

Abstract
From the nucleus, histone deacetylase 4 (HDAC4) regulates a variety of cellular processes, including growth, differentiation, and survival, by orchestrating transcriptional changes. Extracellular signals control its repressive influence mostly through regulating its nuclear-cytoplasmic shuttling. In particular, specific posttranslational modifications such as phosphorylation and caspase-mediated proteolytic processing operate on HDAC4 to promote its nuclear accumulation or export. To understand the signaling properties of this deacetylase, we investigated its cell death-promoting activity and the transcriptional repression potential of different mutants that accumulate in the nucleus. Here we show that, compared to that of other nuclear forms of HDAC4, a caspase-generated nuclear fragment exhibits a stronger cell death-promoting activity coupled with increased repressive effect on Runx2- or SRF-dependent transcription. However, this mutant displays reduced repressive action on MEF2C-driven transcription. Photobleaching experiments and quantitative analysis of the raw data, based on a two-binding-state compartmental model, demonstrate the existence of two nuclear pools of HDAC4 with different chromatin-binding properties. The caspase-generated fragment is weakly bound to chromatin, whereas an HDAC4 mutant defective in 14-3-3 binding or the wild-type HDAC5 protein forms a more stable complex. The tightly bound species show an impaired ability to induce cell death and repress Runx2- or SRF-dependent transcription less efficiently. We propose that, through specific posttranslation modifications, extracellular signals control two distinct nuclear pools of HDAC4 to differentially dictate cell death and differentiation. These two nuclear pools of HDAC4 are characterized by different repression potentials and divergent dynamics of chromatin interaction.
Caspases show the exquisite ability to target important regulators of the cell survival-death decision in order to enhance the apoptosis' verdict (15, 49). Hence, caspase substrates are instrumental to understanding the cellular pathways that govern cell survival and cell death. In principle, caspase substrates can be used as picklocks to open the doors keeping secret important survival pathways.
Histone deacetylases (HDACs) are chief regulators of gene expression as part of transcriptional corepressor complexes that influence chromatin remodeling (12, 20, 24). Histone deacetylation by HDAC promotes chromatin condensation favoring transcriptional repression. In mammals, the different HDACs can be subdivided into four classes based on sequence similarity to three yeast HDACs (14, 19). Based on structural characteristics, class II can be further separated into classes IIa and IIb (26, 36, 51, 55). HDAC4, HDAC5, HDAC7, and HDAC9 belong to class IIa, and they play key roles in tissue growth and differentiation (10, 11). These deacetylases are distinguished by (i) the presence of an amino-terminal regulatory region devoted to binding transcription factors and corepressors, (ii) a carboxy-terminal region that includes the catalytic domain, and (iii) nucleocytoplasmic shuttling in a signal-responsive fashion. In fact, a class II HDAC can enter the nucleus or be exported into the cytoplasm in response to specific signals, thereby resetting the regulatory program. A set of conserved serines, targets of various kinases, are the principal determinants of HDAC localization. Phosphoserines represent docking sites for 14-3-3 chaperone proteins, which escort the HDACs from the nucleus into the cytoplasm, relieving their repressive action (26, 36, 51, 55).
Among class IIa members, nucleocytoplasmic shuttling of HDAC4 also is under caspase regulation. During apoptosis, caspase-3 cleaves HDAC4 and generates an amino-terminal fragment, which accumulates in the nucleus and acts as a transcriptional repressor to trigger apoptosis (28, 37). The proapoptotic activity of HDAC4 also can be manifested independently from caspase maturation. In neuronal cells, apoptotic signals, such as low-potassium or excitotoxic glutamate, promote nuclear accumulation of HDAC4, and its ablation by short interfering RNA treatment suppresses neuronal cell death (4).
Recent studies have revealed that class IIa HDACs, and particularly HDAC4, are part of repressive complexes that orchestrate different posttranslation modifications on targets proteins, including phosphorylation, deacetylation, and sumoylation (18, 31, 36, 42, 58). HDAC4 associates with several transcription factors, including MEF2 family members Runx2 and SRF (13, 36, 50). The transcription factors target HDAC4 and the different associated corepressors to specific promoters on the DNA. Hence, it is possible that the proapoptotic activity of HDAC4 reflects its ability to silence the expression genes critical for survival.
Paradoxically, nuclear accumulation of HDAC4 and the subsequent repressive action on gene expression are critical events in the control of differentiation as well as apoptosis (4, 30, 35, 37). In spite of this, it is not clear how HDAC4 can differentially modulate the expression of genes involved in the control of differentiation or apoptosis. To answer this question, we investigated the proapoptotic activities, the repressive activities, and the nucleocytoplasmic trafficking ability of HDAC4 and mutants that constitutively accumulate in the nucleus. For comparison, we also analyzed HDAC5, a class IIa enzyme highly related to HDAC4 but less susceptible to caspase modulation (37). In particular, analysis of two mutants, one mimicking dephosphorylation at the 14-3-3-binding sites and the other for caspase processing, have led us to propose that different signals, such as differentiation and apoptosis, control two distinct nuclear pools of HDAC4 with peculiar repression activities and divergent dynamics of chromatin interaction.
MATERIALS AND METHODS
Cell culture, transfection, and microinjection.
U2OS osteosarcoma cells and IMR90-E1A fibroblasts (expressing the adenoviral oncoprotein E1A) were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, penicillin, and streptomycin. The transfection of cells was performed using the calcium phosphate precipitation method. Nuclear microinjection was performed using an automated injection system (Zeiss) as previously described (17).
Plasmid constructions.
pFLAG-CMV5 constructs expressing HDAC4 (full length), HDAC4ΔC (amino acids 1 to 289), and HDAC4ΔN (amino acids 289 to 1084) were previously described (37). pFLAGCMV5HDAC4D289A and the construct for the HDAC4 point mutant L1062A were obtained by in vitro mutagenesis using the Gene-Taylor kit (Invitrogen), and full-length pFLAGCMV5-HDAC4 was used as the template. The following primers were used: primer L1062A FW-middle, 5′-CGGTCACCGCCATGGCCTCGGCCTCCGTGGGCGT-3′; primer L1062A RV-middle, 5′-CGAGGCCATGGCGGTGACCGTCTCGGCTTC-3′; primer D289A FW-middle, 5′-AGGTCCGTTGGATGTCACAGCCTCCGCGTGCAG-3′; and primer D289A RV-middle, 5′-TGTGACATCCAACGGACGCTTTTTTAGAGC-3′. A pFLAG-CMV5 construct expressing the HDAC4 mutant TM (HDAC4/TM) was generated by subcloning the HDAC4 fragment obtained from pEGFPC2HDAC4/TM (52).
pEGFP-N1 constructs expressing full-length HDAC4 or HDAC4 mutants were obtained by being subcloned from the respective pFLAGCMV5 constructs described above.
pFLAGCMVHDAC5 was generated by subcloning the NotI/XhoI fragment from pBJ5.1 (21) into pFLAGCMV, while pEGFPN1HDAC5 was generated by PCR with pBJ5.1HDAC5 as the template and the following set of primers: primer FW, 5′-TCACTCGAGATGAACTCTCCCAAC-3′; and primer RV, 5′-CATAAGCTTCAGGGCAGGCTCCTGCTCCAT-3′.
The pEGFP-MEF2C construct was generated by PCR using pcDNA3.1MEF2C (37) as a template and the following set of primers: primer FW, 5′-CATGAAGGATCCATGGGGAGAAAAAAGATTCAG-3′; and primer RV, 5′-CATGACGGATCCAATGTTGCCCATCCTTCAGA-3′. pCMVhOSF2 was used as a template to generate pcDNA3HA-Runx2 by PCR with the following set of primers: FW, 5′-CTCTACAGATCTATGGCATCAAACAGCCTCTTC-3′; and RV, 5′-CCCTACCTCGAGTCAATATGGTCGCCAAACAGATTC-3′.
The pEGFPN1 construct bearing the histone H1.2 was produced by PCR with the template pT7T3PacI histone H1.2 and the following set of primers: FW, 5′-CATCACGAATTCAATGTCCGAGACTGCTCCT-3′; and RV, 5′-TACTACAGATCTAATTTCTTCTTGGGCGCCGC-3′.
All constructs generated were sequenced to check for the respective introduced mutations and deletions and the translating fidelity of the inserted PCR fragments.
Immunofluorescence and photobleaching experiments.
For immunofluorescence assays, cells were fixed with 3% paraformaldehyde in phosphate-buffered saline for 20 min at room temperature and permeabilized with 0.1% Triton X-100. Coverslips were labeled with either anti-FLAG (Sigma) or antihemagglutinin (anti-HA) (Sigma) antibody, followed by Alexa Fluor 546- or 633-conjugated anti-mouse and anti-rabbit secondary antibodies (Molecular Probes). Cells were imaged with a Leica true confocal scanner SP equipped with a 488 λ Ar laser and a 543 to 633 λ HeNe laser.
For fluorescent-recovery-after-photobleaching (FRAP) experiments (8, 39, 45, 47), different green fluorescent protein (GFP) constructs coding for the indicated proteins (2 ng/μl) were microinjected in the nuclei of U2OS or E1A cells grown on glass-bottom dishes (Willco Wells). Two hours later, cells were subjected to FRAP experiments in a 5% CO2 atmosphere at 37°C.
A Planatochromat 63× objective (1.4 numerical aperture) was used. Images were acquired with a 4,187-pinhole Airy disk unit and a 256- by 256-pixel resolution. Five prebleach images were collected at minimum laser power (12%) every 230 ms. Cell bleaching was performed through four impulses of 230 ms at 100% laser power in a small square area (4 μm per side, 20 by 20 pixels). One hundred recovery images were acquired every 230 ms, followed by 60 recovery images at 5-s intervals (laser power, 12%). The fluorescence intensity was measured by MethaMorph 6.0 software. Recovery curves were obtained by subtracting background values and normalizing data according the following formula: I = 100 × T0It/TtI0, where I is intensity, T0 is the total cellular/nuclear intensity during prebleach, Tt is the total cellular/nuclear intensity at time point t, I0 is the intensity in the region of interest during prebleach, and It is the average intensity in the region of interest at time point t (38). This expression accounts for the small loss in total intensity caused by bleach itself.
The recovery kinetics of the bleached area shown are averages of results from at least 10 cells from at least three independent experiments. The setup was tested entirely with fixed cells.
Mathematical model for interpreting FRAP data.
The purpose of a mathematical model for FRAP experiments is to provide a quantitative interpretation of observations in order to gain insight into the dynamics of nuclear proteins undergoing binding-unbinding events. In this work, we have developed a two-binding-state compartmental model including diffusion. Our model is a variant of the compartmental model proposed by Phair and coauthors (39). Basically, the complete two-binding-site system is roughly decomposed into two components, one representing a (relatively fast) diffusion-driven process and a second representing a (slower) reaction process. This approach is borrowed from the methodology of model reduction. A complex system is decomposed into weakly interacting subsystems; less relevant terms in the exact solution of each subsystem are neglected, while the principal components are retained in order to catch the dominant behavior of the overall system. Analogous arguments were exploited by Sprague et al. (47) for a FRAP model based on a reaction-diffusion partial differential equation. Due to space limitations, hereafter we briefly discuss the method, using as a template the model with one binding site for notational simplicity; analogous arguments also are valid in the complete two-binding-site model.
Our starting point is a classical compartmental model for FRAP analysis of a single binding site (8). Let D denote the diffusional transfer coefficient, let kon and koff be the binding and unbinding rate constants, respectively, and let r denote the bleach spot/nuclear area ratio. The functions u0(t) and u1(t) represent the population of diffusing molecules inside and outside the bleach spot at time t, respectively; analogously, v0(t) and v1(t) represent the population of bound molecules inside and outside the bleach spot, respectively. Using matrix-vector format and the dot notation to denote the time derivative, the differential equations describing the dynamics of a FRAP experiment are the following:
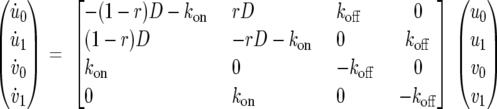 |
The theoretical fluorescence recovery curve is given by F(t) = u0(t) + v0(t). Rather simple computations show that the system converges toward the unique stationary solution
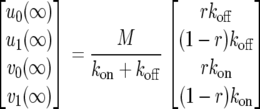 |
where M = u0(0) + u1(0) + v0(0) + v1(0), independent of the initial condition [u0(0), u1(0), v0(0), v1(0)]. In fact, the sum of all compartments is constant over time: u0(t) + u1(t) + v0(t) + v1(t) = M.
Consider the auxiliary (a) functions ai(t) = vi(t)/ui(t), for i = 0.1; then, ai(t)→kon/koff as t→∞. Furthermore, the approximation ai(t) ≈ kon/koff is quite accurate in the trailing phase of FRAP (the turnover regime) and in a much wider range when the time scale of the binding reaction is considerably faster than that of diffusion. In that case, the chemical equilibrium is approached faster than the diffusive equilibrium. Under the assumption that ai(t) is almost constant, straightforward computations lead to the reduced model
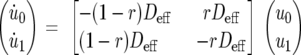 |
where Deff = D/(1 + kon/koff) is the effective diffusion coefficient (47). Consequently, assuming the normalization condition F(∞) = 1, the theoretical FRAP curve is given by F(t) = u0(t) + v0(t) = [1 + ai(t)]u0(t) ≈ 1 − ρ exp(−Deff t) for some constant ρ, depending on the initial conditions. Using a widespread terminology, this kind of dynamics falls into the so-called effective diffusion scenario (46, 47).
On the other hand, consider the auxiliary function b(t) = u0(t)/u1(t). In the long run, b(t)→r/(1 −r). If the reaction is slower than diffusion, the approximation b(t) ≈ r/(1 − r) is rather accurate in a large time interval, since free molecules tend to equilibrate. In this case, we can look at free molecules as a unique, well-mixed pool: U(t) = u0(t) + u1(t), with u0(t) ≈ rU(t) and u1(t) ≈ (1 − r)U(t), where U is the total amount of free molecules. Under this assumption, the basic model reduces to the differential equation
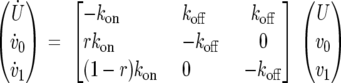 |
Assuming that immediately after the bleaching the molecules are chemically in equilibrium, i.e., a0(0) = a1(0) = kon/koff, and the normalization F(∞) = 1, then the associated FRAP curve is given by F(t) ≈ rU(t) + v0(t) = 1 − ρ exp(−koff t), with ρ depending on the initial conditions. This kind of dynamic is referred to as a reaction-dominant scenario (47). It is worth noting that analogous expressions for F(t), both in the effective diffusion case and in the reaction-dominant case, were obtained by various authors with different FRAP models (46, 47).
In the complete two-binding-site model, the effective diffusion scenario and the reaction-dominant scenario are superposed and pertain, respectively, to two weakly interacting molecular groups (although the relative amounts are unknown in advance), each group roughly corresponding to one of the two binding states. Indeed, as experimentally shown previously (4), the two binding reactions have complementary behaviors. One of them is rather fast and produces short-living complexes, the mean life span of which is often comparable to the time scale of free diffusion, while the other is much slower and more stable. The resulting normalized FRAP curve is a weighted sum of their respective contributions: F(t) = 1 − ρ1 exp(−λ1t) − ρ2 exp(−λ2t).
The exponential terms are discriminated by the inequality λ1 > λ2. The interpretation of the coefficients appearing in the previous formula is the following. λ1 is Deff, the effective diffusion coefficient. With respect to the coefficients of the original model, we have the equation Deff = D/(1 + kon,1/koff,1), where D is the pure diffusion coefficient and kon,1, koff,1 are the binding-unbinding rate constants of the first (faster) binding site. λ2 represents koff,2, the unbinding rate constant of the second (slower) binding site. ρ1 and ρ2 are the relative percentages of bound molecules in each binding state at equilibrium.
After data acquisition, every single FRAP experiment was fitted against the function F(t), described above, by means of a nonlinear least-squares method, giving a value for ρ1,2 and λ1,2 for each cell; the fit was based on the measured values for F(t) in 100 frames after the bleaching, at a rate of 230 ms/frame. Numerical computations were performed with Matlab. Subsequently, we performed a statistical analysis of the resulting values for kon,2 and koff,2 for each cell line.
Quantitative RT-PCR and reporter gene assays.
Cells were transfected using Lipofectamine (Invitrogen). Twenty-four hours posttransfection, cells were harvested and cDNAs were synthesized. Real-time PCR (RT-PCR) was performed using the Bio-Rad iQ5 and SYBR green technology. Data were analyzed by comparative threshold cycle using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and hypoxanthine phosphoribosyltransferase as housekeeping controls. All reactions were done in triplicate. All primer sequences used in this article are available on request.
For luciferase assays, cells were transfected at 30 to 40% confluence with the indicated mammalian expression plasmids. Cells were collected 24 h after transfection, and luciferase activity was measured and normalized for Renilla luciferase activity using the dual-luciferase reporter assay system according to the vendor's instructions (Promega).
Immunoblotting.
Proteins were transferred to 0.2-μm-pore-sized nitrocellulose membranes (Schleicher & Schuelt) using a semidry blotting apparatus (Pharmacia). Membranes were incubated overnight at room temperature with anti-GFP (37), anti-FLAG (Sigma), and anti-HA (Sigma) primary antibodies, followed by peroxidase-conjugated anti-rabbit (KPL) or anti-mouse (Euroclone) secondary antibody. Blots were developed with Super Signal West Pico or Dura, as recommended by the vendor (Pierce).
In vitro binding assays.
Radiolabeled HDAC4 and its mutant derivatives were generated using a T T7-coupled reticulocyte lysate system (Promega). Glutathione S-transferase (GST) fusion proteins immobilized onto glutathione-Sepharose were prepared as previously described (37). Beads were incubated with 200 μl of binding buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 0.05% NP-40, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride) at 4°C for 3 h in the presence of the appropriate amounts of the 35S-labeled proteins. The beads were separated by brief centrifugation and washed four times with washing buffer containing 20 mM HEPES, pH 7.5, 150 mM NaCl, 0.05% NP-40, and 10% glycerol. Beads were boiled in sodium dodecyl sulfate (SDS) sample buffer, and proteins were resolved on an SDS-10% polyacrylamide gel electrophoresis (PAGE) gel.
RESULTS
Subcellular localization of different HDAC4 mutants.
The caspase-cleaved fragment of HDAC4, ΔC (Fig. 1A), accumulates in the nucleus, where it can induce apoptosis (28, 37). Nuclear accumulation is a prerequisite for the induction of cell death, but from the nucleus, HDAC4 also can regulate differentiation (30, 32, 35, 51). To understand the mechanisms that drive HDAC4 toward apoptosis or differentiation, we studied the cell death activity of different nuclear forms of HDAC4, generated by point mutations and deletions. These mutants (Fig. 1A) include the amino-terminal fragment ΔC, originated from the caspase cleavage (28, 37), and the triple mutant TM, which is defective in binding the 14-3-3 proteins (21, 53). In particular, these two mutants mimic posttranslation modifications implicated in the regulation of HDAC4 nucleocytoplasmic shuttling. We also have taken advantage of L1062A, a point mutant defective for CRM1-mediated nuclear export (52); D289A, the caspase-resistant form; and ΔN, the caspase-generated carboxy-terminal fragment of HDAC4, which is exclusively cytosolic (37). These mutants were compared to wild-type (wt) HDAC4 and HDAC5, a class II deacetylase that is highly related to HDAC4.
FIG. 1.

Subcellular localization of HDAC4 and HDAC5 in E1A-transformed IMR90 cells. (A) Schematic representation of HDAC4, HDAC5, and HDAC4 mutants used in this study. Like HDAC5, HDAC4 consists of an N-terminal regulatory region, a catalytic domain (marked “deacetylase”), and a C-terminal NES sequence. Within the N-terminal regulatory region, there are crucial serine residues (i.e., S246, S467, and S632) for phosphorylation and 14-3-3 binding. Also indicated in this region are a transcription factor (TF) docking site, aspartic acid (D) 289 for caspase cleavage, and lysine (K) 559 for sumoylation. (B) Subcellular localization of HDAC4 and HDAC5. Nuclei of E1A-expressing IMR90 cells were microinjected with plasmids expressing HDAC4 or its mutants as GFP fusion proteins, and 2 h later cells were fixed to visualize GFP signals by confocal microscopy. For inhibition of nuclear export, cells were grown for an additional 2 h in the presence of ratjadone A (Ratj.) (5 ng/ml). Approximately 300 cells, derived from three independent experiments, were scored for the quantitative analysis shown in each diagram. Data represent arithmetic means ± standard deviations. PKD, protein kinase D; Nuc, nuclear, cyto, cytosolic.
HDAC4 oligomerizes when ectopically expressed and can form aggregates of different sizes that could interfere with nucleocytoplasmic shuttling. To overcome this problem, the different mutants fused to monomeric GFP were microinjected into the nuclei of IMR90-E1A-transformed cells (E1A cells), and their subcellular localization was analyzed soon after. The subcellular localization was evaluated only in the cells that expressed low levels of the fusion proteins with a homogenous pattern of GFP staining.
As shown in Fig. 1B, all mutants showed the expected subcellular localization. In particular, only ∼10% of the cells showed wt HDAC4 or the D289A mutant exclusively in the nuclear compartment, whereas HDAC5 was exclusively nuclear in ∼80% of the cells. These results suggest that in E1A-transformed IMR90 cells, HDAC4, but not HDAC5, undergoes active nuclear export. Indeed, nuclear accumulation of HDAC4 and mutant D289A was observed following inhibition of the CRM1-dependent nuclear export. Furthermore, the nuclear export signal (NES) mutant L1062A was observed with almost an exclusively nuclear localization (∼100% of cells).
The triple mutant TM showed nuclear localization in ∼70% of the cells, suggesting that nuclear export can still occur, albeit with low efficiency. Surprisingly, the carboxy-terminal fragment ΔN, which lacks the nuclear localization signal (NLS) and the transcription factor-binding site (Fig. 1A), displayed a partial relocalization to the nucleus in the presence of the CRM1 inhibitor (Fig. 1B).
Differences in the apoptotic activity of nuclear HDAC4 mutants.
When we investigated the apoptotic potential of the different mutants, distinctions emerged, particularly among the various nuclear forms of HDAC4 (Fig. 2A). wt HDAC4 triggered apoptosis in ∼19% of the cells. A similar score was obtained for the caspase-uncleavable mutant D289A, indicating that HDAC4 killer activity is independent of the proteolytic processing at Asp289. The ΔC mutant induces cell death in ∼29% of the cells, whereas both L1062A and TM mutants were less potent and triggered apoptosis in ∼16 and ∼12% of the transfected cells, respectively. Spontaneous apoptosis occurring in response to transfection varied between 5 and 10% in IMR90-E1A cells. The different apoptotic potential of the HDAC4 mutants cannot be ascribed to a variation in their expression levels. Indeed, as illustrated in Fig. 2B, all of the HDAC4 mutants were expressed at comparable levels.
FIG. 2.

Apoptotic activity of HDAC4 mutants. (A) β-Gal, HDAC4, and its mutants were transiently expressed as indicated in IMR90-E1A cells using the vector pFLAG-CMV5 (2 μg each). pEGFP-N1 (0.1 μg) was used as the reporter. Forty-eight hours posttransfection, apoptotic cells were scored based on morphology changes. Cells showing a collapsed shape and presenting extensive membrane blebbing were scored as apoptotic. Data represent arithmetic means ± standard deviations for three independent experiments. (B) β-Gal, HDAC4, and its mutants were expressed as described for panel A. Cell lysates were prepared for immunoblotting with anti-Flag and anti-GFP antibodies as indicated. (C) β-Gal, HDAC4, and its mutants were expressed as described for panel A, except that pEGFP-N1 was replaced with pEGFP-caspase-3 (0.2 μg) to assess caspase-3 processing. Cell lysates were prepared for immunoblotting as described for panel B. (D) Densitometric analysis of immunoblots similar to that shown in panel C. The percentage of caspase-GFP maturation was calculated as the ratio between the proform and the cleaved form of caspase-3. Data represent arithmetic means ± standard deviations for three independent experiments.
The differences in the apoptotic response were confirmed by scoring for caspase-3 activation. The caspase-3-GFP fusion was coexpressed with HDAC4 and its different mutants. The appearance of the small fragment of caspase-3 fused to GFP was used as an indicator for proenzyme activation (23). Figure 2C confirms the robust proapoptotic effect of HDAC4/ΔC compared to that of the nuclear L1062A and TM mutants. Densitometric analysis of the caspase-3/GFP-positive bands (Fig. 2D) proved that mutant ΔC promoted ∼35% of caspase-3 processing, whereas the L1062A and TM mutants promoted proenzyme maturation to a much lower extent, ∼11 and ∼6%, respectively.
To confirm that nuclear accumulation of HDAC4 is not sufficient to trigger cell death, we analyzed the cell-death-promoting activity of HDAC5, which, in contrast to HDAC4, is largely nuclear in IMR90-E1A cells and is less susceptible to caspase-3 processing (37). As illustrated in Fig. 3A, HDAC5 showed a weak proapoptotic effect, even less than that of wt HDAC4, which is largely cytosolic. The weak proapoptotic activity of HDAC5 was verified by probing caspase-3-GFP processing (Fig. 3C and D). Less than 10% of caspase-3 was activated in HDCA5-expressing cells. In contrast, more than 30% of caspase-3 activation was observed when the ΔC mutant was expressed. Curiously, HDAC5 and the TM mutant of HDAC4 were similarly impaired in their proapoptotic activities. Taken together, these results demonstrate that the nuclear accumulation of HDAC4 is not sufficient to induce cell death.
FIG. 3.

Analysis of the apoptotic activity of HDAC5 in IMR90-E1A cells. (A) Plasmids expressing β-Gal, HDAC5, HDAC4, and the mutant ΔC were derived from pFLAG-CMV5 and were transfected (2 μg each) into IMR90-E1A cells together with pEGFP-N1 (0.1 μg) as a reporter. The appearance of apoptotic cells was scored 48 h after transfection. Cells with a collapsed morphology and extensive membrane blebbing were scored as apoptotic. Data represent arithmetic means ± standard deviations for three independent experiments. (B) Expression constructs for β-Gal, HDAC5, HDAC4, and mutant ΔC (2 μg) were transfected into E1A cells together with pEGFP-N1 (0.1 μg) as a control for transfection efficiency. Cell lysates were prepared for Western immunoblot analysis with anti-FLAG and anti-GFP antibodies. (C) The transfection was performed as described for panel B, except that pEGFP-N1 was replaced with pEGFP-caspase-3 (0.2 μg) to score caspase-3 processing. Cell lysates were prepared and analyzed as described for panel B. (D) Densitometric analysis of the immunoblots shown in panel C. The percentage of caspase-GFP maturation was calculated as the ratio between the proform and the cleaved caspase. Data represent arithmetic means ± standard deviations for three independent experiments.
Repressive activities of the HDAC4 mutants on MEF2C- or Runx2-dependent transcription.
Members of the MEF2 family are well-known targets of the HDAC4 repressive activity (32, 51, 55). Depending on the cell type, MEF2 members play important roles in the control of differentiation, survival, and apoptosis (22, 32). Hence, we decided to explore whether the variations in the proapoptotic activities of the HDAC4 mutants reflect differences in the magnitude of the MEF2C transcriptional repression. With the exception of the ΔN mutant, which is defective in MEF2C binding, the wt protein and all the tested mutants strongly repressed MEF2C transcription in a dose-dependent manner (Fig. 4A). Most importantly, the ΔC mutant, which shows a strong proapoptotic activity, was less repressive than the nuclear-localized mutant TM, which weakly induces cell death. As verified by immunoblotting, MEF2C and all the tested HDAC4 forms were expressed at comparable levels (data not shown). HDAC5 led to repression with a dose-dependent profile undistinguishable from that of wt HDAC4 or the TM mutant (data not shown). Overall, these experiments indicate that there is no correlation between the strong proapoptotic activity of the caspase-generated HDAC4 fragment and the repression of MEF2C-dependent transcription.
FIG. 4.

Repression of MEF2C- and Runx2-dependent transcription. (A) IMR90-E1A cells were transfected with the 3× MEF2-Luc luciferase reporter (1 μg), the internal control luciferase reporter pRL-CMV (20 ng), pcDNA3.1-HA-MEF2C (1 μg), and increasing amounts (8, 45, 40, and 400 ng) of pFLAG-CMV5 expressing HDAC4 or the indicated mutant. Four hundred nanograms of pFLAG-CMV5 expressing β-Gal was used as a reference, and empty pFLAG-CMV5 was used to normalize the total amount of transfected DNA. Cells were lysed 24 h after transfection. Data represent arithmetic means ± standard deviations for three independent experiments. (B) IMR90-E1A cells were transfected with the 6× OSE2-Luc luciferase reporter (1 μg), pRL-CMV (20 ng), pCMV-Runx2 (1 μg), and increasing amounts (0.12, 0.4, 1.2, and 3.2 μg) of a pFLAG-CMV5-derived vector expressing HDAC4 or the indicated mutant. A volume of 3.2 μg of pFLAG-CMV5 expressing β-Gal was used as a reference, and empty pFLAG-CMV5 was used to normalize the total amount of transfected DNA. Cells were lysed 24 h after transfection. Data represent arithmetic means ± standard deviations for three independent experiments. (C) In vitro binding properties of the different HDAC4 mutants. GST-MEF2C, GST-Runx2, and GST alone (as a control), immobilized on glutathione-Sepharose beads, were incubated with the in vitro-translated products of the indicated HDAC4 constructs. After being washed, proteins bound to the beads were evaluated by SDS-PAGE.
Surprisingly, dose-dependent repressive studies also showed that wt HDAC4, which is exported from the nucleus, repressed MEF2C transcription in a manner similar to that of the nuclearly localized mutants. Since MEF2C can promote the nuclear accumulation of HDAC4 (5), it is possible that in IMR90-E1A cells, once the complex between the deacetylase and the transcription factor has been formed, the activity of specific kinases (55), including Ca2+/calmodulin-dependent protein kinases (CaMKs), is not sufficient to promote HDAC4 nuclear export. Therefore, MEF2C transcription also is efficiently repressed by wt HDAC4.
To address this issue, we investigated the localization of HDAC4 when MEF2C and CaMK were coexpressed in IMR90-E1A cells. Figure S1A in the supplemental material illustrates the triple immunofluorescent assay used to visualize HDAC4, MEF2C, and active CaMKIV or inactive CaMKIV in the same cells. The quantitative analysis (see Fig. S1B in the supplemental material) confirmed that MEF2C provokes the nuclear accumulation of HDAC4. By increasing the level of the active CaMKIV but not that of the inactive kinase, HDAC4 nuclear export is stimulated and MEF2C-dependent transcription is activated (see Fig. S1C in the supplemental material). Hence, these data suggest that CaMKIV levels in E1A cells could dictate the repressive imprinting of HDAC4 on MEF2C.
HDAC4 can bind to various transcription factors and modulate their activities. Runx2 recently has been identified as an important HDAC4 target implicated in osteoblast differentiation (25, 50). Therefore, we investigated whether differences in the repressive activities of the HDAC4 mutants could be observed in the case of Runx2. Dose-dependent repressive studies highlighted that, in contrast to MEF2C, Runx2 was more efficiently repressed by HDAC4/ΔC than HDAC4/TM or the other HDAC4 nuclear forms (Fig. 4B). As verified by immunoblotting, Runx2 and all the tested HDAC4 forms were expressed at comparable levels (data not shown). As already reported (25, 50), when higher doses of the carboxy-terminal fragment of HDAC4 were expressed, residual repression on Runx2 activity was observed. Also in the case of Runx2, HDAC5 showed a dose-dependent repression profile undistinguishable from that of the TM mutant (data not shown).
To determine whether the differential repression activity of HDAC4/ΔC on MEF2C- and Runx2-dependent transcription stems from differences in the binding to the transcription factors, we performed GST fusion protein pulldown experiments.
wt HDAC4 and the indicated mutants (Fig. 4C) were synthesized in vitro and 35S labeled in a TNT-coupled rabbit reticulocyte lysate system. Next, the glutathione beads prebound with GST, GST-MEF2C, or GST-Runx2 were incubated with the radiolabeled HDAC4 proteins. The pulldown experiments confirmed that (i) the amino-terminal region of HDAC4 interacts with the transcription factors, and (ii) the interaction between HDAC4 and Runx2 is weaker than that between HDAC4 and MEF2C. Importantly, HDAC4/ΔC, which represses MEF2C less efficiently than wt HDAC4, strongly interacts with the transcription factor. Overall, these data suggest that the differential repressive influence of the caspase-cleaved HDAC4 fragment on MEF2C and Runx2 with respect to the wt or the TM mutant cannot be ascribed to differences in the binding affinity for the transcription factors.
Repressive activities of HDAC4 mutants on SRF-dependent transcription.
SRF is another MAD-box protein directly or indirectly modulated by HDAC4 (7, 13). SRF binds to the SRE element (CArG box), which is associated with a variety of genes involved in the control of proliferation, survival, and muscle and neuronal functions (9). To substantiate the differential effects of HDAC4 mutants on MEF2C- and Runx2-dependent transcription, we analyzed the repressive activities of HDAC4 mutants on SRF-dependent transcription.
During mouse embryogenesis, SRF plays a pivotal role in controlling cell survival. An important target of the SRF prosurvival activity is the antiapoptotic gene bcl-2 (41). Hence, we explored whether the bcl-2 promoter could be used as a readout to study HDAC4-dependent repression.
Transcription of the bcl-2 gene is under the control of two promoters, P1 and P2, which are subjected to intense regulation (44, 54, 56). In contrast to the mouse bcl-2 gene, only one SRF binding site is present in the P2 promoter of the human bcl-2 (Fig. 5A) (42).
FIG. 5.

Repression of SRF-dependent transcription. (A) Schematic representation of the Bcl-2 promoter and of the fragments used in this study. Arrows depict the transcription starting sites from the P1 and P2 promoters. The binding sites for SRF and some regulatory elements are shown. (B) IMR90-E1A cells were transfected with the LB124, LB335, or LB332 reporter (1 μg) and a pFLAG-CMV5-derived β-Gal expression plasmid (0.4 μg) or a pFLAG-CMV5-based vector expressing HDAC4. A pCGN vector expressing the wt form of SRF (SRF-WT) (1 μg) or its deleted derivative lacking the transactivation domain (SRF-DN) (1 μg) also was cotransfected. pRL-CMV (20 ng) was included to normalize the transfection efficiency. Data represent arithmetic means ± standard deviations for three independent experiments. (C) IMR90-E1A cells were transfected with the LB335-Luc reporter (1 μg), pRL-CMV (20 ng), pCGN-SRF-WT (1 μg), and increasing amounts (0.2, 0.4, 0.8, and 1.6 μg) of a pFLAG-CMV5-based vector expressing HDAC4 and the indicated mutants. A volume of 1.6 μg of pFLAG-CMV5 expressing β-Gal was used as a reference, and empty pFLAG-CMV5 was used to normalize the total amount of transfected DNA. Cells were lysed 24 h after transfection. Data represent arithmetic means ± standard deviations for three independent experiments. (D) In vitro binding properties of the different HDAC4 mutants. GST-SRF and GST alone (as a control), immobilized on glutathione-Sepharose beads, were incubated with the in vitro-translated products of the indicated HDAC4 constructs. After being washed, proteins bound to the beads were evaluated by SDS-PAGE. (E) IMR90-E1A cells were transfected with the LB335-Luc reporter (1 μg); pRL-CMV (20 ng); a pEXV-derived vector for RhoA-V14, RhoA-Rac1-N17, or β-Gal (3 μg each); and a pFLAG-CMV5 plasmid for expression of the HDAC4 mutant ΔC (100 or 400 ng). pFLAG-CMV5 was used to normalize the total amount of transfected DNA. Cells were lysed 24 h after transfection. Data are shown as fold activation with respect to Rac1-N17-expressing cells. Data represent arithmetic means ± standard deviations for three independent experiments. (F) Regulation of bcl-2 mRNA expression by HDAC4/ΔC. Quantitative RT-PCR analysis was performed to quantify bcl-2 mRNAs. pFLAG-CMV expression plasmids encoding the indicated proteins were transfected in E1A cells, and mRNA was isolated 24 h later. Samples were normalized to GAPDH and hypoxanthine phosphoribosyltransferase. Values represent the means from four independent experiments ± standard deviations. (G) pFLAG-CMV5-HDAC4 (2 μg) was cotransfected into IMR90-E1A cells together with pFLAGCMV5-β-Gal, pGDSV7S-Bcl-2, and a pUSE vector expressing the myristylated constitutively active form (A-Akt) or the K179M inactive form (I-Akt) of Akt1, as indicated. pEGFP-N1 (0.1 μg) was used as the reporter. The appearance of apoptotic cells was scored 48 h after transfection. Cells showing a collapsed morphology and with extensive membrane blebbing were scored as apoptotic. Data represent arithmetic means ± standard deviations for three independent experiments.
Luciferase reporter assays were performed to verify the ability of SRF to modulate bcl-2 transcription in human cells. Transcription from the P2 promoter, but not from the P1 promoter, which does not contain a CArG box, was efficiently induced by ectopically expressed SRF (Fig. 5B). Coexpression of HDAC4 significantly repressed SRF-dependent transcription from the bcl-2 P2 promoter. When the entire bcl-2 promoter (P1 plus P2) was evaluated, SRF still was able to activate transcription, albeit with reduced intensity, as previously reported, possibly due to the presence of negative regulative sites (41, 56). HDAC4 also repressed the transcriptional activity of SRF when the entire promoter (P1 plus P2) was used. Finally, the dominant-negative mutant of SRF was unable to stimulate transcription from the bcl-2 promoter.
Next, we compared the repressive action of the different HDAC4 mutants by dose-dependent studies. On SRF, the HDAC4 mutants recapitulate a repression profile quite similar to the one described above for Runx2. Namely, the caspase-cleaved amino-terminal proapoptotic fragment of HDAC4 proved to have the strongest SRF-repressive activity, and the carboxy-terminal fragment still maintains a repressive influence (Fig. 5C). Here again HDAC5 repressed SRF activity in a manner similar to that of the HDAC4/TM mutant (data not shown).
GST pulldown experiments were performed next to evaluate the binding between SRF and HDAC4. Figure 5D shows that SRF, similarly to Runx2, weakly interacts with the amino-terminal region of HDAC4 (compare Fig. 5D to 4C for MEF2C binding).
To corroborate the data on the repressive effect of HDAC4/ΔC on Bcl-2 expression, we evaluated the role of endogenous SRF. Expression of the small GTPase RhoA was used to activate SRF. As shown in Fig. 5E, transcription from the P2 promoter was stimulated when the constitutively active RhoA-V14 was expressed. Furthermore, HDAC4/ΔC repressed RhoA-induced bcl-2 transcription (Fig. 5E). To investigate the physiological relevance, we also assessed the expression levels of endogenous bcl-2 mRNA in E1A cells expressing HDAC4/ΔC, HDAC4/ΔN, or HDAC4/TM or β-galactosidase (β-Gal) using quantitative RT-PCR (Fig. 5F).
HDAC4/ΔC consistently (P < 0.05) repressed Bcl-2 mRNA expression, which was not the case for expression with β-Gal or HDAC4/ΔN. The incomplete repression effect of ΔC probably reflects the limited contribution of the P2 promoter to bcl-2 transcription in E1A cells.
Having suggested a role for an HDAC4 amino-terminal fragment in the control of Bcl-2 expression, we next investigated whether Bcl-2 could efficiently repress apoptosis induced by the ΔC mutant. In E1A cells, we coexpressed ΔC together with bcl-2 or with another antiapoptotic gene, such as Akt, for comparison. Bcl-2 efficiently suppressed ΔC-induced cell death, whereas a constitutively active form of Akt was much less efficient (Fig. 5G). This result implies that the repression of Bcl-2 expression could be relevant in the apoptotic response elicited by ΔC.
FRAP measurement of HDAC4 interaction dynamics in living cells.
Our studies have indicated that caspase processing of HDAC4 has two main effects: (i) it promotes the nuclear accumulation of the amino-terminal fragment, and (ii) it strengthens the repressive influence of HDAC4 on Runx2- and SRF-dependent transcription and alleviates the repression on MEF2C.
In vitro binding experiments have not clarified how this switch in the repressive activities could be achieved. Considering that HDAC4 is part of multiprotein complexes, in vitro studies could be limiting with respect to the in vivo complexity. Hence, to clarify the repressive peculiarities of the different HDAC4 forms, we decided to study their dynamics of interaction with chromatin in vivo.
FRAP largely has been used to investigate dynamic molecular interactions of proteins in living cells, including their binding to chromatin (46). FRAP studies have been instrumental in discovering that chromatin proteins show a high rate turnover on chromatin and that the dynamics of protein binding are responsive to extracellular signals (27, 38, 39, 40).
We performed FRAP studies to evaluate HDAC4 binding to chromatin and to understand if specific posttranslation modifications (i.e., caspase cleavage or dephosphorylation of the 14-3-3-binding sites) could control the turnover of HDAC4 repressive complexes onto chromatin in vivo. To exclude cell type restrictions, experiments were carried out both with E1A cells and with the osteosarcoma cell line U2OS. The different monomeric GFP-fused constructs were expressed by microinjection, and FRAP experiments were performed 2 or 4 h later, at which time cells were treated with ratjadone. Only cells presenting a homogenous distribution of the GFP were selected for the analysis. Typical examples of the cells used in the FRAP studies are shown for each experiment (Fig. 6 and 7). The irreversibility of our experimental settings for photobleaching was verified by analyzing fluorescent recovery in fixed cells (data not shown).
FIG. 6.

Determination of HDAC4 trafficking by FRAP analysis. (A and B) Subcellular localization of HDAC4 in U2OS and IMR90-E1A cells. IMR90-E1A (A) or U2OS (B) cells received nuclear microinjections with plasmid encoding GFP-HDAC4, and 2 h later cells were fixed for microscopic analysis. Subcellular localization of GFP fusion proteins was visualized by confocal microscopy. For inhibition of nuclear export, cells were grown for 2 h in the presence of ratjadone A (5 ng/ml). Approximately 300 cells, from three independent experiments, were scored for the quantitative analysis reported in the diagrams. Data represent arithmetic means ± standard deviations. (C and D) FRAP analysis was performed on IMR90-E1A (C) or U2OS cells (D) expressing GFP, NLS-GFP, or GFP-HDAC4. FRAP on HDAC4 was carried out in the presence or absence of ratjadone A (Ratj) (5 ng/ml). The recovery profiles represent the average profiles of individually photobleached cells, as indicated. The intracellular distribution of different GFP fusion proteins is shown in the upper part. Nuc, nuclear; Cyto, cytoplasmic.
FIG. 7.

FRAP analysis of different nuclear proteins. (A and B) FRAP analysis was performed with IMR90-E1A (A) or U2OS (B) cells expressing GFP-HDAC4, GFP-MEF2C, or GFP-H1.2. FRAP on GFP-HDAC4 was carried out in the presence or absence of ratjadone A (Ratj) (5 ng/ml). The recovery profiles represent the average profiles of individually photobleached cells, as indicated. The distribution of the different GFP fusion proteins is shown in the upper part. (C and D) FRAP analysis was performed with IMR90-E1A (C) or U2OS (D) cells expressing GFP fused with HDAC4, HDAC5, and HDAC4 mutants ΔC, L1062A, and TM. FRAP on HDAC4 was carried out in the presence of ratjadone A (5 ng/ml). The recovery profiles represent the average profiles of individually photobleached cells, as indicated. The distribution of the different GFP proteins is shown in the upper part.
Initially we compared the subcellular localization of wt HDAC4 in the two cell lines. Figure 6A and B illustrate that in both cell lines HDAC4 is actively exported from the nucleus in a CRM1-senstive manner. In the U2OS cell line, ∼95% of cells accumulate HDAC4 exclusively in the cytoplasm, whereas in E1A this percentage decreases to ∼45%. These results suggest that HDAC4 nuclear export is more effective in U2OS than in E1A cells.
Figure 6C depicts the kinetics of wt HDAC4 recovery in the cytoplasm and when it is present in the nucleus, following inhibition of the CRM1-mediated nuclear export. GFP alone and a GFP derivative, which contains an NLS (NLS-GFP), exemplified inert nonbinding proteins. In E1A cells, the time required to reach 80% of the maximal fluorescence intensity recovery for cytosolic HDAC4 (t80) was 5.6 s (Table 1). Although very rapid, this kinetic of recovery was slower than the one showed by GFP, with a t80 of 0.9 s. These data suggest that HDAC4 is subjected to binding events in the cytoplasm and that these interactions are highly dynamic. In FRAP experiments performed after ratjadone treatment of nuclear HDAC4, the observed t80 increased to 7.8 s. Again, this kinetics of recovery is rapid, but compared with the t80 of the NLS-GFP, which is 0.9 s (Table 1), it is evocative of binding events, possibly to chromatin (P < 0.001).
TABLE 1.
Recovery kinetics of wt and mutant HDAC4 in E1A and U2OS cellsa
| Protein |
t80 in E1A cells
|
t80 in U2OS cells
|
||
|---|---|---|---|---|
| Time(s) ± SD | P value | Time(s) ± SD | P value | |
| HDAC4+R | 7.8 ± 1.7 | NA | 4.8 ± 1.6 | NA |
| HDAC4-cyto | 5.6 ± 2.5 | <0.001 | 3.0 ± 1.8 | <0.001 |
| HDAC5 | 21.1 ± 9.6 | <0.001 | 14.2 ± 7.9 | <0.05 |
| MEF2C | 8.1 ± 2.9 | 0.77 | 5.6 ± 2.5 | 0.27 |
| HDAC4/TM | 15.2 ± 2.9 | <0.001 | 12.6 ± 2.3 | <0.001 |
| HDAC4/ΔC | 5.3 ± 1.9 | <0.001 | 3.5 ± 1.6 | <0.05 |
| HDAC4/L1062A | 7.1 ± 2.6 | 0.45 | 5.3 ± 2.0 | 0.45 |
| HDAC4-nucl | 15.6 ± 3.0 | <0.001 | ||
| GFP | 0.9 ± 0.6 | <0.001 | 1.0 ± 0.5 | <0.001 |
| NLS-GFP | 0.9 ± 0.3 | <0.001 | 1.4 ± 0.7 | <0.001 |
| Histone H1.2 | 60.6 ± 18.2 | <0.001 | 78.2 ± 19.2 | <0.001 |
P values were determined by the Student's t test. NA, not applicable; R, ratjadone A; cyto, cytoplasmic.
The results from U2OS cells were quite similar to the ones obtained from E1A cells, even though the recovery curves of nuclear HDAC4 (after ratjadone treatment) display differences (compare Fig. 6C and D), with a faster recovery in U2OS cells. However, in U2OS cells, an increase of the nuclear mobility also was noted for MEF2C (Table 1; also, see below), thus suggesting the existence of a more general difference between the two cell lines.
FRAP experiments reveal two different populations of nuclear HDAC4.
The FRAP studies next were focused to define in detail the nuclear mobility of the different HDAC4 mutants. Initially, we compared the recovery of HDAC4 in ratjadone-treated cells to the recovery of two proteins that directly bind the DNA, although with different dynamics: the transcription factor MEF2C and the linker histone H1, subtype 1.2. Furthermore, for E1A cells, FRAP experiments also were performed with a few of the cells that accumulate HDAC4 in the nucleus in the absence of the block in the nuclear export produced by the ratjadone treatment (HDAC4-nucl).
The resulting FRAP recovery profiles are represented in Fig. 7A. Although H1.2 is the histone most weakly bound to chromatin (48), the interaction is relatively stable compared to that of MEF2C, which shows high mobility, which is common to many transcription factors (39). The t80 for H1.2 was almost 1 min, whereas for MEF2C it was 8.1 s, which is similar to the 7.8 s for HDAC4 in ratjadone-treated cells (the recovery curves are indistinguishable from each other). A similar behavior was observed in U2OS cells; again, the recovery curves for MEF2C and HDAC4 (after ratjadone treatment) are indistinguishable (Fig. 7B).
Importantly, the recovery of the nuclear wt HDAC4 (t80 of 15.6 s), which probably does not bind the 14-3-3 proteins and hence is not phosphorylated in the critical serine residues, is slower than that of nuclear HDAC4 in ratjadone-treated cells.
FRAP experiments performed on HDAC4 mutants and on HDAC5 showed that the recovery curves can be grouped into two distinct clusters. In the first cluster we can group wt HDAC4 after ratjadone treatment, HDAC4 L1064A, and the caspase-cleaved form, HDAC4/ΔC. They are characterized by similar fast recovery kinetics, with t80s from 5.3 s for HDAC4/ΔC to 7.8 s for HDAC4 in ratjadone-treated cells.
HDAC5 and HDAC4/TM can be grouped into the second cluster. They show slow recovery curves, with t80s from 15.2 s for HDAC4/TM to 20.9 s for HDAC5. The existence of two distinct clusters grouping the recovery curves of HDAC5, HDAC4, and its nuclear mutants also was confirmed in U2OS cells (Fig. 7D and Table 1). Importantly, when accumulated in the nucleus of E1A cells in the absence of ratjadone treatment, wt HDAC4 also can be included in the second cluster, as it is distinguished by slow kinetics of recovery (Fig. 7A and Table 1).
FRAP model-based data analysis.
Different mathematical models have been proposed over time for the quantitative analysis of FRAP data. Among them, compartmental models have gained great popularity because of their simplicity in formulation and analysis and their effectiveness in describing observed dynamics. In the present work, we adopt a two-binding-state compartmental model including diffusion. Basically, our model is a variant of the compartmental model proposed by Phair and coauthors (39), to which we have added a diffusion process. Unbound molecules can diffuse to and from the bleach spot; moreover, they eventually can bind into a complex form, but bound molecules are no longer subject to diffusion. A similar model (but for a single binding site and a strip-shaped bleached region) was described previously (8). The model is described by a set of ordinary differential equations with constant coefficients, the forms of which come from standard chemical kinetic principles (8, 39, 47), and contain six parameters: the bleach spot/nuclear area ratio, a diffusion coefficient, and two binding-unbinding association rate constants for each binding site. Still, before putting the model to work, we must face drawbacks due to the complexity and our uncertainties of the underlying phenomena.
Indeed, conventional hypotheses behind FRAP modeling are barely fulfilled in practice, notably, the assumption that immediately after the bleaching no fluorescent molecules are present in the bleach spot. Furthermore, using realistic initial conditions for the differential equations, the theoretical FRAP curve may contain too many terms to be fitted in a reliable way. In fact, coefficients in the closed-form exact solution are very sensitive to uncertainties in dynamic parameters and initial conditions. Hence, data errors and noise may lead to meaningless or unreliable parameter values. Moreover, some terms in the exact solution may have little relevance to the theoretical FRAP curve.
For these reasons, based on our knowledge of the underlying molecular dynamics and on the existing literature, we empirically made a couple of simplifications to the standard compartmental model (see Material and Methods).
Using this approach, the mathematical simulations produced a very good approximation of the experimental data, and the obtained parameters can again be grouped into two distinct clusters. Table 2 summarizes the data obtained for the slow fractions from both E1A and U2OS cell lines.
TABLE 2.
Kinetics properties of wt and mutant HDAC4 in the nuclei of E1A and U2OS cells
| Protein | E1A cells
|
U2OS cells
|
||||
|---|---|---|---|---|---|---|
| koff,2 | Mean residence time(s) | % Bound proteins (±SD) | koff,2 | Mean residence time(s) | % Bound proteins (±SD) | |
| Histone H1.2 | 0.0414 ± 0.029 | 24.1 | 52.0 ± 11.7 | 0.0322 ± 0.009 | 32.0 | 67.1 ± 10.1 |
| HDAC4+R | 0.1282 ± 0.030 | 7.9 | 40.0 ± 8.8 | 0.2041 ± 0.047 | 5.0 | 40.3 ± 9.1 |
| HDAC5 | 0.0851 ± 0.043 | 11.8 | 51.0 ± 12.9 | 0.0888 ± 0.045 | 11.3 | 55.9 ± 8.9 |
| MEF2C | 0.1627 ± 0.073 | 6.1 | 47.5 ± 15.8 | 0.1807 ± 0.062 | 5.5 | 43.4 ± 12.4 |
| HDAC4/TM | 0.1037 ± 0.032 | 9.7 | 59.1 ± 10.6 | 0.0972 ± 0.035 | 10.2 | 51.9 ± 13.0 |
| HDAC4/ΔC | 0.1954 ± 0.088 | 5.1 | 37.1 ± 10.0 | 0.2233 ± 0.086 | 4.5 | 31.4 ± 12.0 |
| HDAC4/L1062A | 0.1161 ± 0.041 | 8.6 | 36.5 ± 13.2 | 0.1584 ± 0.037 | 6.3 | 38.6 ± 11.3 |
Based on the mean residence time on chromatin and the percentage of bound proteins, the first cluster includes HDAC4 in ratjadone-treated cells, HDAC4/ΔC, and HDAC4 L1064A. In E1A and U2OS cells, the mean residence time for these proteins is 5.1 to 8.6 s and 4.5 to 6.3 s, respectively. The percentage of bound protein ranges from 36.5 to 40% in E1A cells and from 31.4 to 40.3% in U2OS cells.
The second cluster includes HDAC4/TM and HDAC5, and both bind chromatin for longer time periods and at higher percentages. The mean residence times for HDAC4/TM and HDAC5 were 9.7 and 11.7 s in E1A cells and 10.2 and 11.3 s in U2OS cells, respectively. The percentage of HDAC4/TM showing strong binding was 59.1 and 51.9% in E1A and U2OS cells, respectively, and for HDAC5 it was 51 and 55.9%, respectively.
Our confidence in the mathematical method is reinforced by the fact that FRAP curves for GFP and NLS-GFP, having a predictably lower affinity for chromatin than other bona fide chromatin-binding proteins used in our experiments, exhibit a negligible reaction-dominant behavior and higher effective diffusion coefficients (data not shown).
In summary, FRAP analysis indicates that specific posttranslational modifications generate different nuclear populations of HDAC4, characterized by different repressive influences and different kinetics of binding to chromatin.
DISCUSSION
The control of HDAC4 subcellular localization plays a pivotal role in modulating gene expression in response to specific signals that regulate cell growth, differentiation, and apoptosis. In many cell types, HDAC4 is continuously exported from the nucleus into the cytoplasm (1, 21, 29, 33, 34, 52, 53, 57, 59). Nucleocytoplasmic shuttling of HDAC4 is regulated by specific signals through different posttranslation modifications, such as phosphorylation and proteolytic processing (Fig. 8). Hence, specific posttranslational modifications of HDAC4 act as molecular switches to directly target cells toward different programs, including growth, differentiation, and apoptosis.
FIG. 8.

Illustration of two different modes of HDAC4 regulation by nucleocytoplasmic shuttling. Specific phosphorylation of HDAC4 by kinases such as CaMKs, PKDs, and c-TAK promotes the binding to 14-3-3 proteins and cytoplasmic localization. Dephosphorylation then promotes nuclear localization (mode 1). HDAC4 also is subject to caspase cleavage, and the N-terminal fragment then translocates to the nucleus for transcriptional repression (mode 2). While mode 1 is reversible, mode 2 is irreversible. Mode 2 may operate during apoptosis, and mode 1 may be important for controlling cell differentiation or other cellular programs. PKD, protein kinase D.
Dephosphorylation and caspase processing of HDAC4 differentially affect its repressive influence.
The aim of this study was to understand the mechanisms through which HDAC4 controls cell death. To tackle this issue, we have taken advantage of a series of HDAC4 mutants. In particular, two HDAC4 nuclear mutants, evocative of posttranslation modifications controlled by extracellular signals, were instrumental for this study (37, 52). One of them is the amino-terminal fragment ΔC, which mimics caspase cleavage, whereas the other one is the TM point mutant, which mimics the dephosphorylation of the 14-3-3 docking sites.
These two mutants diverge profoundly in their ability to elicit apoptosis. The ΔC mutant clearly triggered cell death and caspase activation, whereas the TM point mutant only modestly affected cell survival. These pieces of evidence clearly indicate that nuclear accumulation of HDAC4 per se is not sufficient to trigger apoptosis, as confirmed by the weak proapoptotic effect of HDAC5, which is mostly nuclear.
HDAC4 can target different transcription factors to orchestrate distinct genetic programs (13, 26, 36, 50, 51, 55). Our studies on the repression influence of HDAC4 have been focused on MEF2C, Runx2, and SRF, three well-characterized HDAC4 partners. Dose-dependent analyses have revealed that the caspase-cleaved form ΔC represses Runx2 and SRF activities more efficiently than the TM mutant, which is more powerful against MEF2C. Hence, caspase-dependent processing of HDAC4 seems to be relevant to improve the repression of SRF and Runx2 activities.
All of the investigated transcription factors can be involved in the control of survival. In fact, ectopic expression of dominant-negative forms for these transcription factors promptly induced cell death in our cells (data not shown). Transgenic mice studies (3) have demonstrated that Myc/Runx2-expressing lymphomas show low rates of apoptosis, in contrast to lymphomas induced solely by Myc, which are highly prone to apoptosis. Moreover, the Runx2/Myc combination overcomes the requirement for genetic inactivation of the p53 pathway (3). Runx2 also is involved in the parathyroid hormone-dependent suppression of apoptosis observed in cultured osteoblasts (2).
Under various circumstances, SRF also has been implicated in the control of cell survival (9). In particular, SRF survival activity is critical during early development. An important target of the prosurvival effect of SRF is the antiapoptotic gene bcl-2 (41). We have confirmed that SRF regulates Bcl-2 expression from the P2 promoter in oncogene-transformed cells and demonstrated that HDAC4 represses this activation. Moreover, similarly to Runx2, the caspase-cleaved fragment of HDAC4, the ΔC mutant, was the most efficient repressor of SRF activity.
P1 is the predominant promoter of the bcl-2 gene in B cells, whereas in neuronal cells bcl-2 transcription is preferentially initiated from the P2 promoter (44). Previous studies showed that nuclear accumulation of HDAC4 in neuronal cells plays a critical role during apoptosis induced by specific stimuli (4). In this study, we have demonstrated the ability of HDAC4 to repress bcl-2 transcription and the antagonizing role of Bcl-2 against HDAC4-induced cell death. Together, these observations point to Bcl-2 as a relevant target of HDAC4 proapoptotic activity.
HDAC4 may repress SRF transcription by direct binding (13) and/or indirectly through the interaction with SRF partners such as myocardin (7). We have not investigated this point. In spite of this, we noted that (i) SRF, like Runx2 and in contrast to MEF2C, is weakly bound by HDAC4, and (ii) the carboxy-terminal fragment of HDAC4 represses SRF and Runx2 but not MEF2C. In conclusion, large bodies of evidence indicate that the repressive influence of HDAC4 on SRF and Runx2 is based on mechanisms that diverge from those involved in the repression of MEF2C.
Mechanisms for nucleocytoplasmic trafficking of HDAC4.
FRAP has been extensively used to measure the dynamics of binding of nuclear proteins to chromatin in living cells (27, 38, 39, 40). We have applied this technique to characterize the mobility of HDAC4 and to understand the differences in repressive imprinting of caspase-cleaved fragment compared to that of other nuclear forms of HDAC4.
HDAC4 transiently binds chromatin, and an immobile fraction of the enzyme cannot be found. To provide a quantitative interpretation of the FRAP data, we have developed a two-binding-state compartmental model including diffusion. Our confidence in the described model relies mainly on the excellent fit of the function F(t) to our experimental data (the fit error is well below the level of data noise) and is supported by the fact that FRAP curves for GFP and NLS-GFP have a predictably low affinity with respect to GFP fusions with chromatin-binding proteins, exhibit small values for ρ2, and exhibit high values for λ1 in our experiments, thus leading to the conclusion of an essentially diffusion-driven behavior.
By applying the developed mathematical model, we propose that HDAC4 binds to chromatin with a mean residence time in the order of 4 to 12 s, depending on the introduced mutation. This result is in agreement with previous reports describing transient binding and short residence times for many chromatin-associated proteins (27, 38, 39, 40, 46). In our experimental settings, we have analyzed cells with the lowest levels of GFP fusion expression and with a homogenous distribution of the fluorescence. This selection made us conclude that the described kinetics of recovery illustrate the binding of HDAC4 to chromatin but not to nonchromatin compartments resulting from aggregate formation. This conclusion is confirmed by the fast kinetics of recovery.
Short residence times imply that an incessant flow of the repressive complexes is required to obtain a stable silencing of a genomic site. As a consequence, gene expression can simply be reactivated by competing for binding during dissociation of the HDAC4-containing complexes with specific activator complexes. Hence, transient interactions with chromatin are emerging as the prerequisite for an accurate governing of transcription (39).
The analysis of our FRAP data revealed that two different populations of HDAC4, in terms of mobility, exist in the nuclear compartment. The caspase-cleaved form HDAC4/ΔC, the mutant in the NES sequence, and wt HDAC4 are highly mobile in CRM1-inhibited cells. In contrast, HDAC4/TM, defective in 14-3-3 binding, HDAC5, and nuclear HDAC4, in the few cells where it can be observed in the absence of nuclear export inhibitors, are less mobile. The mathematical fitting of our data to a two-binding-site compartmental FRAP model (8, 39) confirmed these observations. In particular, the two nuclear mutants that recapitulate posttranslational modifications (HDAC4/ΔC and HDAC4/TM) differ profoundly for the time of binding to chromatin (5.1 to 4.5 s and 9.7 to 10.2 s in E1A and U2OS cells, respectively) and for the percentage of protein implicated in the binding at a certain time (37 to 31% and 59 to 52% in E1A and U2OS cells, respectively).
The HDAC4/ΔC mutant is characterized by the absence of a large region of the protein important for the interaction with other corepressors (51, 55). Hence, it is plausible that the differences evidenced by FRAP experiments reflect the formation of different repressive complexes on the DNA. We exclude the possibility that the different mobilities are due to the reduced mass of HDAC4/ΔC, because the diffusion constant and the recovery time for FRAP change extremely slowly as a function of mass (43). Furthermore, the L1062A mutant and wt HDAC4 each have a mass similar to that of HDAC4/TM, but they show fast FRAP recoveries.
FRAP experiments also provide an explanation for the divergences in the repressive influences of HDAC4/ΔC and HDAC4/TM. The carboxy-terminal region of HDAC4 contributes, possibly through the recruitment of other corepressors such as SMRT/N-CoR/HDAC3 (16, 51, 55), to the strengthening of the interactions between the deacetylase and specific genomic regions. Since HDAC4/TM is more potent than HDAC4/ΔC in repressing MEF2C, a more stable interaction with chromatin could be necessary to efficiently repress MEF2C transcription. On the other hand, the more transient binding of the caspase-generated HDAC4 fragment to chromatin could make it more available for interactions with other repression complexes, targeting transcription factors such as Runx2 and SRF, which are important in the induction of apoptosis. A transient binding could be an absolute requirement because of the weaker interaction observed in vitro between HDAC4 and SRF or Runx2 compared to that between HDAC4 and MEF2C.
The understanding of the highly dynamic behavior of the L1062A mutant is an issue not fully resolved. In fact, this mutant is not a strong inducer of apoptosis. To discuss this point, it is important that, when mutated in the NES or under the influence of ratjadone, HDAC4 is accumulated in the nucleus and is accumulated in complex with the 14-3-3 proteins. Although HDAC4 bound to the 14-3-3 proteins could interact with DNA more dynamically, it is also committed for nuclear exporting. Hence, we speculate that nuclear HDAC4 in complex with 14-3-3 (which provides competence for export) cannot repress Runx2 and SRF transcription, as is the case with the amino-terminal fragment. However, it is evident that further studies need to be undertaken to resolve this issue.
Nevertheless, the FRAP experiments on the L1062A mutant describe a new mechanism through which the 14-3-3 chaperones can modulate HDAC class IIa functions (6, 51, 55). Conformational changes operated by 14-3-3 chaperone proteins could regulate the time of residence of HDAC4 on chromatin.
In conclusion, this study sheds new light on regulation of the HDAC4 repressive potential and provides novel insights into its nuclear dynamics related to apoptotic and differentiation signals.
Supplementary Material
Acknowledgments
We thank Linda M. Boxer (Stanford University) for Bcl-2 promoter fragments and Ron Prywes (Columbia University) and Annick Harel-Bellan (CNRS Villejuif) for SRF expression plasmids. We thank G. Karsenty (Columbia University), G. Tell (Università di Udine), and M. Montacino (Universidad de Concepcion) for Runx2 expression plasmids and reporters, A. R. Means (Duke University) for CaMIV kinase-expressing vectors, and S. Goruppi (Tufts University) for Akt plasmids. We also thank E. di Centa (Università di Udine) for help with DNA sequencing.
This work was supported by grants from AIRC (Associazione Italiana Ricerca sul Cancro), MIUR, and Regione FVG.
Footnotes
Published ahead of print on 16 July 2007.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Backs, J., K. Song, S. Bezprozvannaya, S. Chang, and E. N. Olson. 2006. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J. Clin. Investig. 116:1853-1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bellido, T., A. A. Ali, L. I. Plotkin, Q. Fu, I. Gubrij, P. K. Roberson, R. S. Weinstein, C. A. O'Brien, S. C. Manolagas, and R. L. Jilka. 2003. Proteasomal degradation of Runx2 shortens parathyroid hormone-induced anti-apoptotic signaling in osteoblasts. A putative explanation for why intermittent administration is needed for bone anabolism. J. Biol. Chem. 278:50259-50272. [DOI] [PubMed] [Google Scholar]
- 3.Blyth, K., F. Vaillant, L. Hanlon, N. Mackay, M. Bell, A. Jenkins, J. C. Neil, and E. R. Cameron. 2006. Runx2 and MYC collaborate in lymphoma development by suppressing apoptotic and growth arrest pathways in vivo. Cancer Res. 66:2195-2201. [DOI] [PubMed] [Google Scholar]
- 4.Bolger, T. A., and T. P. Yao. 2005. Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J. Neurosci. 25:9544-9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borghi, S., S. Molinari, G. Razzini, F. Parise, R. Battini, and S. Ferrari. 2001. The nuclear localization domain of the MEF2 family of transcription factors shows member-specific features and mediates the nuclear import of histone deacetylase 4. J. Cell Sci. 114:4477-4483. [DOI] [PubMed] [Google Scholar]
- 6.Bridges, D., and G. B. Moorhead. 2004. 14-3-3 Proteins: a number of functions for a numbered protein. Sci. STKE 242:re10. [DOI] [PubMed]
- 7.Cao, D., Z. Wang, C. L. Zhang, J. Oh, W. Xing, S. Li, J. A. Richardson, D. Z. Wang, and E. N. Olson. 2005. Modulation of smooth muscle gene expression by association of histone acetyltransferases and deacetylases with myocardin. Mol. Cell. Biol. 25:364-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrero, G., D. McDonald, E. Crawford, G. de Vries, and M. J. Hendzel. 2003. Using FRAP and mathematical modeling to determine the in vivo kinetics of nuclear proteins. Methods 29:14-28. [DOI] [PubMed] [Google Scholar]
- 9.Chai, J., and A. S. Tarnawski. 2002. Serum response factor: discovery, biochemistry, biological roles and implications for tissue injury healing. J. Physiol. Pharmacol. 53:147-157. [PubMed] [Google Scholar]
- 10.Chang, S., T. A. McKinsey, C. L. Zhang, J. A. Richardson, J. A. Hill, and E. N. Olson. 2004. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol. Cell. Biol. 24:8467-8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang, S., B. D. Young, S. Li, X. Qi, J. A. Richardson, and E. N. Olson. 2006. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell 126:321-334. [DOI] [PubMed] [Google Scholar]
- 12.Cress, W. D., and E. Seto. 2000. Histone deacetylases, transcriptional control, and cancer. J. Cell Physiol. 184:1-16. [DOI] [PubMed] [Google Scholar]
- 13.Davis, F. J., M. Gupta, B. Camoretti-Mercado, R. J. Schwartz, and M. P. Gupta. 2003. Calcium/calmodulin-dependent protein kinase activates serum response factor transcription activity by its dissociation from histone deacetylase, HDAC4. Implications in cardiac muscle gene regulation during hypertrophy. J. Biol. Chem. 278:20047-20058. [DOI] [PubMed] [Google Scholar]
- 14.de Ruijter, A. J., A. H. van Gennip, H. N. Caron, S. Kemp, and A. B. van Kuilenburg. 2003. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 370:737-749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fischer, U., R. U. Janicke, and K. Schulze-Osthoff. 2003. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. 10:76-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischle, W., F. Dequiedt, M. J. Hendzel, M. G. Guenther, M. A. Lazar, W. Voelter, and E. Verdin. 2002. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell 9:45-57. [DOI] [PubMed] [Google Scholar]
- 17.Fontanini, A., R. Chies, E. L. Snapp, M. Ferrarini, G. M. Fabrizi, and C. Brancolini. 2005. Glycan-independent role of calnexin in the intracellular retention of Charcot-Marie-tooth 1A Gas3/PMP22 mutants. J. Biol. Chem. 280:2378-2387. [DOI] [PubMed] [Google Scholar]
- 18.Gregoire, S., and X. J. Yang. 2005. Association with class IIa histone deacetylases upregulates the sumoylation of MEF2 transcription factors. Mol. Cell. Biol. 25:2273-2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gregoretti, I. V., Y. M. Lee, and H. V. Goodson. 2004. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J. Mol. Biol. 338:17-31. [DOI] [PubMed] [Google Scholar]
- 20.Grozinger, C. M., and S. L. Schreiber. 2002. Deacetylase enzymes: biological functions and the use of small-molecule inhibitors. Chem. Biol. 9:3-16. [DOI] [PubMed] [Google Scholar]
- 21.Grozinger, C. M., and S. L. Schreiber. 2000. 2000. Regulation of histone deacetylase 4 and 5 transcriptional activity by 14-3-3-dependent cellular localization. Proc. Natl. Acad. Sci. USA 97:7835-7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heidenreich, K. A., and D. A. Linseman. 2004. Myocyte enhancer factor-2 transcription factors in neuronal differentiation and survival. Mol. Neurobiol. 29:155-166. [DOI] [PubMed] [Google Scholar]
- 23.Henderson, C. J., E. Aleo, A. Fontanini, R. Maestro, G. Paroni, and C. Brancolini. 2005. Caspase activation and apoptosis in response to proteasome inhibitors. Cell Death Differ. 12:1240-1254. [DOI] [PubMed] [Google Scholar]
- 24.Henderson, C., and C. Brancolini. 2003. Apoptotic pathways activated by histone deacetylase inhibitors: implications for the drug-resistant phenotype. Drug Resist. Updat. 6:247-256. [DOI] [PubMed] [Google Scholar]
- 25.Kang, J. S., T. Alliston, R. Delston, and R. Derynck. 2005. Repression of Runx2 function by TGF-β through recruitment of class II histone deacetylases by Smad3. EMBO J. 244:2543-2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khochbin, S., A. Verdel, C. Lemercier, and D. Seigneurin-Berny. 2001. Functional significance of histone deacetylase diversity. Curr. Opin. Genet. Dev. 11:162-166. [DOI] [PubMed] [Google Scholar]
- 27.Laricchia-Robbio, L., T. Tamura, T. Karpova, B. L. Sprague, J. G. McNally, and K. Ozato. 2005. Partner-regulated interaction of IFN regulatory factor 8 with chromatin visualized in live macrophages. Proc. Natl. Acad. Sci. USA 102:14368-14373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu, F., M. Dowling, X. J. Yang, and G. D. Kao. 2004. Caspase-mediated specific cleavage of human histone deacetylase 4. J. Biol. Chem. 279:34537-34546. [DOI] [PubMed] [Google Scholar]
- 29.Liu, Y., W. R. Randall, and M. F. Schneider. 2005. Activity-dependent and -independent nuclear fluxes of HDAC4 mediated by different kinases in adult skeletal muscle. J. Cell Biol. 168:887-897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu, J., T. A. McKinsey, C. L. Zhang, and E. N. Olson. 2000. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol. Cell 6:233-244. [DOI] [PubMed] [Google Scholar]
- 31.Ma, K., J. K. Chan, G. Zhu, and Z. Wu. 2005. Myocyte enhancer factor 2 acetylation by p300 enhances its DNA binding activity, transcriptional activity, and myogenic differentiation. Mol. Cell. Biol. 25:3575-3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKinsey, T. A., C. L. Zhang, and E. N. Olson. 2002. MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem. Sci. 27:40-47. [DOI] [PubMed] [Google Scholar]
- 33.McKinsey, T. A., C. L. Zhang, and E. N. Olson. 2000. Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc. Natl. Acad. Sci. USA 97:14400-14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McKinsey, T. A., C. L. Zhang, and E. N. Olson. 2001. Identification of a signal-responsive nuclear export sequence in class II histone deacetylases. Mol. Cell. Biol. 21:6312-6321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKinsey, T. A., C. L. Zhang, J. Lu, and E. N. Olson. 2000. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408:106-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miska, E. A., C. Karlsson, E. Langley, S. J. Nielsen, J. Pines, and T. Kouzarides. 1999. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J. 18:5099-5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paroni, G., M. Mizzau, Henderson, G. Del Sal, C. Schneider, and C. Brancolini. 2004. Caspase-dependent regulation of histone deacetylase 4 nuclear-cytoplasmic shuttling promotes apoptosis. Mol. Biol. Cell 15:2804-2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Phair, R. D., and T. Misteli. 2000. High mobility of proteins in the mammalian cell nucleus. Nature 404:604-609. [DOI] [PubMed] [Google Scholar]
- 39.Phair, R. D., P. Scaffidi, C. Elbi, J. Vecerova, A. Dey, K. Ozato, D. T. Brown, G. Hager, M. Bustin, and T. Misteli. 2004. Global nature of dynamic protein-chromatin interactions in vivo: three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol. Cell. Biol. 24:6393-6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schaaf, M. J., L. Willetts, B. P. Hayes, B. Maschera, E. Stylianou, and S. N. Farrow. 2006. The relationship between intranuclear mobility of the NF-κB subunit p65 and its DNA binding affinity. J. Biol. Chem. 281:22409-22420. [DOI] [PubMed] [Google Scholar]
- 41.Schratt, G., U. Philippar, D. Hockemeyer, H. Schwarz, S. Alberti, and A. Nordheim. 2004. SRF regulates Bcl-2 expression and promotes cell survival during murine embryonic development. EMBO J. 23:1834-1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shalizi, A., B. Gaudilliere, Z. Yuan, J. Stegmuller, T. Shirogane, Q. Ge, Y. Tan, B. Schulman, J. W. Harper, and A. Bonni. 2006. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science 311:1012-1017. [DOI] [PubMed] [Google Scholar]
- 43.Siggia, E. D., J. Lippincott-Schwartz, and S. Bekiranov. 2000. Diffusion in inhomogeneous media: theory and simulations applied to whole cell photobleach recovery. Biophys. J. 79:1761-1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith, M. D., E. A. Ensor, R. S. Coffin, L. M. Boxer, and D. S. Latchman. 1998. Bcl-2 transcription from the proximal P2 promoter is activated in neuronal cells by the Brn-3a POU family transcription factor. J. Biol. Chem. 273:16715-16722. [DOI] [PubMed] [Google Scholar]
- 45.Snapp, E. L., R. S. Hegde, M. Francolini, F. Lombardo, S. Colombo, E. Pedrazzini, N. Borgese, and J. Lippincott-Schwartz. 2003. Formation of stacked ER cisternae by low affinity protein interactions. J. Cell Biol. 163:257-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sprague, B. L., and J. G. McNally. 2005. FRAP analysis of binding: proper and fitting. Trends Cell Biol. 15:84-91. [DOI] [PubMed] [Google Scholar]
- 47.Sprague, B. L., R. L. Pego, D. A. Stavreva, and J. G. McNally. 2004. Analysis of binding reactions by fluorescence recovery after photobleaching. Biophys. J. 86:3473-3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Th'ng, J. P., R. Sung, M. Ye, and M. J. Hendzel. 2005. H1 family histones in the nucleus. Control of binding and localization by the C-terminal domain. J. Biol. Chem. 280:27809-27914. [DOI] [PubMed] [Google Scholar]
- 49.Timmer, J. C., and G. S. Salvesen. 2006. Caspase substrates. Cell Death Differ. 14:66-72. [Epub ahead of print.] [DOI] [PubMed] [Google Scholar]
- 50.Vega, R. B., K. Matsuda, J. Oh, A. C. Barbosa, X. Yang, E. Meadows, J. McAnally, C. Pomajzl, J. M. Shelton, J. A. Richardson, G. Karsenty, and E. N. Olson. 2004. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 119:555-566. [DOI] [PubMed] [Google Scholar]
- 51.Verdin, E., F. Dequiedt, and H. G. Kasler. 2003. Class II histone deacetylases: versatile regulators. Trends Genet. 19:286-293. [DOI] [PubMed] [Google Scholar]
- 52.Wang, A. H., and X. J. Yang. 2001. Histone deacetylase 4 possesses intrinsic nuclear import and export signals. Mol. Cell. Biol. 21:5992-6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang, A. H., M. J. Kruhlak, J. Wu, N. R. Bertos, M. Vezmar, B. I. Posner, D. P. Bazett-Jones, and X. J. Yang. 2000. Regulation of histone deacetylase 4 by binding of 14-3-3 proteins. Mol. Cell. Biol. 20:6904-6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiang, H., J. Wang, and L. M. Boxer. 2006. Role of the cyclic AMP response element in the bcl-2 promoter in the regulation of endogenous Bcl-2 expression and apoptosis in murine B cells. Mol. Cell. Biol. 26:8599-8606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang, X. J., and S. Gregoire. 2005. Class II histone deacetylases: from sequence to function, regulation, and clinical implication. Mol. Cell. Biol. 25:2873-2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Young, R. L., and S. J. Korsmeyer. 1993. A negative regulatory element in the bcl-2 5′-untranslated region inhibits expression from an upstream promoter. Mol. Cell. Biol. 13:3686-3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao, X., A. Ito, C. D. Kane, T. S. Liao, T. A. Bolger, S. M. Lemrow, A. R. Means, and Y. P. Yao. 2001. The modular nature of histone deacetylase HDAC4 confers phosphorylation-dependent intracellular trafficking. J. Biol. Chem. 276:35042-35048. [DOI] [PubMed] [Google Scholar]
- 58.Zhao, X., T. Sternsdorf, T. A. Bolger, R. M. Evans, and T. P. Yao. 2005. Regulation of MEF2 by histone deacetylase 4- and SIRT1 deacetylase-mediated lysine modifications. Mol. Cell. Biol. 25:8456-8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou, X., V. M. Richon, A. H. Wang, X. J. Yang, R. A. Rifkind, and P. A. Marks. 2000. Histone deacetylase 4 associates with extracellular signal-regulated kinases 1 and 2, and its cellular localization is regulated by oncogenic Ras. Proc. Natl. Acad. Sci. USA 97:14329-14333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.