

Abstract
In situ hybridization is an important tool for analyzing gene expression and developing hypotheses about gene functions. The discovery of hundreds of microRNA (miRNA) genes in animals has provided new challenges for analyzing gene expression and functions. The small size of the mature miRNAs (∼20-24 nucleotides in length) presents difficulties for conventional in situ hybridization methods. However, we have developed a modified in situ hybridization method for detection of mammalian miRNAs in tissue sections, based upon the use of RNA oligonucleotide probes in combination with highly specific wash conditions. Here we present detailed procedures for detection of miRNAs in tissue sections or cultured cells. The methods described can utilize either nonradioactive hapten-conjugated probes that are detected by enzyme-coupled antibodies, or radioactively labeled probes that are detected by autoradiography. The ability to visualize miRNA expression patterns in tissue sections provides an additional tool for the analyses of miRNA expression and function. In addition, the use of radioactively labeled probes should facilitate quantitative analyses of changes in miRNA gene expression.
Keywords: microRNAs, gene expression, nervous system, brain, development, embryos, fluorescein, mouse, rat, human
1. Introduction
miRNAs (miRNAs) are short endogenous RNAs, ∼20-24 nucleotide (nt) long, that were first identified in the nematode C. elegans, and subsequently found in most animals and plants [for review see 1, 2, 3]. MiRNAs have been shown to function as post-transcriptional negative regulators of gene expression in a variety of systems [reviewed in 4, 5]. There are about 500 known miRNAs in humans, although prediction algorithms suggest that the total number of miRNAs could be substantially larger [6], and experimental approaches suggest that new miRNAs still remain to be identified [7, 8]. Thus, miRNAs appear likely to be a major component of gene regulation. MiRNA synthesis is a multiple step process (reviewed in [1, 2, 9]). MiRNAs are initially synthesized as longer primary RNA transcripts, and some miRNAs are clustered in genomes, where they can be transcribed as polycistronic primary transcripts. The nuclear microprocessor complex, which includes the endonuclease Drosha, recognizes secondary structures in primary transcripts and cleaves the primary transcripts to release short stem-loop miRNA precursors (∼65 nt long), which are then exported to the cytoplasm. In the cytoplasm, the Dicer endonuclease cleaves a stem-loop precursor to release both the mature miRNA and its complementary strand. In most cases, one strand (arm) of the stem-loop precursor accumulates as a stable miRNA after Dicer processing, while the complementary strand (referred to as the “*” strand) is degraded. Mature miRNAs are incorporated into ribonucleoprotein (RNP) complexes that contain a member of the Argonaute protein family as well as other proteins, and these RNP complexes function as negative regulators of mRNA translation and/or mRNA level (reviewed in [10]).
For several years after the discovery that large numbers of miRNAs were present in animals, a major limitation for understanding miRNA function was the difficulty in determining detailed spatial expression patterns for specific miRNAs. However, two approaches for detection of miRNAs by in situ hybridization (ISH) have been developed recently. Plasterk and colleagues have used an ISH method based on Locked Nucleic Acid (LNA) oligonucleotide probes to detect mature miRNAs in whole mounts of zebrafish and mouse embryos [11, 12]. The LNA-based ISH methodology has been applied to whole-mount embryos from a variety of vertebrate species, including chicken and medaka [13, 14], and for the analysis of adult human brain sections [15] as well as mouse tissue sections [16, 17]. Independently, we have developed a miRNA ISH method based upon RNA oligonucleotide probes and have used this method for the detection of mature miRNAs in tissue sections from embryonic and adult mice [18], as well as from rat and human brains (this article) and zebrafish (our unpublished observations). We have also detected miRNAs by ISH in cultured cells [18]. The LNA-based miRNA ISH method derives a high degree of sequence specificity from the base-pairing properties of LNA probes. In contrast, we have used high-stringency wash conditions based on tetramethylammonium chloride (TMAC) in combination with RNase A treatment to remove unhybridized probe to generate highly sequence specific conditions for miRNA ISH with RNA probes. Both methods appear to generate similar results based on the comparison of published expression patterns. One potential advantage of RNA probe based ISH is that the use of TMAC washes allows probes of different sequence compositions (with potentially different melting temperatures) to be processed under identical conditions [19, 20].
Although miRNAs are generated by processing from longer precursor and primary transcripts that contain the same miRNA sequence, miRNA ISH using either RNA or LNA probes appears to primarily detect mature miRNA [12, 18]. Most likely, the discrimination between mature miRNA and precursor or primary transcript RNAs reflects the accumulation of mature miRNAs to high levels in cells, relative to mRNAs [21], and the location of the target miRNA sequences within a stem-loop secondary structure in miRNA precursors and primary transcripts, which seems likely to reduce access of probes for hybridization during ISH. It also seems possible that miRNAs base-paired with a target mRNA in RNP complexes are less accessible to hybridization during ISH, in which case miRNA ISH may preferentially detect mature miRNAs that are not bound to a target mRNA, but this possibility has not been tested.
To date, most published studies on miRNA ISH have used probes labeled with non-radioactive haptens (digoxigenin or fluorescein). Bound hapten-labeled probes are detected by histochemical enzymatic reactions after application of alkaline phosphatase (AP) -conjugated anti-hapten antibodies. For RNA oligonucleotide probes, we have used fluorescein as a hapten, since it is convenient to commercially synthesize RNA oligonucleotides with a 5'-end fluorescein. Nonradioactive ISH methods are rapid and permit the precise localization of mRNA or miRNA expression to specific cells or even cellular compartments, as well as facilitate whole-mount analyses, but these methods are less amenable to quantitative analysis of miRNA levels. However, ISH with radioactively labeled probes allows accurate semi-quantitative or quantitative comparisons of mRNA expression levels [22, 23] using autoradiographic methods (e.g. x-ray film, photographic emulsions, phosphorimagers). As described below, we have modified our miRNA ISH method to allow the use of 33P 5' end-labeled RNA probes for the detection of miRNAs in tissue sections. This approach should allow comparisons of miRNA levels among different cells or structures (e.g. brain nuclei). In the following sections, we provide detailed methods for detection of miRNAs by ISH using RNA oligonucleotide probes labeled with either a fluorescein hapten or 33P.
2. Experimental Design considerations
2.1 Probe Design and Controls
In most cases, we have used synthetic RNA oligonucleotide probes with 20 nt of complementarity to a specific target miRNA, and the conditions presented were optimized for this probe size. However, probes with 21 or 22 nt complementary to a target miRNA appear to function similarly [18]. Most of our fluorescein-labeled RNA probes also include two additional nucleotides at the 5' end that do not match the target miRNA. These bases are not required for probe function, but they were included to potentially improve access of the anti-fluorescein antibody to the 5' end fluorescein. Since the TMAC-based wash conditions are not sensitive to sequence composition, the choice of which 20 nt sequence within a miRNA longer than 20 nt should be used for probe generation appears to be arbitrary. Since many miRNAs are part of gene families that are composed of miRNAs that differ at only one or a few positions, all probe designs should be compared against miRBase (http://microrna.sanger.ac.uk/) [24] to identify potential unanticipated miRNA matches. We have observed that our conditions can prevent significant hybridization of an RNA oligonucleotide probe with two mismatches to an abundant miRNA [18], and in some cases it may be possible to distinguish miRNAs that differ by a single nucleotide near the center of the miRNA. In general, if it is desirable to distinguish between closely related miRNAs, probes should be positioned on the target miRNAs so as to maximize the number of mismatches with non-target miRNA(s), and so that potential duplexes between probe and non-target miRNA(s) will be as short as possible (to reduce cross-hybridization). However, the short length of miRNAs limits the opportunities for probe specificity optimization. In addition, it is unlikely that any miRNA ISH procedure will be able to distinguish between miRNAs that differ by only a single nucleotide at one end (e.g. miR-128a and miR-128b).
Several different types of control RNA oligonucleotide probes are possible. To confirm the sequence specificity of ISH, control probes can include mutations that create mismatches with the target miRNA at 1 to 3 internal positions [11, 18]. It is important to avoid mismatch mutations that would allow a probe to hybridize to other members of a miRNA gene family unintentionally. To detect probe trapping or other types of sequence-independent probe binding that can lead to elevated background, a reversed or scrambled sequence RNA probe can be used (after comparison to miRBase to check for unanticipated targets). One limitation of reversed or scrambled sequence controls is that it is not easy to verify that they are functional probes, since they have no endogenous target. An alternative is to use a probe that detects a known miRNA which is not expressed in the tissue of interest (or which is expressed in a distinct pattern within the tissue of interest). In this case, the integrity and function of the control probe can be confirmed by ISH using the appropriate tissue. When comparing ISH using control probes to ISH with probes for miRNAs of interest, it is important to use identical processing for all probes, including the duration of AP staining or autoradiography. In all cases, comparing and contrasting a probe-specific hybridization signal with the non-specific hybridization signal from one or more control probes provides the user with some level of confidence in the anatomical pattern of gene expression observed. Beyond probe sequence design, we also rely upon anatomical replication of hybridization results within and across tissue sections/slides. Multiple tissue sections are placed per microscope slide, and if the anatomy is replicated in these tissue sections, then replicate hybridization signals should be detected in each section/slide, suggesting that the hybridization results are representative of cells expressing the miRNA being evaluated. Similarly if there is symmetry of gene expression (e.g. right and left brain hemispheres), one would predict that hybridization results would display similar anatomical symmetry thereby demonstrating an anatomical consistency of spatial expression patterns.
It is also possible to compare the expression patterns of mature miRNAs determined by miRNA ISH with the expression patterns of the corresponding miRNA primary transcripts, as detected by conventional ISH methods [18]. The expression of the mature miRNA and the primary transcript should overlap, although identical miRNAs can be encoded by multiple genes, potentially complicating ISH analysis of the primary transcripts. In addition, recent observations indicate that miRNA maturation or stability may be regulated independently of primary transcript synthesis [17, 25, 26], so that the mature miRNA expression pattern may not match the primary transcript pattern in all cases.
2.2 RNA oligonucleotide Probe Preparation
RNA oligonucleotide probes can be custom synthesized commercially (Invitrogen, Dharmacon). For nonradioactive ISH, RNA oligonucleotides are synthesized with a 5' end fluorescein modification. In most cases, gel purification of fluorescein probes as described below is essential to reduce background on tissue sections (apparently from unincorporated fluorescein-labeled nucleotide). For radioactively-labeled ISH, RNA oligonucleotides are custom synthesized without any 5' end modification (5' hydroxyl) and are enzymatically labeled at the 5' end with 33P using T4 DNA Kinase, as described below. Note that 5' fluorescein-modified RNA oligonucleotides cannot be enzymatically labeled with T4 DNA Kinase.
2.3.1. Gel purification of fluorescein-labeled RNA oligonucleotides
Load 20 μg of fluorescein-labeled RNA oligonucleotide onto an 18% polyacrylamide/TBE gel. After electrophoresis, identify the RNA oligonucleotide band by fluorescence from the incorporated fluorescein (using a standard UV transilluminator). Isolate the highest molecular weight band. Cut out RNA in smallest band possible and transfer to 100μl of diethyl pyrocarbonate (DEPC)-treated 1X phosphate buffered saline (PBS; see section 3 for descriptions of solutions). Crush gel into as small pieces as possible and transfer to a microfuge tube. Incubate overnight at 37° C. Centrifuge at ∼11,000g for 15 min and collect supernatant. To precipitate probe: add 1/10 volume DEPC-treated 3M NaOAc, 2.5 volumes 100% ethanol, and store at −20° C for 1 hr. Centrifuge at ∼11,000g for 15 min. Remove ethanol and allow pellet to air dry. Pellet will be visibly yellow. Resuspend in 10μl of RNase-free water and store at −20° C. Integrity of the purified probe can be confirmed by gel elecrophoresis using a small amount of the purified probe on an 18% polyacrylamide/TBE gel (the unpurified probe can be used for size comparison).
2.3.2. 5' end labeling of RNA oligonucleotides with 33P
Incubate 5-10 pmol of RNA oligonucleotide and ∼100 μCi of 1000 Ci/mmol 33P-γATP with T4 DNA Kinase according to the enzyme manufacture's recommendations (typically 30-45 minutes at 37° C). 33P-labeled RNA oligonucleotides are purified from unincorporated 33P-γATP using P6 BioRad Spin columns according to the manufacture's directions. Typical incorporation percentages are ∼50%. Since incorporation rates are similar across different RNA oligonucleotides, we routinely use ∼0.5 − 1.0 × 106 cpm of labeled RNA oligonucleotide per 40 μl of hybridization buffer.
2.4. Preparation of tissue sections for miRNA ISH
As with conventional mRNA ISH, tissue can be fixed at the time of collection by immersion or perfusion, or fresh-frozen tissue sections can be used. Embryos or small pieces of tissue can be fixed by overnight immersion in 4% paraformaldehyde (PFA) in PBS at 4° C. It is better to fix large adult tissues such as brain by perfusion with PFA. After fixation, tissues are rinsed in 1X PBS, dehydrated in 15% sucrose until the tissue sinks, and embedded in OCT (Fisher). Tissue sections (12 μm) are cut using a cryostat and transferred to SuperFrost/plus slides (Fisher). Slides are stored at −20° C until ISH. Alternately, for fresh-frozen tissue, tissues are collected as quickly as possible and immediately frozen in dry ice-chilled isopentane baths (−35° C). Frozen tissues are then stored at −80° C until sectioning. Frozen tissue sections (10-14 μm thick) are thaw mounted onto clean, SuperFrost microscope slides and stored at −80° C until ISH.
2.5. Preparation of Cultured Cells for miRNA ISH
Cells are plated on poly-L-Lysine and laminin-coated microscope slides. This coating helps retain cells during the ISH processing steps. Microscope slides are coated with poly-L-Lysine(10μg/ml) in 1X PBS for 3.5 hrs, followed by air drying in a laminar flow hood. Slides are subsequently rinsed three times with distilled water and stored at at 4°C (for up to two weeks). Prior to use, slides are coated with mouse Laminin (2μg/ml, Invitrogen) overnight, then washed with 1X PBS twice prior to plating cultured cells onto the slides in appropriate media. At the desired time point, cells are washed with 1X D-PBS (Invitrogen) for 5 min and fixed in PFA for 20 min. Fixed cells are rinsed three times with DEPC-treated 1X PBS and stored in DEPC-treated 1X PBS at 4°C until in situ hybridization (Cells can be stored for a few days, but we have not systematically tested whether the signal deteriorates with storage). Process for miRNA ISH as described for tissue sections below, but omit proteinase K and RNase A as indicated.
2.6. Maintenance of an RNase-free work place
RNase contamination of reagents or slide processing labware prior to hybridization can compromise any RNA in situ hybridization procedure. To ensure that all glassware, reagent bottles, tubes, slide holders, etc. are RNase free, either purchase RNase-free products (sterile disposable tubes) or clean with commercial solutions designed to inactivate RNases (e.g. RNase Away, Ambion). Similarly, maintenance of an RNase-free work environment can be vital to the generation of maximal in situ hybridization results. See [27] for additional information.
3. Preparation of solutions and reagents
3.1 DEPC-treatment of solutions
Where indicated, treat solutions with DEPC to inhibit RNases. Add DEPC to 0.1% final concentration, mix, and leave overnight at room temperature in a fume hood. Autoclave solutions the next day. Note that solutions that contain Tris cannot be treated with DEPC. We set aside a stock of Tris for use only with RNA.
3.2. PFA (4% Paraformaldehyde in 1X PBS, 50 ml)
Microwave 47.5 ml of DEPC-treated water 30 sec. Add 2 g PFA. Mix and heat with stirring. Add 50 μl of 10N NaOH. Keep at 68° with mixing until PFA dissolves completely. Add 40 μl of concentrated HCl. Add 2.5 ml 20X PBS (DEPC treated). pH should be ∼7.4. Filter the solution using a 0.45μm filter. This solution can be stored at 4 °C for up to three days. Caution: handle paraformaldehyde in a fume hood.
3.3 20X PBS (1 L)
| NaCl | 160.0 g |
| KCl | 4.0 g |
| Na2HPO4 | 28.8 g |
| KH2PO4 | 4.8 g |
Dissolve in 800 ml of water. Adjust the pH to 7.4 with HCl. Adjust the volume to one liter with water. Treat with DEPC.
3.4 Proteinase K (PK) Buffer
50mM Tris (pH 7.5) (stock 1M; cannot be DEPC treated)
5mM EDTA (stock 0.5 M ; DEPC-treated)
3.5 Proteinase K
From Roche. Dilute 66 μl of 7.5 mg/ml stock in 50ml of PK buffer just prior to use, for final concentration of 10 μg/ml.
3.6 1M Triethanolamine, pH 8.0 (10X stock)
Add 66.5 ml Triethanolamine and 20 ml concentrated HCl to 413.5 ml DEPC-treated water.
3.7 Acetic Anhydride
From Sigma (Catalog No. A6404).
3.8 Hybridization Solution (50 ml)
| Final concentration | Amount of stock solution to make 50 ml |
| 50% formamide | 25 ml formamide |
| 5X SSC | 12.5ml of 20 X SSC (see below) |
| 0.3 mg/ml yeast RNA (ICN) | 0.3 ml of 50 mg/ml stock in DEPC-treated water |
| 100 μg/ml heparin (Sigma) | 100 μl of 50 mg/ml stock in DEPC-treated water |
| 1X Denhardt's Solution | 0.5 ml of 100X stock (see below) |
| 0.1% Tween 20 | 0.5 ml of 10% Tween 20 in DEPC-treated water |
| 0.1% CHAPS | 0.5 ml of 10% CHAPS in DEPC-treated water |
| 5 mM EDTA | 0.5 ml of 0.5 M EDTA (pH 8.0) in DEPC-treated water |
| 3 μl/ml DNA random primer | 150 μl of stock (see below) |
3.9 20X SSC (1Liter) pH 7.0
| NaCl | 175.3 g |
| Sodium Citrate | 88.2 g |
Dissolve in 800ml distilled water. Adjust pH to 7.0 with HCl.
DEPC (0.1%) treat solution overnight in chemical hood. Autoclave solution next morning.
3.9 100X Denhardt's Solution
2% BSA (ICN 810661)
2% Polyvinylpyrrolidone (PVP-40)
2% Ficoll 400
Make a slurry in DEPC-treated water and then dilute to final volume. Filter sterilize and store in aliquots at −20° C.
3.10 DNA random primers
The inclusion of random 12-mer DNA oligonucleotides (custom synthesized by Invitrogen) in the hybridization solution reduces background, presumably by blocking nonspecific binding of probes. Resuspend oligonucleotides in DEPC-treated water at 0.1 nmole/μl and store in aliquots at −20° C.
3.11 RNase A Solution (10 μg/ml)
200 μl of 10 mg/ml RNase A stock (Roche) in 200 ml of 2X SSC solution (10 μg/ml).
3.12 3M TMAC solution (200 ml)
| 5 M Tetramethylammonium chloride | 120 ml |
| 10% Tween 20 | 4 ml |
| 1 M Tris pH 8.0 | 10 ml |
Add DEPC-treated water to 200 ml.
3.13 PBT Solution
1X PBS with 0.1% Triton X-100.
3.14 Sheep serum
Normal Sheep Serum (Jackson ImmunoResearch Laboratories Inc.).
3.15 Anti-fluorescein antibody
Anti-fluorescein-AP (Roche 1426338). Pre-absorb the antibody [28] before use to reduce background. To produce 1ml pre-absorbed antibody add ∼50 μl volume of embryo powder to 0.5 ml 1X PBT and incubate at 70 °C for 30 min. Vortex for 10 min. Cool mixture on ice. Add 5 μl sheep serum + 1μl antibody. Mix for 1 hr at 4°C. Centrifuge at 14,000 rpm for 4 minutes. Collect supernatant and dilute to 1 ml of 1X PBT/20% sheep serum, for a final antibody concentration of 1:1,000. Store at 4°C. (Pre-absorb antibody ONLY on the day of usage)
3.16 AP Buffer
100 mM Tris (pH 9.5)
50 mM MgCl2
100 mM NaCl
0.1% Tween 20
Add sterile water to final volume. The AP staining reaction should generate a purple precipitate on sections. If precipitate color is brown, check the pH of the AP buffer
3.17 AP substrate solution (50 ml)
| Nitro-Blue Tetrazolium Chloride (NBT, Roche) | 37.5 μl of 100 mg/ml stock |
| 5-Bromo-4-Chloro-3′-Indolyphosphate (BCIP, Roche) | 350 μl of 50 mg/ml stock |
Dilute in 50 ml of AP buffer just prior to use and mix well. Final concentrations are NBT: 75 μg/ml and BCIP: 350 μg/ml.
4. miRNA ISH Procedure
All steps are performed at room temperature unless indicated otherwise. This method is based in part on ISH methods for riboprobes described in [28].
4.1. Day 1
4.1.1a. Option 1: For sections of immersion or perfusion fixed tissues
Slides are removed from storage at −20°C and air dried at 37°C for 30 min. Slides are then placed into 4% paraformaldehyde in PBS (made fresh) for 20 min in a fume hood. Continue at step 4.1.2.
4.1.1b. Option 2: For sections of fresh frozen tissues
Sections are removed from the −80° C freezer and immediately placed into 4% paraformaldehyde in PBS (made fresh) for 20 min at room temperature in a fume hood.
4.1.2.
Wash slides two times in 1X PBS for 10 min each.
4.1.3.
Treat the slides with 10 μg/ml proteinase K for 6 min at room temperature. This step is omitted when performing ISH on cultured cells
Note: different batches of proteinase K must be titrated for best results. The proteinase K concentration and duration of this step were optimized on E14.5 mouse embryo sections. The same conditions are effective for sections from other embryonic ages, as well as sections of adult brain and retina. However, depending on fixation conditions and/or tissue age, it may be possible to increase ISH signals for some sections by optimizing the concentration of proteinase K (or the duration of the Proteinase K treatment).
4.1.4.
Wash slides in 1X PBS for 10 min.
4.1.5.
Fix slides in 4% paraformaldehyde in PBS for 15 min in fume hood.
4.1.6.
Rinse slides in DEPC-treated water.
4.1.7.
Acetic anhydride treatment: make 0.1 M TEA by diluting 20 ml 1M Triethanolamine (TEA) (pH 8.0) in 180 ml DEPC-treated water. In fume hood, add 0.5 ml acetic anhydride to 0.1 M TEA and make certain that acetic anhydride is well dispersed. Immediately submerge slides in this solution for 5 min. Repeat this step with a fresh dilution of acetic anhydride in TEA for 5 min.
4.1.8.
Wash slides in 1X PBS for 10 min.
4.1.9.
Pre-hybridization: For each slide, 200 μl of hybridization solution is added into a Secure-Seal hybridization chamber (RPI Corp. Cat no. 248867) with the gasket side up. Then the slide is carefully inverted onto the chamber such that the sections face the solution and the gasket forms a seal along the edge of the slide. Avoid bubbles. Alternately, it possible to add pre-hybrization solution directly onto the slides (covering the sections) and then use conventional coverslips or Lifterslips (Eric Scientific). Incubate 2.5 hrs at 37°C (in incubator or in a covered dish in a water bath). If coverslips/Lifterslips are used, the slides must be incubated in a sealed humidified chamber (cover bottom with filter paper soaked in 50% formamide and place slides on plastic supports above filter paper).
4.1.10a. Option 1: hybridization with fluorescein-labeled RNA oligonucleotide probes
Pre-warm hybridization solution with probe at 37°C for 10min before applying to slides. Remove hybridization chamber and discard the pre-hybridization solution. Add 200 μl of hybridization solution containing 1 μg/ml RNA oligonucleotide probe and re-apply Secure-Seal hybridization chamber as described for pre-hybridization above. Continue at step 4.1.11.
4.1.10b. Option 2: hybridization with radioactively-labeled RNA oligonucleotide probes
Pre-warm hybridization solution with probe at 37°C for 10min before applying to slides. Remove gasket/coverslip and discard pre-hybridization solution. Dilute 0.5 − 1.0 × 106 cpm of 5'-33P-labeled RNA oligonucleotide probe into 40-50 μl of hybridization buffer (For two rodent brain sections, 40-50 μl of hybridization buffer is typically sufficient.). Add solution over sections and apply coverslip/LifterSlips as described in 4.1.9). When radioactive probes are used, employ appropriate precautions for handling radioactive materials at this and subsequent steps.
4.1.11.
Hybridize overnight at 37° C in incubator or in a covered dish in a water bath (If coverslips/LifterSlips are used, slides must be incubated in a humidified chamber as described 4.1.9.). Note: the precise hybridization temperature is important; higher hybridization temperatures may decrease signal substantially.
4.2. DAY 2
4.2.1.
Remove probe. Wash in 2X SSC for 15 min at 37° C. Used fluorescein-labeled probes can be saved and re-used. Store used probes at −20° C.
4.2.2.
RNase A treatment: incubate slides in 10 μg/ml RNase A in 2X SSC for 30 min at 37°C. The RNase A treatment is not required for in situs on cultured cells. Note: the concentration of RNase A may need to be titrated depending on the batch and/or supplier.
4.2.3.
Rinse in 2X SSC twice for 15 min each.
4.2.4.
High-stringency TMAC washes: dilute 3 M TMAC + 0.2% Tween 20 in 50 mM Tris (pH 8.0) to make TMAC wash solution. TMAC is toxic: employ appropriate precautions when handling. Pre-warm the TMAC wash solution to 54° C prior to adding slides. Wash slides twice for 5 min each at 54° C, then wash slides once for 10 min at 54° C. These steps are performed in a dish in a shaking water bath. Note: the precise temperature and duration of these washes is critical, as is pre-warming the wash solution. Measure the bath temperature to confirm the temperature.
4.2.5a. Detection of radioactively-labeled probes
after TMAC washes, rinse slides once in 2x SSC for 10 min, then dehydrate the slides through a graded series of 50% to 100% ethanol. Allow slides to air dry, then expose to X-ray film (BioMax MR film, Kodak). Exposure times can vary from 18 h to 20 days, depending upon the relative abundance of each miRNA within tissue areas/regions, and must be determined empirically. High resolution autoradiographic images can be collected using photographic emulsions with potentially longer exposure times (again this must be empirically determined) followed by photographic development in standard photographic development solutions.
4.2.5b. Detection of fluorescein-labeled probes
after TMAC washes, perform the additional washing and antibody-based detection steps starting at 4.2.6.
4.2.6.
Rinse slides twice in 1X PBT for 10 min each.
4.2.7.
Blocking: incubate slides in 1X PBT/20% sheep serum for ∼1hr to reduce non-specific antibody binding.
4.2.8.
Add 200 μl of the pre-absorbed 1:1000 diluted anti-fluorescein antibody in PBT/20% sheep serum (see 3.15) to each slide. Use the Secure-Seal hybridization chambers as described for pre-hybridization (see 4.1.9). Incubate for 4hrs at room temperature.
4.2.9.
Wash slides in 1X PBT for 10 min.
4.2.10.
Wash slides in 1X PBT overnight at 4° C.
4.3. DAY 3
4.3.1.
Wash slides in AP buffer twice for ∼5 min each.
4.3.2.
Detection: remove last AP buffer wash and add AP substrate solution (make just prior to use). Signal on sections will be purple and may take 3-10 hours (or more) to develop at room temperature, depending on the probe. Probes complementary to abundant miRNAs (e.g. miR-124a) usually produce detectable signal within the first hour. When long staining times are necessary, it may improve staining to change the AP substrate solution every 3-5 hours (In addition, if the AP substrate solution becomes purple or pink it should be changed.). For all miRNA probes that we have tested, the observed signal appears cytoplasmic, not nuclear. To temporarily halt the AP reaction, the AP substrate solution can be replaced with AP buffer without NBT/BCIP (However, strong signals may continue to slowly intensify). Staining can be restarted by returning sections to the AP substrate solution. Staining is performed with the slides laid flat in plastic dishes (see section 4.5.), so that the formation of the AP precipitate can be monitored under a stereomicroscope. Minimize exposure of sections to bright light while staining; avoid shaking or mixing AP substrate solution while staining as this can inhibit precipitate formation. While it is possible to increase the rate of AP staining by using higher temperatures (30°-37° C), this may decrease the signal-to-noise ratio and is not recommended for most probes.
4.3.3.
After staining reaction is complete, wash slides twice in 1X PBS for 10 min each.
4.3.4.
Fix slides in 4% paraformaldehyde for 10 min (or longer) to completely and permanently halt the AP staining reaction. Rinse in 1X PBS.
4.3.5.
Slides can be stored without coverslips in 1X PBS at 4° C indefinitely. For analysis of staining, semi-permanently mount coverslips on slides with Aqua-Poly/mount (Polysciences, Inc. #18606). Coverslips mounted with Aqua-Poly/mount can be removed from sections by soaking in water if necessary. Slides with mounted coverslips can be stored at room temperature.
4.4.
For examples of miRNA detection using these procedures see Figure 1 (fluorescein-hapten labeled probes) and Figure 2 (33P-labeled probes).
Figure 1.
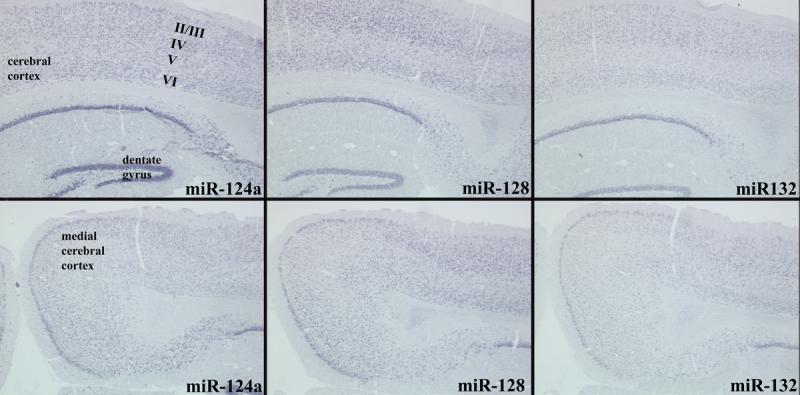
Detection of miRNAs miR-124a, miR-128a/b, and miR-132 in adult mouse brain sections. ISH was performed on fixed tissue with fluorescein-hapten labeled probes as described in the text. A-C show comparable regions of cerebral cortex and hippocampus/dentate gyrus. D-F show the medial cerebral cortex. Numbers in A indicate approximate positions of cortical layers II-VI. All three miRNAs label numerous neurons in similar patterns.
Figure 2.

Detection of miR-124a in rat and human brain sections using 33P-labeled RNA probes. A 21-nt RNA oligonucleotide complementary to miR-124a or a control 21-nt RNA oligonucleotide that contained three internal nucleotide mismatches to miR-124a were 5'-end labeled using T4 DNA kinase and γ33PATP. Approximately 1.0 × 106 cpm of labeled probe were hybridized to adult brain sections from rat (miR-124a probe: A, B; mismatch probe: E, F) or human (miR-124a probe: C, D; mismatch probe: G, H) and processed for ISH as described in the text. Slides were air dried and exposed to BioMax MR film (Kodak) for 18 hours at room temperature prior to standard development. Images were collected on a flat bed scanner at 600 dpi. Prominent hybridization signals are present in distinct layers of cerebral cortex in both rat and human tissue sections with miR-124a probe (black arrows in A-D) but not with the miR-124a mismatch probe. Signal is also visible in distinct layers of the hippocampus. Note the intense labeling in the dentate gyrus (white arrowhead in A, B), with lower hybridization signals in other subfields of the hippocampus.
4.5. Slide containers used for processing
Steps 4.1.1-4.1.5 and steps 4.3.3-4.3.4 are performed with slides held vertically in glass Coplin Jars with screw-on lids (#08-813E, Fisher Scientific) which hold five pairs of slides back to back. Steps 4.1.6-4.1.8, 4.2.1-4.2.6, and 4.2.9-10 are done with slides in black polyoxymethylene-poly-acetal plastic containers/slide racks (#441800000/#441820000, Bel-Art Products) which hold 25 slides in a horizontal position. Steps 4.3.1-2 are performed in Quadriperm disposable plastic dishes (T-2896-1, ISC BioExpress) which hold four slides placed flat in individual wells.
5. Potential problems and troubleshooting
5.1. Tissue and fixation problems
Although the described miRNA in situ procedure works with tissue fixed in PFA under a variety of conditions, as well as with fresh frozen tissue, we have observed low signal strength with some tissue preparations. Retinas or other thin tissues may have substantially reduced signals if allowed to remain in fixative for long periods of time (e.g. overnight). We suggest fixation times of ∼1 hr for mouse retinas and similar tissues. If signals are weak, we recommend testing shorter fixation times. Different ages or types of tissues, or different fixation conditions may require adjustment of the duration of Proteinase K treatment (or adjustment of the Proteinase K concentration) for optimal signal (4.1.3). Excessive Proteinase K treatment leads to poor tissue morphology (sections may tear or become detached from slides during processing), while inadequate Proteinase K treatment may reduce the in situ hybridization signal.
5.2. Temperature control
Temperature variations can increase nonspecific background and/or reduce probe-derived signals. It is important to pre-warm each solution prior to transferring slides into it. The temperature of the TMAC washes (4.2.4) is especially critical. If the wash temperature is too high, signal will be reduced, while if the temperature is too low, it will reduce specificity [19].
5.3. Sections on the same slide have different signal/background intensity
If slides are processed with the long axis oriented vertically during temperature critical steps (e.g. in Coplin Jars), we have sometimes seen differences in signal between sections on the same slide. We believe this reflects vertical temperature gradients within the container (especially if the container temperature is controlled by partial immersion in a water bath). We recommend all temperature controlled steps be performed with the long axis of the slide horizontal or flat for more uniform temperature control. It is also possible to have poor staining or high background on a section if it is covered by an air bubble during the hybridization, pre-hybrization, or antibody incubation steps.
5.4. High nonspecific background
The use of unpurified fluorescein-labeled probes may lead to high background signal levels, most likely from contaminating fluorescein-labeled nucleotides that were not incorporated into the oligonucleotide probe during synthesis. Gel purification of probes is strongly recommended (see 2.3.1). Using concentrations of probe greater than listed in 4.1.10 also may increase background in some cases.
5.5. Little or no probe-derived signal
Potential causes for a lack of signal include degradation of the probe (e.g. because of RNase contamination during probe purification), RNase contamination at other steps (see section 2.6), or problems with the RNase A treatment (4.2.2). Although rare, occasional batches of RNase A may contain contaminating ribonucleases that can degrade dsRNA. Boiling the RNase A [27] or testing a different RNase A lot can resolve this issue. Excessive levels of RNase A also can reduce the signal, so it may be necessary to titrate the amount of RNase A used. For radioactive probes, the possibility of inefficient end labeling should also be considered. Inefficient labeling of commercially synthesized fluorescein-labeled RNA oligonucleotide probes appears to be rare.
6. Concluding remarks
The major advantage of ISH for detecting miRNA expression is the ability to readily and precisely determine sites of miRNA expression, with resolution at the single cell level, or even at subcellular levels when using non-radioactive probes. The use of radioactively-labeled probes should permit better quantitative assessments of changes in miRNA levels. The large numbers of animal miRNAs and their presence in various tissues suggest that miRNA expression analyses will continue to provide insights into numerous biological processes, including development, adult organ function, and pathologies such as cancer [3, 26, 29].
Acknowledgements
We thank Ethan Ebner and Zhaoping Qin for technical assistance with radioactive ISH procedures. We thank Jenn-Yah Yu, Melissa Tippens, Kwan-Ho Chung, Paresh Patel and Anne Vojtek for helpful discussions. D.L.T. was funded by a grant from the Wilson Medical Research Foundation. R.C.T. was supported by HD043828-02S1 and DK068059-02.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bartel DP. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.He L, Hannon GJ. Nat Rev Genet. 2004;5:522–31. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 3.Mattick JS, Makunin IV. (Spec No 1).Hum Mol Genet. 2005;14:R121–32. doi: 10.1093/hmg/ddi101. [DOI] [PubMed] [Google Scholar]
- 4.Valencia-Sanchez MA, Liu J, Hannon GJ, Parker R. Genes Dev. 2006;20:515–24. doi: 10.1101/gad.1399806. [DOI] [PubMed] [Google Scholar]
- 5.Kloosterman WP, Plasterk RH. Dev Cell. 2006;11:441–50. doi: 10.1016/j.devcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 6.Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I. Cell. 2006;126:1203–17. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 7.Berezikov E, Thuemmler F, van Laake LW, Kondova I, Bontrop R, Cuppen E, Plasterk RH. Nat Genet. 2006;38:1375–7. doi: 10.1038/ng1914. [DOI] [PubMed] [Google Scholar]
- 8.Berezikov E, van Tetering G, Verheul M, van de Belt J, van Laake L, Vos J, Verloop R, van de Wetering M, Guryev V, Takada S, van Zonneveld AJ, Mano H, Plasterk R, Cuppen E. Genome Res. 2006;16:1289–98. doi: 10.1101/gr.5159906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murchison EP, Hannon GJ. Curr Opin Cell Biol. 2004;16:223–9. doi: 10.1016/j.ceb.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 10.Tolia NH, Joshua-Tor L. Nat Chem Biol. 2007;3:36–43. doi: 10.1038/nchembio848. [DOI] [PubMed] [Google Scholar]
- 11.Kloosterman WP, Wienholds E, de Bruijn E, Kauppinen S, Plasterk RH. Nat Methods. 2006;3:27–9. doi: 10.1038/nmeth843. [DOI] [PubMed] [Google Scholar]
- 12.Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, de Bruijn E, Horvitz HR, Kauppinen S, Plasterk RH. Science. 2005;309:310–1. doi: 10.1126/science.1114519. [DOI] [PubMed] [Google Scholar]
- 13.Darnell DK, Kaur S, Stanislaw S, Konieczka JK, Yatskievych TA, Antin PB. Dev Dyn. 2006;235:3156–65. doi: 10.1002/dvdy.20956. [DOI] [PubMed] [Google Scholar]
- 14.Ason B, Darnell DK, Wittbrodt B, Berezikov E, Kloosterman WP, Wittbrodt J, Antin PB, Plasterk RH. Proc Natl Acad Sci U S A. 2006;103:14385–9. doi: 10.1073/pnas.0603529103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nelson PT, Baldwin DA, Kloosterman WP, Kauppinen S, Plasterk RH, Mourelatos Z. Rna. 2006;12:187–91. doi: 10.1261/rna.2258506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ryan DG, Oliveira-Fernandes M, Lavker RM. Mol Vis. 2006;12:1175–84. [PubMed] [Google Scholar]
- 17.Wulczyn FG, Smirnova L, Rybak A, Brandt C, Kwidzinski E, Ninnemann O, Strehle M, Seiler A, Schumacher S, Nitsch R. Faseb J. 2006 doi: 10.1096/fj.06-6130com. [DOI] [PubMed] [Google Scholar]
- 18.Deo M, Yu JY, Chung KH, Tippens M, Turner DL. Dev Dyn. 2006;235:2538–48. doi: 10.1002/dvdy.20847. [DOI] [PubMed] [Google Scholar]
- 19.Wood WI, Gitschier J, Lasky LA, Lawn RM. Proc Natl Acad Sci U S A. 1985;82:1585–8. doi: 10.1073/pnas.82.6.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jacobs KA, Rudersdorf R, Neill SD, Dougherty JP, Brown EL, Fritsch EF. Nucleic Acids Res. 1988;16:4637–50. doi: 10.1093/nar/16.10.4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lim LP, Lau NC, Weinstein EG, Abdelhakim A, Yekta S, Rhoades MW, Burge CB, Bartel DP. Genes Dev. 2003;17:991–1008. doi: 10.1101/gad.1074403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwak SP, Patel PD, Thompson RC, Akil H, Watson SJ. Endocrinology. 1993;133:2344–50. doi: 10.1210/endo.133.5.8404687. [DOI] [PubMed] [Google Scholar]
- 23.Lu XY, Bagnol D, Burke S, Akil H, Watson SJ. Horm Behav. 2000;37:335–44. doi: 10.1006/hbeh.2000.1584. [DOI] [PubMed] [Google Scholar]
- 24.Griffiths-Jones S. Methods Mol Biol. 2006;342:129–38. doi: 10.1385/1-59745-123-1:129. [DOI] [PubMed] [Google Scholar]
- 25.Obernosterer G, Leuschner PJ, Alenius M, Martinez J. RNA. 2006;12:1161–7. doi: 10.1261/rna.2322506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM. Genes Dev. 2006;20:2202–7. doi: 10.1101/gad.1444406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sambrook J, MacCallum P, Russell D. 2001.
- 28.Birren SJ, Lo L, Anderson DJ. Development. 1993;119:597–610. doi: 10.1242/dev.119.3.597. [DOI] [PubMed] [Google Scholar]
- 29.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR. Nature. 2005;435:834–8. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]