

Abstract
Small molecules can provide valuable tools to investigate virus biology. We developed a chemical screening approach to identify small molecule inhibitors of poorly understood, pre-early gene expression steps in herpes simplex virus infection, using green fluorescent protein fused to an early protein. Our assay identified ouabain, a cardiac glycoside. Ouabain reversibly decreased viral yield by 100-fold without affecting cellular metabolic activity in an overnight assay. The antiviral potencies of other cardiac glycosides correlated with their potencies against the known target of these compounds, the cellular sodium potassium ATPase. Ouabain had a reduced effect if added 8 hours post infection. It did not inhibit viral attachment or entry, but did reduce the expression of viral immediate early and early genes by at least 5 fold. Collectively, these results implicate a cellular target that was hitherto not considered important for a stage of HSV replication prior to viral gene expression.
Keywords: Herpes simplex virus, chemical screening, ouabain, cardiac glycosides, sodium-potassium ATPase, attachment, entry, capsid trafficking, uncoating, immediate-early gene expression
Introduction
Perturbation of biological systems with small molecule inhibitors has shed much light on the function of those systems in their uninhibited states. This is especially true for the replication of viruses, where pharmacological research has yielded greater understanding of the function of viral gene products and viral interactions with the host cell, in addition to clinically useful antiviral drugs.
Herpes simplex virus (HSV) is a clinically significant human pathogen (reviewed in Roizman et al., 2007). The process of HSV infection begins with attachment to a cellular receptor, followed by entry into the cytoplasm via one of a number of host cell type dependent pathways (reviewed in Spear, 2004), trafficking of the viral nucleocapsid to the nucleus on the cellular microtubule network (Sodeik et al., 1997), and uncoating of the virion at the nuclear pore to insert the viral DNA and trans-activators into the nucleus (Batterson et al., 1983). Immediate early and early rounds of gene expression occur, permitting replication of the viral genome and maximal expression of late gene products (Honess and Roizman, 1974). Progeny virions are then assembled and egress from the host cell. The stage of HSV DNA replication is relatively well understood, thanks in part to a large number of known pharmacological interventions which target this event (reviewed in Coen and Schaffer, 2003). Comparatively less is known about other stages in HSV infection, especially events prior to DNA replication. More could be learned about these earlier events with the identification of small molecule inhibitors that target them.
Towards this end, we designed a cell-based screening approach using a virus that expresses a reporter with early kinetics. We previously reported the construction and characterization of a recombinant HSV-1 ICP8-GFP, which expresses the Green Fluorescent Protein (GFP) fused to the viral single stranded DNA binding protein, ICP8 (Taylor et al., 2003). ICP8, a viral early gene product, is expressed prior to DNA replication, and its expression indicates that the prior steps of viral infection have already occurred successfully. We hypothesized that this virus could be used as a reporter in a screen to identify compounds that inhibit viral replication at a stage prior to expression of this early protein. Our screen and subsequent analysis identified ouabain, a well characterized inhibitor of the cellular sodium-potassium ATPase (reviewed in Xie and Cai, 2003), as an inhibitor of HSV replication after entry and before immediate early gene expression.
Results
Design and execution of a chemical screening approach
We designed a screen using a high content screening microscope at the Harvard Institute for Cell and Chemical Biology (ICCB). We used this assay to examine the ICCB’s known bioactive compounds library, a selection of 480 compounds with previously described biological activities. Vero cells were plated in black, clear-bottomed 384 well plates and incubated overnight at 37°C, yielding confluent monolayers of cells. Compounds were then robotically added to individual wells in duplicate at room temperature prior to infection. ICP8-GFP virus was added to the drug-containing media at a multiplicity of infection (MOI) of 20, and infection was allowed to proceed at 37°C for 18 hours. At the end of this incubation, cells were fixed with paraformaldehyde and treated with Hoechst 33258 to stain nuclear DNA. The screening microscope was used to record images of Hoechst and GFP fluorescence and amounts of fluorescence were quantified using image analysis software. Compounds that reduced the amount of GFP fluorescence by at least one standard deviation relative to the mean GFP signal for all compounds screened were scored as potential hits. From the 480 compound library, 17 compounds met this criterion. The images of the infected cells treated with these potential hits were then examined for signal from the Hoechst stain as a preliminary assessment of cytotoxicity. This eliminated twelve compounds that reduced the amount of Hoechst signal by at least one standard deviation relative to the mean DNA signal for all compounds screened. The remaining 5 compounds, approximately 1% of compounds screened, were ordered for secondary testing. Of these five, four compounds either caused visually apparent toxicity or did not cause an obvious reduction in GFP signal.
The fifth compound, ouabain, caused an obvious reduction in the level of GFP expressed by infected cells that was visually apparent without software quantification (Figure 1, compare panels B and D), and did not cause toxicity as qualitatively measured by examining the cells under light microscopy and reduction of signal in the Hoechst images (Figure 1, compare panels A and C). We selected ouabain for further study.
Figure 1.

Representative fluorescence images of the effect of ouabain on ICP8-GFP. The left column shows signal from the Hoechst 33258 DNA stain and the right column displays the same field in the GFP channel. The top two rows (panels A–D) show signal for Vero cells infected with ICP8-GFP at an MOI of 10 for 18 hours. Cells in the top row (panels A and B) were mock treated, while cells in the middle row (panels C and D) were treated with 14 μM ouabain. The bottom row (panels E and F) shows signal from mock infected cells.
Dose response of ouabain inhibition of GFP fluorescence
We conducted infections in the presence of a range of concentrations of ouabain to quantify the potency and efficacy of ouabain’s inhibition of GFP fluorescence resulting from infection with the ICP8-GFP virus (Figure 2). The concentration of ouabain that reduced ICP8-GFP signal by 50% in this assay was between 14 and 140 nM. Treatment with ≥140 nM ouabain reduced nuclear GFP fluorescence produced by infected cells by five-fold, down to a level which corresponded to the amount of autofluorescent signal observed from untreated, uninfected cells.
Figure 2.
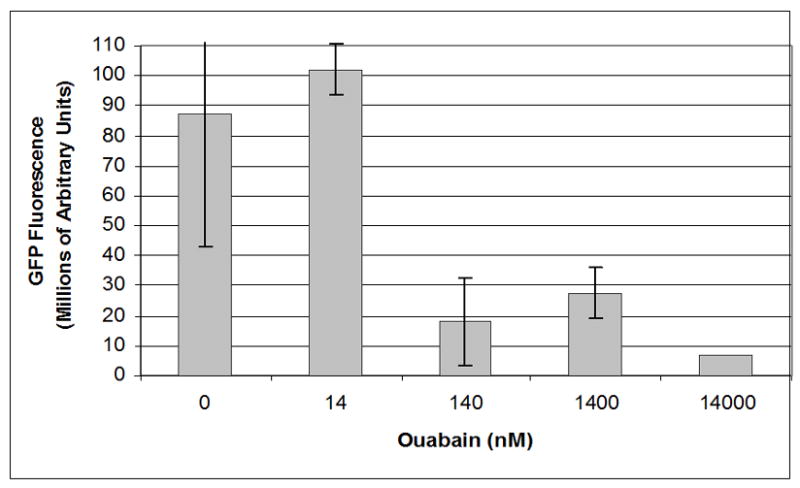
Dose response curve for effect of ouabain on GFP fluorescence. Images were analyzed with Metamorph image analysis software to quantify levels of GFP fluorescence detected after infection with ICP8-GFP virus at an MOI of 20 for 18 hours in the presence of a range of concentrations of ouabain.
Anti-HSV activity of ouabain
Ouabain was assessed for effects on viral replication using wild-type HSV-1 strain KOS. Concentrations of ouabain ≥100nM fully inhibited the formation of HSV-1 plaques on a monolayer of Vero cells after 72 hours. A small number of rounded but still adherent cells, and dead floating cells, were visible in treated, but not untreated, cells after this length of exposure, and this effect was similar in the presence or absence of virus. As an alternative to the plaque assay, a single cycle growth experiment was performed to determine whether ouabain reduced viral yield. In this assay, the concentration of drug that reduced yield by 90% (IC90) was 40 nM, and concentrations of 100 nM or higher consistently reduced viral yield by 100 to 10,000 fold (Figure 3).
Figure 3.

Effect of ouabain doses on viral yield. Cells were treated or mock treated with a range of concentrations of ouabain and simultaneously infected with wild type HSV-1 at an MOI of 10. Viral yield was determined by titration at 24 hours post infection.
A cell viability experiment was conducted to assess whether the observed reductions in GFP expression and viral yield during overnight treatments were caused by cytotoxicity, such as we had observed during the longer duration treatment necessary for plaque formation. Confluent Vero cells were treated with a range of concentrations of ouabain for 24 hours, and cellular metabolic activity was measured by cleavage of a tetrazolium salt, WST-1 (Figure 4). No dose-dependent decrease in metabolic activity compared to untreated cells was observed. This indicated that the antiviral effects of ouabain in the yield assay were not associated with cytotoxicity.
Figure 4.

Lack of cytotoxicity of ouabain. Vero cells were treated with a range of concentrations of ouabain and cell metabolism was measured with a WST-1 assay. The number of metabolically active cells remaining in each well at the time of measurement was determined by comparison to a standard curve, and compared to a mock treated control.
Antiviral effect of other sodium-potassium ATPase inhibitors
Ouabain is a well-characterized inhibitor of the cellular sodium potassium ATPase (reviewed in Xie and Cai, 2003), which is a clinically relevant drug target. As a result, detailed structure-activity relationship data on ouabain and a variety of other cardiac glycosides have been previously reported (Pullen et al., 2004). A selection of inhibitors of the sodium potassium pump was tested for effects on viral yield (Table 1). All compounds tested reduced viral yield by 100-fold at sufficient dosage. The antiviral potency of all of the plant-derived inhibitors correlated with their reported potencies against the pump (Pullen et al., 2004), though bufalin, an inhibitor derived from toad venom, was slightly more potent against viral yield than ouabain despite being slightly less potent against the sodium potassium pump.
Table 1.
Antiviral effect compared to inhibition of the sodium potassium ATPase
| Compound | Drug concentration that reduces Na/K ATPase activity by 50% | Drug concentration that reduces viral yield by 90% |
|---|---|---|
| Ouabain | 23 nM | 40 nM |
| Bufalin | 53 nM | 15 nM |
| Digitoxin | 78 nM | 70 nM |
| Digoxin | 120 nM | 150 nM |
| Strophantidin | 180 nM | 500 nM |
| Ouabagenin | 640 nM | 2500 nM |
Vero cells were infected with wild type HSV at an MOI of 10, and treated with a range of concentrations of the indicated inhibitors of the sodium potassium pump. Viral yield was determined by titration at 24 hours post infection. The right column contains the concentration of each drug that was sufficient to reduce viral yield by 90%. The middle column contains previously reported concentrations of drug that inhibited ATPase activity of cardiac membrane extracts by 50% (Pullen et al., 2004).
Kinetics of the anti-viral effect of ouabain
Next, we performed an experiment to determine if the antiviral effect of ouabain was reversible. Cells that were treated with 100 nM ouabain overnight prior to infection in the absence of drug produced similar levels of virus (2.2 × 109 PFU/ml) to cells that were not treated with any drug (3.6 × 109 PFU/ml). Thus, ouabain did not cause irreversible damage to cells during an overnight treatment. To determine whether ouabain had an irreversible effect when present during viral replication, Vero cells were infected with wild type HSV at an MOI of 10 in the presence of 100 nM ouabain, and infection was allowed to proceed. At a series of time points post infection, the medium was removed and replaced with drug-free media. At 24 hours post infection, all samples were harvested and titrated to determine viral yield (Figure 5). No decrease in viral yield was observed when drug was removed up to 8 hours post infection.
Figure 5.
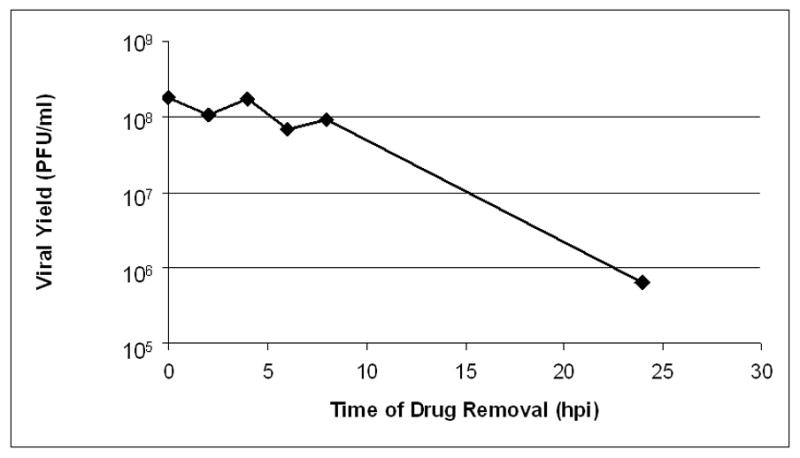
Reversibility of ouabain treatment. Cells were treated with 100 nM ouabain and infected with wild type HSV-1 at an MOI of 10. At the indicated times post-infection, the drug containing medium was removed and infection was allowed to proceed in drug-free media. Viral yield was determined by titration at 24 hours post infection.
We then performed an experiment in which Vero cells were infected with wt HSV-1 at an MOI of 10, and 100 nM ouabain was added at a series of times post-infection. Output virus was harvested at 24 hours post infection and titrated to determine viral yield (Figure 6). The results revealed a clear time-dependent trend, with increased viral yield as ouabain was added later in infection. Viral yield was reduced by 1000 fold when drug was added coincident with infection, but addition of drug at 8 hpi reduced yield by 45 fold, indicating that a substantial portion of ouabain’s antiviral effect occurs prior to 8 hours post infection.
Figure 6.
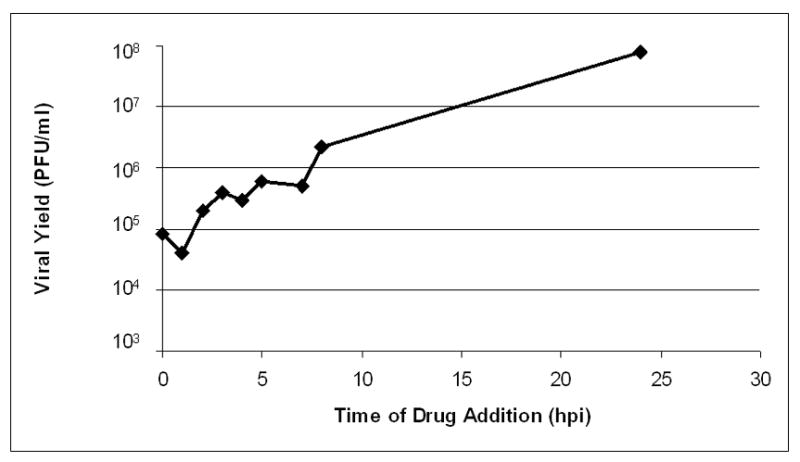
Effect of delayed ouabain addition on viral yield. Cells were infected with wild type HSV-1 at an MOI of 10 and treated with 100 nM ouabain at the indicated times post infection. Viral yield was determined by titration at 24 hours post infection.
Ouabain does not meaningfully inhibit attachment or entry
In order to investigate the stage of the viral replication cycle inhibited by ouabain, we performed attachment and entry assays. Using a standard assay for attachment (MacLean, 1998, Shogan et al, 2006), Vero cells and HSV were pre-chilled to 4°C. The chilled cells were infected with approximately 150 PFU of HSV in the presence of ouabain on ice for 1 hour to permit attachment. Unbound virus and drug were washed off and infection was allowed to proceed for 48 hours. This assay was repeated three times, testing concentrations including 10 nM (a concentration that did not reduce viral yield), 100 nM, 1 μM, and 10 μM. No effect on plaque formation was observed at any of these concentrations. In a representative experiment, untreated cells produced 152 plaques while cells treated with 100 nM ouabain, a concentration that reduces viral yield by 100 to 10,000 fold, produced 146 plaques. To assess effects on attachment at higher MOI,, cells were infected with virus at an MOI of 10 on ice for 1 hour in the presence or absence of 100 nM ouabain. This permitted viral attachment to cells without viral entry. Cells were then washed to remove the drug and any un-attached virions. Fresh, drug free media was then added, and infection was allowed to proceed at 37°C for 24 hours. Yields from the treated culture (1.3 × 108 PFU/ml) were similar to untreated (1.2 × 108 PFU/ml). Thus, in both the standard attachment assay and the high MOI attachment assay, treatment with ouabain had little or no effect.
To determine if pre-attached virus was able to enter cells in the presence of drug, a standard entry assay was performed (MacLean, 1998, Shogan et al, 2006). Chilled, untreated cells were infected with approximately 300 PFU of chilled virus and left on ice for 1 hour to permit attachment. Unbound virus was washed off and entry was allowed to proceed at 37°C for 1 hour in the presence of 10 μM ouabain, a dose 100-fold above the dose that reduces viral yield by 100–10,000 fold. Drug was washed out, and cells were treated with acidic buffer to neutralize any remaining extracellular virus. Infection was then allowed to continue at 37°C in drug-free media for 48 hours. In this assay, the untreated cells produced 272 plaques while the treated cells produced 245 plaques. Thus treatment with ouabain did not substantially reduce the number of viral plaques in this entry assay. To assess effects on entry at higher MOI,, we allowed HSV-1 to attach to cells on ice for 1 hour at an MOI of 10 in the absence of drug. Infected cells were then moved to 37°C for 1 hour in the presence or absence of 100 nM ouabain, a concentration which reduces viral yield by 100 to 10,000 fold (as verified in a parallel assay). At the end of one hour, all cells were treated with an acidic buffer to inactivate remaining extracellular virus. Infection then proceeded in drug-free media for 24 hours. Untreated cells produced a yield of 9.0 × 106 PFU/ml and treated cells produced a yield of 9.6 × 106 PFU/ml. Thus, in both the standard entry assay and the high MOI entry assay, pre-attached virus was able to enter cells in the presence of drug.
Ouabain inhibits expression of immediate early and early genes
The screening assay showed that expression of the ICP8-GFP early gene marker was reduced by 5-fold, which was the limit of detection, in the presence of ouabain. To confirm and extend this finding, we assessed expression of the viral immediate early protein ICP27 by Western blot analysis. Vero cells were infected with wt HSV-1 at an MOI of 10 in the presence or absence of 100 nM ouabain. At 6 hours post infection, cells were lysed and processed for SDS-PAGE and Western Blotting using H1113, a monoclonal antibody against ICP27 (Figure 7). A specific band for ICP27 was visible in the infected cell lysate (lanes 1–4), and absent in uninfected cell lysate (lane 5) and lysates from ouabain treated infected cells (lane 6). Comparison of the lysate from ouabain treated infected cells with a dilution series of infected cell lysate (lanes 2–4) permitted an estimate that the reduction was at least 8 fold. The blot was stripped and probed with anti-actin antibody, which showed that the lysates contained similar amounts of protein (Figure 7, bottom panel, lanes 1, 5, and 6). Thus, ouabain acts at or prior to the expression of immediate early proteins.
Figure 7.
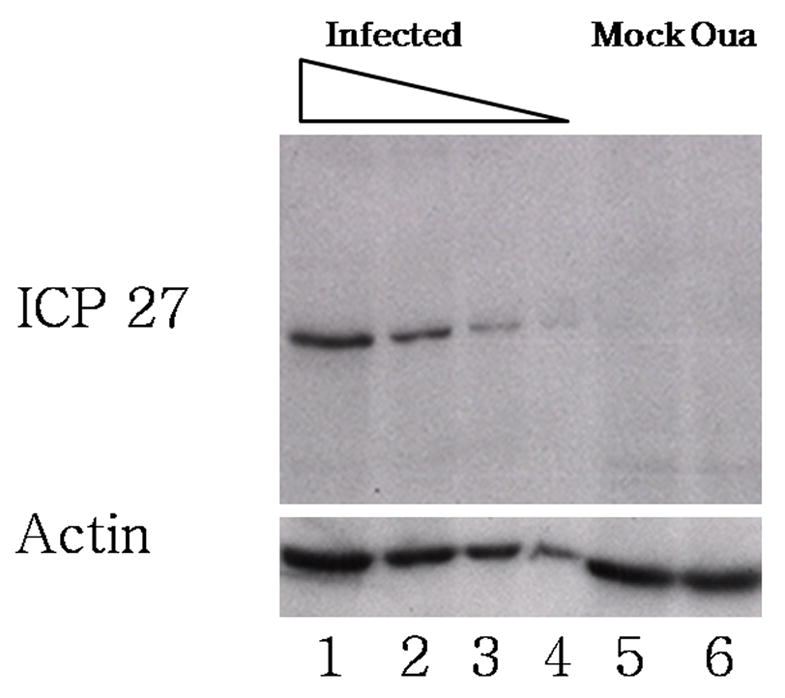
Effect of ouabain on ICP27 expression. Cells were infected or mock infected in the presence or absence of ouabain. Protein lysates were harvested at 6 hours post infection, and ICP27 was detected by western blot using a monocolonal antibody against ICP27 (top panel). Wells 1–4 contain a series of two-fold dilutions of infected, untreated cell lysate into loading buffer (lane 1 contains undiluted sample, and lane 4 contains a 1:8 dilution). Lane 5 contains undiluted lysate from mock infected cells and lane 6 contains undiluted lysate from infected, ouabain treated cells. The blot was stripped and reprobed using a monoclonal antibody against β-actin (bottom panel).
Discussion
We set out to test an approach for chemical screening targeted at a specific stage in the HSV replication cycle. The approach identified a novel inhibitor of HSV replication that acts at an early step in viral replication soon after entry and before immediate-early gene expression. The identification of ouabain as an inhibitor is intriguing as there were few previous results that suggested a role for a Na/K pump in HSV replication.
Temporally controlled reporters are a viable method for identifying stage-specific anti-viral inhibitors
Our screening approach took advantage of the temporal regulation of HSV replication to isolate compounds with specificity for stages prior to the expression of viral early genes. This cell-based approach also assured that the compounds would be relevant to viral infection. This is not the first such cell-based infection assay for inhibitors of HSV replication. One group, van Zeijl et al. (2000), conducted a screen using an automated ELISA assay to determine viral yield, and another group, Boulware et al. (2001), placed the lacZ reporter under the control of the promoter for the viral glycoprotein C, which is expressed late in infection. However, to our knowledge, our screen is the first attempt to specifically exclude inhibitors of viral DNA replication and later steps. The identification of ouabain from our screen is proof in principle that our screening approach has the potential to identify novel compounds that inhibit specific stages in the replication of HSV.
The screening approach was not without limitations. The initial screen had an undesirably high hit rate, primarily because the readout was the loss of expression of a protein. Cytotoxic compounds, and those which are general inhibitors of translation, will prevent expression of the GFP marker used in this assay without shedding any useful insight on mechanisms of viral infection. For this reason, we were greatly concerned about cytotoxicity as a source of true, but uninteresting, positive hits. A larger scale screen using this assay would have to cope with this issue, either by improved sensitivity or additional secondary assays to confirm hits and remove cytotoxic compounds. However, even without adjustments to the assay conditions, advances in screening equipment such as microscopes with higher resolution and sensitivity might help decrease the number of false or uninteresting positives detected by the screen. This might also help lower the initial hit rate and thus facilitate the secondary screening process.
Despite these limitations, we were able to implicate a cellular component that had not been strongly considered for a role in HSV replication. There was no expectation a priori that the sodium potassium pump would be involved in steps of infection prior to early gene expression, so it is unlikely that it would have been selected as a hypothetical candidate for investigation in an in-vitro screening assay. Screening in live cells with whole virus ultimately allows the greatest diversity of potential hits, and the results of our study have validated this approach.
Ouabain inhibits a pre-gene expression step in HSV infection
Ouabain reduced HSV replication in Vero cells by over 100 fold in yield reduction assays. Data from the attachment and entry assays, as well as the result that drug can be removed at up to 8 hours post infection without reducing viral yield, argue strongly that ouabain acts at a stage after fusion with the plasma membrane. Data from the ICP8-GFP assay and the Western blot analysis shows reduction of expression of viral immediate early and early genes by at least 5-fold. Neither cellular nuclear morphology, as observed via Hoechst staining, nor cellular metabolic activity, as measured by the WST cleavage assay was affected by ouabain treatment for the length of time used in these assays. In addition, the effects of ouabain were reversible when drug was removed up to 8 hours post infection, or after an overnight pre-treatment. Collectively, these data suggest that ouabain is inhibiting one of the stages of the viral life cycle that the screen was intended to target, and that its effect is not primarily due to gross cytotoxicity. We also observed a decrease in viral yield in the single cycle growth assay even if drug was added at 8 hours post infection, after viral DNA replication should have occurred. This additional effect may be due to cytotoxicity, or due to a specific effect on a later stage of herpes viral replication. One possible late stage target is egress, as ouabain has been reported to inhibit fusion of cells infected with synctial mutant strains of HSV-1 (Baghian and Kousoulas, 1993). We endeavored to limit the time of ouabain exposure in subsequent experiments to better separate effects on the early stages of viral replication from any later effects.
Ouabain has been previously stated to have an anti-viral effect (Baghian and Kousoulas, 1993), but to our knowledge data to support this conclusion and examine its mechanism have not been reported. The results of our experiments suggest that ouabain is targeting either transport of the viral capsid to the nucleus, uncoating of the capsid to allow translocation of the viral genome, processes that allow viral proteins such as VP16 through the nuclear pore, or more directly, expression of viral genes. Interestingly, it has been reported that depolymerization of the microtubule network with nocodazole, which disrupts transport of the capsid to the nucleus, reduces expression of a reporter expressed with early kinetics by 80–90% (Mabit et al., 2002).
Empirical observations aside, the molecular mechanism of ouabain’s antiviral effects are not entirely clear. We were not able to generate resistant viral mutants to ouabain (not shown). This is consistent with the hypothesis that the antiviral effect is due to inhibition of a cellular target, but could also be explained by inhibition of both a cellular and a viral target, or inhibition of both an early viral target and also a later stage such as egress, either of which would make it more difficult to develop resistance. The experiments with other cardiac glycosides, which indicated that antiviral potency for all but bufalin correlated with potency for ouabain’s known cellular receptor, support the argument that the antiviral effect is due to the compounds’ inhibition of the sodium potassium pump. Bufalin, while structurally similar to the plant-derived cardiac glycosides, is derived from toad venom and has been reported to poison topoisomerase II (Pastor and Cortes, 2003). Host topoisomerase II has been shown to act on progeny HSV genomes (Ebert et al., 1994) and an inhibitor of topoisomerase II has been reported to reduce viral yield by greater than 50% (Hammarsten et al., 1996), so this activity could explain the higher than expected potency of bufalin against viral yield compared to ouabain. The sodium potassium pump is also necessary for proper cellular calcium homeostasis and is active in cellular signaling pathways, (reviewed in Xie and Cai, 2003), presenting a wide array of pathways that could explain its antiviral effect. Studies to determine how inhibition of an ion pump leads to an effect on a specific stage of HSV infection are ongoing.
Materials and Methods
Cells and viruses
Vero (African green monkey kidney) cells were obtained from the American Type Culture Collection and grown and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% newborn calf serum (2% during infection where not otherwise noted) and 1% penicillin-streptomycin (Invitrogen) at 37°C and 5% CO2. HSV type 1 (HSV-1) wild-type strain KOS was propagated in Vero cells. The ICP8-GFP virus was derived from KOS 1.1 as previously described (Taylor et al., 2003). ICP8-GFP was grown in V529 cells, a Vero line that contains the UL5 and UL29 genes, in order to complement the partial replication defect of the ICP8-GFP virus.
Chemicals
The ICCB known bioactive compound library consists of 480 small molecules with previously observed biologic activity. The entire library as a set is commercially available from Biomol, and is fully annotated online at the ICCB website (http://iccb.med.harvard.edu/screening/compound_libraries/bioactives_collection2.htm). The pilot screen used stocks stored in DMSO at concentrations of 5 mg/ml (on average, approximately 10 mM) at −20 °C in dessicated storage containers. Ouabain (Biomol) and other inhibitors of the sodium potassium pump (VWR) were suspended at 10 mM in DMSO and stored at 4°C. Hoechst 33258 (Sigma) was suspended in water at a concentration of 10 mg/ml.
Screening Assay
The assay was conducted in duplicate using black walled, clear-bottomed 384-well cell culture plates (Corning) filled using a Matrix Wellmate plate filler. Vero cells were seeded at a concentration of 3,750 cells per well in these plates and incubated overnight at 37°C. To reduce background, phenol red-free, glutamine-free DMEM (Cellgro) supplemented with 2 mM glutamine, 2% NCS, and 1% penicillin-streptomycin was used. Plates were fully confluent at the time of compound addition to maximize the number of infectable cells in each well. Compounds from the library were added to the media (estimated working concentration 30 μM) by a custom pin-transfer robot built for the ICCB. The Vero cells were then infected with ICP8-GFP virus at an MOI of 20, which was experimentally determined to be sufficient to detect GFP signal in 70–80% of the cells at 18 hours post infection. (The MOI of this infection was determined on the complementing V529 cells. Although the virus is impaired for DNA replication in wild type Vero cells, it was deemed appropriate to use titers from the complementing cell line because the readout of the assay occurs prior to the block.) At 18 hours post-infection, cells were fixed and stained for 30 min by addition of 8% paraformaldehyde in PBS, including 1:5000 of a 10 mg/ml Hoechst 33258 stock solution added just prior to treatment, directly to the infected cells in drug-containing media. The plates were aspirated with a 24-channel aspirating wand (VP Scientific) and washed twice with PBS. Plates were filled with PBS, sealed with foil seals (Corning), and stored at 4°C prior to screening.
A Discovery-1 high content screening microscope (Universal Imaging) was programmed to visualize fluorescent signal from the Hoechst stain, automatically focus on cell nuclei, and record an image. The cells were then illuminated at 488 nm to produce signal from GFP, and a second image was taken in the same focal plane as the nuclei. Metamorph image analysis software (Molecular Devices) was then used to quantify the signal. This software identifies the nuclei as objects, measures the corresponding areas of the GFP channel images, and calculates a total amount of signal recorded from the objects in each of the two channels. The mean and standard deviation for all samples, including both duplicates, were determined. The individual wells were then compared to these aggregate numbers to identify potential hits and cytotoxic compounds. A compound was scored as a potential hit if both of the two duplicates had GFP signal reduced by one standard deviation or more below the mean for all compounds screened. Visual inspection of the plates revealed that compounds that reduced the amount of Hoechst signal detected by one standard deviation or more in either of the two duplicates were causing visible changes in nuclear structure or number. Therefore, this standard was adopted for rejection of potential hits due to cytotoxicity.
Yield reduction assay
All yield reduction assays were conducted on Vero cells using wild-type strain KOS at an MOI of 10 in cell culture tubes, in DMEM containing 2% serum and 1% penicillin-streptomycin. Where not otherwise noted, drugs were added simultaneously with virus and media was not changed. Virus was harvested at 24 hours post infection by freeze thawing the entire culture tube three times, followed by titration on Vero cells.
WST cytotoxicity assay
WST-1 was obtained from Roche and was used according to the manufacturer’s instructions. Cells were plated on black 96 well plates to replicate the conditions used in the screening and yield assays. Cells were treated with a range of concentrations of ouabain for 24 hours. WST-1 was added to each well for 30 min at the recommended 1:10 dilution and the plate was left in a 37 °C incubator with CO2 for 1 hour to allow for reduction by cells. This reaction produced colored product proportional to the number of metabolically active cells in the well. A standard curve was generated by plating 2-fold dilutions of a 6 × 106 cells/ml solution immediately prior to the assay, which had a linear range of approximately 1500 to 200,000 cells per well. The absorbance produced by the experimental samples corresponded to a number of cells (approximately 80,000 cells/well) that lay within the linear range of the standard curve. Absorbance at 440 nm was quantified in a Wallac 1420 plate reader.
Attachment assay
The effects of compounds on attachment were assessed using a previously described assay (modified in Shogan et al, 2006 from a standard protocol in MacLean, 1998). In this assay (standard assay of attachment), Vero cell monolayers were prechilled to 4 degrees, and infected with approximately 150–300 PFU of wild type HSV-1 strain KOS in the presence or absence of compounds for 1 hour on ice. We also performed a high MOI assay of attachment, in which Vero cells in cell culture tubes were infected at an MOI of 10 in the presence or absence of compounds on ice for one hour. This incubation is long enough to permit the majority of the inoculum to attach to cells (B. Shogan, A.W.D., and D.M.C., unpublished results). In both assays, cells were washed with PBS three times, to remove compounds and unattached virus, and overlaid with drug-free medium containing 1.2% methylcellulose in the standard assay, or unsupplemented drug free medium in the high MOI assay. For the standard assay, infection was allowed to proceed at 37° for 48 hours, after which cells were fixed and stained for plaque counting. Virus from the high MOI assay was harvested at 24 hours post infection by freeze thawing the entire culture tube three times, followed by titration on Vero cells. As a control to show that entry does not occur during the attachment step in this assay, cells with attached virus were treated with acidic glycine as described for the entry assay below, and no plaques were observed. Known inhibitors of viral attachment such as heparin inhibit plaque formation in this assay (L. Kruse, B. Shogan, E. Macari, A.W.D., and D.M.C., unpublished results and Shogan et al, 2006).
Entry assay
The effects of compounds on entry were assessed using a previously described assay (modified in Shogan et al, 2006 from a standard protocol in MacLean, 1998). In this assay (standard assay of entry), Vero cell monolayers were prechilled to 4 degrees, and infected with approximately 150–300 PFU of wild type HSV-1 strain KOS in drug-free medium for 1 hour on ice in minimal volume to permit attachment. We also performed a the high MOI assay of entry, in which Vero cells in cell culture tubes were infected at an MOI of 10 on ice for one hour. In both assays, cells were washed three times with chilled PBS to remove unattached virus, and overlaid with medium containing compounds. Entry was allowed to proceed at 37° for one hour, an incubation which is long enough to permit the majority of pre-attached virus to enter cells (L. Kruse, B. Shogan, E. Macari, A.W.D., and D.M.C., unpublished results), and then 1 ml of acidic glycine was added to cells to inactivate remaining extracellular virus. After 20 seconds of treatment at room temperature, cells were washed three times with medium to return the pH to neutral. For the standard assay, cells were then overlaid with medium containing 1.2% methylcellulose, incubated at 37°C for 2 days, and fixed and stained to count plaques. We have previously shown that a phosphorothioate oligonucleotide inhibits entry in this assay (Shogan et al, 2006). Virus from the high MOI assay was harvested at 24 hours post infection by freeze thawing the entire culture tube three times, followed by titration on Vero cells.
Western blot analysis
Confluent 60 mm dishes of Vero cells were mock infected or infected with wild type HSV-1 strain KOS for 6 hours in the presence or absence of 100 nM ouabain, added simultaneously with virus. After 6 hours, cells were washed three times with 3 ml of PBS for 30 seconds at room temperature. Protein lysates were harvested by addition of 350 μl of 2x Laemmli sample buffer (Bio-Rad) supplemented with 0.7 M β-mercaptoethanol (Bio-Rad). Lysates were boiled for 5 minutes, frozen, and boiled again immediately prior to loading on 10% SDS-polyacrylamide gels. Infected cell lysate samples with and without ouabain were diluted in Laemmli buffer to generate standard curves for quantifying amounts of protein present.
For immunodetection of the ICP27 protein, membranes were blocked overnight in TBST (20 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1% Tween 20) supplemented with 5% milk. H1113 mouse monocolonal antibody (Virusys) against ICP27 was added to the blocking solution at 1:5000 for 1 hour at room temperature. Blots were washed three times for 5 minutes each in TBST and goat anti-mouse HRP-conjugated secondary antibody (Southern Biotech) was added in 1:5000 TBST. Blots were washed again three times for 5 minutes each in TBST and enhanced chemiluminescence reagents were used as directed by the manufacturer (Pierce). Blots were stripped using Restore Western Stripping Buffer (Bio-Rad) for 1 hour at room temperature, followed by re-blocking overnight and reprobing as described above with 1:5000 anti-actin monoclonal antibody (Abcam) in blocking solution as a loading control.
Acknowledgments
We gratefully acknowledge the staff of the ICCB for help and expertise in conducting the screen. In particular, Caroline Shamu, Nicola Tolliday and Su Chiang helped with the initial design, Stewart Rudnicki and Katrina Schulberg supervised the automation of the screening assay and pin transfer of compounds, Jon Hoyt assisted with the use of the microscope, and Andrew Lach helped with statistical analysis of the screen results. We also thank members of the Coen lab for advice and support. This work was supported by National Institutes of Health Grants ROI AI 26077 to D.M.C. and AI 063106 to D.M.K.
Note added in proof After this paper appeared online, we were made aware of a recent paper (Hartley, C., Hartley, M., Pardoe, I., Knight, A., 2006. Iconic contra-viral therapy (ICVT): a new approach to the treatment of DNA virus infections. Arch. Virol. 151, 2495-2501) that reported inhibition of HSV plaque formation by the cardiac glycoside digoxin.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Works Cited
- Baghian A, Kousoulas KG. Role of the Na+, K+ pump in herpes simplex type 1-induced cell fusion: melittin causes specific reversion of syncytial mutants with the syn1 mutation to Syn+ (wild-type) phenotype. Virology. 1993;196:548–556. doi: 10.1006/viro.1993.1510. [DOI] [PubMed] [Google Scholar]
- Batterson W, Furlong D, Roizman B. Molecular genetics of herpes simplex virus. VIII. further characterization of a temperature-sensitive mutant defective in release of viral DNA and in other stages of the viral reproductive cycle. J Virol. 1983;45:397–407. doi: 10.1128/jvi.45.1.397-407.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulware SL, Bronstein JC, Nordby EC, Weber PC. Identification and characterization of a benzothiophene inhibitor of herpes simplex virus type 1 replication which acts at the immediate early stage of infection. Antiviral Res. 2001;51:111–125. doi: 10.1016/s0166-3542(01)00147-4. [DOI] [PubMed] [Google Scholar]
- Coen DM, Schaffer PA. Antiherpesvirus drugs: a promising spectrum of new drugs and drug targets. Nat Rev Drug Discov. 2003;2:278–288. doi: 10.1038/nrd1065. [DOI] [PubMed] [Google Scholar]
- Ebert SN, Subramanian D, Shtrom SS, Chung IK, Parris DS, Muller MT. Association between the p170 form of human topoisomerase II and progeny viral DNA in cells infected with herpes simplex virus type 1. J Virol. 1994;68:1010–1020. doi: 10.1128/jvi.68.2.1010-1020.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarsten O, Yao X, Elias P. Inhibition of topoisomerase II by ICRF-193 prevents efficient replication of herpes simplex virus type 1. J Virol. 1996;70:4523–4529. doi: 10.1128/jvi.70.7.4523-4529.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honess RW, Roizman B. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J Virol. 1974;14:8–19. doi: 10.1128/jvi.14.1.8-19.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabit H, Nakano MY, Prank U, Saam B, Dohner K, Sodeik B, Greber UF. Intact microtubules support adenovirus and herpes simplex virus infections. J Virol. 2002;76:9962–9971. doi: 10.1128/JVI.76.19.9962-9971.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclean CA. HSV entry and spread. In: Brown SM, MacLean AR, editors. Herpes simplex virus protocols. Humana Press; Totowa, N.J: 1998. pp. 9–18. [Google Scholar]
- Pastor N, Cortes F. Bufalin influences the repair of X-ray-induced DNA breaks in Chinese hamster cells. DNA Repair (Amst) 2003;2:1353–1360. doi: 10.1016/j.dnarep.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Pullen MA, Brooks DP, Edwards RM. Characterization of the neutralizing activity of digoxin-specific Fab toward ouabain-like steroids. J Pharmacol Exp Ther. 2004;310:319–325. doi: 10.1124/jpet.104.065250. [DOI] [PubMed] [Google Scholar]
- Roizman B, Knipe D, Whitley R. Herpes Simplex Viruses. In: Knipe D, Howley P, Griffin D, Lamb R, Martin M, Roizman B, Straus S, editors. Fields Virology. 5. Lippincott, Williams & Wilkins; Philadelphia, PA: 2007. pp. 2501–2601. [Google Scholar]
- Shogan B, Kruse L, Mulamba GB, Hu A, Coen DM. Virucidal activity of a GT-rich oligonucleotide against herpes simplex virus mediated by glycoprotein B. J Virol. 2006;80:4740–4747. doi: 10.1128/JVI.80.10.4740-4747.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodeik B, Ebersold MW, Helenius A. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J Cell Biol. 1997;136:1007–21. doi: 10.1083/jcb.136.5.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear PG. Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol. 2004;6:401–410. doi: 10.1111/j.1462-5822.2004.00389.x. [DOI] [PubMed] [Google Scholar]
- Taylor TJ, McNamee EE, Day C, Knipe DM. Herpes simplex virus replication compartments can form by coalescence of smaller compartments. Virology. 2003;309:232–247. doi: 10.1016/s0042-6822(03)00107-7. [DOI] [PubMed] [Google Scholar]
- van Zeijl M, Fairhurst J, Jones TR, Vernon SK, Morin J, LaRocque J, Feld B, O’Hara B, Bloom JD, Johann SV. Novel class of thiourea compounds that inhibit herpes simplex virus type 1 DNA cleavage and encapsidation: resistance maps to the UL6 gene. J Virol. 2000;74:9054–9061. doi: 10.1128/jvi.74.19.9054-9061.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Cai T. Na+-K+-ATPase-mediated signal transduction: from protein interaction to cellular function. Mol Interv. 2003;3:157–168. doi: 10.1124/mi.3.3.157. [DOI] [PubMed] [Google Scholar]