

Abstract
Alzheimer’s disease presents morphologically with senile plaques, primarily made of extracellular amyloid-β (Aβ) deposits, and neurofibrillary lesions, which consist of intracellular aggregates of hyperphosphorylated tau protein. To study the in vivo induction of tau pathology, dilute brain extracts from aged Aβ-depositing APP23 transgenic mice were intracerebrally infused in young B6/P301L tau transgenic mice. Six months after the infusion, tau pathology was induced in the injected hippocampus but also in brain regions well beyond the injection sites such as the entorhinal cortex and amygdala, areas with neuronal projection to the injection site. No or only modest tau induction was observed when brain extracts from aged nontransgenic control mice and aged tau-depositing B6/P301L transgenic mice were infused. To further study Aβ-induced tau lesions B6/P301L tau transgenic mice were crossed with APP23 mice. Although Aβ deposition in double-transgenic mice did not differ from single APP23 transgenic mice, double-transgenic mice revealed increased tau pathology compared to single B6/P301L tau transgenic mice predominately in areas with high Aβ plaque load. The present results suggest that both extract-derived Aβ species and deposited fibrillary Aβ can induce the formation of tau neurofibrillary pathology. The observation that infused Aβ can trigger the tau pathology in the absence of Aβ deposits provides an explanation for the discrepancy between the neuroanatomical location of Aβ deposits and the development and spreading of tau lesions in Alzheimer’s disease brain.
Alzheimer’s disease (AD) is a neurodegenerative disorder that is pathologically characterized by senile plaques, primarily composed of extracellular deposits of amyloid-β (Aβ) peptide, and neurofibrillary tangles, composed of intracellular aggregates of hyperphosphorylated tau protein.1 The amyloid cascade hypothesis of AD pathogenesis holds that accumulation of polymerized forms of Aβ in the brain is an early and critical event that triggers a cascade of events leading to hyperphosphorylation and somatodendritic segregation of tau and formation of neurofibrillary tangles.2 Consistent with this view, previous in vivo studies have revealed an interaction between Aβ and tau pathology in vulnerable brain regions.3,4,5
Previously we have shown that intracerebral injection of dilute Aβ-containing human and murine brain extracts induces cerebral amyloidosis in young, predepositing amyloid precursor protein (APP23) transgenic mice.6 In the present study we examined whether the same Aβ-containing brain extracts would also induce tau pathology in young B6/P301L mutated tau transgenic mice and thus confirm and expand previous studies in which synthetic Aβ was intracerebrally infused in tau transgenic mice.4 However, in contrast to this latter study in which a high concentration of fibrillary synthetic Aβ was injected and gave rise to amyloid deposits in the host at the injection site, the Aβ in the brain extract was at low concentration and did not form amyloid deposits in the injected host.6 To complement this exogenous induction of tau pathology, we also crossbred B6/P301L tau transgenic mice with APP23 transgenic mice to study the induction of tau pathology by (genetic) expression of APP/Aβ. A similar cross breeding strategy has previously been reported to induce tau pathology in selected brain regions.3 Our results suggest that both the infusion of an Aβ-containing extract and the deposition of fibrillary Aβ can induce the formation of neurofibrillary pathology.
Materials and Methods
Transgenic Mice
Heterozygous B6/P301L transgenic mice and nontransgenic wild-type littermate controls were used. B6/P301L mice were obtained by backcrossing the original P301L transgenic mice (JNPL3 mice)7 with C57BL/6J mice for at least seven generations. JNPL3 mice express the shortest four-repeat (4R0N) tau with the P301L mutation under control of the mouse prion promoter. B6/P301L transgenic mice were also crossbred with APP23 transgenic mice expressing human KM670/671NL mutant APP.8
Stereotaxic Surgery
Host mice were anesthetized with a mixture of ketamine (10 mg/kg body weight) and xylazine (20 mg/kg body weight) in saline. Unilateral stereotaxic injections of 2.5 μl of brain extracts were placed with a 5-μl Hamilton syringe into the right neocortex (A/P, −2.5 mm from bregma; L, −2.0 mm; D/V, −0.8 mm) and hippocampus (A/P, −2.5 mm from bregma; L, −2.0 mm; D/V, −1.8 mm). Injection speed was 1.25 μl/minute, and the needle was kept in place for an additional 3 minutes before it was slowly withdrawn. The surgical area was cleaned with sterile saline, and the incision was sutured. Operated animals were monitored weekly in their cages for signs of behavioral changes. No seizure activity was apparent during or after the infusion. All experiments were in compliance with protocols approved by the local animal care and use committee.
Preparation of Brain Extract
Brain extracts were prepared by first dissecting out the neocortex of a 24-month-old APP23 transgenic female mouse (APP23 extract) and an aged matched female nontransgenic littermate control. Brain extract was also prepared from the brainstem and diencephalon of a 21.5-month-old B6/P301L transgenic female mouse (B6/P301L extract). Tissue samples were frozen and stored at −80°C until use. The tissue pieces were then homogenized at 10% (w/v) in sterile phosphate-buffered saline (PBS), vortexed, sonicated for 5 seconds, and centrifuged at 3000 × g for 5 minutes as previously described.6 The supernatant was recovered and immediately frozen (−80°C).
Western Blot Analysis of Brain Homogenates
To determine Aβ levels in the brain homogenates,9 samples were diluted 1 to 4 in sample buffer [0.48 mol/L Bis-Tris, 0.21 mol/L Bicine, 1.32% (w/v) sodium dodecyl sulfate, 20% (w/v) sucrose, 3.33% (v/v) 2-mercaptoethanol, 0.0053% (w/v) bromophenol blue]. Samples were then subjected to 10% Bicine-Tris 8 mol/L urea sodium dodecyl sulfate-polyacrylamide gel electrophoresis.10 Synthetic Aβ1-40 and 1-42 (Bachem, Bubendorf, Switzerland) were used as controls. Proteins were transferred onto a polyvinylidene difluoride membrane and probed with monoclonal antibody 6E10 specific to human Aβ (diluted 1:500; Covance, Emeryville, CA). The secondary antibody was horseradish peroxidase-conjugated goat anti-mouse IgG (Chemicon, Temecula, CA). Bands were visualized using SuperSignal (Pierce, Rockford, IL) and developed onto Kodak X-OMAT AR film (Eastman-Kodak, Rochester, NY).
To determine tau levels in the brain homogenates, samples were dissolved in sample buffer and run on 10% sodium dodecyl sulfate-polyacrylamide Tris-tricine gels. Proteins were electrophoretically transferred onto a polyvinylidene difluoride membrane and probed with monoclonal antibody HT7 (diluted 1:1000, recognizes amino acids 159 to 163 of tau phosphorylated and nonphosphorylated epitopes; Pierce). The secondary antibody was horseradish peroxidase-conjugated sheep anti-rabbit IgG (NA931; Amersham-Biosciences, Uppsala, Sweden). Bands were visualized using SuperSignal and developed onto Kodak X-OMAT AR film.
Histology and Immunohistochemistry
Host mice were deeply anesthetized, and they were perfused transcardially with 4% paraformaldehyde in sodium phosphate buffer (pH 7.4) and processed for paraffin embedding. Immunohistological stainings were done on 5-μm coronal paraffin sections throughout areas of interest (see quantification), according to standard procedures.11 The following antibodies were used: monoclonal AT8 antibody (1:1000; Innogenetics, Gent, Belgium) specific to tau phosphorylated at ser-202 and thr-205; polyclonal anti-Aβ antibody NT12 (1:1000; courtesy of P. Paganetti, Basel, Switzerland) or DW6 (1:1000; courtesy of D. Walsh, Dublin, UK); polyclonal antibody to GFAP (1:20,000; Sigma Aldrich, St. Louis, MO); and polyclonal antibody to Iba-1 (1:1000; Wako, Neuss, Germany). Secondary antibodies were obtained from Vector Laboratories, Burlingame, CA (Vectastain ABC kits), for peroxidase staining. In addition sections were stained histologically with silver-impregnation by the Gallyas protocol.12
Stereological Assessment of Tau Deposition
For selection of sampling sites for the estimation of tau-immunopositive area fraction in the brain regions of interest stereological sampling rules were applied.13 Every 72nd section through the hippocampus (from positions AP −1.0 mm to AP −4.0 mm from bregma), entorhinal cortex (AP −2.5 mm to −4.0 mm), sensorimotor cortex (AP −1.0 mm to AP −3.5 mm), amygdala (AP −1.0 mm to AP −3.5 mm), and brainstem (AP −4.0 mm to AP −5.0 mm) was immunostained with AT8. Therefore, eight to nine sections through the hippocampus, four to five through the entorhinal cortex, six to seven through the sensorimotor cortex and amygdala, and two to three through the brain stem were analyzed per animal. The brain regions were delineated at low magnification according to cytoarchitectural criteria.14 Within a single section, a systematic random sampling scheme was performed to capture video images for measurement of AT8-positive staining (percent area). All images were captured with an ×40 objective using a Zeiss Axioskop microscope (Zeiss, Göttingen, Germany) with a motorized x-y-z stage coupled to a Sony CCD-IRIS color camera. To analyze the individual images for AT8-positive burden, 24-bit color images (20 to 50 images per brain region) were converted to 8-bit gray scale images by using conversion algorithms associated with Adobe Photoshop CS (Adobe, San Jose, CA). Calculation of the percent area occupied by tau-immunopositive area fraction was performed using the public domain NIH ImageJ. Statistical analysis included analysis of variance and posthoc testing with the help of Statistica 5.0 (StatSoft, Tulsa, OK).
Stereological Assessment of Amyloid Deposition
To assess the burden of Aβ deposition in brains, we have used the same stereological sampling as described above for the assessment of AT8-positive deposits. Briefly, every 144th section through the hippocampus (from positions AP −1.0 mm to AP −4.0 mm from bregma), neocortex (AP −1.0 mm to AP −4.0 mm), amygdala (AP −1.0 mm to AP −3.5 mm), and brain stem (AP −4.0 mm to AP −5.5 mm) was immunostained with DW6. Therefore, four to five sections through the hippocampus and neocortex, three to four through the amygdala, and one to two through the brain stem were analyzed per animal. Using a systematic random sampling scheme, images were captured with a ×20 objective and converted to 8-bit gray scale for calculating the percent area occupied by Aβ-immunoreactive pixels. Statistical analysis included analysis of variance and posthoc testing with the help of Statistica 5.0.
Results
Tau Pathology in P301L Tau Transgenic Mice on C57BL/6J Background
On a mixed C57BL/DBA2/SW background, heterozygous P301L transgenic female mice develop tau pathology between 7 to 11 months of age, whereas disease onset in males is between 11 and 15 months.3,7 To minimize immunological consequences and to minimize genetic variability in brain extract infusion studies and cross-breeding (see below), P301L transgenic mice were backcrossed to a C57BL/6J genetic background for at least seven times (thereafter termed B6/P301L mice; see Materials and Methods). Surprisingly, B6/P301L mice revealed a substantial delay in the development of tau pathology compared to the original P301L mice.
In female B6/P301L mice intracellular tau deposition (assessed with phosphorylation-dependent AT8 antibody) was not apparent in a group of 11- to 12-month-old mice (mean age, 11.5 ± 0.3 months; n = 5; indicated is the SEM) (Figure 1, A–E). At ∼18 months of age, female B6/P301L mice became symptomatic and were unable to spread their hindlimbs when lifted by the tail. Within 1 to 3 months of disease onset, mice showed weight loss, and there were signs of dystonic posture. At that stage another group of mice (mean age, 19.8 ± 0.6 months; n = 6) was sacrificed and analyzed. Neuropathologically, similar to the original report of P301L mice on the mixed background,7 a substantial amount of hyperphosphorylated tau protein accumulated in affected neurons of the spinal cord, brain stem nuclei, and also of the hippocampus, entorhinal cortex, and amygdala (Figure 1, F–J). In the spinal cord, amygdala and brain stem (Figure 1, I and J), the majority (∼80 to 90%) of these tau lesions were Gallyas-positive similar to neurofibrillary tangles found in AD brain. These changes were followed by axonal degeneration in descending fiber tracts of the spinal cord and by neurogenic atrophy in skeletal muscles (not shown), all overall similar as described previously in P301L mice on a mixed genetic background.7
Figure 1.

Tau pathology in heterozygous P301L tau transgenic mice on the genetic C57BL6/J background (B6/P301L). A and F: Overview picture taken from an AT8-stained section shows the distribution of tau pathology in female B6/P301L mice at 12 months (A) and 21 months (F) of age. B–E and G–J are high-magnification pictures taken from A and F and reveal in the 21-month-old mice but not the 12-month-old mice a substantial amount of tau lesions in the hippocampus (B, G; top middle box in A and F), lateral entorhinal cortex (C, H; bottom right box in A and F), amygdala (D, I), and deep mesencephalic nucleus (E, J; top left box in A and F). Insets in I and J show Gallyas-positive tangles, indicating that a large subset of the accumulating tau proteins form insoluble aggregates. All panels have same magnification. Scale bar = 100 μm.
Male heterozygous B6/P301L mice did not show any clinical symptoms up to 30 months of age. Consistently, a group of 27-month-old B6/P301L mice (26.5 ± 1.1 months; n = 6) revealed only little tau pathology in the spinal cord and brainstem with no signs of tau lesions in other areas of the brain.
Characterization of Murine Brain Extracts
Brain homogenates were prepared from the neocortex of aged, Aβ-depositing APP23 mice and an aged-matched nontransgenic control littermate (Figure 2, A and B). Western blotting of the APP23 brain extract revealed Aβ levels of ∼1 to 10 ng/μl with more Aβ1-40 compared to Aβ1-42 (Figure 2D) consistent with previous findings.6,9 As an additional control, an extract from the brain stem and diencephalon of an aged B6/P301L transgenic mouse was used. By immunohistochemistry and Western blotting, the presence of tau lesions and of the shortest 4-repeat human tau isoform was demonstrated (Figure 2, C and D).
Figure 2.

Histological and biochemical analysis of the injected brain extracts. A: Aβ-immunostaining of neocortex of a 24-month-old APP23 transgenic mouse, similar to the one from which the extract was prepared, reveals extensive cerebral amyloid deposition. B: No amyloid was found in a nontransgenic age-matched littermate control brain. C: AT8-immunostaining of the brainstem of a female 21-month-old B6/P301L transgenic mouse, similar to the one from which the tau extract was prepared, reveals numerous tau-positive deposits. All panels have the same magnification. D: Left: Immunoblotting with antibody 6E10 specific to human Aβ. Lane 1, synthetic Aβ1-40 and 1-42 (1 ng/μl each); lane 2, APP23 brain extract; lane 3, nontransgenic APP23 littermate control. Right: Western blotting analysis with HT7 antibody specific to human tau. Lane 1, AD brain extract reveals the various human tau isoforms (arrowheads); lane 2, B6/P301L brain extract shows the transgenic 4R0N human isoform. Scale bar = 50 μm.
Intracerebral Injection of Aβ-Containing Brain Extract Induces Tau Pathology in B6/P301L Tau Transgenic Mice
Extracts were unilaterally injected into the right hippocampus and overlying somatosensory cortex of young (5- to 6-month-old) heterozygous B6/P301L female mice. All mice were analyzed 6 months later, ie, when host mice were 11 to 12 months of age. At that age, uninjected B6/P301L female mice have not yet developed significant tau pathology (Figure 1, A–E).
Intracerebral injection of the Aβ-rich APP23 extract induced robust deposition of tau throughout the injected hippocampus (assessed by phosphorylation-dependent AT8 antibody; Figure 3, A and B). In contrast injection of nontransgenic brain extract or PBS did not induce any tau lesions (Figure 3, C and E). Modest tau pathology was also induced with the B6/P301L extract, but this was not statistically significant (Figure 3D and Figure 4A). APP23 extracts injected into nontransgenic mice did not induce any tau deposits (Figure 3F), indicating that the expression of human tau was necessary for the induction of tau lesions by the Aβ-containing extracts. All these observations were supported by quantitative analysis (Figure 4A).
Figure 3.

Intracerebral injection of Aβ-containing mouse brain extract induces tau pathology in the hippocampus of B6/P301L tau transgenic mice. Young 5- to 6-month-old B6/P301L transgenic mice and control littermates were injected unilaterally in the hippocampus and overlying neocortex with various brain extracts and analyzed 6 months later. A: Overview picture taken from an AT8-stained section shows the pattern of induced tau deposits throughout the injected hippocampus but also entorhinal cortex (asterisk) of a B6/P301L transgenic mouse injected with the APP23 brain extract (the injected site is the right one in the panel). B: The injected hippocampal area, box in A, is shown in higher magnification. C: Injection with the nontransgenic control extract did not induce tau pathology in the hippocampus. D and E: Modest induction of tau pathology was seen with the B6/P301L extract (D), whereas no induced tau deposits were observed after PBS injection (E). F: When Aβ-containing APP23 extract was injected into a nontransgenic B6/P301L mouse, no tau lesions were induced. Scale bar = 100 μm.
Figure 4.
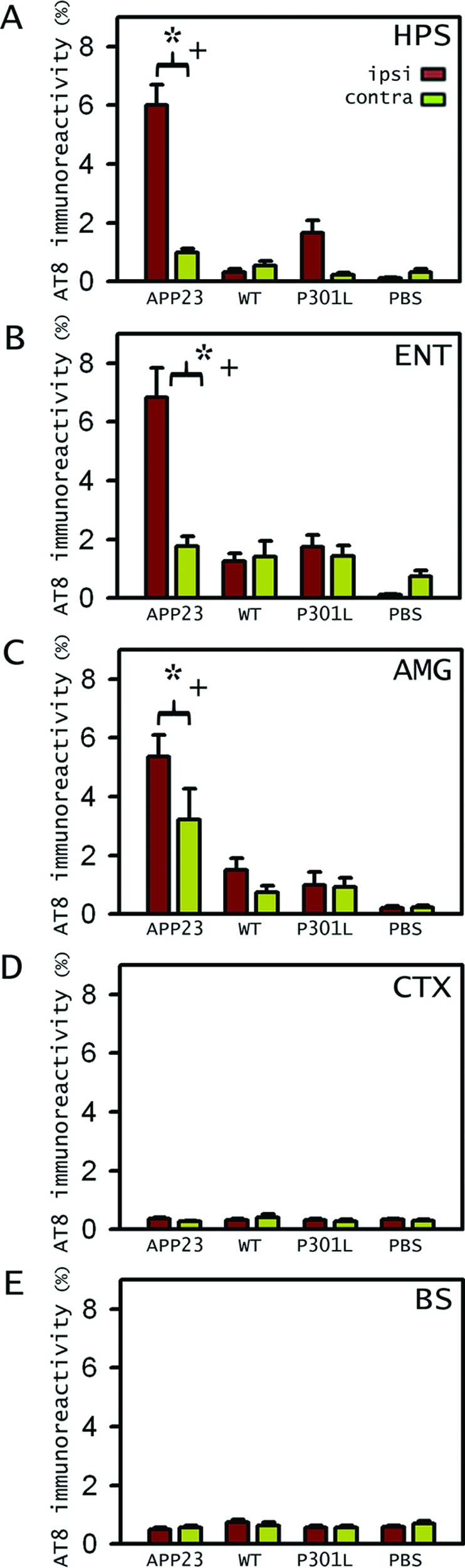
Tau pathology induction in various regions in B6/P301L tau tg mice. A: Stereological analysis of percent area occupied by AT8-positive staining (two-way analysis of variance, extract × hemisphere) in the hippocampus (HPS) confirms the significant induction of tau deposition by the APP23 extract compared to PBS (+n = 5/group; P < 0.0001; Newmann-Keuls posthoc tests; indicated is SEM) and compared to the contralateral site (*P < 0.0001). Tau induction also occurred in some brain regions away from the injection site, such as the entorhinal cortex (ENT) (B) and the amygdala (AMG) (C). Analysis again revealed significant induction of the APP23 extract compared to PBS (+n = 5/group, P < 0.0001) and to the contralateral site (*P < 0.05), which for the amygdala also showed some induction compared to PBS (P < 0.001). D and E: No induced tau pathology was found in the sensorimotor cortex (CTX) (D) and brain stem (BS) (E) (n = 5/group; P > 0.05). APP23, extract from an aged APP23 mouse brain; WT, wild-type nontransgenic extract; P301L, extract from an aged B6/P301L mouse brain.
No tau lesions were observed around the injection site of the neocortex. Tau pathology, however, was induced in brain regions farther away from the injection sites, such as the entorhinal cortex and the amygdala, but not brain stem (Figure 3A and Figure 4, B–E). In most regions tau induction appeared confined to the injected hemisphere. However, in the amygdala the induction, albeit to a lesser degree, also occurred contralateral to the injection site (Figure 4C). Immunostaining for Aβ at any time after the infusion of the Aβ-containing extracts did not reveal any Aβ deposition (data not shown), consistent with previous studies.6 Gliosis was limited to the needle track and was similar among all mice independent of whether they were injected with PBS or brain extracts.
Increased Tau Pathology in Double APP23 × B6/P301L Tau Transgenic Mice
B6/P301L tau transgenic mice were crossed with APP23 transgenic mice. Before amyloid deposition, double-transgenic B6/P301L × APP23 mice did not exhibit increased tau pathology compared to the single transgenic B6/P301L littermate control mice (results not shown). However, at the time of significant amyloid deposition, a robust increase of tau pathology (assessed by phosphorylation-dependent AT8 antibody) in the entorhinal cortex, hippocampus, and to a lower extent in the amygdala but not in brain stem regions was noted in the double-transgenic B6/P301L × APP23 mice compared to the single transgenic B6/P301L control mice. This was true for the female double-transgenic mice (21.6 ± 0.9 months old; data not shown) and particularly appreciable in male double-transgenic mice (27.6 ± 0.7 months old; Figure 5, A and C) because single male B6/P301L tau mice do not develop tau pathology up to this age (Figure 5B). In the entorhinal cortex and hippocampus, a subset (∼50%) of these induced tau lesions were Gallyas-positive (Figure 5I). Stereological analysis of the amyloid load did not reveal any differences between double B6/P301L × APP23 and single transgenic APP23 male mice. Notably, however, there was a great region-specific variability of amyloid deposition with neocortex and hippocampus revealing the highest amyloid load, followed by the amygdala and the brain stem, with the latter showing virtually no amyloid deposition (Figure 5D). Tau lesions were typically induced in the vicinity of congophilic plaques in the entorhinal cortex and hippocampus (Figure 5, E and F); however, in the amygdala there was a clear separation with induced tau lesions in the basolateral part and amyloid deposition in the dorso- and ventrolateral parts (Figure 5H). In areas not prone to develop tau pathology in B6/P301L mice, such as the piriform and somatosensory cortex, there was no or only modest tau pathology induction in the double-transgenic mice despite a high plaque load (Figure 5G). In all mice congophilic amyloid deposits were surrounded by a robust activation of microglia and astroglia (not shown) similar to previous results in APP23 mice.8,15
Figure 5.

Induced tau lesions in B6/P301L × APP23 mice. A and B: AT8-stained coronal sections show tau pathology in the entorhinal cortex and adjacent CA1 field of an aged male 28-month-old double-transgenic B6/P301L × APP23 mouse (A) whereas no pathology was observed in a male age-matched B6/P301L mouse (B). C: Stereological analysis of tau immunoreactivity (AT8 antibody) in the entorhinal cortex (ENT), hippocampus (HPS), and amygdala (AMG) revealed tau induction in B6/P301L × APP23 transgenic animals (n = 7) compared to both single transgenic B6/P301L mice (n = 7) and to APP23 littermates (n = 4) (one-way analysis of variance for genotype: *ENT, P < 0.01; HPS, P < 0.05; AMG, P < 0.001; indicated is SEM) but not in the brain stem. D: Stereological analysis of amyloid load in the neocortex (CTX), hippocampus (HPS), amygdala (AMG), and brain stem (BS) revealed no difference between the double-transgenic mice and the single APP23 transgenic littermates in any of the brain region analyzed (P > 0.05). E–H: High magnification of an adjacent section to A double immunostained for tau (AT8) and Aβ shows the distribution of tau pathology in close vicinity of amyloid deposits in the entorhinal cortex (E), and hippocampus (F) (see also boxed areas in A). In the piriform cortex (G), and dorso- and ventrolateral amygdala (asterisks in H), no significant induction of tau pathology was observed despite robust amyloid load. Notably in the basolateral part of the amygdala, substantial induction of tau lesions were observed (arrowheads in H) although no significant amyloid deposition was apparent in this area. E–G have the same magnification. I: Gallyas silver staining shows that many of the induced tau deposits in the entorhinal cortex were of fibrillary nature. Scale bars = 75 μm.
Discussion
The present study was undertaken to confirm and expand previous in vivo studies demonstrating a link between Aβ and the induction of neurofibrillary pathology.3,4 We have shown that tau pathology can be induced in the brain of young B6/P301L tau transgenic mice by a single injection of a dilute Aβ-containing extract of a transgenic APP23 mouse brain, but not by PBS, nontransgenic control extract, or extract from a transgenic B6/P301L mouse brain with tau lesions. These observations suggest that Aβ is key to an effective induction of the tau pathology.
A similar induction of tau pathology, albeit only in the amygdala, has been described after the infusion of synthetic Aβ1-42 into the somatosensory cortex and hippocampus of a tau transgenic mouse model.4 However, several important differences between the two studies emerge. 1) Gotz and colleagues4 injected fibrillary synthetic Aβ at a high concentration (1 μg/μl), and the injected Aβ was readily detectable as amyloid and persisted at the injection sites. In contrast, a highly dilute extract, with a 100- to 1000-fold lower Aβ concentration (1 to 10 ng/μl) was used in the present study. 2) Gotz and colleagues4 used as host a mouse model (albeit with the same P301L mutation) that develops tau pathology earlier (5 to 6 months of age) than the B6/P301L mouse model used in the present study. This may explain the induction of tau lesions already 3 weeks after the injection whereas in the present study analysis was done 6 months after infusion. 3) Gotz and colleagues4 reported that the induced tau lesions were Gallyas-positive whereas the majority of the induced tau lesions in the present study were Gallyas-negative and of pretangle stage. Again, this difference may be explained by the difference in susceptibility of developing neurofibrillary lesions of the two transgenic hosts or the concentration of the injected Aβ.
We have previously shown that extracts identical to the APP23 extracts have potent Aβ-amyloid-inducing activity whereas synthetic Aβ did not reveal similar activity.6 From these results we suggested that the amyloid-inducing activity in brain represents a brain-specific conformation or modification of Aβ that is different from synthetic Aβ. Thus from this latter study and the work of Gotz and colleagues,4 a dissociation of the Aβ-amyloid-inducing activity and tau lesion-inducing activity of Aβ species could be speculated, ie, synthetic Aβ is sufficient for the induction of the tau pathology but not for the induction of Aβ-amyloid.6 However, because of the above mentioned differences between the present study and that of Gotz and colleagues,4 the conclusion that synthetic Aβ and Aβ-containing brain extract are equally potent inducers of tau pathology appears premature. Thus, to rigorously test the hypothesis of a dissociation of the amyloid-inducing activity of Aβ from the neurofibrillary-inducing activity of Aβ, future studies will be necessary in which brain extract versus synthetic Aβ will be tested in the same transgenic model.
The observation that tau lesions were induced not only at the hippocampal injection site, but also in remote areas such as entorhinal cortex and amygdala, and at the contralateral sites might be explained by the axonal projections of these brain regions to the injection site.4,16 Various Aβ species are known to interact with synapses and to affect adversely their structure and function,17 which in turn may lead to retrograde signaling, damage and induction of tau lesions in the remote cell bodies. It is also possible that Aβ is taken up and transported retrogradely to the soma where Aβ interacts with tau and induces tau phosphorylation and aggregation.18,19,20 Independent of the exact mechanism, the idea of synaptic damage with retrogradely induced tau lesions might at least partly explain why induction of tau lesions was found in the entorhinal cortex and amygdala but not in the brain stem, which exhibits only diffuse projection to the injection sites. The lack of induction of tau pathology in the somatosensory cortex may simply be a result of low susceptibility of this region to form tau lesions in such tau transgenic mice.4,7,21 In addition, or alternatively to such a retrograde model of tau pathology propagation, the infused Aβ may have been transported via blood and/or via interstitial fluid pathways to the contralateral site and remote areas.22,23
Tau pathology was also induced in B6/P301L transgenic mice when crossed with APP23 mice, confirming a previous study in which the same tau transgenic mouse model, albeit on a mixed genetic background, was crossed with Tg2576 transgenic mice.3 Although in the infusion experiment, exogenously applied Aβ species have triggered the tau pathology, in the double B6/P301L × APP23 transgenic mice, it may be argued that the tau lesions were initiated by an intraneuronal (indirect) interaction of tau and Aβ, because both the Thy1 promoter (in case of the APP23 mice) and PrP promoter (in case of the B6/P301L mice) are expressed to a great extent in similar neuronal populations.24,25 However, because the induction of the tau lesions appeared concomitant with the appearance of significant cerebral amyloidosis, it is more likely that the extracellular deposition of amyloid is the trigger of the neurofibrillary pathology.
The idea of a local induction of the tau pathology by the amyloid deposits is supported by the current region-specific analysis of tau pathology induction versus amyloid load. The most robust induction of tau pathology was in the entorhinal cortex, followed by hippocampus, amygdala, and brain stem. Consistently, we found the highest plaque load in the entorhinal cortex, followed by the hippocampus, the amygdala, and brain stem. However, consistent with previous findings,3 neurofibrillary tangle-bearing neurons were not necessarily in close vicinity of the amyloid deposits exemplified in the basolateral amygdala, with a robust induction of tau pathology but virtually no amyloid deposition. These observations suggest that in the double-transgenic mice tau induction in areas without amyloid aggregates may be triggered from remote areas with amyloid deposits similar to the suggested mechanism with the Aβ-containing brain extracts.
Although the infusion experiment strongly suggests Aβ species as being the trigger of the induced tau pathology in remote areas, the mechanism of local tau induction in the crossed mice is more open for speculation. Congophilic amyloid deposition is always accompanied by a strong neuroinflammatory response15,26,27 and microglia activation has recently been linked to the progression of tau pathology in transgenic mice.28 It has also been shown that congophilic plaques induce local axonal dystrophy and synaptic abnormalities,16,29 which in turn may promote tau aggregation.
In conclusion, our results suggest that Aβ can induce neurofibrillary pathology by different mechanisms. In particular the observation that infused Aβ can trigger the tau pathology in the absence of amyloid deposits provides an explanation for the discrepancy between the neuroanatomical location of amyloid deposits, or absence thereof, and the development and spreading of tau lesions in AD brain.
Acknowledgments
We thank Michael Calhoun (Tübingen, Germany) for helpful discussions and P. Paganetti (Basel, Switzerland) and D. Walsh (Dublin, Ireland) for the generous donation of antibodies.
Footnotes
Address reprint requests to Mathias Jucker, the Department of Cellular Neurology, Hertie-Institute for Clinical Brain Research, University of Tübingen, Otfried-Müller Str.27, D-72076 Tübingen, Germany. E-mail: mathias.jucker@uni-tuebingen.de.
Supported by the Alzheimer Association (grant ZEN-06-27341 to M.J.), the German National Genome Network (grant NGFN2 to M.J.), the German Competence Network in Degenerative Dementias (grant BMBF-01GI0705 to M.J.), and the National Institutes of Health (grant R01-NS46355 to J.L.).
T.B. and F.C. contributed equally to this study.
References
- Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD, Danner S, Abramowski D, Sturchler-Pierrat C, Burki K, van Duinen SG, Maat-Schieman ML, Staufenbiel M, Mathews PM, Jucker M. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci. 2004;7:954–960. doi: 10.1038/nn1302. [DOI] [PubMed] [Google Scholar]
- Wiltfang J, Smirnov A, Schnierstein B, Kelemen G, Matthies U, Klafki HW, Staufenbiel M, Huther G, Ruther E, Kornhuber J. Improved electrophoretic separation and immunoblotting of beta-amyloid (A beta) peptides 1-40, 1-42, and 1-43. Electrophoresis. 1997;18:527–532. doi: 10.1002/elps.1150180332. [DOI] [PubMed] [Google Scholar]
- Bondolfi L, Calhoun M, Ermini F, Kuhn HG, Wiederhold KH, Walker L, Staufenbiel M, Jucker M. Amyloid-associated neuron loss and gliogenesis in the neocortex of amyloid precursor protein transgenic mice. J Neurosci. 2002;22:515–522. doi: 10.1523/JNEUROSCI.22-02-00515.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallyas F. Silver staining of Alzheimer’s neurofibrillary changes by means of physical development. Acta Morphol Acad Sci Hung. 1971;19:1–8. [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Paxinos G. New York: Academic Press,; The Mouse Brain in Stereotaxic Coordinates. 2001 [Google Scholar]
- Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol. 1999;154:1673–1684. doi: 10.1016/S0002-9440(10)65423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phinney AL, Deller T, Stalder M, Calhoun ME, Frotscher M, Sommer B, Staufenbiel M, Jucker M. Cerebral amyloid induces aberrant axonal sprouting and ectopic terminal formation in amyloid precursor protein transgenic mice. J Neurosci. 1999;19:8552–8559. doi: 10.1523/JNEUROSCI.19-19-08552.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Busciglio J, Lorenzo A, Yeh J, Yankner BA. Beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron. 1995;14:879–888. doi: 10.1016/0896-6273(95)90232-5. [DOI] [PubMed] [Google Scholar]
- Walker LC, Callahan MJ, Bian F, Durham RA, Roher AE, Lipinski WJ. Exogenous induction of cerebral beta-amyloidosis in betaAPP-transgenic mice. Peptides. 2002;23:1241–1247. doi: 10.1016/s0196-9781(02)00059-1. [DOI] [PubMed] [Google Scholar]
- Guo JP, Arai T, Miklossy J, McGeer PL. Abeta and tau form soluble complexes that may promote self aggregation of both into the insoluble forms observed in Alzheimer’s disease. Proc Natl Acad Sci USA. 2006;103:1953–1958. doi: 10.1073/pnas.0509386103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, Atzori C, Migheli A, Crowther RA, Ghetti B, Spillantini MG, Goedert M. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci. 2002;22:9340–9351. doi: 10.1523/JNEUROSCI.22-21-09340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, May PC, O’Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, DeMattos RB, Holtzman DM. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci. 2003;23:8844–8853. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 2005;28:202–208. doi: 10.1016/j.tins.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13:159–163. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci USA. 1999;96:14088–14093. doi: 10.1073/pnas.96.24.14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder AK, Ermini F, Bondolfi L, Krenger W, Burbach GJ, Deller T, Coomaraswamy J, Staufenbiel M, Landmann R, Jucker M. Invasion of hematopoietic cells into the brain of amyloid precursor protein transgenic mice. J Neurosci. 2005;25:11125–11132. doi: 10.1523/JNEUROSCI.2545-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Tsai J, Grutzendler J, Duff K, Gan WB. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci. 2004;7:1181–1183. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]