

Summary
In this work we demonstrate a differentiation-induced up-regulation of the expression of plasma membrane Ca2+ATPase (PMCA) isoforms being present in various gastric/colon cancer cell types. We found PMCA1b as the major isoform in non-differentiated cancer cell lines, whereas the expression level of PMCA4b was significantly lower. Cell differentiation initiated with short chain fatty acids (SCFAs) and trichostatin A, or spontaneous differentiation of post-confluent cell cultures resulted in a marked induction of PMCA4b expression, while only moderately increased PMCA1b levels. Up-regulation of PMCA4b expression was demonstrated both at the protein and mRNA levels, and closely correlated with the induction of established differentiation markers. In contrast, the expression level of the Na+/K+-ATPase or that of the sarco/endoplasmic reticulum Ca2+ATPase 2 protein did not change significantly under these conditions. In membrane vesicles obtained from SCFA-treated gastric/colon cancer cells a marked increase in the PMCA-dependent Ca2+ transport activity was observed, indicating a general increase of PMCA function during the differentiation of these cancer cells.
Because various PMCA isoforms display distinct functional characteristics, we suggest that up-regulated PMCA expression, together with a major switch in PMCA isoform pattern may significantly contribute to the differentiation of gastric/colon cancer cells. The analysis of PMCA expression may provide a new diagnostic tool for monitoring the tumor phenotype.
Keywords: gastric/colon carcinoma, Ca2+ homeostasis, differentiation induction, plasma membrane Ca2+ATPase, gene expression
1. Introduction
Plasma membrane Ca2+ATPases (PMCAs) play an important role in the regulation of cellular Ca2+ signaling and the control of cell activation (for review see [1]). Binding of various ligands to their receptors in the plasma membrane leads to the intracellular production of D-myo-inositol-1,4,5-tris-phosphate (IP3) and consequent release of Ca2+ from the endoplasmic reticulum (ER). Ca2+ release from the ER and ensuing Ca2+ influx from the extracellular space through store-operated Ca2+ channels lead to increased cytosolic Ca2+ concentration and to the activation of various Ca2+ - and/or Ca2+-calmodulin-dependent enzymes [2, 3]. As PMCAs transport Ca2+ ions from the cytosol into the extracellular space, by decreasing cytosolic free Ca2+ level these enzymes are essential for the control of the magnitude of Ca2+ transients, and for the termination of a Ca2+-dependent cell activation event. PMCA activity also determines the cytosolic Ca2+ concentration of a cell at the resting state. As PMCAs are themselves activated by the Ca2+-calmodulin complex, PMCA activity is involved in the control of cellular Ca2+ oscillations, as well [4, 5].
Mammalian PMCAs are encoded by four genes (PMCA1-4 or ATP2B1-4), and tissue- and development-specific alternative splicing of the primary transcripts of these genes generates a multitude of PMCA isoforms. PMCA1 and PMCA4 are expressed in virtually all tissues, whereas PMCA2 and PMCA3 are primarily found in some specialized tissue/cell types [6-8]. Okunade et al. [9] studied the relative importance of PMCA1 and PMCA4. They developed and analyzed mice carrying null mutations in these PMCA genes and proposed the major housekeeping role for PMCA1, but not for PMCA4. Thus, it appears that the PMCA1 isoform fulfills housekeeping roles essential for cellular Ca2+ signaling and/or homeostasis, whereas other PMCA isoforms serve specialized, tissue- or cell type-specific functions.
The PMCA variants identified so far in normal gastric and colon tissues are encoded by the PMCA1 and PMCA4 genes. The presence of PMCA2 and PMCA3 gene products has not been detected in these tissue types, even by the most sensitive techniques (for details see [8]). PMCA4 codes for the full-length PMCA4b isoform and the C-terminally truncated PMCA4a splice variant. Biochemically, PMCA4b is characterized by: i) low basal activity in the absence of calmodulin; ii) slow activation by the Ca2+-calmodulin complex and iii) slow inactivation of the calmodulin-activated pump [10-12]. For PMCA1 biochemical data are limited due to the difficulties encountered during the cloning and expression of its cDNA [13, 14]. The affinity of PMCA1 for calmodulin appeared to be similar to that of PMCA4, but it has higher affinity for ATP and a higher susceptibility to degradation by calpain [15]. Further studies are required to understand why cells co-express PMCA1 and PMCA4, and how these pumps contribute to the cellular Ca2+ signaling and homeostasis.
The intestinal/colonic epithelium is in a constant state of renewal. Cells proliferate and become differentiated as they migrate from the base of the crypts towards the surface. Alterations of the tightly regulated balance between the highly proliferative/less differentiated and the non-proliferative/highly differentiated states may lead to hyperplasia, benign (polyps) or malignant tumors. The pivotal role of Ca2+ in the pathophysiology of intestinal/colonic epithelium is well documented (for review see [16]). Increasing cytosolic free Ca2+ concentration has been observed during the ontogeny of intestinal epithelium [17], moreover, the differentiation-inducing effect of increased cytosolic Ca2+ levels has also been published [18]. In addition, the regulation of colonic epithelial cell proliferation and differentiation by the extracellular Ca2+ concentration predicts a chemopreventive action of Ca2+ on colon tumorigenesis [18-20]. Whereas data in the literature suggest a cross-talk between Ca2+ homeostasis and the control of epithelial differentiation, the exact mechanisms of action of Ca2+ and proteins involved in Ca2+ homeostasis in colon carcinogenesis remain to be clarified.
Formerly, we observed that the Ca2+ homeostasis of the ER is remodeled during the differentiation of gastric and colon carcinoma cells, and showed that, although normal colonic epithelium expresses the sarco/endoplasmic reticulum Ca2+ATPase 3 (SERCA3) proteins abundantly, the expression of these enzymes is strongly decreased in colon cancer cells. SERCA3 expression could be induced by short chain fatty acids (SCFAs) that are physiological differentiation inducers present in the gut lumen due to fermentation of dietary fibers by the colonic flora. In addition to the modulated SERCA expression pattern detected during the drug-induced differentiation of various gastric/colon adenocarcinoma cell lines, enhanced SERCA3 expression accompanied also the spontaneous differentiation of several colon cancer cell lines in post-confluent cultures without any drug treatment [21, 22].
Gene expression can be controlled through deacetylation of histones. A class of agents, the histone deacetylase (HDAC) inhibitors inhibit that process and are accepted to be potent differentiation inducers [23]. Treatment of tumor cells with HDAC inhibitors therefore can contribute to the re-expression of suppressed genes and can accelerate cell differentiation [24], and these agents are promising drugs that are currently in early phase of clinical trials [25, 26]. In our present study, we used SCFA-type, as well as structurally unrelated HDAC inhibitors to induce cancer cell differentiation.
As the biochemical activity, as well as the transcriptional regulation of various proteins involved in cellular Ca2+ homeostasis (pumps, channels and Ca2+ binding proteins) are modulated by Ca2+, these proteins function in a tightly interconnected manner. To further characterize this homeostatic matrix, in the present work we investigated the expression pattern and the function of plasma membrane Ca2+ATPases in various gastric and colon cancer cell lines, and studied the effect of cell differentiation on their expression and activity. Implication of PMCAs in the differentiation of various tissue/cell types has already been published [27-33]. Our studies, focused on gastric/colon carcinomas, indicate that the major PMCA isoform expressed in these cancer cell lines is PMCA1b, whereas PMCA4b is expressed only at a reduced level. Differentiation induction resulted in markedly enhanced PMCA4b expression, whereas PMCA1b expression was only slightly increased. These data provide further evidence for the essential housekeeping function of PMCA1, but not PMCA4, and show, for the first time, that PMCA gene expression is regulated in an isoform-dependent manner during the differentiation of gastric/colon cancer cells. Changes in the relative abundance of PMCA1 and PMCA4 detected during the differentiation of gastric and colon adenocarcinoma cell lines towards a more mature phenotype correlate well with previous data on the co-existence of PMCA1 and 4 mRNAs in normal gastric/colon tissues (reviewed in [8]). Our observations taken together with earlier data on SERCA expression indicate that the expression of several key enzymes involved in cellular Ca2+ homeostasis (i.e.: SERCA-, as well as PMCA-type Ca2+ pumps) is anomalous in malignant cells. This points at a more general defect of the Ca2+ homeostasis of these cells, reflected by the loss of the expression of proteins involved in Ca2+-dependent functions of normally differentiated cells.
2. Materials and methods
2.1. Cell Culture
The epithelial HeLa, the kidney fibroblast-like COS-7, the gastric carcinoma KATO-III, as well as the DLD-1, HT-29, LS-174T and Caco-2 colon carcinoma cell lines were obtained from the ATCC (Manassas, VA, USA). All cell lines, exept HT-29 cells, were cultured according to the instructions of the ATCC with the modifications that the RPMI-1640-based media contained Glutamax-I (alanyl-glutamine) in addition to 2 mM L-glutamine, and that media were supplemented with 100 units/ml penicillin and 100 μg/ml streptomycin (GibcoBRL Life Technologies Ltd, Paisley, UK). HT-29 cells were cultured in DMEM (High Glucose, GibcoBRL Life Technologies Ltd, Paisley, UK) supplemented with 10% heat-inactivated fetal calf serum, 100 units/ml penicillin and 100 μg/ml streptomycin. All cell lines were maintained at 37°C in a humidified atmosphere containing 5% CO2.
2.2. Induction of Cell Differentiation
Exponentially growing cells were trypsinized and seeded into 21 cm2 cell culture Petri dishes at a density of 4-8 × 104 cells/cm2 for the DLD-1, HT-29 and LS-174T cell lines, and at 0.5-1 × 104 cells/cm2 for Caco-2 cells. When cells reached 50-70% confluency as determined by microscopic examination (day 1-3 post-plating, depending on the rate of growth), medium was renewed and drugs were added from concentrated stock solutions. To avoid the attachment and for uniformity of cultures, KATO-III cells that grow in a semiadherent manner in cell culture flasks were seeded for treatments into 58 cm2 sterile bacteriological Petri dishes at an initial density of 1-2 × 105 cells/ml.
For drug-induced cell differentiation, short chain fatty acids (butyric acid and valeric acid; purchased from Sigma-Aldrich Kft., Hungary) were neutralized by dissolving in 7.5% sodium bicarbonate solution at a concentration of 0.9 M, sterile filtered and kept at −20°C. Trichostatin A (obtained from Sigma-Aldrich Kft., Hungary) was added to the cell cultures from concentrated stock solutions made in dimethyl sulfoxide (DMSO). The final concentration of DMSO vehicle did not exceed 0.1%, was included in control experiments and did not interfere with the assays. Untreated control cells were harvested in the exponential phase of non-confluent growth.
The human Caco-2 colon adenocarcinoma cell line spontaneously undergoes differentiation in post-confluent cultures. After reaching confluency, the initially rapidly growing cells stop to proliferate and display structural, biochemical and functional characteristics close to a mature enterocytic phenotype [34]. Post-confluent Caco-2 cell cultures were re-fed by renewal of medium every two days.
2.3. Gel Electrophoresis, Electrotransfer and Immunostaining
After drug treatments as indicated in Figures for the KATO-III, DLD-1, HT-29 and LS-174T cell lines, or at different stages of post-confluency for the Caco-2 cell line, the cells were quickly washed twice with 0.15 M NaCl, precipitated with ice-cold 6% trichloroacetic acid (TCA) overnight at 4°C and centrifuged for 15 min at 12000 × g at 4°C. The TCA-precipitated total cellular protein pellets were then dissolved in modified Läemmli-type sample buffer containing 90 mM Tris-HCl, pH 7.9, 2% SDS, 10% glycerol, 5 mM EDTA, 125 mg/ml urea, 100 mM dithiothreitol (DTT), 0.02% bromophenol blue [35], and supplemented before use with 0.1 mM phenylmethyl-sulfonyl-fluoride (PMSF) and 10 μg/ml leupeptin. The protein concentration of the samples was determined by the modified Lowry method using bovine serum albumin (BSA) as standard. Total cell lysates were electrophoresed on 6%, 7.5% or 9% Läemmli-type SDS-polyacrylamide gels and electrotransferred onto polyvinylidene fluoride (PVDF)-membranes (Bio-Rad, Richmond, CA, USA). Transfer of proteins onto PVDF-membranes was controlled occasionally by AmidoBlack staining. Saturation of membrane sheets and immunostaining were performed as described in our earlier works [36]. 1:2000 dilution of the pan-anti-PMCA (5F10) and the anti-SERCA2 (IID8), 1:1000 dilution of the anti-PMCA4 (JA9) and 1:1500 dilution of the anti-PMCA4b (JA3) monoclonal antibodies (ascites) were used. The IID8 antibody [37] was from Affinity BioReagents, Neshanic Station, NJ, USA. The 5F10, JA9 and JA3 antibodies were obtained as described earlier [38, 39]. For the detection of SERCA3 expression, the purified PL/IM430 monoclonal antibody [40] was used at a final concentration of about 1-2 μg/ml. Immunostaining for the detection of Na+/K+-ATPase, carcinoembryonic antigen (CEA) and of dipeptidyl-peptidase IV (DPP-IV) was performed using the anti-Na+/K+-ATPase, clone no.: XVIF9-G10 (BIOMOL), the anti-CEA, clone no.: C6G9 (Sigma-Aldrich) and the anti-DPP-IV, clone no.: HBB 3/775/42 monoclonal antibodies at 1:1000, 1:1500 and 1:800 dilutions, respectively [22, 41]. Antibody binding on PVDF-membranes was detected using the enhanced chemiluminescence (ECL) Western-blot reagents from Amersham International, UK. The luminograms were scanned using a Bio-Rad Gel Doc densitometer.
2.4. Quantitative Evaluation of the Differentiation-induced Changes in PMCA Expression
To estimate the fold increases in PMCA1b and PMCA4b protein expression during in vitro differentiation induction performed on the various gastric/colon carcinoma cell lines, the following protocol was established. Increasing amounts (2-60 μg) of total cellular protein from untreated and Na+-valerate- or Na+-butyrate-treated KATO-III and DLD-1 cells, or from pre-confluent and day 22 post-confluent Caco-2 cells were analyzed by Western-blotting using either the 5F10 pan-anti-PMCA (for PMCA1b), or the PMCA4b-specific JA3 monoclonal antibodies. Luminograms were scanned and quantitative evaluation was performed separately for both the PMCA1b and PMCA4b isoforms using the Bio-Rad Gel Doc densitometer and the Quantity One version 4.4 software. We determined sample points where luminescent signal intensities for treated and untreated control cells were equal. The ratios of corresponding loaded total protein quantities were used to calculate fold increase values for the expression of PMCA1b and 4b isoforms in the samples of the differentiated cells.
2.5. Isolation of Mixed Microsomal Membranes
Microsomal membranes from control, 3 mM Na+-valerate- or 3 mM Na+-butyrate-treated KATO-III cells were prepared as described in [42] for HL-60 cells. Isolation of microsomal membranes from control, 3 mM Na+-valerate- or 2 mM Na+-butyrate-treated DLD-1 cells, as well as from COS-7 cells transiently transfected with human PMCA4a cDNA according to the procedure published in [43], was performed as described by Enyedi et al. [44] with the following modifications: cells were washed with ice-cold phosphate-buffered saline (PBS) solution, then harvested in the same medium containing 0.1 mM PMSF, 6 μg/ml aprotinin, 2.2 μg/ml leupeptin and 1 mM EGTA, pH 7.4. Cells were sedimented and then resuspended in an ice-cold hypotonic solution containing 10 mM Tris-HCl, pH 7.4, 1 mM MgCl2, 0.5 mM EGTA, 4 μg/ml aprotinin, 2 μg/ml leupeptin and 4 mM DTT. After lysis, homogenization and two centrifugation steps (for 30 min at 2000 × g at 4°C, then for 60 min at 105 × g at 4°C) the final pellet (the 105 × g membrane fraction) was resuspended in a solution of 0.25 M sucrose, 0.15 M KCl, 10 mM Tris-HCl, pH 7.4, 2 mM DTT, 20 μM CaCl2, and the membrane suspension was stored in liquid N2.
2.6. Ca2+ Transport Assay
Ca2+ uptake by microsomal membrane vesicles prepared from control, Na+-valerate- or Na+-butyrate-treated KATO-III and DLD-1 cells was carried out as described previously [45, 46] in a reaction mixture containing 100 mM KCl, 25 mM N-tris-(hydroxymethyl) methyl-2-aminoethanesulfonic acid-triethanolamine (TES-TEA), pH 7.2, 7 mM MgCl2, 40 mM KH2PO4/K2HPO4, pH 7.2, 200 nM thapsigargin (to block SERCA-driven Ca2+ uptake), 4 μg/ml oligomycin (to block mitochondrial ATP synthesis), 100 μM CaCl2 (labeled with 45Ca2+, 45 GBq/mmol, Amersham International, UK) and 90 μM EGTA to obtain 8.1 μM free Ca2+ concentration. The free Ca2+ was calculated with MaxChelator (http://www.stanford.edu/~cpatton/maxc.html). Calmodulin at 20 μg/ml (1174 nM) concentration was also added to the reaction mixture to obtain maximal PMCA-driven Ca2+ uptake. Ca2+ uptake was initiated by the addition of 5 mM ATP and measured for 5 min. The concentration of the microsomal membrane proteins during the transport measurements was 200 μg/ml for KATO-III, and 100 μg/ml for the DLD-1 cells, respectively. The reaction was terminated by rapid filtration of the microsomes using Millipore membrane filters (0.45 μm pore size) and counting was performed as described in [47].
2.7. RNA Isolation and Reverse Transcription
Following the appropriate treatments, cells were lysed in Trizol reagent (GibcoBRL Life Technologies Ltd, Paisley, UK) and total cellular RNA was isolated according to the manufacturer’s protocol. Total cDNA was prepared by reverse transcription with random hexamers using 1μg of total RNA (denatured for 10 min at 65°C) in a reaction volume of 20 μl with 200 units of Moloney murine leukemia virus (MMLV) reverse transcriptase (Promega, Madison, WA) in the buffer supplied, 40 units of RNase inhibitor, 0.5 mM dNTP and 100 ng of random hexamer primers (obtained from Promega, Madison, WA) at 25°C for 10 min, 37°C for 45 min and 95°C for 5 min.
2.8. Relative mRNA Quantification
1 μl of total cDNA was amplified using the LightCycler FastStart DNA Master SYBR Green I kit (Roche, Mannheim, Germany) with 2 mM MgCl2, 0.5 μM amplification primers for the PMCA4b (forward: 5’-ccgtatccagactcagatcaaa-3’; reverse: 5’-gaaagccttgtcaggattttcc-3’) and 0.2 μM amplification primers for the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene (forward: 5’-gaaggtgaaggtcggagtca-3’; reverse: 5’-gacaagcttcccgttctcag-3’). The PCR program consisted of an initial denaturation step at 95°C for 10 min, followed by 45 cycles of denaturation at 95°C for 10 sec, annealing at 60°C for 10 sec and extension at 72°C for 30 sec. Detection of SYBR Green fluorescence (F1 fluorescence) was performed at 60°C in each cycle. For relative mRNA quantification, the second derivative maximum function of the LightCycler software (Roche, Mannheim, Germany) was used. Fluorescence curves were analyzed and crossing thresholds (crossing point, CP, the cycle number at which the fluorescence signal rises above background) were determined for each sample. The relative PMCA4b (target) and GAPDH (reference or housekeeping) mRNA quantities were calculated by comparing the actual CP values to standard curves. To obtain standard curves, 5× serial dilutions of a total cellular cDNA sample were prepared, amplified for the target and control genes, and CP values were plotted as a function of the relative concentrations of the serially diluted cDNA samples. To obtain normalized PMCA4b mRNA quantities in the differently treated samples, ratios of the relative mRNA concentration values for PMCA4b and GAPDH were calculated. The GAPDH CP value fluctuations never exceeded 1 cycle throughout our experiments.
2.9. Statistical Analysis
Data in this work correspond to three or more independent experiments and are presented either as a representative experiment or the mean ± S.D. as specified in the Figure captions. Statistical significance was determined by Student’s t-test.
3. Results
3.1. PMCA Expression Pattern in Gastric and Colon Cancer Cell Lines
Plasma membrane Ca2+ATPase isoforms (PMCAs) were identified and their expression levels were studied in gastric (KATO-III) and various colon (DLD-1, Caco-2, LS-174T and HT-29) cancer cell lines. We analyzed total cellular proteins of exponentially growing cells for overall PMCA (Fig. 1, upper panel), PMCA4 (Fig. 1, lower panel) and PMCA4b (Fig. 1, middle panel) expression. It has been reported that the mRNA of PMCA1b, 4b and 4a isoforms are expressed in gastric/colon tissues (for review see [8]). Therefore, a mixture of PMCA isoforms was used to mark the position of these proteins. For Western-blot analysis of the PMCA isoform pattern and to estimate the relative expression level of the PMCA isoforms in various cancer cell lines, first we used the pan-anti-PMCA 5F10 antibody (Fig. 1, upper panel). As published earlier, the region of the 5F10 epitope (residues 719-738) is highly conserved in all PMCA isoforms [48], thus this antibody probably recognizes all PMCAs with the same affinity. Although PMCA1b is the major PMCA isoform expressed at comparable levels in the gastric/colon cancer cell lines, expression of PMCA4b was also detected at a lower level, as shown in Fig. 1. However, expression of the PMCA4a protein could not be seen in any of the cancer cell lines probed with the pan-PMCA4-specific JA9 antibody (Fig. 1, lower panel). As the presence of PMCA2 and 3 isoforms has not been reported in normal gastric/colonic epithelium, we did not extend our studies to these PMCA pumps.
Fig. 1.

Expression of PMCA-type Ca2+ pumps in gastric and colon cancer cell lines. Equal amounts (30 μg/lane) of total cellular proteins from various gastric (KATO-III) and colon (DLD-1, Caco-2, LS-174T and HT-29) cancer cell lines were loaded for SDS-PAGE, electroblotted and immunostained for: overall PMCA expression using the 5F10 (upper panel); PMCA4b expression using the JA3 (middle panel) and for PMCA4 (4a and 4b) expression using the JA9 (lower panel) monoclonal antibodies. 5 μg of microsomal membrane protein from the epithelial HeLa cells was mixed with 25 ng of microsomal membrane protein from COS-7 cells transfected with the PMCA4a cDNA. This mixed sample was loaded onto the gel to mark the position of the PMCA1b, 4a and 4b isoforms within a single sample.
PMCA1b is the major PMCA isoform in the gastric/colon cancer cell lines. The expression of PMCA4b, but not that of the PMCA4a protein could also be detected in these cells at a lower level.
3.2. PMCA4b Expression Markedly Increases during Short Chain Fatty Acid-induced Differentiation of Gastric/Colon Cancer Cell Lines
The malignant phenotype is associated with inhibition of cell differentiation, and pharmacologically induced differentiation of the malignant cells constitutes the basis of several experimental, as well as clinically accepted routine anticancer therapeutic protocols [49, 50].
We treated the KATO-III, DLD-1, HT-29 and LS-174T cancer cell lines with short chain fatty acids (SCFAs), Na+-valerate or Na+-butyrate. These are well-known differentiation-inducing agents [51] and butyrate is physiologically present in the gut lumen. Fig. 2 presents a concentration dependence of changes in PMCA expression pattern during Na+-valerate or Na+-butyrate treatments of the four cell types. Fig. 3 shows a time course of PMCA expression during valerate- or butyrate-induced differentiation of the KATO-III, DLD-1 and LS-174T cells. SCFA treatments led to a marked up-regulation of PMCA4b expression and to a less pronounced induction of PMCA1b expression in a concentration-dependent manner, in the low millimolar range of the SCFAs (Fig. 2A-D). In parallel, the expression of the SERCA2 housekeeping endoplasmic reticulum Ca2+ATPase isoform slightly decreased, or did not change significantly, depending on treatments and cell types. In KATO-III cells (Fig. 2A) differentiation was monitored by the induction of SERCA3 (endomembrane-associated Ca2+ATPase) expression [22]. In DLD-1, HT-29 and LS-174T colon cancer cells (Fig. 2B-D) the differentiation was confirmed by the enhanced expression of carcinoembrionic antigen (CEA) and/or the dipeptidyl-peptidase IV (DPP-IV) enzyme (data not shown). CEA expression increased at lower concentrations of SCFA than PMCA expression probably due to different mechanisms involved in the regulation of the expression of the two genes.
Fig. 2.
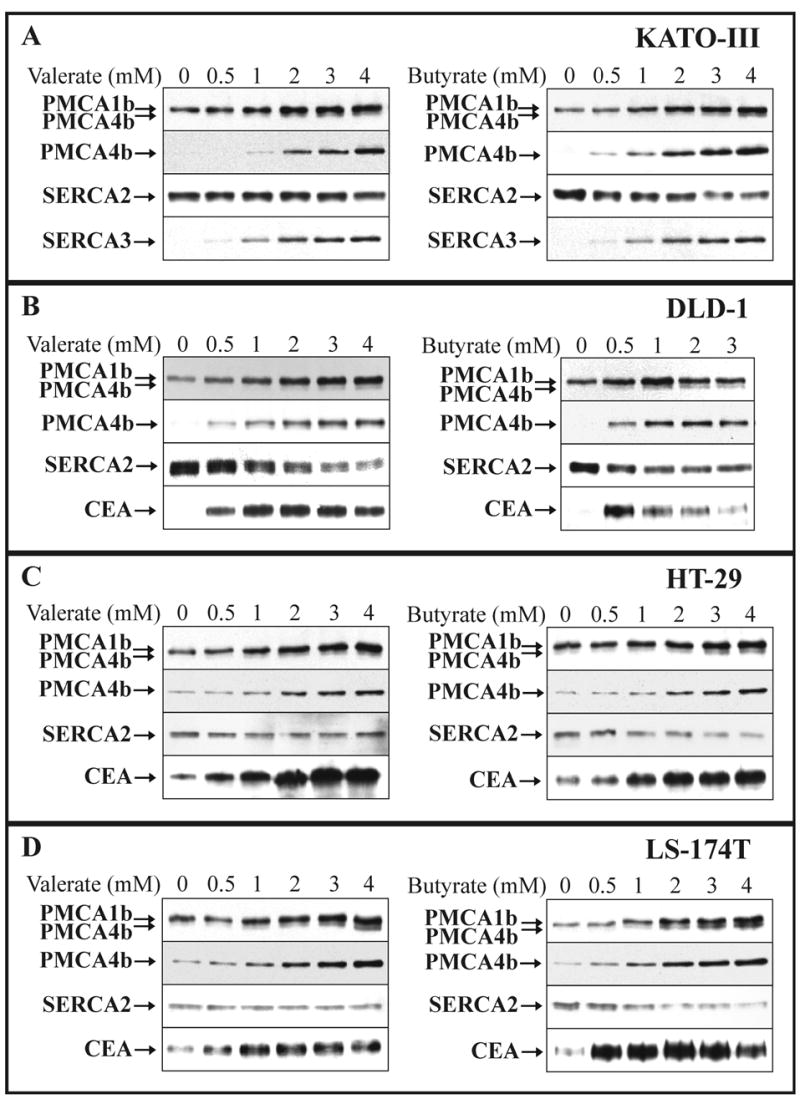
SCFA-induced modulation of PMCA expression in various gastric/colon cancer cell lines: concentration dependence.
KATO-III (A), DLD-1 (B), HT-29 (C) and LS-174T (D) cells were cultured in the presence of various concentrations of Na+-valerate or Na+-butyrate, as indicated on the panels. Total cellular protein lysates were prepared from all treatments at day 3 for KATO-III, DLD-1 and HT-29, and at day 2 for LS-174T cells. Equal amounts of cellular proteins (from 20 to 50 μg/lane, depending on the cell lines and the antibody used for immunostaining) were loaded onto the SDS-polyacrylamide gels, electroblotted and immunostained for overall PMCA (5F10) and PMCA4b (JA3) expressions. The expression of the ubiquitous SERCA2 pump, and that of various established differentiation markers (CEA and/or SERCA3) were monitored in parallel.
Treatment of the four cancer cell lines with these differentiation-inducing SCFAs led to the marked up-regulation of PMCA4b expression and to a less pronounced induction of PMCA1b expression.
Fig. 3.

SCFA-induced modulation of PMCA expression in various gastric/colon cancer cell lines: time course.
KATO-III (A), DLD-1 (B) and LS-174T (C) cells were cultured in the presence of 3 mM Na+-valerate or Na+-butyrate, as indicated on the panels. Total cellular lysates were prepared from all treatments at the indicated time points and equal amounts of cellular proteins (from 20 to 30 μg/lane, depending on the cell lines and the antibody used for immunostaining) were analyzed for overall PMCA (5F10), PMCA4b (JA3) and for Na+/K+-ATPase expressions. Induction of PMCA4b expression could be detected as early as day 0.5-1, and reached a plateau-phase after 1-2 days of SCFA-treatment in LS-174T cells, and after 4-5 days of SCFA-treatment in KATO-III and DLD-1 cells.
Next, we followed changes in the expression of PMCAs in the presence of 3 mM Na+-valerate (left panels) or 3 mM Na+-butyrate (right panels) for up to 3-5 days depending on cell types. Total cellular lysates were analyzed for overall PMCA, PMCA4b and the plasma membrane marker Na+/K+-ATPase expression (Fig. 3A-C). Induction of PMCA4b expression could be detected as early as day 0.5-1. PMCA4b expression reached a plateau-phase after 1-2 days of SCFA treatment in the mucus-secreting LS-174T cells and after 4-5 days of SCFA treatment in gastric KATO-III and enterocytic DLD-1 cells. A concomitant, but less pronounced up-regulation of PMCA1b expression was also detected, however, the expression of the Na+/K+-ATPase did not change significantly or rather slightly decreased during SCFA treatment.
In order to evaluate the SCFA-induced up-regulation of PMCA expression in KATO-III and DLD-1 cell lines (Table 1), different quantities of total cellular proteins were loaded onto the SDS-polyacrylamide gel from cells cultured in the absence or in the presence of Na+-valerate or Na+-butyrate (see details in the “Materials and methods”). Western-blot analysis revealed an about 5-9 times increase in PMCA4b expression, significantly higher than that of PMCA1b (about 1.5-3 times) during SCFA-induced cell differentiation in both cell lines.
Table 1.
SCFA-induced modulation of PMCA1b and PMCA4b expressions in KATO-III gastric and DLD-1 colon cancer cells: quantitative evaluation
| Cell line | Treatment | PMCA1b expression (fold increase) | PMCA4b expression (fold increase) |
|---|---|---|---|
| KATO-III | 3 mM Na+-valerate (day 5) | 1.7 ± 0.6 (n=10) | 5.7 ± 1.9 (n=18) |
| 3 mM Na+-butyrate (day 4) | 2.1 ± 0.5 (n=15) | 8.5 ± 1.7 (n=10) | |
| DLD-1 | 3 mM Na+-valerate (day 5) | 2.9 ± 1.0 (n=16) | 5.4 ± 0.6 (n=21) |
| 3 mM Na+-butyrate (day 6) | n.d.* | 6.0 ± 1.0 (n=16) |
KATO-III and DLD-1 cells were cultured in the absence or in the presence of 3 mM Na+-valerate or Na+-butyrate for the times indicated in the Table. Cells were TCA-precipitated and Western-blot analysis of the samples followed by the evaluation of the luminograms was performed as described in the “Materials and methods”. The data represent the fold increases of the PMCA1b and PMCA4b expressions at protein level in the differently treated, differentiated cells (means ± S.D. of n ≥10 from at least two independent treatments). The expression levels of the PMCA1b and 4b proteins in untreated cells were used as control values for quantifications.
Data are not shown because of the negligible changes in the PMCA1b expression due to 6 days 3 mM Na+-butyrate treatment of DLD-1 cells.
3.3. PMCA4b mRNA Expression Is Highly Up-regulated during SCFA-induced Cell Differentiation
Fig. 4A shows that after 4 days treatment the relative quantities of PMCA4b transcripts, normalized to the GAPDH mRNA, increased 10- and 14-fold in KATO-III cells in response to the differentiation-inducing agents 3 mM Na+-valerate or 3 mM Na+-butyrate, respectively. SCFA-induced differentiation of DLD-1 cells resulted in a 15-25-fold increase in the normalized PMCA4b mRNA quantities after a 5 days treatment (Fig. 4B). Data obtained for PMCA4b transcripts are in good correlation with the increased PMCA4b protein levels detected in SCFA-treated KATO-III and DLD-1 cells (Figs. 2 and 3).
Fig. 4.

Up-regulated PMCA4b mRNA expression in SCFA-treated KATO-III and DLD-1 cells.
KATO-III gastric (A) and DLD-1 colon (B) cancer cells were treated with 3 mM Na+-valerate or 3 mM Na+-butyrate for 4 or 5 days, respectively. Untreated cells were used as controls. Real-time PCR analysis was conducted by using DNA templates obtained following reverse transcription of mRNAs from SCFA-treated and untreated cells, and PMCA4b-specific oligonucleotide primers. As internal control, GAPDH was also amplified from the same RNA preparations. Normalized PMCA4b mRNA/GAPDH mRNA ratios were calculated by using crossing point values and separate calibration curves for the two amplicons.
The bars in panels (A) and (B) represent the means ± S.D. of three independent determinations of a representative experiment. Statistical significance is denoted by **p<0.01.
The SCFA-induced differentiation resulted in strong induction of PMCA4b mRNA expression in both cell lines.
3.4. Modulation of PMCA-driven Ca2+ Transport Function by SCFA-induced Cell Differentiation
To study whether the modulated PMCA expression during SCFA-induced cell differentiation could have functional consequences on cellular Ca2+ transport, we investigated PMCA-driven active Ca2+ transport into microsomal membrane vesicles obtained either from KATO-III gastric (Fig. 5A), or from DLD-1 colonic epithelial cells (Fig. 5B) treated for 5 days with Na+-valerate or Na+-butyrate. Microsomal membrane vesicles from untreated KATO-III or DLD-1 cells were used for control measurements. Maximal PMCA-driven Ca2+ transport activities were measured at saturating Ca2+ (8.1 μM free Ca2+) and calmodulin (1174 nM) concentrations in the presence of thapsigargin that blocks Ca2+ transport driven by SERCAs. A 2- and a 3-fold increase in the PMCA-driven vesicular Ca2+ transport activity were detected in 3 mM Na+-valerate- and in 3 mM Na+-butyrate-treated KATO-III cells, respectively. In DLD-1 cells treated with either 3 mM valerate or 2 mM butyrate, an approximately 2-fold increase in PMCA-driven vesicular Ca2+ transport activity could be detected. These data show that differentiation induction modulates not only PMCA gene expression, but also PMCA activity in various gastric/colon cancer cell lines.
Fig. 5.
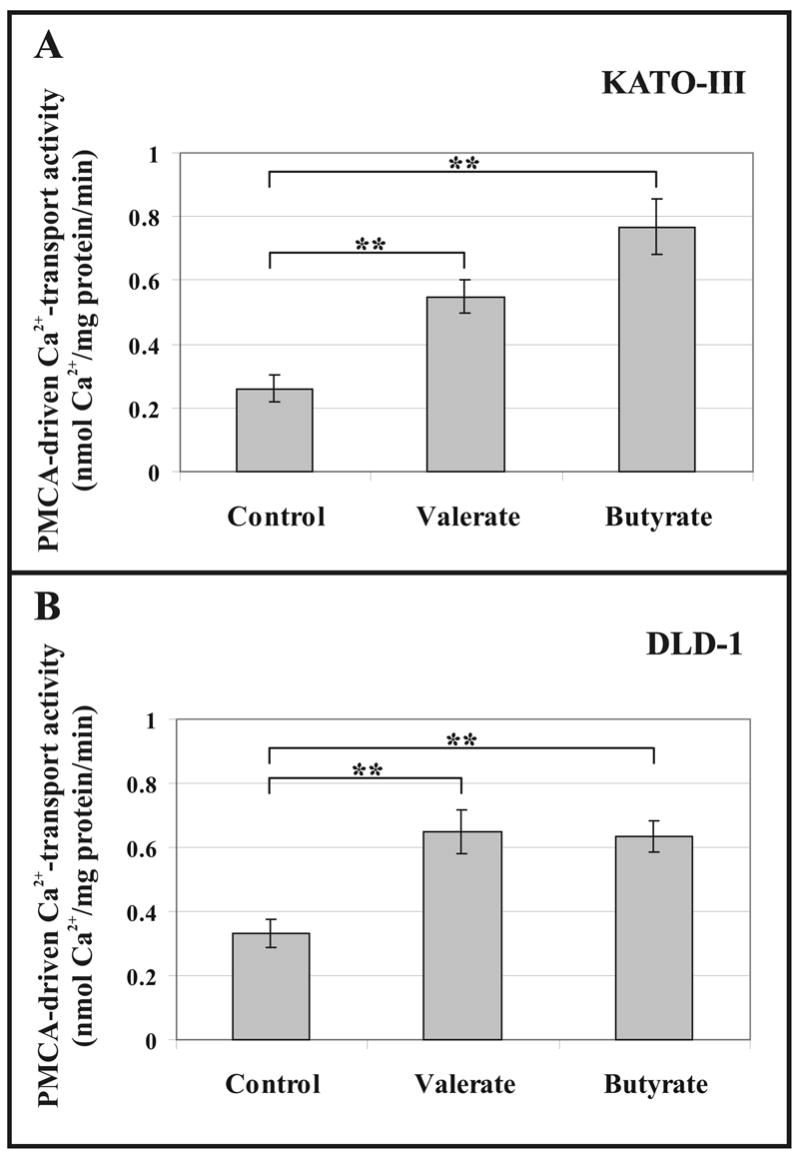
Up-modulated PMCA-driven Ca2+ transport activities of microsomal membranes from SCFA-treated KATO-III and DLD-1 cells.
KATO-III cells (A) were treated for 5 days with 3 mM Na+-valerate or 3 mM Na+-butyrate. DLD-1 cells (B) were treated for 5 days with 3 mM Na+-valerate or 2 mM Na+-butyrate. Untreated cells were used as controls. Maximal PMCA-driven Ca2+ transport activities were determined for each microsomal membrane preparation at saturating Ca2+ (8.1 μM free Ca2+) and calmodulin (1174 nM) concentrations.
The bars in panels (A) and (B) represent the means ± S.D. of 6-9 determinations from three independent experiments. Statistical significance is denoted by **p<0.01.
SCFA-induced differentiation resulted in up-regulated PMCA-driven Ca2+ transport in both cell lines.
3.5. Growth Inhibition per se Does Not Result in Induction of PMCA Expression in KATO-III Cells
As shown in Fig. 6A, Na+-butyrate possessed higher growth arrest inducing potency for KATO-III cells than did Na+-valerate (about 77% growth inhibition at 3 mM Na+-butyrate and about 51% growth inhibition at 3 mM Na+-valerate at day 3 of treatments). When the cells were cultured for 3 days without SCFAs in serum-free culture medium containing 0.5% bovine serum albumin (BSA), an approximately 54% growth inhibition could be detected, very similar to that observed at 3 days with 3 mM Na+-valerate treatment. In Fig. 6B we show that growth inhibition induced by serum withdrawal did not affect PMCA expression. This indicates that the increased PMCA4b expression was specifically induced by SCFAs.
Fig. 6.
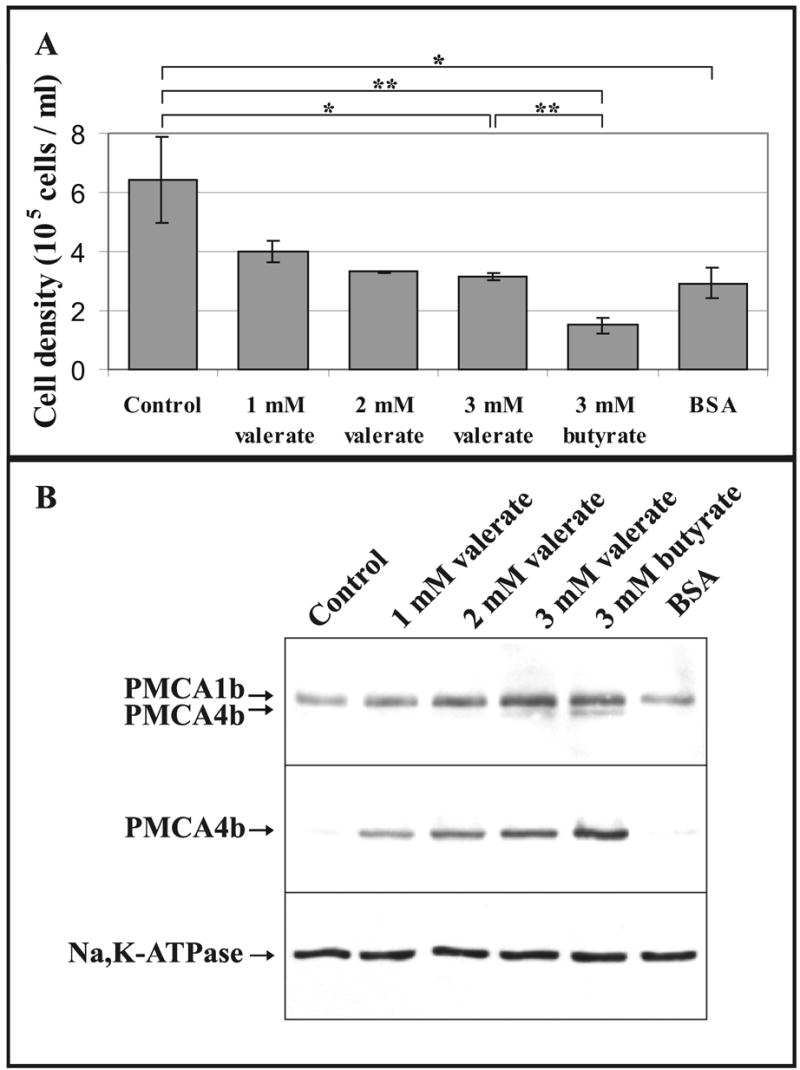
PMCA4b induction by SCFAs is not linked to their growth inhibitory effect. KATO-III cells, at an initial density of 1 × 105 cells/ml, were cultured in control conditions, or in the presence of 1mM, 2mM or 3 mM Na+-valerate or 3 mM Na+-butyrate, or were cultured without SCFAs in serum-free culture medium containing 0.5% bovine serum albumin (BSA).
A, Cell densities detected at day 3. The bars represent the means ± S.D. of 2-3 determinations from at least two independent experiments. Statistical significance is denoted by when *p<0.05 or **p<0.01. At 1 mM and 2 mM Na+-valerate treatments we had only 2 determinations being not sufficient to calculate the statistical significance.
B, Western-blotting of total cellular lysates prepared from all treatments at day 3. Equal amounts of cellular proteins were analyzed for overall PMCA (5F10), PMCA4b (JA3) and Na+/K+-ATPase expressions.
Growth inhibition induced by serum withdrawal was without any effect on PMCA expression, whereas PMCA4b induction could be obtained by various SCFAs with or without growth inhibition.
3.6. Enhanced PMCA4b Expression during Trichostatin A-induced Differentiation of Gastric/Colon Cancer Cell Lines
We tested, if the induction of PMCA expression occurred also when the differentiation of various gastric/colon cancer cell lines was induced with highly specific HDAC inhibitors structurally different from the SCFAs. We treated the KATO-III, DLD-1 and LS-174T cancer cell lines with trichostatin A, a well characterized HDAC inhibitor [52], and investigated the changes in the PMCA expression pattern. In Fig. 7 the concentration dependence, while in Fig. 8 the time course of changes in PMCA expression are presented. As shown in Fig. 7A-C, trichostatin A induced up-regulation of PMCA4b expression in a concentration-dependent manner in the 10-400 nanomolar range depending on cell types. The expression of PMCA1b increased only slightly. In parallel experiments the enhancement in PMCA4b expression appeared to be more pronounced when the cells were cultured in the presence of 3 mM Na+-butyrate than during treatments with the most effective concentrations of trichostatin A (Fig. 7). As shown in Fig. 8A-C, induction of PMCA4b expression could be detected as early as day 0.5-1, and it reached a plateau-phase after 1-2 days when the cells were treated with 250-300 nM trichostatin A. PMCA1b expression did not change significantly and the expression of the Na+/K+-ATPase decreased slightly during these treatments.
Fig. 7.

Trichostatin A-induced modulation of PMCA expression in various gastric/colon cancer cell lines: concentration dependence.
KATO-III (A), DLD-1 (B) and LS-174T (C) cells were treated with various concentrations of trichostatin A or 3 mM Na+-butyrate as indicated on the panels. Total cellular lysates were prepared from all treatments at day 3 for KATO-III, at day 4 for DLD-1 and at day 2 for LS-174T cells. Equal amounts of cellular proteins (from 20 to 30 μg/lane, depending on cell types and the antibody used for immunostaining) were analyzed for overall PMCA (5F10) and PMCA4b (JA3) expressions.
Treatment of the three cancer cell lines with the HDAC inhibitor trichostatin A led to a marked up-regulation of PMCA4b expression.
Fig. 8.

Trichostatin A-induced modulation of PMCA expression in various gastric/colon cancer cell lines: time course.
KATO-III (A), DLD-1 (B) and LS-174T (C) cells were treated with the indicated concentrations of trichostatin A for 4 days (KATO-III and DLD-1 cells) or for 3 days (LS-174T cells). Total cellular lysates were prepared at the indicated time points and equal amounts of cellular proteins (from 20 to 30 μg/lane, depending on cell types and the antibody used for immunostaining) were analyzed for overall PMCA (5F10), PMCA4b (JA3) and Na+/K+-ATPase expressions.
The time course of the trichostatin A-induced up-regulation of PMCA4b expression reached a plateau-phase at days 1-2 for KATO-III and LS-174T cells and at days 2-3 for DLD-1 cells.
3.7. Induction of PMCA Expression during Spontaneous Differentiation of Caco-2 Cells in Post-confluent Cultures
Finally, we analyzed differentiation-induced changes in the PMCA expression pattern in a model system that differs substantially from those described above. The Caco-2 human colon adenocarcinoma cell line undergoes spontaneous differentiation and acquires a mature polarized enterocytic phenotype when cultured in post-confluent cultures without any additional treatment [34, 53]. Caco-2 cells constitute a widely used model of enterocytic differentiation and function. The cells were allowed to reach confluency and cultured further for more than 3 weeks. Cells were harvested at different time points and lysed for both Western-blot (Fig. 9A-B), and real-time PCR analysis (Fig. 9C). The expression of PMCA isoforms and of the Na+/K+-ATPase, as well as that of two established markers of enterocytic differentiation, CEA and DPP-IV [41, 53, 54] was monitored. As shown in Fig. 9A, a marked and gradual induction of PMCA4b expression could be detected that reached a plateau-phase after approximately 3 weeks of post-confluency. This PMCA4b induction followed a time course very similar to that of DPP-IV, an enzyme located in the apical plasma membrane of the enterocytes. However, a less pronounced increase appeared in the PMCA1b expression. Interestingly, this change in PMCA1b expression was manifested in the early phase of post-confluency and did not change significantly in the later period of differentiation. The differentiation of Caco-2 cells was also reflected by the marked enhancement in CEA expression. The differentiation induction resulted in a slight decrease in the expression of the plasma membrane marker Na+/K+-ATPase. Only a very small increase in the SERCA2 expression, but a significant induction in the expression of the SERCA3 proteins could be detected in accordance with our earlier results in [22] (data not shown). When PMCA4b protein expression was quantified at 3 weeks of post-confluency (for details see “Materials and methods”), an approximately 8-fold increase was obtained (Fig. 9B) as compared to the PMCA4b expression in pre-confluent Caco-2 cells. In parallel, only an approximately 3-fold increase in the PMCA1b expression could be detected. As shown in Fig. 9C, when differentiation of Caco-2 cells was induced in post-confluent cultures, the relative quantities of PMCA4b mRNA gradually increased after the cell culture achieved confluency. In this setting, a pre-confluent cell culture was used as control, and samples were collected at days 3, 7, 15 and 31 after confluency. At day 3 a small (1.7-fold), and at days 7 and 15, gradually larger (5- and 9-fold) increases in quantities of PMCA4b mRNA were detected with a plateau-like effect, i.e. no further increase was measured at day 31. The PMCA4b mRNA content of the differentiating Caco-2 cells reached the plateau-phase approximately 1 week earlier (after 2 weeks of post-confluency) than the PMCA4b protein (after 3 weeks of post-confluency, Fig. 9A). The dissimilarity observed in the kinetics of the induction of PMCA4b mRNA and the PMCA4b protein could be partly due to the higher sensitivity of the real-time PCR when compared to that of the Western-blotting. In addition, the turnover rates of the mRNA and the corresponding protein may differ, as well. We conclude that up-regulation of PMCA4b expression due to spontaneous differentiation of Caco-2 cells is manifested both at the mRNA and protein levels as it has also been shown in our earlier work for the SERCA3 gene in this adenocarcinoma cell line [22].
Fig. 9.
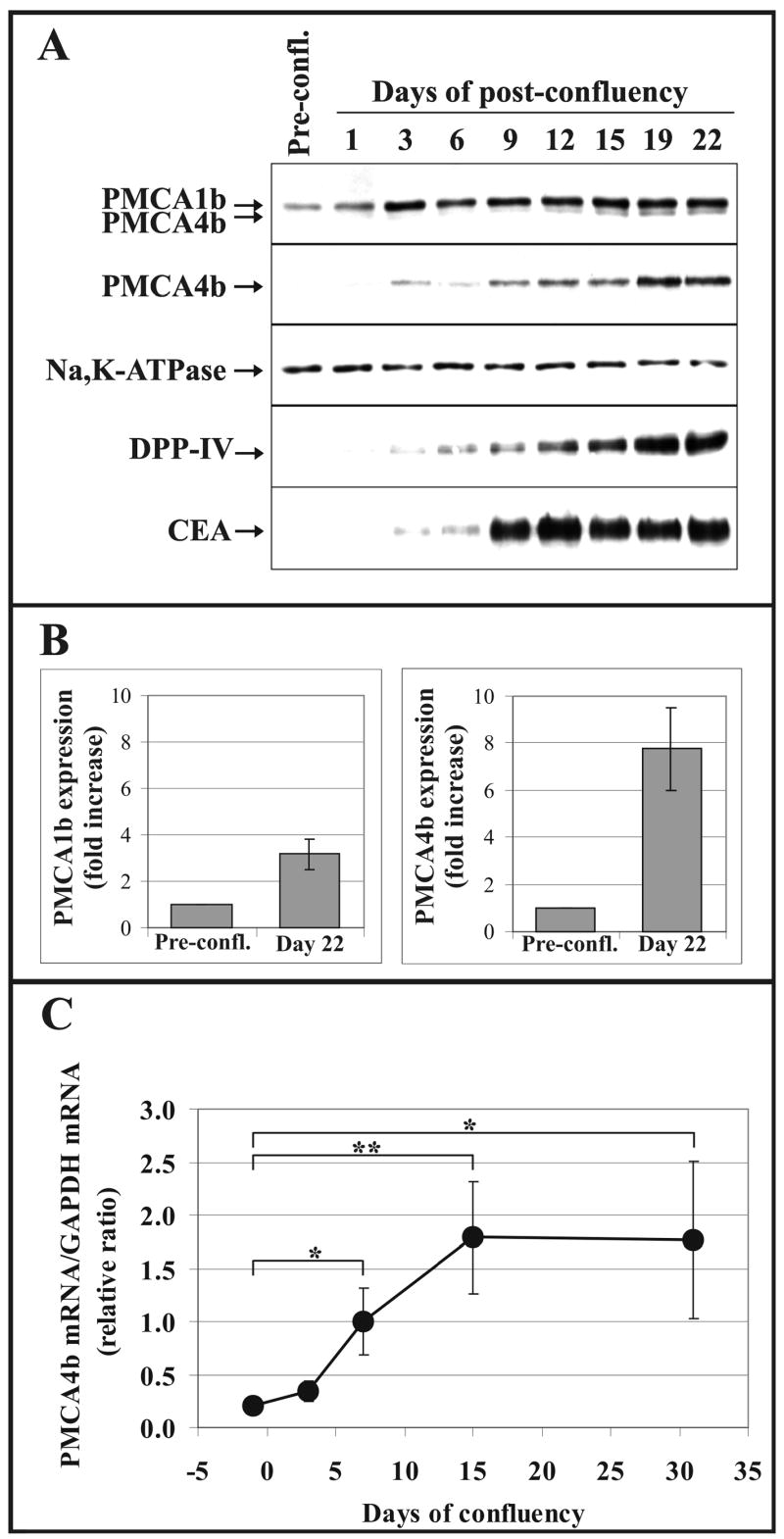
Modulated PMCA expression in differentiating Caco-2 colon cancer cells. Caco-2 cells were allowed to reach confluency and cultured in post-confluency for more than 3 weeks. Cells were harvested at different time points and lysed for both Western-blot analysis (A and B) and real-time PCR analysis (C).
A, Western-blot analysis of the expression of PMCA isoforms by using the pan-anti-PMCA (5F10) and the PMCA4b-specific (JA3) monoclonal antibodies. The expression of Na+/K+-ATPase and two established markers of enterocytic differentiation (DPP-IV and CEA) were monitored in parallel. Equal amounts of total cellular protein harvested at the indicated time points were loaded onto SDS-polyacrylamide gels (from 20 μg to 50 μg, depending on the antibody used for immunostaining).
B, The bars represent the fold increases of the PMCA1b and PMCA4b protein expressions in differentiated Caco-2 cells at day 22 of post-confluency (means ± S.D. of n ≥15 from at least two independent experiments). The expression levels of PMCA1b and 4b in pre-confluent cultures of Caco-2 cells were used as control values for quantifications.
C, Real-time PCR analysis was conducted with PMCA4b-specific primers using: equal amounts of total cDNA templates obtained following reverse transcription of mRNAs from pre-confluent Caco-2 cells or from cells cultured in post-confluency for the time points indicated. As internal control, GAPDH was simultaneously amplified from the same cDNA templates. Normalized PMCA4b/GAPDH mRNA ratios were calculated by using crossing point values and separate calibration curves for the two amplicons, and plotted as a function of time elapsed from confluency. Day 0 is when confluency is reached.
The data in this panel represent the means ± S.D. of at least three separate determinations of a representative experiment. Statistical significance is denoted by when *p<0.05 or **p<0.01. Strong induction of PMCA4b expression was observed both at the protein and mRNA levels during culturing Caco-2 cells in post-confluent conditions that allows differentiation of cells toward an enterocytic phenotype.
4. Discussion
In this work we report for the first time on differentiation-induced modulation of the plasma membrane Ca2+ transport ATPase (PMCA) expression pattern in various colon and gastric carcinoma cells. We show that the dominant PMCA isoform expressed in gastric/colon adenocarcinoma cell lines is PMCA1b, and that PMCA4b is also expressed but at a strongly suppressed level. Differentiation induced by various treatments such as histone deacetylase inhibition or during post-confluent growth resulted in the preferential induction of PMCA4b expression in a wide selection of cancer cell lines.
PMCAs are considered as key molecular components of cellular Ca2+ homeostasis and signaling, and consequently of various cell functions. By extruding Ca2+ from the cytosol into the extracellular space, these transport proteins shape the Ca2+ signals and thus participate in the fine-tuning of the Ca2+-dependent cellular responses. The nearly 30 members of this Ca2+ ATPase family, with their developmentally regulated and tissue/cell type-specific expression pattern fulfill a wide range of important cell biological functions [1, 8, 55-57]. Among the mammalian PMCAs encoded by four genes, PMCA1 and PMCA4 are expressed in virtually all tissue/cell types [8]. However, Okunade and co-workers proposed an essential housekeeping role for the PMCA1, but not for PMCA4 [9]. This is based on the fact, that the PMCA4 null mutant mice did not exhibit embryonic lethality and appeared outwardly normal. In contrast to PMCA4 ablation, loss of both copies of the PMCA1 gene caused embryonic lethality. One can conclude that cells equipped with PMCA1 as the only PMCA pump preserve their viability, whereas PMCA4 alone is not sufficient for cell survival.
The PMCA variants detected so far at the mRNA level in normal gastrointestinal tissues are encoded by the PMCA1 and PMCA4 genes (for review see [8]). The relative abundance of the PMCA1 and PMCA4 mRNAs along the gastrointestinal tract is known to depend on location along the tract: PMCA1 is predominant in the mucosa of the small intestine and is the most abundant in duodenum, while PMCA4 is predominant in colon [58, 59]. Therefore, the present work does not analyze the relative expression of the various PMCA isoforms in normal gastric/colonic mucosa. However, we could demonstrate the presence of PMCA1b and PMCA4b mRNAs in samples of normal colonic crypts, as well as that of the PMCA1b and PMCA4b proteins in normal colon tissue sections in accordance with the literature (data not shown). In a former publication, distribution of the Ca2+ATPase activity and the correlated ATP-dependent Ca2+ transport activity was examined along the villus-crypt axis in basolateral membrane preparations of rat duodenum [60]. The Ca2+ATPase and ATP-dependent Ca2+ transport activities were low in the young, undifferentiated cells in the crypt base, while these activity values increased progressively towards the villus tip where terminally differentiated epithelial cells are located. In that paper, however, the authors did not specify the molecular identity of the corresponding Ca2+-transport ATPase(s).
Among the consequences of the in vitro differentiation of the adenocarcinoma cell lines of gastric (KATO-III), colon enterocytic (DLD-1, HT-29 and Caco-2) or colon mucus-secreting phenotypes (LS-174T) we observed marked increases of PMCA4b expression both at mRNA and protein levels (Figs. 2-4, 7-9 and Table 1). However, differentiation induction of these cancer cells was accompanied by slighter modifications in PMCA1b expression. Differentiation-induced changes in PMCA protein expression resulted also in a marked increase in PMCA-driven Ca2+ transport activity of membrane vesicles obtained from SCFA-treated gastric/colon cancer cells, as compared to untreated control cells (Fig. 5). Following various differentiation-inducing treatments, the level of PMCA4b protein expression became nearly as high as that of the PMCA1b protein (see butyrate-treated KATO-III cells in Figs. 3 and 7 or butyrate-treated LS-174T cells in Figs. 2 and 3), as estimated by Western-blotting using the pan-anti-PMCA 5F10 antibody. Butyrate-treatments were somewhat more effective for induction of PMCA4b expression than valerate-treatments in all examined adenocarcinoma cell lines (Figs. 2-4 and 6, Table 1). Differentiation induction with trichostatin A resulted in a less pronounced PMCA4b induction than treatments with Na+-butyrate (Fig. 7) or Na+-valerate (data not shown), probably due to differences in either the stability of these differentiation-inducing agents in the culture media, or the mechanism of their actions in the cells. Indeed, trichostatin A is known to modulate gene expression through inhibition of HDACs [52] while the mechanism of action of Na+-butyrate and Na+-valerate is more complex, and includes not only HDAC inhibition [51], but also interactions with other intracellular targets, such as serine-threonine protein phosphatases [61] or serine-threonine protein kinases [62].
Comparative biochemical analysis of various native and transfected PMCA isoforms indicates that a major distinctive characteristic of the PMCA4b isoform resides in the slow activation kinetics of the enzyme by the Ca2+/calmodulin complex and in the equally slow dissociation of calmodulin from the enzyme. PMCA4b thus displays delayed calmodulin-dependent activation (at rising Ca2+ concentrations) and slow inactivation (at decreasing Ca2+ concentrations) [10-12, 43]. PMCA4b may therefore confer specific kinetic characteristics to a Ca2+ transient. Because of the difficulties encountered during the cloning and expression of PMCA1 cDNA [13, 14], biochemical data are fairly limited for this PMCA pump. Thus, it is currently difficult to hypothesize how PMCA1 shapes the Ca2+ transients during signaling events. Very recently, Liu and co-workers presented data for the distinct roles of PMCA1 and PMCA4 isoforms in the Ca2+ homeostasis of bladder smooth muscle from wild-type and various PMCA gene-ablated mice [63]. They proposed that PMCA1 is involved in overall Ca2+ clearance, while PMCA4 plays an essential role in cytosolic Ca2+ increase and the contractile response to carbachol receptor-mediated signal transduction pathway.
As reviewed by Lipskaia and Lompré [64], alterations in temporal kinetics of Ca2+ signaling can control cell growth and proliferation. Changes of Ca2+ signaling and handling control also muscle development [65], neuronal precursor differentiation [66, 67], osteoblast growth and differentiation [68]. Increasing cytosolic free Ca2+ concentration has been observed during the ontogeny of intestinal epithelium [17], moreover, gradual modulation of cellular Ca2+ handling during the maturation of epithelial cells has also been observed ex vivo in isolated normal colonic crypts [69]. Higher resting cytosolic Ca2+ levels and diminished ER Ca2+ content were reported in butyrate-treated gastric cancer cells (KATO-III) when compared to less differentiated untreated control cells [22]. Because of the wide scale of proteins involved in cellular Ca2+ handling, it is impossible to assign the differentiation-induced changes observed in cellular Ca2+ signaling to a single protein species, such as for example a PMCA isoenzyme. However, the modulation of the expression pattern of various Ca2+ATPases, as shown for PMCA4b in the present work, may be relevant in this context.
The participation of several PMCA isoforms including PMCA4b in noncovalent multi-protein complexes is now well documented, and numerous interacting partners have been identified [70-80]. Several of these interactions result in the modulation by PMCA of the function or activity of the interacting partners and appear to be specific when compared to other PMCA isoforms. PMCA4 has also been observed to display a polarized expression pattern in kidney epithelial cells [81], and to communicate with mitochondria located in close proximity to the plasma membrane [82]. Finally, PMCA4 has recently been published to be specifically associated with lipid rafts in synaptic plasma membranes [83], whereas the PMCA1, 2 and 3 proteins were found in other membrane domains. Taken together these observations show, that despite their shared basic biochemical activity (i.e.: Ca2+/calmodulin-dependent Ca2+ extrusion), various PMCA isoforms may also be involved in distinct cell type-specific functions via participating in functionally integrated multi-protein complexes. Variations of the interacting partners in these complexes provide a further level of complexity in the physiological functions of the PMCA isoenzymes. Such a functional specialization of PMCAs may be, among others, the basis of the existence of the tissue-type dependent expression of various PMCA isoforms and, as shown in this work, of the preferential induction of PMCA4b expression that leads to a major switch in PMCA isoform pattern during cell differentiation in gastric/colon carcinomas.
Modulations of PMCA expression have been observed during neuronal [27, 28] and retinal [29] maturation, in myogenic [30], megakaryoblastoid [31], osteoblast-like [32] and trophoblast-like [33] differentiation models, as well as in cornea [84], in lactating mammary tissue [85] and oral and breast cancer [86, 87]. In addition, marked induction of the expression of the SERCA3 endoplasmic reticulum Ca2+ATPases has been shown earlier during megakaryocytopoiesis [88], during cell differentiation in various leukemia [21, 42] and gastric/colon cancer cells ([21, 22, 89]. When taken together with data shown on PMCA expression in our present work, these observations indicate that the remodeling of cellular Ca2+ homeostasis during differentiation is a phenomenon that concerns Ca2+ sequestration in the ER, as well as Ca2+ extrusion across the plasma membrane. This points at an integrative regulation of the remodeling of cellular Ca2+ homeostasis during differentiation, and suggests that the defects observed in the Ca2+ homeostasis of cancer cells (such as deficient expression of SERCA3 and PMCA4b isoenzymes) are part of a more general anomaly, probably linked to the differentiation blockage observed in malignant cells. These results extend further previous observations reported on rearrangements of the intracellular Ca2+ homeostasis of the androgen-dependent LNCaP prostate cancer cell line [90] and during the neuroendocrine-differentiation of prostate cancer cells [91], as well.
It has been previously described that pharmacological modulation of Ca2+ homeostasis by SERCA inhibitors or Ca2+ ionophores can affect cell survival, proliferation and apoptosis in various cell types [92, 93]. Furthermore, such treatments can induce or enhance cell differentiation in Friend erythroleukemia cells [94], in myeloid leukemia [95] and colon cancer [89]. This indicates that modulation of cellular Ca2+ homeostasis during differentiation is not a simple passive consequence of the differentiation process. Rather, a cross-talk exists between Ca2+ signaling and various cellular signaling systems that control cell survival, proliferation, differentiation and apoptosis. An additional argument in support of this idea is the involvement of various PMCA isoforms both in cell proliferation [64, 96, 97] and apoptosis [9, 76, 98, 99]. Suppressed proliferation of breast cancer MCF-7 cells or induced apoptosis in vascular smooth muscle cells via antisense-mediated PMCA inhibition were published earlier [97, 99].
In summary, although the precise identification of isoform-specific PMCA functions during differentiation requires further work, our observations clearly demonstrate differentiation-induced and isoform-specific changes in PMCA gene expression in gastric/colon cancer cells. We conclude that PMCA expression is deficient in colon and gastric carcinoma, and that the modulation of PMCA expression and function constitutes an integral part of the cellular signaling network set in motion during differentiation. Thus, the analysis of PMCA expression may be a useful new diagnostic tool for monitoring the tumor phenotype. As it is well documented that deregulation of Ca2+ homeostasis and signaling is associated with the pathophysiology of numerous tissue types, such as intestinal/colonic epithelium, among the mechanisms involved in cellular Ca2+ handling, PMCA isoenzymes together with SERCA proteins (for review see [21]) may serve as potential targets for the pharmacological modulation of carcinogenesis.
Acknowledgments
We are grateful to Professor N. Crawford (London, UK) for providing the PL/IM 430 hybridoma cells. We thank Dr. K. Pászty (Membrane Research Group of the Hungarian Academy of Sciences, Budapest, Hungary) for her help in expression of PMCA4a cDNA in COS-7 cells. We are indebted to Vera Homolya (National Medical Centre, Budapest, Hungary) for her help in the real-time PCR analysis. We wish to express special thanks to Klára Bánhidi, Csongorné Horváth and Ildikó Bakonyi (National Medical Centre, Budapest, Hungary) for their excellent technical assistance. Finally, we thank Prof. B. Sarkadi (National Medical Centre, Budapest, Hungary) for his support of the work presented in this paper.
This work was supported in part by Hungarian Academy of Sciences grants OTKA T046814 (to T.K.) and OTKA T049476 (to Á.E.), by the NKTH/KPI grant F-21/05 (to T.K.), by the Ministére des Affaires Étrangeres, France, BALATON N° 11046NA (to B.P.), by the INSERM (Institut de la Santé et de la Recherche Médicale), France (to B.P. and J.E.), by the Association pour la Recherche sur le Cancer, France, La Ligue Contre le Cancer, France (to B.P.) and by the NIH (GM28835) (to J.T.P).
Abbreviations
- PMCA
plasma membrane Ca2+ATPase
- SERCA
sarco/endoplasmic reticulum Ca2+ATPase
- ER
endoplasmic reticulum
- 5F10
pan-anti-PMCA monoclonal antibody
- JA9
anti-PMCA4 monoclonal antibody
- JA3
anti-PMCA4b monoclonal antibody
- PL/IM 430
pan-anti-SERCA3 monoclonal antibody
- IID8
pan-anti-SERCA2 monoclonal antibody
- CEA
carcinoembryonic antigen
- DPP-IV
dipeptidyl-peptidase IV
- SCFA
short chain fatty acid
- HDAC
histone deacetylase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- BSA
bovine serum albumin
- RT
reverse transcriptase
- CP
crossing point
- TCA
trichloroacetic acid
- PMSF
phenylmethyl-sulfonyl-fluoride
- TES-TEA
N-Tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid-triethanolamine
- PVDF
polyvinylidene fluoride
- ECL®
enhanced chemiluminescence
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Strehler EE, Treiman M. Calcium pumps of plasma membrane and cell interior. Curr Mol Med. 2004;4:323–335. doi: 10.2174/1566524043360735. [DOI] [PubMed] [Google Scholar]
- 2.Berridge MJ. Capacitative calcium entry. Biochem J. 1995;312(Pt 1):1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parekh AB, Penner R. Store depletion and calcium influx. Physiol Rev. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- 4.Penniston JT, Enyedi A. Modulation of the plasma membrane Ca2+ pump. J Membr Biol. 1998;165:101–109. doi: 10.1007/s002329900424. [DOI] [PubMed] [Google Scholar]
- 5.Bautista DM, Hoth M, Lewis RS. Enhancement of calcium signalling dynamics and stability by delayed modulation of the plasma-membrane calcium-ATPase in human T cells. J Physiol. 2002;541:877–894. doi: 10.1113/jphysiol.2001.016154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stauffer TP, Guerini D, Carafoli E. Tissue distribution of the four gene products of the plasma membrane Ca2+ pump. A study using specific antibodies. J Biol Chem. 1995;270:12184–12190. doi: 10.1074/jbc.270.20.12184. [DOI] [PubMed] [Google Scholar]
- 7.Guerini D. The significance of the isoforms of plasma membrane calcium ATPase. Cell Tissue Res. 1998;292:191–197. doi: 10.1007/s004410051050. [DOI] [PubMed] [Google Scholar]
- 8.Strehler EE, Zacharias DA. Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol Rev. 2001;81:21–50. doi: 10.1152/physrev.2001.81.1.21. [DOI] [PubMed] [Google Scholar]
- 9.Okunade GW, Miller ML, Pyne GJ, et al. Targeted ablation of plasma membrane Ca2+-ATPase (PMCA) 1 and 4 indicates a major housekeeping function for PMCA1 and a critical role in hyperactivated sperm motility and male fertility for PMCA4. J Biol Chem. 2004;279:33742–33750. doi: 10.1074/jbc.M404628200. [DOI] [PubMed] [Google Scholar]
- 10.Caride AJ, Penheiter AR, Filoteo AG, Bajzer Z, Enyedi A, Penniston JT. The plasma membrane calcium pump displays memory of past calcium spikes. Differences between isoforms 2b and 4b. J Biol Chem. 2001;276:39797–39804. doi: 10.1074/jbc.M104380200. [DOI] [PubMed] [Google Scholar]
- 11.Caride AJ, Filoteo AG, Penheiter AR, Paszty K, Enyedi A, Penniston JT. Delayed activation of the plasma membrane calcium pump by a sudden increase in Ca2+: fast pumps reside in fast cells. Cell Calcium. 2001;30:49–57. doi: 10.1054/ceca.2001.0212. [DOI] [PubMed] [Google Scholar]
- 12.Penheiter AR, Caride AJ, Enyedi A, Penniston JT. Tryptophan 1093 is largely responsible for the slow off rate of calmodulin from plasma membrane Ca2+ pump 4b. J Biol Chem. 2002;277:17728–17732. doi: 10.1074/jbc.M111608200. [DOI] [PubMed] [Google Scholar]
- 13.Liu BF, Xu X, Fridman R, Muallem S, Kuo TH. Consequences of functional expression of the plasma membrane Ca2+ pump isoform 1a. J Biol Chem. 1996;271:5536–5544. doi: 10.1074/jbc.271.10.5536. [DOI] [PubMed] [Google Scholar]
- 14.Adamo HP, Verma AK, Sanders MA, et al. Overexpression of the erythrocyte plasma membrane Ca2+ pump in COS-1 cells. Biochem J. 1992;285(Pt 3):791–797. doi: 10.1042/bj2850791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guerini D, Pan B, Carafoli E. Expression, purification, and characterization of isoform 1 of the plasma membrane Ca2+ pump: focus on calpain sensitivity. J Biol Chem. 2003;278:38141–38148. doi: 10.1074/jbc.M302400200. [DOI] [PubMed] [Google Scholar]
- 16.Kirchhoff P, Geibel JP. Role of calcium and other trace elements in the gastrointestinal physiology. World J Gastroenterol. 2006;12:3229–3236. doi: 10.3748/wjg.v12.i20.3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Black BL, Rogers JO. Development of Ca2+ homeostasis in epithelial cells from embryonic and neonatal intestine. Am J Physiol. 1992;263:G371–379. doi: 10.1152/ajpgi.1992.263.3.G371. [DOI] [PubMed] [Google Scholar]
- 18.Lipkin M, Reddy B, Newmark H, Lamprecht SA. Dietary factors in human colorectal cancer. Annu Rev Nutr. 1999;19:545–586. doi: 10.1146/annurev.nutr.19.1.545. [DOI] [PubMed] [Google Scholar]
- 19.Kallay E, Kifor O, Chattopadhyay N, et al. Calcium-dependent c-myc proto-oncogene expression and proliferation of Caco-2 cells: a role for a luminal extracellular calcium-sensing receptor. Biochem Biophys Res Commun. 1997;232:80–83. doi: 10.1006/bbrc.1997.6225. [DOI] [PubMed] [Google Scholar]
- 20.Chakrabarty S, Radjendirane V, Appelman H, Varani J. Extracellular calcium and calcium sensing receptor function in human colon carcinomas: promotion of E-cadherin expression and suppression of beta-catenin/TCF activation. Cancer Res. 2003;63:67–71. [PubMed] [Google Scholar]
- 21.Papp B, Brouland JP, Gelebart P, Kovacs T, Chomienne C. Endoplasmic reticulum calcium transport ATPase expression during differentiation of colon cancer and leukaemia cells. Biochem Biophys Res Commun. 2004;322:1223–1236. doi: 10.1016/j.bbrc.2004.08.030. [DOI] [PubMed] [Google Scholar]
- 22.Gelebart P, Kovacs T, Brouland JP, et al. Expression of endomembrane calcium pumps in colon and gastric cancer cells. Induction of SERCA3 expression during differentiation. J Biol Chem. 2002;277:26310–26320. doi: 10.1074/jbc.M201747200. [DOI] [PubMed] [Google Scholar]
- 23.Gabrielli BG, Johnstone RW, Saunders NA. Identifying molecular targets mediating the anticancer activity of histone deacetylase inhibitors: a work in progress. Curr Cancer Drug Targets. 2002;2:337–353. doi: 10.2174/1568009023333818. [DOI] [PubMed] [Google Scholar]
- 24.Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1:287–299. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 25.Lindemann RK, Gabrielli B, Johnstone RW. Histone-deacetylase inhibitors for the treatment of cancer. Cell Cycle. 2004;3:779–788. [PubMed] [Google Scholar]
- 26.Kelly WK, Marks PA. Drug insight: Histone deacetylase inhibitors--development of the new targeted anticancer agent suberoylanilide hydroxamic acid. Nat Clin Pract Oncol. 2005;2:150–157. doi: 10.1038/ncponc0106. [DOI] [PubMed] [Google Scholar]
- 27.Usachev YM, Toutenhoofd SL, Goellner GM, Strehler EE, Thayer SA. Differentiation induces up-regulation of plasma membrane Ca(2+)-ATPase and concomitant increase in Ca(2+) efflux in human neuroblastoma cell line IMR-32. J Neurochem. 2001;76:1756–1765. doi: 10.1046/j.1471-4159.2001.00169.x. [DOI] [PubMed] [Google Scholar]
- 28.Kip SN, Gray NW, Burette A, Canbay A, Weinberg RJ, Strehler EE. Changes in the expression of plasma membrane calcium extrusion systems during the maturation of hippocampal neurons. Hippocampus. 2006;16:20–34. doi: 10.1002/hipo.20129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Renteria RC, Strehler EE, Copenhagen DR, Krizaj D. Ontogeny of plasma membrane Ca2+ ATPase isoforms in the neural retina of the postnatal rat. Vis Neurosci. 2005;22:263–274. doi: 10.1017/S0952523805223027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hammes A, Oberdorf-Maass S, Jenatschke S, et al. Expression of the plasma membrane Ca2+-ATPase in myogenic cells. J Biol Chem. 1996;271:30816–30822. doi: 10.1074/jbc.271.48.30816. [DOI] [PubMed] [Google Scholar]
- 31.Paszty K, Kovacs T, Lacabaratz-Porret C, et al. Expression of hPMCA4b, the major form of the plasma membrane calcium pump in megakaryoblastoid cells is greatly reduced in mature human platelets. Cell Calcium. 1998;24:129–135. doi: 10.1016/s0143-4160(98)90080-x. [DOI] [PubMed] [Google Scholar]
- 32.Stains JP, Weber JA, Gay CV. Expression of Na(+)/Ca(2+) exchanger isoforms (NCX1 and NCX3) and plasma membrane Ca(2+) ATPase during osteoblast differentiation. J Cell Biochem. 2002;84:625–635. [PubMed] [Google Scholar]
- 33.Moreau R, Daoud G, Masse A, Simoneau L, Lafond J. Expression and role of calcium-ATPase pump and sodium-calcium exchanger in differentiated trophoblasts from human term placenta. Mol Reprod Dev. 2003;65:283–288. doi: 10.1002/mrd.10303. [DOI] [PubMed] [Google Scholar]
- 34.Pinto M, Robine-Leon S, Appay MD, Kedinger M, Triadou N. Enterocyte-like differentiation and polarization of the human colon carcinoma cell line Caco-2. Biol Cell. 1983;47:323–330. [Google Scholar]
- 35.Heim R, Iwata T, Zvaritch E, et al. Expression, purification, and properties of the plasma membrane Ca2+ pump and of its N-terminally truncated 105-kDa fragment. J Biol Chem. 1992;267:24476–24484. [PubMed] [Google Scholar]
- 36.Kovacs T, Felfoldi F, Papp B, et al. All three splice variants of the human sarco/endoplasmic reticulum Ca2+-ATPase 3 gene are translated to proteins: a study of their co-expression in platelets and lymphoid cells. Biochem J. 2001;358:559–568. doi: 10.1042/0264-6021:3580559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jorgensen AO, Arnold W, Pepper DR, Kahl SD, Mandel F, Campbell KP. A monoclonal antibody to the Ca2+-ATPase of cardiac sarcoplasmic reticulum cross-reacts with slow type I but not with fast type II canine skeletal muscle fibers: an immunocytochemical and immunochemical study. Cell Motil Cytoskeleton. 1988;9:164–174. doi: 10.1002/cm.970090208. [DOI] [PubMed] [Google Scholar]
- 38.Borke JL, Caride A, Verma AK, Penniston JT, Kumar R. Plasma membrane calcium pump and 28-kDa calcium binding protein in cells of rat kidney distal tubules. Am J Physiol. 1989;257:F842–849. doi: 10.1152/ajprenal.1989.257.5.F842. [DOI] [PubMed] [Google Scholar]
- 39.Borke JL, Minami J, Verma A, Penniston JT, Kumar R. Monoclonal antibodies to human erythrocyte membrane Ca++-Mg++ adenosine triphosphatase pump recognize an epitope in the basolateral membrane of human kidney distal tubule cells. J Clin Invest. 1987;80:1225–1231. doi: 10.1172/JCI113196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hack N, Wilkinson JM, Crawford N. A monoclonal antibody (PL/IM 430) to human platelet intracellular membranes which inhibits the uptake of Ca2+ without affecting the Ca2+ +Mg2+-ATPase. Biochem J. 1988;250:355–361. doi: 10.1042/bj2500355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hauri HP, Sterchi EE, Bienz D, Fransen JA, Marxer A. Expression and intracellular transport of microvillus membrane hydrolases in human intestinal epithelial cells. J Cell Biol. 1985;101:838–851. doi: 10.1083/jcb.101.3.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Launay S, Gianni M, Kovacs T, et al. Lineage-specific modulation of calcium pump expression during myeloid differentiation. Blood. 1999;93:4395–4405. [PubMed] [Google Scholar]
- 43.Caride AJ, Elwess NL, Verma AK, et al. The rate of activation by calmodulin of isoform 4 of the plasma membrane Ca(2+) pump is slow and is changed by alternative splicing. J Biol Chem. 1999;274:35227–35232. doi: 10.1074/jbc.274.49.35227. [DOI] [PubMed] [Google Scholar]
- 44.Enyedi A, Verma AK, Filoteo AG, Penniston JT. A highly active 120-kDa truncated mutant of the plasma membrane Ca2+ pump. J Biol Chem. 1993;268:10621–10626. [PubMed] [Google Scholar]
- 45.Enyedi A, Verma AK, Filoteo AG, Penniston JT. Protein kinase C activates the plasma membrane Ca2+ pump isoform 4b by phosphorylation of an inhibitory region downstream of the calmodulin-binding domain. J Biol Chem. 1996;271:32461–32467. doi: 10.1074/jbc.271.50.32461. [DOI] [PubMed] [Google Scholar]
- 46.Enyedi A, Elwess NL, Filoteo AG, Verma AK, Paszty K, Penniston JT. Protein kinase C phosphorylates the “a” forms of plasma membrane Ca2+ pump isoforms 2 and 3 and prevents binding of calmodulin. J Biol Chem. 1997;272:27525–27528. doi: 10.1074/jbc.272.44.27525. [DOI] [PubMed] [Google Scholar]
- 47.Sarkadi B, Szasz I, Gardos G. Characteristics and regulation of active calcium transport in inside-out red cell membrane vesicles. Biochim Biophys Acta. 1980;598:326–338. doi: 10.1016/0005-2736(80)90010-3. [DOI] [PubMed] [Google Scholar]
- 48.Caride AJ, Filoteo AG, Enyedi A, Verma AK, Penniston JT. Detection of isoform 4 of the plasma membrane calcium pump in human tissues by using isoform-specific monoclonal antibodies. Biochem J. 1996;316(Pt 1):353–359. doi: 10.1042/bj3160353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spira AI, Carducci MA. Differentiation therapy. Curr Opin Pharmacol. 2003;3:338–343. doi: 10.1016/s1471-4892(03)00081-x. [DOI] [PubMed] [Google Scholar]
- 50.Gilbert J, Gore SD, Herman JG, Carducci MA. The clinical application of targeting cancer through histone acetylation and hypomethylation. Clin Cancer Res. 2004;10:4589–4596. doi: 10.1158/1078-0432.CCR-03-0297. [DOI] [PubMed] [Google Scholar]
- 51.Della Ragione F, Criniti V, Della Pietra V, et al. Genes modulated by histone acetylation as new effectors of butyrate activity. FEBS Lett. 2001;499:199–204. doi: 10.1016/s0014-5793(01)02539-x. [DOI] [PubMed] [Google Scholar]
- 52.Monneret C. Histone deacetylase inhibitors. Eur J Med Chem. 2005;40:1–13. doi: 10.1016/j.ejmech.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 53.Hauck W, Stanners CP. Control of carcinoembryonic antigen gene family expression in a differentiating colon carcinoma cell line. Caco-2, Cancer Res. 1991;51:3526–3533. [PubMed] [Google Scholar]
- 54.Darmoul D, Lacasa M, Baricault L, et al. Dipeptidyl peptidase IV (CD 26) gene expression in enterocyte-like colon cancer cell lines HT-29 and Caco-2. Cloning of the complete human coding sequence and changes of dipeptidyl peptidase IV mRNA levels during cell differentiation. J Biol Chem. 1992;267:4824–4833. [PubMed] [Google Scholar]
- 55.Guerini D, Coletto L, Carafoli E. Exporting calcium from cells. Cell Calcium. 2005;38:281–289. doi: 10.1016/j.ceca.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 56.Carafoli E, Brini M. Calcium pumps: structural basis for and mechanism of calcium transmembrane transport. Curr Opin Chem Biol. 2000;4:152–161. doi: 10.1016/s1367-5931(99)00069-1. [DOI] [PubMed] [Google Scholar]
- 57.Prasad V, Okunade GW, Miller ML, Shull GE. Phenotypes of SERCA and PMCA knockout mice. Biochem Biophys Res Commun. 2004;322:1192–1203. doi: 10.1016/j.bbrc.2004.07.156. [DOI] [PubMed] [Google Scholar]
- 58.Howard A, Legon S, Walters JR. Human and rat intestinal plasma membrane calcium pump isoforms. Am J Physiol. 1993;265:G917–925. doi: 10.1152/ajpgi.1993.265.5.G917. [DOI] [PubMed] [Google Scholar]
- 59.Freeman TC, Howard A, Bentsen BS, Legon S, Walters JR. Cellular and regional expression of transcripts of the plasma membrane calcium pump PMCA1 in rabbit intestine. Am J Physiol. 1995;269:G126–131. doi: 10.1152/ajpgi.1995.269.1.G126. [DOI] [PubMed] [Google Scholar]
- 60.van Corven EJ, Roche C, van Os CH. Distribution of Ca2+-ATPase, ATP-dependent Ca2+-transport, calmodulin and vitamin D-dependent Ca2+-binding protein along the villus-crypt axis in rat duodenum. Biochim Biophys Acta. 1985;820:274–282. doi: 10.1016/0005-2736(85)90121-x. [DOI] [PubMed] [Google Scholar]
- 61.Cuisset L, Tichonicky L, Jaffray P, Delpech M. The effects of sodium butyrate on transcription are mediated through activation of a protein phosphatase. J Biol Chem. 1997;272:24148–24153. doi: 10.1074/jbc.272.39.24148. [DOI] [PubMed] [Google Scholar]
- 62.Russo GL, Della Pietra V, Mercurio C, et al. Down-regulation of protein kinase CKII activity by sodium butyrate. Biochem Biophys Res Commun. 1997;233:673–677. doi: 10.1006/bbrc.1997.6515. [DOI] [PubMed] [Google Scholar]
- 63.Liu L, Ishida Y, Okunade G, Pyne-Geithman GJ, Shull GE, Paul RJ. Distinct Roles of PMCA Isoforms in Ca2+-Homeostasis of Bladder Smooth Muscle: Evidence from PMCA Gene-Ablated Mice. Am J Physiol Cell Physiol. 2006 doi: 10.1152/ajpcell.00313.2006. [DOI] [PubMed] [Google Scholar]
- 64.Lipskaia L, Lompre AM. Alteration in temporal kinetics of Ca2+ signaling and control of growth and proliferation. Biol Cell. 2004;96:55–68. doi: 10.1016/j.biolcel.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 65.Berchtold MW, Brinkmeier H, Muntener M. Calcium ion in skeletal muscle: its crucial role for muscle function, plasticity, and disease. Physiol Rev. 2000;80:1215–1265. doi: 10.1152/physrev.2000.80.3.1215. [DOI] [PubMed] [Google Scholar]
- 66.Ciccolini F, Collins TJ, Sudhoelter J, Lipp P, Berridge MJ, Bootman MD. Local and global spontaneous calcium events regulate neurite outgrowth and onset of GABAergic phenotype during neural precursor differentiation. J Neurosci. 2003;23:103–111. doi: 10.1523/JNEUROSCI.23-01-00103.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spitzer NC, Root CM, Borodinsky LN. Orchestrating neuronal differentiation: patterns of Ca2+ spikes specify transmitter choice. Trends Neurosci. 2004;27:415–421. doi: 10.1016/j.tins.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 68.Zayzafoon M. Calcium/calmodulin signaling controls osteoblast growth and differentiation. J Cell Biochem. 2006;97:56–70. doi: 10.1002/jcb.20675. [DOI] [PubMed] [Google Scholar]
- 69.Lindqvist SM, Sharp P, Johnson IT, Satoh Y, Williams MR. Acetylcholine-induced calcium signaling along the rat colonic crypt axis. Gastroenterology. 1998;115:1131–1143. doi: 10.1016/s0016-5085(98)70084-8. [DOI] [PubMed] [Google Scholar]
- 70.Schuh K, Uldrijan S, Gambaryan S, Roethlein N, Neyses L. Interaction of the plasma membrane Ca2+ pump 4b/CI with the Ca2+/calmodulin-dependent membrane-associated kinase CASK. J Biol Chem. 2003;278:9778–9783. doi: 10.1074/jbc.M212507200. [DOI] [PubMed] [Google Scholar]
- 71.DeMarco SJ, Strehler EE. Plasma membrane Ca2+-atpase isoforms 2b and 4b interact promiscuously and selectively with members of the membrane-associated guanylate kinase family of PDZ (PSD95/Dlg/ZO-1) domain-containing proteins. J Biol Chem. 2001;276:21594–21600. doi: 10.1074/jbc.M101448200. [DOI] [PubMed] [Google Scholar]
- 72.DeMarco SJ, Chicka MC, Strehler EE. Plasma membrane Ca2+ ATPase isoform 2b interacts preferentially with Na+/H+ exchanger regulatory factor 2 in apical plasma membranes. J Biol Chem. 2002;277:10506–10511. doi: 10.1074/jbc.M111616200. [DOI] [PubMed] [Google Scholar]
- 73.Kim E, DeMarco SJ, Marfatia SM, Chishti AH, Sheng M, Strehler EE. Plasma membrane Ca2+ ATPase isoform 4b binds to membrane-associated guanylate kinase (MAGUK) proteins via their PDZ (PSD-95/Dlg/ZO-1) domains. J Biol Chem. 1998;273:1591–1595. doi: 10.1074/jbc.273.3.1591. [DOI] [PubMed] [Google Scholar]
- 74.Goellner GM, DeMarco SJ, Strehler EE. Characterization of PISP, a novel single-PDZ protein that binds to all plasma membrane Ca2+-ATPase b-splice variants. Ann N Y Acad Sci. 2003;986:461–471. doi: 10.1111/j.1749-6632.2003.tb07230.x. [DOI] [PubMed] [Google Scholar]
- 75.Chen J, McLean PA, Neel BG, Okunade G, Shull GE, Wortis HH. CD22 attenuates calcium signaling by potentiating plasma membrane calcium-ATPase activity. Nat Immunol. 2004;5:651–657. doi: 10.1038/ni1072. [DOI] [PubMed] [Google Scholar]
- 76.Armesilla AL, Williams JC, Buch MH, et al. Novel functional interaction between the plasma membrane Ca2+ pump 4b and the proapoptotic tumor suppressor Ras-associated factor 1 (RASSF1) J Biol Chem. 2004;279:31318–31328. doi: 10.1074/jbc.M307557200. [DOI] [PubMed] [Google Scholar]
- 77.Williams JC, Armesilla AL, Mohamed TM, et al. The sarcolemmal calcium pump, alpha-1 syntrophin and neuronal nitric oxide synthase are part of a macromolecular protein complex. J Biol Chem. 2006;281:23341–23348. doi: 10.1074/jbc.M513341200. [DOI] [PubMed] [Google Scholar]
- 78.Buch MH, Pickard A, Rodriguez A, et al. The sarcolemmal calcium pump inhibits the calcineurin/nuclear factor of activated T-cell pathway via interaction with the calcineurin A catalytic subunit. J Biol Chem. 2005;280:29479–29487. doi: 10.1074/jbc.M501326200. [DOI] [PubMed] [Google Scholar]
- 79.Rimessi A, Coletto L, Pinton P, Rizzuto R, Brini M, Carafoli E. Inhibitory interaction of the 14-3-3{epsilon} protein with isoform 4 of the plasma membrane Ca(2+)-ATPase pump. J Biol Chem. 2005;280:37195–37203. doi: 10.1074/jbc.M504921200. [DOI] [PubMed] [Google Scholar]
- 80.Sgambato-Faure V, Xiong Y, Berke JD, Hyman SE, Strehler EE. The Homer-1 protein Ania-3 interacts with the plasma membrane calcium pump. Biochem Biophys Res Commun. 2006;343:630–637. doi: 10.1016/j.bbrc.2006.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kip SN, Strehler EE. Characterization of PMCA isoforms and their contribution to transcellular Ca2+ flux in MDCK cells. Am J Physiol Renal Physiol. 2003;284:F122–132. doi: 10.1152/ajprenal.00161.2002. [DOI] [PubMed] [Google Scholar]
- 82.Frieden M, Arnaudeau S, Castelbou C, Demaurex N. Subplasmalemmal mitochondria modulate the activity of plasma membrane Ca2+-ATPases. J Biol Chem. 2005;280:43198–43208. doi: 10.1074/jbc.M510279200. [DOI] [PubMed] [Google Scholar]
- 83.Sepulveda MR, Berrocal-Carrillo M, Gasset M, Mata AM. The plasma membrane Ca2+-ATPase isoform 4 is localized in lipid rafts of cerebellum synaptic plasma membranes. J Biol Chem. 2006;281:447–453. doi: 10.1074/jbc.M506950200. [DOI] [PubMed] [Google Scholar]
- 84.Talarico EF, Jr, Kennedy BG, Marfurt CF, Loeffler KU, Mangini NJ. Expression and immunolocalization of plasma membrane calcium ATPase isoforms in human corneal epithelium. Mol Vis. 2005;11:169–178. [PubMed] [Google Scholar]
- 85.Reinhardt TA, Filoteo AG, Penniston JT, Horst RL. Ca(2+)-ATPase protein expression in mammary tissue. Am J Physiol Cell Physiol. 2000;279:C1595–1602. doi: 10.1152/ajpcell.2000.279.5.C1595. [DOI] [PubMed] [Google Scholar]
- 86.Saito K, Uzawa K, Endo Y, et al. Plasma membrane Ca2+ ATPase isoform 1 down-regulated in human oral cancer. Oncol Rep. 2006;15:49–55. [PubMed] [Google Scholar]
- 87.Lee WJ, Roberts-Thomson SJ, Monteith GR. Plasma membrane calcium-ATPase 2 and 4 in human breast cancer cell lines. Biochem Biophys Res Commun. 2005;337:779–783. doi: 10.1016/j.bbrc.2005.09.119. [DOI] [PubMed] [Google Scholar]
- 88.Lacabaratz-Porret C, Launay S, Corvazier E, Bredoux R, Papp B, Enouf J. Biogenesis of endoplasmic reticulum proteins involved in Ca2+ signalling during megakaryocytic differentiation: an in vitro study. Biochem J. 2000;350(Pt 3):723–734. [PMC free article] [PubMed] [Google Scholar]
- 89.Brouland JP, Gelebart P, Kovacs T, Enouf J, Grossmann J, Papp B. The loss of sarco/endoplasmic reticulum calcium transport ATPase 3 expression is an early event during the multistep process of colon carcinogenesis. Am J Pathol. 2005;167:233–242. doi: 10.1016/S0002-9440(10)62968-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vanden Abeele F, Skryma R, Shuba Y, et al. Bcl-2-dependent modulation of Ca(2+) homeostasis and store-operated channels in prostate cancer cells. Cancer Cell. 2002;1:169–179. doi: 10.1016/s1535-6108(02)00034-x. [DOI] [PubMed] [Google Scholar]
- 91.Vanoverberghe K, Vanden Abeele F, Mariot P, et al. Ca2+ homeostasis and apoptotic resistance of neuroendocrine-differentiated prostate cancer cells. Cell Death Differ. 2004;11:321–330. doi: 10.1038/sj.cdd.4401375. [DOI] [PubMed] [Google Scholar]
- 92.Apati A, Janossy J, Brozik A, Bauer PI, Magocsi M. Calcium induces cell survival and proliferation through the activation of the MAPK pathway in a human hormone-dependent leukemia cell line, TF-1. J Biol Chem. 2003;278:9235–9243. doi: 10.1074/jbc.m205528200. [DOI] [PubMed] [Google Scholar]
- 93.Denmeade SR, Isaacs JT. The SERCA pump as a therapeutic target: making a “smart bomb” for prostate cancer. Cancer Biol Ther. 2005;4:14–22. doi: 10.4161/cbt.4.1.1505. [DOI] [PubMed] [Google Scholar]
- 94.Schaefer A, Magocsi M, Stocker U, Kosa F, Marquardt H. Early transient suppression of c-myb mRNA levels and induction of differentiation in Friend erythroleukemia cells by the [Ca2+]i-increasing agents cyclopiazonic acid and thapsigargin. J Biol Chem. 1994;269:8786–8791. [PubMed] [Google Scholar]
- 95.Launay S, Gianni M, Diomede L, Machesky LM, Enouf J, Papp B. Enhancement of ATRA-induced cell differentiation by inhibition of calcium accumulation into the endoplasmic reticulum: cross-talk between RAR alpha and calcium-dependent signaling. Blood. 2003;101:3220–3228. doi: 10.1182/blood-2002-09-2730. [DOI] [PubMed] [Google Scholar]
- 96.Husain M, Jiang L, See V, et al. Regulation of vascular smooth muscle cell proliferation by plasma membrane Ca(2+)-ATPase. Am J Physiol. 1997;272:C1947–1959. doi: 10.1152/ajpcell.1997.272.6.C1947. [DOI] [PubMed] [Google Scholar]
- 97.Lee WJ, Robinson JA, Holman NA, McCall MN, Roberts-Thomson SJ, Monteith GR. Antisense-mediated Inhibition of the plasma membrane calcium-ATPase suppresses proliferation of MCF-7 cells. J Biol Chem. 2005;280:27076–27084. doi: 10.1074/jbc.M414142200. [DOI] [PubMed] [Google Scholar]
- 98.Paszty K, Verma AK, Padanyi R, Filoteo AG, Penniston JT, Enyedi A. Plasma membrane Ca2+ATPase isoform 4b is cleaved and activated by caspase-3 during the early phase of apoptosis. J Biol Chem. 2002;277:6822–6829. doi: 10.1074/jbc.M109548200. [DOI] [PubMed] [Google Scholar]
- 99.Sasamura S, Furukawa K, Shiratori M, Motomura S, Ohizumi Y. Antisense-inhibition of plasma membrane Ca2+ pump induces apoptosis in vascular smooth muscle cells. Jpn J Pharmacol. 2002;90:164–172. doi: 10.1254/jjp.90.164. [DOI] [PubMed] [Google Scholar]