extremely similar in this context, but that the 11 residue deletion in the bS lipid-binding domain lead to a substantial destabilization of the C-terminal half of the second of the two helices of the micelle-bound proteins. Because the micelle-bound state of the synucleins is proposed to mimic the lipid-bound state, which in turn is thought to mediate the normal function of these proteins, we interpreted our observations to mean that functional differences between aS and gS would be determined primarily by the sequence differences in the C-terminal tails of the proteins, which are thought to mediate protein-protein interactions. In contrast, the C-terminal tails of aS and bS are more similar, and functional differences between the latter two proteins may be determined by the observed structural and dynamic differences in their lipid-bound domains.
The function of aS revolves around the regulation of neurotransmitter-carrying synaptic vesicles, suggesting that modulations of this function could potentially play a role in PD. Nevertheless, the appearance of aS amyloid fibrils in the hallmark Lewy body deposits of PD 21; 22; 23, combined with the increased aggregation rates observed in vitro for PD-linked aS mutants 39; 40; 41; 42; 43, has focused much attention on the potential role of aS aggregation in PD. Here, therefore, we have compared the structural characteristics of aS, bS and gS in their isolated free states, which have typically been used as the initiation point for comparative in vitro aggregation studies of these proteins.
Structural properties of free aS
The free state of aS has been extensively characterized previously by us and others. It was shown to contain residual helical structure in the N-terminal lipid-binding domain, and to prefer more extended structures in the acidic C-terminal tail 15. Free aS was also shown to be more compact than would be expected for an ideal random coil conformation 14; 72, and to populate conformations in which the C-terminal tail makes transient contacts with the NAC region of the protein, as well as with the very N-terminal region of the protein 44; 45. Based on observations of residual amphipathic helical structure in aS, we originally suggested that intramolecular contacts involving the NAC region might act to occlude this hydrophobic region of the protein and retard intermolecular interactions leading to aggregation 46. This hypothesis was subsequently modified to invoke interactions between the C-terminal tail and the NAC region as being key to retarding the aggregation of the protein 44; 45. More recently, comparisons of measured and predicted RDCs in free aS have been interpreted to reflect the presence of long-range contacts within the protein 67; 68. Although the idea that long-range contacts within aS may retard the aggregation of the protein has proven popular, a different model proposed that compact conformations of the protein would actually favor amyloid fibril formation 73.
Residual secondary structure in bS and gS
Both bS and gS have been shown to aggregate more slowly than aS in vitro, although the difference is much greater for bS 14; 54. Therefore, structural studies of free bS and gS provide an opportunity to determine which structural properties exert the greatest influence on the aggregation propensities of the synucleins. Our results indicate that residual secondary structure in gS, as judged by chemical shifts (Figure 3), is quite similar to that previously observed in aS 15; 46. Recently, a comparison of the secondary structure preferences of gS and aS, based on NMR chemical shifts, concluded that gS has a somewhat increased preference for helical structure in the NAC domain of the proteins 16. Such a difference is not readily apparent in our Cα chemical shift data. This may be due either to slight differences in experimental conditions between the studies, or possibly to the use of a more sophisticated chemical shift analysis algorithm by Marsh et al. to tease out secondary structure preferences. We have generally found that Cα shifts alone are the most reliable indicator of weak helical propensity, as they are least sensitive to nearest neighbor effect, which can be difficult to correct for. Thus, based on our data, if there are differences in residual helical structure between aS and gS, they must be quite small.
In contrast to gS, residual structure within free bS is found to differ substantially from that of aS. In particular, the helical propensity of bS is clearly reduced between residues 66 and 83 when compared with that of aS. This region subsumes the 11-residue deletion within the bS sequence, which corresponds to aS residues 72 to 83. Interestingly, the reduction of the helical propensity of bS accurately predicts the destabilization of helical structure that is observed in the micelle-bound state of bS (Figure 4). Thus, residual helical structure in all three synucleins appears to accurately reflect well-formed helical structure in their micelle-bound states. Since the helical structures of micelle-bound gS and aS are very similar 74, this further supports our conclusion that residual structure in the N-terminal domains of the free states of aS and gS are highly similar as well.
The C-terminal domains of all three synucleins exhibit chemical shifts that are indicative of a general preference for more extended conformations. In aS, small Cα secondary shifts of oscillating sign persist for nearly 20 residues past the end of the lipid-binding domain, but the remainder of the C-terminal tail exhibits two distinct negative stretches in the data 46, which correspond fairly well with the two 16-residue imperfect repeats in the tail region 12. For bS, the negative secondary shifts begin immediately after the end of the lipid-binding domain at position 84, and remain substantially negative throughout the C-terminal tail, indicating that this region of bS is likely more extended than the corresponding region of aS. gS has a considerably shorter C-terminal tail than aS or bS, and exhibits only a short region of negative secondary shifts near the very C-terminus, indicating that the tail of gS samples more random conformations than that of either aS or bS.
Transient long-range structure in bS and gS
PRE measurements provide an effective means for detecting long-range interactions in poorly structured proteins 62; 63. Using this methodology, we have confirmed previous reports 44; 45 of apparent contacts between the C-terminal tail of aS and the NAC region as well as the N-terminus of the protein (Figure 6). Our data also illustrate the compact nature of the free state of aS in the vicinity of both N-terminal domain spin label sites (positions 20 and 85). Given the hypothesis that long-range contacts involving the C-terminal tail of aS occlude intermolecular interaction sites, it might be expected that such interactions may exist in bS and gS as well, since these proteins are better protected from aggregation. Instead our data show no evidence in either protein for contacts between the C-terminal tail region and sites within the N-terminal lipid-binding domain. Indeed, for bS, the introduction of a spin-label at position 114 leads to a broadening profile that closely reproduces that which would be expected for an idealized random coil, supporting the chemical shift indications for a highly extended ensemble of conformations in this region. For gS, the broadening distribution in the tail deviates slightly from the predicted random coil behavior on the N-terminal side of the label, consistent with the chemical shift indications for less extended structure. For both bS and gS, labeling within the N-terminal lipid-binding domain provides evidence that this domain is more compact than a model random coil, as was observed for aS. However, some lipid-binding domain contacts between the very N-terminal regions and the NAC-corresponding regions of bS and gS appear to be unique to these family members and absent in aS.
The absence of long-range contacts between the N- and C-terminal domains of bS and gS would suggest that any intermolecular interactions sites within these proteins should be more exposed than in aS, favoring aggregation reactions. For bS, the 11-residue deletion within the NAC region removes a number of apolar residues, potentially reducing the self-affinity of this region, and perhaps explaining the low aggregation propensity of this variant, even in the absence of intramolecular protection by the C-terminal tail. For gS, the NAC region is largely intact, but three glycine residues in this region (G67, G84 and G86) are altered to glutamate in this family member. Thus, the introduction of negative charges may explain the decreased ability of this region to mediate intermolecular contacts, and may also explain the lack of contacts with the C-terminal tail, since the latter is highly negatively charged. The Gly to Glu substitutions would also be consistent with the reported increase in the helical character of the gS NAC region 16, despite our own inability to detect this increase.
Because RDCs observed for aS were interpreted to reflect the presence of long-range contacts within aS, we measured RDC values under our conditions for all three synucleins (Figure 7). Because RDCs in denatured proteins have also been found to reflect local secondary structure propensities 66, we compared the measured RDCs with the corresponding Cα secondary shifts. We find a qualitative correlation between the two measurements, and especially within the C-terminal tails of all three synucleins. Given this correspondence, we believe it likely that the observed RDCs in the C-terminal tails arise primarily from the preferential alignment of locally extended chain segments, and that there is no clear need to invoke the presence of any long-range contacts to explain these RDC data, although we can not rule out a contribution from such effects. Interestingly, however, we note that the RDCs we observe for aS are not in precise agreement with those previously reported 45. In the C-terminal tail, we do observe two regions of significant negative RDCs, but the magnitudes we measure are not as high as those previously reported. Furthermore, at the N-terminus of aS, we observe relatively small RDCs in contrast to the larger values previously seen 45. In fact, our aS RDC values appear to agree quite well with the RDC values predicted in the absence of any long-range contacts 67.
The discrepancy between our RDC measurements and those previously reported may be due to differences in experimental conditions, as well as to the use of different alignment media. Nevertheless, under our experimental conditions, we are able to detect transient long-range contacts in aS using PRE measurements, but these contacts do not appear to be reflected in the RDC measurements, which as stated agree nicely with predictions based on the absence of such contacts. Thus, we believe that considerable care must be taken before RDC measurements can be used as a reliable indicator of long-range contacts in disordered synucleins or other proteins.
Interactions of aS, bS and gS
Both bS and gS have been reported to inhibit the aggregation of aS. To determine whether this effect may reflect direct interactions, we used NMR chemical shift mapping to look for such interactions. Our data do not indicate any direct interaction between aS and bS or gS in their monomeric free states under our conditions. Therefore, the mechanism by which bS and gS retard aS aggregation likely involves interactions between non-monomeric forms of the proteins, or is perhaps mediated by some other effect. One possibility is that the high sequence homology between aS, bS and gS allows bS or gS to interact with synuclein fibrils or oligomers in a fashion that blocks further addition of aS monomers. Studies of the inhibition of mouse aS aggregation by human aS lead to similar suggestions 75 and a similar mechanism has also recently been indicated for the inhibitory effect of the Alzheimer’s disease peptide Aβ40 on the aggregation of Aβ42 76. Alternately, it is possible that the inhibitory effects of bS and gS are not mediated by specific interactions. Synucleins have been reported to function as protein chaperones 77, yet given their highly disordered nature, they seem unlikely to do so in any conventional sense. Possibly, high concentrations of aS, bS or gS may instead act as chemical chaperones in the same way as a number of individual amino acids are able to 78.
Because polycations do interact with aS and enhance its aggregation, we sought to determine if they also bind to bS and gS. Using chemical shift mapping, we showed (Figure 9) that poly-L-lysine and poly-L-arginine bind to the C-terminus of bS in a fashion quite similar to aS binding. Polycation interactions with gS appeared to be weaker, as a much lesser degree of chemical shift perturbation is evident at comparable ratios of protein to polycation. This may reflect the shorter length, and lesser negative charge, of the gS tail. Although the effects of polycation binding on the aggregation of bS and gS have not been reported, it has been shown that metal binding, and in particular copper binding, to bS catalyzes amyloid fibril formation by this protein. We and others have shown that copper binds to negatively charged side chains in the C-terminal tail of aS, as well as to the N-terminal amino group and to His 50 79; 80, and it can therefore be expected that copper will bind to the same sites in the C-terminal tail of bS that mediate polycation binding. Thus, we would predict that polycations would also catalyze amyloid fibril formation by bS. Polycation binding to the C-terminal tail of aS was proposed to catalyze the aggregation of aS by perturbing long-range contacts between the C-terminal tail and the NAC region of the protein. In the case of bS, we have demonstrated the absence of any long-range interactions between its C-terminal tail and its N-terminal lipid-binding domain. Thus, copper or polycation binding to bS can not induce aggregation by relieving such intramolecular contacts. Instead, we propose that cation binding to the C-terminal tail of bS neutralizes the highly negative charge of the tail, and therefore reduces electrostatic repulsion between bS monomers, facilitating the aggregation of the protein. We furthermore propose that the same mechanism likely underlies the effects of polycation and metal binding on aS aggregation. This mechanism readily accounts for the available data without requiring any invocation of effects of cation binding on long-range structure within aS or bS.
Finally, lowering the pH of aS, bS or gS samples from 7.4 to 6.0 resulted in spectral changes in the C-terminal tails of all three synucleins which resemble those induced by polycations (Figure 8 and Figure 9), suggesting that at lower pH the C-terminal tails of all three proteins may form interactions with cationic species. The most likely candidates for such species are groups that acquire a greater positive charge upon the drop in pH, which should include the primary amino group of each protein as well as any histidine residues. Because gS lacks histidines but still exhibits a comparable effect, the primary amino groups are likely to play an important role.
Figure 8.

Weighted average of chemical shift changes in the ¹H- 15N HSQC spectra for 15N-labeled aS (A), bS (B) and gS (C) upon going from pH 7.4 to pH 6.0 (bars) and upon pH changes accompanying the addition of five-fold molar excess unlabeled bS to 15N-labeled aS (A), five-fold molar excess unlabeled aS to 15N-labeled bS (B), and five-fold molar excess unlabeled aS to 15N-labeled gS (C) (solid red lines). The weighted average of the ¹H and 15N chemical shifts of a given residue was calculated as [(Δδ²NH + Δδ²N/25)/2]1/2 104.
Thus, lower pH may induce or enhance interactions between the acidic C-terminal tails of all three synucleins and their N-terminal amino groups, which would be consistent with the more compact nature of all three proteins at low pH 14. The relation of such contacts to those observed using PRE experiments in aS at pH 7.4 is unclear at present. The latter experiments also suggest C-terminal to N-terminal contacts, and lowering the pH may simply strengthen these interactions, which in the case of aS may occur at the expense of C-terminal to NAC contacts. However, since lower pH is known to enhance the aggregation of aS 14, any pH-induced long-range C-terminal to N-terminal contacts are unlikely to be protective against, and instead may facilitate aggregation. If their formation reduces C-terminal to NAC contacts, it remains possible that such a shift in long-range transient structure acts to increase the exposure of the NAC region, and thereby increases the likelihood of aS aggregation. Low pH also enhances gS aggregation, but a dramatic effect on bS aggregation was not observed 14. Although no C-terminal to N-terminal or C-terminal to NAC contacts are detected using PRE experiments in bS or gS at pH 7.4, low pH-induced C-terminal to N-terminal contacts could act to alter other contacts such as those observed within the lipid-binding domains of each protein.
Conclusions
Our results reveal a number of differences between the free states of aS, bS and gS. In particular, residual secondary structure in bS is strongly affected by the 11-residue deletion in the NAC region of this family member, resulting in the truncation of residual helical structure at position 65, the same location where the highly helical structure of the micelle-bound state of bS becomes destabilized. In contrast, for both aS and gS residual helical structure persists through the N-terminal lipid-binding domain. Thus, residual secondary structure in the synucleins is found to be a good predictor of well-formed structure in folded states of these proteins, as has been previously observed for a number of other systems 81; 82; 83; 84; 85; 86; 87. The C-terminal tails of both bS and gS, like that of aS, populate more highly extended conformations, but the extended region of bS is longer than that of either aS or gS, beginning immediately after the end of the lipid-binding domain. For both aS and gS less extended structure persists beyond the end of the lipid-binding domain, possibly reflecting the presence of an ordered, but non-helical segment following the helical domains of both micelle-bound proteins 20; 61.
RDC measurements from weakly aligned synuclein samples corroborate the analysis of residual secondary structure in the proteins, clearly reflecting the extended nature of the C-terminal tails of all three proteins. However, the residual helical structure in the lipid-binding synuclein domains, which closely tracks the highly helical folded structure of the proteins, does not lead to the expected sign reversal of the measured RDC values. Such behavior was also observed for certain regions, such as the G-helix, of acid-denatured apomyoglobin 66, and may reflect either compact pre-helical conformations or a mixture of transiently helical and extended conformation. Further investigation will be required to resolve this matter. The RDC data for all three synucleins appear to be adequately accounted for by local structural preferences, and there is no clear need to invoke long-range structure to account for the RDCs. In support of this view, PRE data show that neither bS nor gS possess the transient long-range structure observed in aS, yet the RDC data for all three proteins are qualitatively and to some extend quantitatively similar.
The absence of long-range contacts between the C-terminal tails and the lipid-binding domains of bS and gS would suggest that these proteins should aggregate more rapidly than aS if such contacts indeed retard aggregation, yet bS and gS are reported to aggregate more slowly than aS. Although sequence alterations within the NAC region of bS and gS may in part account for their reduced aggregation, this observation raises questions regarding the importance of the interactions of the C-terminal tail of aS in aS aggregation. It is known that truncation of aS enhances the fibril forming propensity of the protein 88; 89; 90. However, this observation can be easily accounted for by the highly negative charge of the aS tail, which should disfavor intermolecular interactions through charge repulsion. Polycation binding to aS is also known to enhance its aggregation 69; 70 and was proposed to do so by perturbing interactions between the C-terminal tail and the NAC region 71. Yet we do not observe evidence for such an effect in our data, and furthermore cation-binding to the C-terminal tail of bS can also enhances the aggregation of this protein, for which we have now demonstrated the absence of any long-range interactions involving the C-terminal tail. Thus it seems more likely that the primary effect of cation- or polycation-binding on synuclein aggregation may be attributed to charge neutralization of the C-terminal tails of these proteins, and any effect on long-range structure is likely to be secondary.
Although bS and gS lack long-range structure involving their C-terminal tails at pH 7.4, our data show that their lipid-binding domains are relatively compact. Thus, it remains possible that the arrangement of transient contacts within the lipid-binding domains of the free synucleins plays an important role in modulating the availability of intermolecular interactions sites for nucleating aggregation reactions. Indeed, our data suggest the existence of lipid-binding domain contacts that are unique to bS and gS, and also indicate that pH changes have the potential to alter such contacts. Nevertheless, in light of our observations that bS and gS are if anything less compact than aS, and possess either comparable or reduced residual helical structure, it seems most likely that the primary origin of the different in vitro aggregation rates of the synucleins lies in the effects of amino acid substitutions on the local aggregation-determining properties, such as overall charge, hydrophobicity, and secondary structure propensity 91, of their primary sequences.
Methods
Materials
Recombinant aS, bS and gS were expressed in E. coli BL21 (DE3) using plasmid constructs kindly provided by Dr. Peter Lansbury (Department of Neurology, Harvard Medical School). To produce isotopically labeled proteins for NMR studies, saturated overnight LB-kanamycin cultures were used to inoculate M9 minimal media made with uniformly labeled 13C-glucose and/or 15N-ammonium chloride. Cultures were grown at 37 °C to an A600 nm of 0.5–0.6 at which point protein expression was induced with 1 mM IPTG. Cells were harvested by centrifugation 4 hours post-induction. Proteins were purified using a protocol identical to that developed during previous studies of aS 15 involving ion-exchange and reverse-phase chromatography and were stored at −20 °C after lyophilization.
Circular Dichroism
Circular dichroism (CD) spectra were measured on an AVIV 62 DS spectrometer equipped with a sample temperature controller. Far-UV CD spectra were monitored from 190 to 260 nm at 10 °C using final protein concentrations of 1 mg/ml with a path length of 0.2 mm, response time of 1 s, and scan speed of 50 nm/min. Protein concentrations were measured using absorption at 280 nm.
NMR
NMR measurements were performed on samples dissolved in 100 mM NaCl, 10 mM Na2HPO4, pH 7.4 in 90%/10% H2O/D2O as previously described for aS 15. NMR spectra were acquired at 10 °C using either a 600 MHz Varian instrument (Weill Cornell) or 800 or 900 MHz Bruker instruments (New York Structural Biology Center). Pairs of HNCACB/CBCACONH and HNCACO/HNCO triple resonance experiments were collected for each protein to enable sequence-specific backbone and Cβ resonance assignments. Secondary chemical shifts were calculated using the random coil shifts determined using hexapeptides in 1 M urea at pH ~5.0 58. We have found that the use of these random coil shifts yields the most self consistent results for measurements under our conditions, as judged by fewer sporadic large secondary shifts, and a greater degree of contiguity within the data 86; 92. Longitudinal (R1) and transverse (R2) relaxation rates for the backbone 15N nuclei were recorded using relaxation times of t1 = 10, 30, 80, 160, 640, 1280, and 1800 ms and t2 = 20, 34, 66, 85, 106, 130, 136, 154 and 178 ms. R2 data were measured using a pulse sequence employing a Carr Purcell Meiboom Gill pulse train with a 900 µs delay between π pulses. To estimate noise levels, duplicate spectra were recorded for t = 10, 80, and 640 ms (t1 spectra) and t = 66, and 178 ms (t2 spectra). R1 and R2 relaxation rates were determined by fitting resonance heights as a function of the relaxation delay time using NMRview 93. 15N-(¹H) steady-state heteronuclear NOE data were obtained as the ratio of peak heights in paired spectra collected with and without proton saturation during the relaxation delay of 5 s. The experimental uncertainty was estimated using the standard deviation of four pairs of spectra. All NMR data were processed with NMRPipe 94 and analyzed using NMRView 93.
Residual Dipolar Coupling
Polyacrylamide gels were prepared with an acrylamide concentration CA of 7.7 % (w/v), and bisacrlyamide cross-linker concentration CB of 0.4 %. Polymerization was initiated by addition of 1% (w/v) ammonium persulfate and 0.1 % (v/v) N,N,N_-tetramethylethylenediamine immediately prior to casting in a 5.4 mm diameter teflon cylinder. After polymerization, gels were washed with distilled water with gentle agitation for three hours to remove extra unreacted acrylamide. Then, gels were cut into approximately 1.3 cm lengths and dialyzed with the buffer used for NMR experiments with gentle agitation for three hours. The gels were then soaked in 0.3 mM protein solution overnight for NMR experiments. The soaked gels were pushed into open ended NMR tubes stretching the gels using a special apparatus (NewEra Enterprises, Inc) as described 95. NMR spectra were acquired at 10 °C. The solvent deuterium quadrupolar splitting was used to monitor the alignment. The 15N-¹H dipolar coupling was measured using the HNCO based IPAP experiment 96. The 15N-¹H residual dipolar couplings were calculated as the difference between scalar couplings measured in an isotropic sample and total couplings measured in an aligned sample.
Paramagnetic Relaxation Enhancement
Since there are no cysteine residues in any of the three synucleins, site-directed mutagenesis was used to substitute selected residues with cysteine. E20, A85, E110, P120, and A140 of aS were selected for site-directed mutagenesis. In bS, E20, A74, E104, P114, and A134 were selected for cysteine substitution, and in gS, E20, A85, E110, P120, and D127 were selected. The purified mutant proteins were labeled with 1-oxyl-2,2,5,5,-tetramethylpyrroline-3-methyl-methanethiosulfonate (MTSL, Toronto Research Chemicals) as described 97. Paramagnetic relaxation enhancement was measured by collecting ¹H-15N HSQC spectra using 0.25 mM protein samples at 10 °C. For control samples, DTT was added to the MTSL spin-labeled sample to reduce the nitroxide spin label from the protein. The intensities of cross peaks in the ¹H-15N HSQC spectra of both the spin labeled and reduced samples were measured and their ratio calculated.
To calculate theoretical PRE curves, the distance between each residue and the spin label attachment point was calculated assuming a Gaussian distribution of the root mean square end-to-end distance for a random walk polypeptide 98; 99 according to:
| 1) |
where r is the end-to-end distance between a given residue and the spin label site, n is the number of chain links, l is the link length of the chain, taken to be 3.8 Å 100, and α is the cosine of bond-angle supplements for the freely rotating chain model, which was set to a value of 0.8 based on experimentally determined estimates of statistical segment lengths in poly-L-alanine 99.
The paramagnetic contribution to the transverse relaxation rate, R2P was calculated from:
| 2) |
where r is the distance between each residue and the spin label, K is 1.23 × 10–32 cm6s−2, ωH is the Larmor frequency of a proton and τC is the effective correlation time for synucleins (estimated at 4 ns based on NMR relaxation measurements) 101. Subsequently, the peak intensity ratio between the paramagnetic (IP) and the diamagnetic (ID) states was calculated from:
| 3) |
where R2D is the transverse relaxation rate in the diamagnetic state, which was set to 4 s−1, the approximate average R2 obtained by NMR for synucleins and t is the duration of the INEPT delays (4 ms) in HSQC pulse sequence 102.
Chemical shift mapping
For studies of the interaction of aS with its homologues, ten-fold concentrated solutions of unlabeled bS or gS were titrated into aS solutions to achieve final concentrations of 35 uM or 70 uM 15N-labeled aS in 100 mM NaCl, 10 mM Na2HPO4, pH 7.4, 90%/10% H2O/D2O. 15N-¹H HSQC spectra were collected at 1:0, 1:1, 1:3 and 1:5 molar ratios of 15N-labeled aS to unlabeled bS or gS at 10 °C. Samples for the inverse experiments were prepared similarly by adding unlabeled aS to achieve final concentrations of 35 µM or 70 µM 15N-labeled bS or gS at the same molar ratios of labeled to unlabeled proteins as above. For studies of synuclein family member interactions with polycations, ¹H-15N HSQC spectra were collected for 70 µM 15N-labeled aS, bS, and gS in the presence of 100 µg/ml poly-L-lysine or 50 µg/ml poly-L-argine (Sigma-Aldrich).
Supplementary Material
Acknowledgements
This work was supported by NIH/NIA grant AG019391, the Irma T. Hirschl Foundation, and a gift from Herbert and Ann Siegel (to D.E.). We thank Dr. Peter Lansbury (Harvard Medical School) for the kind gift of expression vectors and Trudy Ramlall and Carla Rospigliosi for technical assistance. D.E. is a member of the New York Structural Biology Center, supported by NIH grant GM66354
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakajo S, Tsukada K, Omata K, Nakamura Y, Nakaya K. A new brain-specific 14-kDa protein is a phosphoprotein. Its complete amino acid sequence and evidence for phosphorylation. Eur J Biochem. 1993;217:1057–1063. doi: 10.1111/j.1432-1033.1993.tb18337.x. [DOI] [PubMed] [Google Scholar]
- 3.Akopian AN, Wood JN. Peripheral nervous system-specific genes identified by subtractive cDNA cloning. J Biol Chem. 1995;270:21264–21270. doi: 10.1074/jbc.270.36.21264. [DOI] [PubMed] [Google Scholar]
- 4.Clayton DF, George JM. The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998;21:249–254. doi: 10.1016/s0166-2236(97)01213-7. [DOI] [PubMed] [Google Scholar]
- 5.Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994;345:27–32. doi: 10.1016/0014-5793(94)00395-5. [DOI] [PubMed] [Google Scholar]
- 6.Ji H, Liu YE, Jia T, Wang M, Liu J, Xiao G, Joseph BK, Rosen C, Shi YE. Identification of a breast cancer-specific gene, BCSG1, by direct differential cDNA sequencing. Cancer Res. 1997;57:759–764. [PubMed] [Google Scholar]
- 7.Nakajo S, Shioda S, Nakai Y, Nakaya K. Localization of phosphoneuroprotein 14 (PNP 14) and its mRNA expression in rat brain determined by immunocytochemistry and in situ hybridization. Brain Res Mol Brain Res. 1994;27:81–86. doi: 10.1016/0169-328x(94)90187-2. [DOI] [PubMed] [Google Scholar]
- 8.Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T. The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- 9.Lavedan C, Leroy E, Dehejia A, Buchholtz S, Dutra A, Nussbaum RL, Polymeropoulos MH. Identification, localization and characterization of the human gamma-synuclein gene. Hum Genet. 1998;103:106–112. doi: 10.1007/s004390050792. [DOI] [PubMed] [Google Scholar]
- 10.Buchman VL, Hunter HJ, Pinon LG, Thompson J, Privalova EM, Ninkina NN, Davies AM. Persyn, a member of the synuclein family, has a distinct pattern of expression in the developing nervous system. J Neurosci. 1998;18:9335–9341. doi: 10.1523/JNEUROSCI.18-22-09335.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:11282–11286. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nielsen MS, Vorum H, Lindersson E, Jensen PH. Ca2+ binding to alpha-synuclein regulates ligand binding and oligomerization. J Biol Chem. 2001;276:22680–22684. doi: 10.1074/jbc.M101181200. [DOI] [PubMed] [Google Scholar]
- 13.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 14.Uversky VN, Li J, Souillac P, Millett IS, Doniach S, Jakes R, Goedert M, Fink AL. Biophysical properties of the synucleins and their propensities to fibrillate: inhibition of alpha-synuclein assembly by beta- and gamma-synucleins. J Biol Chem. 2002;277:11970–11978. doi: 10.1074/jbc.M109541200. [DOI] [PubMed] [Google Scholar]
- 15.Eliezer D, Kutluay E, Bussell R, Jr, Browne G. Conformational properties of alpha-synuclein in its free and lipid- associated states. J Mol Biol. 2001;307:1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 16.Marsh JA, Singh VK, Jia Z, Forman-Kay JD. Sensitivity of secondary structure propensities to sequence differences between alpha- and gamma-synuclein: implications for fibrillation. Protein Sci. 2006;15:2795–2804. doi: 10.1110/ps.062465306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 18.Bussell R, Jr, Eliezer D. A structural and functional role for 11-mer repeats in alpha-synuclein and other exchangeable lipid binding proteins. J Mol Biol. 2003;329:763–778. doi: 10.1016/s0022-2836(03)00520-5. [DOI] [PubMed] [Google Scholar]
- 19.Chandra S, Chen X, Rizo J, Jahn R, Sudhof TC. A broken alpha - helix in folded alpha -Synuclein. J Biol Chem. 2003;278:15313–15318. doi: 10.1074/jbc.M213128200. [DOI] [PubMed] [Google Scholar]
- 20.Sung YH, Eliezer D. Secondary structure and dynamics of micelle bound beta- and gamma-synuclein. Protein Sci. 2006;15:1162–1174. doi: 10.1110/ps.051803606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 22.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc Natl Acad Sci U S A. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- 24.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease [see comments] Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 25.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 26.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 27.Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destee A. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 28.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 29.George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995;15:361–372. doi: 10.1016/0896-6273(95)90040-3. [DOI] [PubMed] [Google Scholar]
- 30.Jenco JM, Rawlingson A, Daniels B, Morris AJ. Regulation of phospholipase D2: selective inhibition of mammalian phospholipase D isoenzymes by alpha- and beta-synucleins. Biochemistry. 1998;37:4901–4909. doi: 10.1021/bi972776r. [DOI] [PubMed] [Google Scholar]
- 31.McDermott M, Wakelam MJ, Morris AJ. Phospholipase D. Biochem Cell Biol. 2004;82:225–253. doi: 10.1139/o03-079. [DOI] [PubMed] [Google Scholar]
- 32.Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 33.Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- 35.Liu S, Ninan I, Antonova I, Battaglia F, Trinchese F, Narasanna A, Kolodilov N, Dauer W, Hawkins RD, Arancio O. alpha-Synuclein produces a long-lasting increase in neurotransmitter release. Embo J. 2004;23:4506–4516. doi: 10.1038/sj.emboj.7600451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KL, Orrison B, Chen A, Ellis CE, Paylor R, Lu B, Nussbaum RL. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha- synuclein. J Neurosci. 2002;22:8797–8807. doi: 10.1523/JNEUROSCI.22-20-08797.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yavich L, Tanila H, Vepsalainen S, Jakala P. Role of alpha-synuclein in presynaptic dopamine recruitment. J Neurosci. 2004;24:11165–11170. doi: 10.1523/JNEUROSCI.2559-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larsen KE, Schmitz Y, Troyer MD, Mosharov E, Dietrich P, Quazi AZ, Savalle M, Nemani V, Chaudhry FA, Edwards RH, Stefanis L, Sulzer D. Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci. 2006;26:11915–11922. doi: 10.1523/JNEUROSCI.3821-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 40.Giasson BI, Uryu K, Trojanowski JQ, Lee VM. Mutant and wild type human alpha-synucleins assemble into elongated filaments with distinct morphologies in vitro. J Biol Chem. 1999;274:7619–7622. doi: 10.1074/jbc.274.12.7619. [DOI] [PubMed] [Google Scholar]
- 41.Narhi L, Wood SJ, Steavenson S, Jiang Y, Wu GM, Anafi D, Kaufman SA, Martin F, Sitney K, Denis P, Louis JC, Wypych J, Biere AL, Citron M. Both familial Parkinson's disease mutations accelerate alpha-synuclein aggregation. J Biol Chem. 1999;274:9843–9846. doi: 10.1074/jbc.274.14.9843. [DOI] [PubMed] [Google Scholar]
- 42.Choi W, Zibaee S, Jakes R, Serpell LC, Davletov B, Crowther RA, Goedert M. Mutation E46K increases phospholipid binding and assembly into filaments of human alpha-synuclein. FEBS Lett. 2004;576:363–368. doi: 10.1016/j.febslet.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 43.Greenbaum EA, Graves CL, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, Axelsen PH, Giasson BI. The E46K mutation in alpha-synuclein increases amyloid fibril formation. J Biol Chem. 2005;280:7800–7807. doi: 10.1074/jbc.M411638200. [DOI] [PubMed] [Google Scholar]
- 44.Dedmon MM, Lindorff-Larsen K, Christodoulou J, Vendruscolo M, Dobson CM. Mapping long-range interactions in alpha-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J Am Chem Soc. 2005;127:476–477. doi: 10.1021/ja044834j. [DOI] [PubMed] [Google Scholar]
- 45.Bertoncini CW, Jung YS, Fernandez CO, Hoyer W, Griesinger C, Jovin TM, Zweckstetter M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc Natl Acad Sci U S A. 2005;102:1430–1435. doi: 10.1073/pnas.0407146102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bussell R, Jr, Eliezer D. Residual structure and dynamics in Parkinson's disease-associated mutants of alpha-synuclein. J Biol Chem. 2001;276:45996–46003. doi: 10.1074/jbc.M106777200. [DOI] [PubMed] [Google Scholar]
- 47.Heise H, Hoyer W, Becker S, Andronesi OC, Riedel D, Baldus M. Molecular-level secondary structure, polymorphism, and dynamics of full-length alpha-synuclein fibrils studied by solid-state NMR. Proc Natl Acad Sci U S A. 2005;102:15871–15876. doi: 10.1073/pnas.0506109102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Der-Sarkissian A, Jao CC, Chen J, Langen R. Structural organization of alpha-synuclein fibrils studied by site-directed spin labeling. J Biol Chem. doi: 10.1074/jbc.M305266200. [DOI] [PubMed] [Google Scholar]
- 49.Lavedan C, Buchholtz S, Auburger G, Albin RL, Athanassiadou A, Blancato J, Burguera JA, Ferrell RE, Kostic V, Leroy E, Leube B, Mota-Vieira L, Papapetropoulos T, Pericak-Vance MA, Pinkus J, Scott WK, Ulm G, Vasconcelos J, Vilchez JJ, Nussbaum RL, Polymeropoulos MH. Absence of mutation in the beta- and gamma-synuclein genes in familial autosomal dominant Parkinson's disease. DNA Res. 1998;5:401–402. doi: 10.1093/dnares/5.6.401. [DOI] [PubMed] [Google Scholar]
- 50.Lincoln S, Crook R, Chartier-Harlin MC, Gwinn-Hardy K, Baker M, Mouroux V, Richard F, Becquet E, Amouyel P, Destee A, Hardy J, Farrer M. No pathogenic mutations in the beta-synuclein gene in Parkinson's disease. Neurosci Lett. 1999;269:107–109. doi: 10.1016/s0304-3940(99)00420-6. [DOI] [PubMed] [Google Scholar]
- 51.Brighina L, Okubadejo NU, Schneider NK, Lesnick TG, de Andrade M, Cunningham JM, Farrer MJ, Lincoln SJ, Rocca WA, Maraganore DM. Beta-synuclein gene variants and Parkinson's disease: A preliminary case-control study. Neurosci Lett. 2007;420:229–234. doi: 10.1016/j.neulet.2007.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Galvin JE, Uryu K, Lee VM, Trojanowski JQ. Axon pathology in Parkinson's disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc Natl Acad Sci U S A. 1999;96:13450–13455. doi: 10.1073/pnas.96.23.13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ohtake H, Limprasert P, Fan Y, Onodera O, Kakita A, Takahashi H, Bonner LT, Tsuang DW, Murray IV, Lee VM, Trojanowski JQ, Ishikawa A, Idezuka J, Murata M, Toda T, Bird TD, Leverenz JB, Tsuji S, La Spada AR. Beta-synuclein gene alterations in dementia with Lewy bodies. Neurology. 2004;63:805–811. doi: 10.1212/01.wnl.0000139870.14385.3c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Biere AL, Wood SJ, Wypych J, Steavenson S, Jiang Y, Anafi D, Jacobsen FW, Jarosinski MA, Wu GM, Louis JC, Martin F, Narhi LO, Citron M. Parkinson's disease-associated alpha-synuclein is more fibrillogenic than beta- and gamma-synuclein and cannot cross-seed its homologs. J Biol Chem. 2000;275:34574–34579. doi: 10.1074/jbc.M005514200. [DOI] [PubMed] [Google Scholar]
- 55.Yamin G, Munishkina LA, Karymov MA, Lyubchenko YL, Uversky VN, Fink AL. Forcing nonamyloidogenic beta-synuclein to fibrillate. Biochemistry. 2005;44:9096–9107. doi: 10.1021/bi048778a. [DOI] [PubMed] [Google Scholar]
- 56.Hashimoto M, Rockenstein E, Mante M, Mallory M, Masliah E. beta-Synuclein inhibits alpha-synuclein aggregation: a possible role as an anti-parkinsonian factor. Neuron. 2001;32:213–223. doi: 10.1016/s0896-6273(01)00462-7. [DOI] [PubMed] [Google Scholar]
- 57.Park JY, Lansbury PT., Jr Beta-synuclein inhibits formation of alpha-synuclein protofibrils: a possible therapeutic strategy against Parkinson's disease. Biochemistry. 2003;42:3696–3700. doi: 10.1021/bi020604a. [DOI] [PubMed] [Google Scholar]
- 58.Wishart DS, Bigam CG, Holm A, Hodges RS, Sykes BD. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J Biomol NMR. 1995;5:67–81. doi: 10.1007/BF00227471. [DOI] [PubMed] [Google Scholar]
- 59.Spera S, Bax A. Empirical correlation between protein backbone conformation and Cα and Cβ nuclear magnetic resonance chemical shifts. J. Am. Chem. Soc. 1991;113:5490–5492. [Google Scholar]
- 60.Wishart DS, Sykes BD, Richards FM. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J Mol Biol. 1991;222:311–333. doi: 10.1016/0022-2836(91)90214-q. [DOI] [PubMed] [Google Scholar]
- 61.Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human alpha-synuclein. J Biol Chem. 2005;280:9595–9603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- 62.Gillespie JR, Shortle D. Characterization of long-range structure in the denatured state of staphylococcal nuclease. I. Paramagnetic relaxation enhancement by nitroxide spin labels. J Mol Biol. 1997;268:158–169. doi: 10.1006/jmbi.1997.0954. [DOI] [PubMed] [Google Scholar]
- 63.Gillespie JR, Shortle D. Characterization of long-range structure in the denatured state of staphylococcal nuclease. II. Distance restraints from paramagnetic relaxation and calculation of an ensemble of structures. J Mol Biol. 1997;268:170–184. doi: 10.1006/jmbi.1997.0953. [DOI] [PubMed] [Google Scholar]
- 64.Shortle D, Ackerman MS. Persistence of native-like topology in a denatured protein in 8 M urea. Science. 2001;293:487–489. doi: 10.1126/science.1060438. [DOI] [PubMed] [Google Scholar]
- 65.Louhivuori M, Paakkonen K, Fredriksson K, Permi P, Lounila J, Annila A. On the origin of residual dipolar couplings from denatured proteins. J Am Chem Soc. 2003;125:15647–15650. doi: 10.1021/ja035427v. [DOI] [PubMed] [Google Scholar]
- 66.Mohana-Borges R, Goto NK, Kroon GJ, Dyson HJ, Wright PE. Structural characterization of unfolded states of apomyoglobin using residual dipolar couplings. J Mol Biol. 2004;340:1131–1142. doi: 10.1016/j.jmb.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 67.Bernado P, Bertoncini CW, Griesinger C, Zweckstetter M, Blackledge M. Defining long-range order and local disorder in native alpha-synuclein using residual dipolar couplings. J Am Chem Soc. 2005;127:17968–17969. doi: 10.1021/ja055538p. [DOI] [PubMed] [Google Scholar]
- 68.Cho MK, Kim HY, Bernado P, Fernandez CO, Blackledge M, Zweckstetter M. Amino acid bulkiness defines the local conformations and dynamics of natively unfolded alpha-synuclein and tau. J Am Chem Soc. 2007;129:3032–3033. doi: 10.1021/ja067482k. [DOI] [PubMed] [Google Scholar]
- 69.Antony T, Hoyer W, Cherny D, Heim G, Jovin TM, Subramaniam V. Cellular polyamines promote the aggregation of alpha-synuclein. J Biol Chem. 2003;278:3235–3240. doi: 10.1074/jbc.M208249200. [DOI] [PubMed] [Google Scholar]
- 70.Goers J, Uversky VN, Fink AL. Polycation-induced oligomerization and accelerated fibrillation of human alpha-synuclein in vitro. Protein Sci. 2003;12:702–707. doi: 10.1110/ps.0230903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fernandez CO, Hoyer W, Zweckstetter M, Jares-Erijman EA, Subramaniam V, Griesinger C, Jovin TM. NMR of alpha-synuclein-polyamine complexes elucidates the mechanism and kinetics of induced aggregation. Embo J. 2004;23:2039–2046. doi: 10.1038/sj.emboj.7600211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morar AS, Olteanu A, Young GB, Pielak GJ. Solvent-induced collapse of alpha-synuclein and acid-denatured cytochrome c. Protein Sci. 2001;10:2195–2199. doi: 10.1110/ps.24301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem. 2001;276:10737–10744. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- 74.Sung Y, Eliezer D. Secondary structure and dynamics of micelle-bound beta- and gamma-synuclein. Prot Sci. 2006 doi: 10.1110/ps.051803606. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rochet JC, Conway KA, Lansbury PT., Jr Inhibition of fibrillization and accumulation of prefibrillar oligomers in mixtures of human and mouse alpha-synuclein. Biochemistry. 2000;39:10619–10626. doi: 10.1021/bi001315u. [DOI] [PubMed] [Google Scholar]
- 76.Yan Y, Wang C. Abeta40 protects non-toxic Abeta42 monomer from aggregation. J Mol Biol. 2007;369:909–916. doi: 10.1016/j.jmb.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 77.Lee D, Paik SR, Choi KY. Beta-synuclein exhibits chaperone activity more efficiently than alpha-synuclein. FEBS Lett. 2004;576:256–260. doi: 10.1016/j.febslet.2004.08.075. [DOI] [PubMed] [Google Scholar]
- 78.Yancey PH, Clark ME, Hand SC, Bowlus RD, Somero GN. Living with water stress: evolution of osmolyte systems. Science. 1982;217:1214–1222. doi: 10.1126/science.7112124. [DOI] [PubMed] [Google Scholar]
- 79.Sung YH, Rospigliosi C, Eliezer D. NMR mapping of copper binding sites in alpha-synuclein. Biochim Biophys Acta. 2006;1764:5–12. doi: 10.1016/j.bbapap.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 80.Rasia RM, Bertoncini CW, Marsh D, Hoyer W, Cherny D, Zweckstetter M, Griesinger C, Jovin TM, Fernandez CO. Structural characterization of copper(II) binding to alpha-synuclein: Insights into the bioinorganic chemistry of Parkinson's disease. Proc Natl Acad Sci U S A. 2005;102:4294–4299. doi: 10.1073/pnas.0407881102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Alexandrescu AT, Abeygunawardana C, Shortle D. Structure and dynamics of a denatured 131-residue fragment of staphylococcal nuclease: a heteronuclear NMR study. Biochemistry. 1994;33:1063–1072. doi: 10.1021/bi00171a004. [DOI] [PubMed] [Google Scholar]
- 82.Eliezer D, Yao J, Dyson HJ, Wright PE. Structural and dynamic characterization of partially folded states of apomyoglobin and implications for protein folding. Nat Struct Biol. 1998;5:148–155. doi: 10.1038/nsb0298-148. [DOI] [PubMed] [Google Scholar]
- 83.Yao J, Chung J, Eliezer D, Wright PE, Dyson HJ. NMR structural and dynamic characterization of the acid-unfolded state of apomyoglobin provides insights into the early events in protein folding. Biochemistry. 2001;40:3561–3571. doi: 10.1021/bi002776i. [DOI] [PubMed] [Google Scholar]
- 84.Yi Q, Scalley-Kim ML, Alm EJ, Baker D. NMR characterization of residual structure in the denatured state of protein L. J Mol Biol. 2000;299:1341–1351. doi: 10.1006/jmbi.2000.3816. [DOI] [PubMed] [Google Scholar]
- 85.Lacy ER, Filippov I, Lewis WS, Otieno S, Xiao L, Weiss S, Hengst L, Kriwacki RW. p27 binds cyclin-CDK complexes through a sequential mechanism involving binding-induced protein folding. Nat Struct Mol Biol. 2004;11:358–364. doi: 10.1038/nsmb746. [DOI] [PubMed] [Google Scholar]
- 86.Eliezer D, Barre P, Kobaslija M, Chan D, Li X, Heend L. Residual structure in the repeat domain of tau: echoes of microtubule binding and paired helical filament formation. Biochemistry. 2005;44:1026–1036. doi: 10.1021/bi048953n. [DOI] [PubMed] [Google Scholar]
- 87.Barre P, Eliezer D. Folding of the repeat domain of tau upon binding to lipid surfaces. J Mol Biol. 2006;362:312–326. doi: 10.1016/j.jmb.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 88.Crowther RA, Jakes R, Spillantini MG, Goedert M. Synthetic filaments assembled from C-terminally truncated alpha-synuclein. FEBS Lett. 1998;436:309–312. doi: 10.1016/s0014-5793(98)01146-6. [DOI] [PubMed] [Google Scholar]
- 89.Murray IV, Giasson BI, Quinn SM, Koppaka V, Axelsen PH, Ischiropoulos H, Trojanowski JQ, Lee VM. Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry. 2003;42:8530–8540. doi: 10.1021/bi027363r. [DOI] [PubMed] [Google Scholar]
- 90.Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid- like cross-beta conformation. Proc Natl Acad Sci U S A. 2000;97:4897–4902. doi: 10.1073/pnas.97.9.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003;424:805–808. doi: 10.1038/nature01891. [DOI] [PubMed] [Google Scholar]
- 92.Eliezer D. Characterizing residual structure in disordered protein States using nuclear magnetic resonance. Methods Mol Biol. 2007;350:49–67. doi: 10.1385/1-59745-189-4:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Johnson BA, Blevins RA. NMRView: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 94.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes [see comments] J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 95.Chou JJ, Gaemers S, Howder B, Louis JM, Bax A. A simple apparatus for generating stretched polyacrylamide gels, yielding uniform alignment of proteins and detergent micelles. J Biomol NMR. 2001;21:377–382. doi: 10.1023/a:1013336502594. [DOI] [PubMed] [Google Scholar]
- 96.Permi P, Rosevear PR, Annila A. A set of HNCO-based experiments for measurement of residual dipolar couplings in 15N, 13C, (2H)-labeled proteins. J Biomol NMR. 2000;17:43–54. doi: 10.1023/a:1008372624615. [DOI] [PubMed] [Google Scholar]
- 97.Bussell R, Jr, Ramlall TF, Eliezer D. Helix periodicity, topology, and dynamics of membrane-associated alpha-synuclein. Protein Sci. 2005;14:862–872. doi: 10.1110/ps.041255905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lietzow MA, Jamin M, Jane Dyson HJ, Wright PE. Mapping long-range contacts in a highly unfolded protein. J Mol Biol. 2002;322:655–662. doi: 10.1016/s0022-2836(02)00847-1. [DOI] [PubMed] [Google Scholar]
- 99.Cantor CR, Schimmel R. Biophysical Chemistry. New York: W. H. Freeman and Co.; 1980. [Google Scholar]
- 100.Zagrovic B, Pande VS. Structural correspondence between the alpha-helix and the random-flight chain resolves how unfolded proteins can have native-like properties. Nat Struct Biol. 2003;10:955–961. doi: 10.1038/nsb995. [DOI] [PubMed] [Google Scholar]
- 101.Kosen PA. Spin labeling of proteins. Methods Enzymol. 1989;177:86–121. doi: 10.1016/0076-6879(89)77007-5. [DOI] [PubMed] [Google Scholar]
- 102.Battiste JL, Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- 103.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Grzesiek S, Stahl SJ, Wingfield PT, Bax A. The CD4 determinant for downregulation by HIV-1 Nef directly binds to Nef. Mapping of the Nef binding surface by NMR. Biochemistry. 1996;35:10256–10261. doi: 10.1021/bi9611164. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.