

Abstract
To investigate the nature of the chemical determinants in DNA required for nonspecific binding and bending by proteins we have created a novel DNA in which inosine–5-methylcytosine and 2,6-diaminopurine–uracil base pairs are substituted for normal base pairs in a defined DNA sequence. This procedure completely switches the patterns of the base pair H bonding and attachment of exocyclic groups. We show that this DNA binds a histone octamer more tightly than normal DNA but, surprisingly, does not alter the orientation of the sequence on the surface of the protein. However, in general, the addition or removal of DNA exocyclic groups reduces or increases, respectively, the affinity for the histone octamer. The average incremental change in binding energy for a single exocyclic group is ≈40 J/mol. The orientation of the DNA in core nucleosomes also is sensitive to the number and nature of the exocyclic groups present. Notably, substitution with the naturally occurring cytosine analogue, 5-methylcytosine, shifts the preferred rotational position by 3 bp, whereas incorporating 2,6-diaminopurine shifts it 2 bp in the opposite direction. These manipulations potentially would alter the accessibility of a protein recognition sequence on the surface of the histone octamer. We propose that exocyclic groups impose steric constraints on protein-induced DNA wrapping and are also important in determining the orientation of DNA on a protein surface. In addition, we consider the implications of the selection of A-T and G-C base pairs in natural DNA.
The local deformation of DNA by DNA-bending proteins can substantially exceed the normal conformational fluctuations of DNA free in solution. Although the interactions that determine the local sequence-dependent conformation of free DNA are relatively well understood (1–7), the nature of the chemical constraints that limit its further deformation have not been explored extensively. The nucleosome core particle in which the central 125 bp of DNA are wrapped in 1.6 superhelical turns about the histone octamer (8, 9) provides a good system to study this problem. In this particle both the major and minor grooves are compressed on the inside of the wrapped DNA and widened on the outside. A major determinant of the rotational placement of the DNA in the nucleosome is the periodic occurrence of short A/T-rich sequences in helical phase and of short G/C-rich sequences in the opposite phase. These short sequences favor local conformations with narrow and wide minor grooves, respectively, and thus together facilitate the tight wrapping of DNA (10–16).
To investigate the nature of other chemical determinants that constrain the wrapping of DNA around the histone octamer we chose a prokaryotic DNA sequence derived from the Escherichia coli tyrT promoter. Previous studies have shown that the histone octamer occupies a preferred rotational position on this DNA (10). We modified this DNA by replacing the naturally occurring bases with analogues either lacking or containing additional exocyclic groups by using a PCR-based procedure (Fig. 1) (17, 18). The modified tyrT sequence then was assembled into nucleosome particles (np) by exchange with unlabeled long chromatin (10). The PCR-based approach already has been used effectively to study the effects of modifying the exocyclic groups on the binding of DNA by the E. coli FIS protein and Drosophila HMG-D (17, 18), whereas other studies investigated the effect of exocyclic group modification on the binding of the Bacillus subtilis phage SP01 protein TF1 (19, 20).
Figure 1.

Structural formulae of the eight different purine⋅pyrimidine base pairs between the natural (A, T, G, C) and exocyclic-modified (U, I, D, M) bases used. A, T, G, and C indicate, respectively, 2′-deoxyadenosine, 2′-deoxythymidine, 2′-deoxyguanosine, and 2′-deoxycytidine, and U, I, D, and M indicate 2′-deoxyuridine, 2′-deoxyinosine, 2,6-diaminopurine-2′-deoxyriboside, and 2′-deoxy-5-methylcytidine. The deoxyribose is represented by a thick bar corresponding to its bonding at positions N9 and N1 of purines and pyrimidines, respectively. The 5-methyl and 2-amino exocyclic groups are shown in bold.
METHODS
Preparation and Radiolabeling of Modified tyrT DNA Fragments.
The protocol previously used for the synthesis of substituted DNA species by PCR amplification (17) was modified as follows. The primers used were 5′-GTTACCTTTAATCCGTTACG and 5′-GGGCTCGGGAACCCCCACCA, the 5′ termini of which were phosphorylated by using ATP and [γ-32P]ATP, respectively. A minimal amount of both primers and template DNA (5 pmol instead of 50 pmol and 0.5 fmol instead of 10 fmol, respectively) typically were used in the PCR amplification. Optimal annealing temperatures were selected between 39° and 35°C. Twenty percent glycerol and 3 mM MgCl2 (final concentrations) were added to the Taq provided buffer (Perkin–Elmer or Promega) for the synthesis of the substituted DNAs that contained inosine. The PCR products were purified by native PAGE run at 5 V/cm overnight, and, finally, the DNAs were stored at less than 10,000 Cerenkov cpm/μl. The specific activity of the PCR product was calculated.
Assembly of Nucleosome Particles.
For binding studies 5 ng (typically 3,000 Cerenkov cpm) of labeled specific DNA (160 bp) was mixed with 1.4 μg of H1-stripped long chromatin and different amounts of bulk core nucleosomal DNA (146 bp) as competitor DNA at 1 M NaCl. The salt was then gradually diluted to 0.1 M in steps of 0.1 M salt by using HE buffer (20 mM Hepes/0.1 mM EDTA, pH 7.5) plus 0.01% Nonidet P-40 as dilution buffer. Assembly analyses of np were carried out by 5% PAGE in the same HE buffer and at the same temperature. Typically the gel was run at 6 mA for approximately 6 hr. A salt dilution protocol (10) was used for preparation of nucleosomal particles for hydroxyl-radical footprinting. Labeled tyrT DNA fragment [0.5 μg (1.5–4 × 106 Cerenkov cpm)] was mixed with 5 μg of nucleosomal core particles (ncp) in 1 M NaCl/Hepes buffer (HE buffer without EDTA) and in the absence of competitor DNA. ncp were dialyzed against HE buffer plus 0.1 M NaCl before use. H1-stripped long chromatin, bulk core nucleosomal DNA, and ncp were purified from chicken erythrocytes as described (21, 22). The apparent difference between the free-energy change on binding of the DNA containing modified nucleotides (m) with that of unmodified DNA (N) at the defined experimental conditions (ΔΔG°m−N) was calculated using the formula ΔΔG°m−N ≈ −RT ln (α°m/α°N), where R is the gas constant, T is the absolute temperature, and α° is the apparent dissociation constant at the defined experimental conditions. α° values were calculated as ratios of background subtracted densities in the np complex to corresponding densities in unbound DNA as reported in previous studies (14). Integrated band densities were quantified by using Molecular Dynamics PhosphorImager and software.
Hydroxyl-Radical Cleavage.
The conditions for hydroxyl-radical cleavage used in the experiment shown (Fig. 4) were as follows: 100 μM (NH4)2Fe(SO4)2, 200 μM EDTA, 0.012% H2O2, 1 mM sodium ascorbate, 80 mM NaCl, 17.5 mM Hepes (pH 7.5), up to 65 nM specific DNA, plus 65 μg/ml of chicken erythrocyte ncp in samples containing specific DNA as assembled np. The cleavage was performed at 4°C for 30 min for assembled np and 5 min for naked DNA. The reaction was stopped with thiourea and EDTA both at a final concentration of 10 mM, glycerol was added to 5%, and the samples were loaded onto a submerged 0.7% agarose gel containing HE buffer to purify the bona fide assembled np from free DNA and possible aggregates and to remove reaction salts. Gel electrophoresis was at 4°C, 3 V/cm for typically 14 hr until the free 160 bp DNA had migrated about 15 cm.
Figure 4.


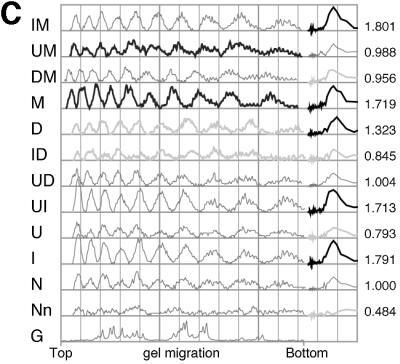
Exocyclic groups affect the rotational positioning of nucleosomal particles (nps) in vitro. Nps were assembled onto 5′ labeled tyrT DNA fragments as described. All the samples were then subjected to cleavage by hydroxyl radical (24, 25), and the assembled nps with labeled specific DNA were purified by agarose gel electrophoresis. Then, DNAs were extracted and aliquots from the same sample were loaded onto different denaturing polyacrylamide gels. Typically, at least three np assembly reactions were performed for each DNA. Representative gel images are shown in A and B. The gel shown in A Left was a 6% polyacrylamide gel containing 8.3 M urea/1× TBE and contains 25% formamide to avoid band compression and thus facilitate the estimation of shifts in the rotational settings assumed by the different substituted DNAs on the histone octamer. All the other gels shown lacked formamide and also were cast by using wedge spacers (Bio-Rad) to highlight the periodicity and corresponding amplitudes of the hydroxyl-radical cleavage pattern. The nucleotide substitutions are specified as in Figs. 1 and 2. Images in A and B were spliced together using the full gel (A Left), a relevant part of gels (B), or a relevant lane from the same gel (A Right). For ease of comparison some substituted DNA species are included in more than one gel. In particular, the gel images in A and B are ordered to show the differences between substituted DNAs that adopt opposite rotational settings [D and M in lanes 8 and 9 (A) and ID and MU in lanes 12 and 13 (B); see also C]. n indicates the cleavage pattern of free DNA, and G indicates marker tracks showing the positions of guanine nucleotides in the sequence by using Maxam and Gilbert reaction. The asterisk in B indicates an artifactual band not generated by hydroxyl-radical cleavage. C shows scans of the samples shown in A Left. On the right-hand side of C the traces of the main harmonic of the Fourier transform are shown adjacent to the corresponding scan. The maximum amplitude values for each DNA from the scan shown were normalized to that of the unsubstituted DNA and are shown on the right-hand ordinate. The scale on the abscissa is in arbitrary units used by the geltrak program and is related to the gel-migration distance (26).
RESULTS
Addition of Exocyclic Group of DNA Reduces and Their Removal Increases the Affinity for Histone Octamer.
To determine whether substitutions with modified nucleotides influenced the affinity of the DNA molecule for the histone octamer, a series of titration experiments was carried out in the presence of different concentrations of competitor DNA (Fig. 2). These experiments revealed two major trends. First, we observed a progressive change in affinity with increasing extents of substitution (Fig. 3). Second, at each level of substitution, removal of exocyclic groups from either the major or minor grooves increased, and their addition decreased, affinity compared with normal DNA. For example, replacement of all G residues with inosine (I DNA) and of all T residues with uridine (U DNA) increased affinity. By contrast, addition of an extra exocyclic group into either groove by replacement of all A residues by 2,6-diaminopurine (D DNA) or of all C residues by 5-methyl cytosine (M DNA) decreased affinity. The relative affinities of the different singly substituted DNA molecules for the histone octamer fall in the order: I DNA ∼ U DNA > normal DNA > M DNA > D DNA. The same qualitative pattern also was observed for all the doubly substituted DNA species with maximal differences for the substitutions that respectively removed (UI DNA) or added (DM DNA) all exocyclic groups in the major or minor grooves. In every case the dominant effect on binding was whether an exocyclic group was added or removed. Neither the location of the exocyclic substitution, i.e., major or minor groove, nor any consequent change in the pattern of hydrogen bonding affected the affinity to any substantial degree.
Figure 2.

Addition of DNA exocyclic groups reduces and their removal increases the affinity of the tyrT-containing DNA fragment for histone octamer as determined by a competitor titration experiment using bandshift assays. 5′ end-labeled 160-bp tyrT DNA fragments (17) containing natural or modified bases (Fig. 1) were produced according to a PCR amplification procedure (described in ref. 17 with modifications) designed to minimize the concentration of end-labeled specific DNA in the assembling mixture and to increase its specific activity for binding studies and hydroxyl-radical footprinting, respectively. Nucleosome particles (nps) were assembled on labeled specific DNA by using a salt-dilution protocol (10) with H1-stripped long chromatin acting as a histone octamer donor and different amounts of bulk core-nucleosomal DNA as competitor DNA. Representative gels of the reconstitution of core particles at 22°C from all 16 substituted variants of tyrT are shown. Letters above gels refer to the type and number of substitutions present (see Fig. 1 for reference and abbreviations). Corresponding to numbers 1, 2, 3, 4, 5, and 6 below the gels, 11, 8, 5, 2, 1, and 0 μg of competitor DNA were added before salt dilution to a constant amount of labeled specific DNA and H1-stripped long chromatin. A control experiment also was carried out by salt dilution on DNA alone (lane 0). Positions of 160-bp tyrT DNA fragment either as complexed with histone octamer (np) or unbound DNA (free) are indicated. N indicates unsubstituted DNA.
Figure 3.

Difference between the free-energy change (ΔΔGm−N°) at 22°C for binding of the substituted (m) and unsubstituted (N) DNA species to the histone. The number of bases substituted is plotted on the abscissa (i.e., 0 substitution: unmodified DNA). Abbreviations for modified DNAs are as for Fig. 1. The median values of the mono-, di-, and trisubstituted DNA species also are plotted (thick line). Data from at least three gels for each DNA were averaged to determine the free-energy difference.
The difference in binding energy between DNA lacking exocyclic groups (UI DNA) and DNA containing a full complement of exocyclic groups (DM DNA) is approximately 11.5 kJ/mol, corresponding to an ≈100-fold change in affinity. Thus, on average the addition of a single exocyclic group decreases the binding energy by roughly 40 J/mol per exocyclic group. We emphasize that this value represents an average of substitutions throughout the tyrT DNA molecule and we think it likely that, in a similar manner to substitutions affecting the affinity of HMG-D (18), the incremental affinity on substitution will be position-dependent. In comparison, Widlund et al. (23) found a range of approximately 5 kJ/mol between normal DNA and a TATA repeat DNA that forms highly stable nucleosome core particles, whereas Shrader and Crothers (14, 15) measured a difference of 8.4 kJ/mol between bulk nucleosomal DNA and their best artificial nucleosome-positioning sequence (TG5). We note that the binding energy of the unsubstituted tyrT DNA is 14.3 kJ/mol lower than the similar TG8 sequence (15).
Exocyclic Groups Affect the Rotational Positioning of Nucleosome Particles.
We then determined the effects of the different substitutions on the rotational positioning of the DNA in the nucleosome core particle. In these experiments the DNA bound to histone octamers was exposed to cleavage by hydroxyl radicals (Fig. 4). We first confirmed that the unsubstituted tyrT DNA adopted a preferred rotational setting on the histone octamer (Fig. 4A). However, detailed analysis of the modulated pattern of hydroxyl-radical cleavage suggested that the core particles contained similar amounts of two settings separated by ≈2 bp. Removal of the 5-methyl group from the major groove (U DNA) did not change the preferred rotational position relative to normal DNA. However, removal of the 2-amino group from the minor groove in both the single-substituted (I DNA) and the double-substituted UI DNA preferentially selected one of the rotational settings of the unsubstituted DNA. In contrast, addition of a 2-amino group in the minor groove (D DNA) changed the rotational positioning by a 2- to 3-bp shift toward the 5′ end compared with unsubstituted DNA. In a similar manner to the addition of an exocyclic group to the minor groove, addition of a pyrimidine 5-methyl group to the major groove (M DNA) also changed the rotational positioning compared with normal DNA. However, in contrast to D DNA, the rotational position of M DNA is shifted 3 bp toward the 3′ end relative to unsubstituted DNA. In other words, the rotational settings of the M- and D-substituted DNAs are in opposite phases. With IM DNA, where every residue in the major groove has a 5-methyl group and no 2-amino groups are found in the minor groove, the rotational setting is similar to that for M DNA. Simply relocating the methyl groups within the major groove, as in UM DNA, also resulted in a similar, preferred rotational position to that for M DNA, although irregularities in the apparent helical repeat alter the relative phases in certain positions (see Fig. 4B, toward the top of the gel). We note, however, that substitutions that remove exocyclic groups from either D- or M-substituted DNA on the opposite groove (i.e., I versus M and U versus D) shift the preferred rotational setting back slightly toward that adopted on unmodified DNA, whereas substitutions that remove those groups on the same groove (i.e., I versus D and U versus M) maintain the phase opposition observed for D and M with a small shift of ≈1 bp in the 5′ direction (Fig. 4 A and C).
These experiments show that removal of the exocyclic groups has only slight effects on the rotational positioning of the DNA molecules. However, addition of extra exocyclic groups, by incorporation of D or M into the DNA molecules, alters the rotational positioning significantly. To determine which of these groups had a dominant effect on the rotational positioning we incorporated both these modified nucleotides into the DNA and observed (Fig. 4 A and C) that the major setting adopted by DM DNA was the same as for M incorporation alone. The triple- and tetra-substituted DNA species showed slight variations in rotational setting, especially between the triple-substituted species substituted with UIM or UID and UDM or IDM. Notably, the rotational setting of the tetra-substituted DNA was essentially the same as that of the unsubstituted DNA (Fig. 4B). We emphasize that the effects of different substitutions on rotational positioning strongly depend on the context of the substitution. For example, whereas the single M and D substitutions result in significant and opposite shifts in rotational position, both M and D substitution in the context of U + I substitution (yielding the triple UIM and UID substitutions) have little effect on rotational positioning (Fig. 4B).
Exocyclic Group Changes the Amplitude of Hydroxyl-Radical Cleavage Profile.
In addition to their effects on the rotational setting the exocyclic groups also changed the amplitude of the periodic modulations of hydroxyl-radical cleavage. We have quantitated this amplitude by Fourier transformation (Fig. 5) and find that certain substitutions, notably I, UI, M, MI, and D increase this quantity very substantially whereas others, especially U, DI, and DM, decrease it even though a preferred position is still discernible. We note that the increase in amplitude consequent on I substitution also is apparent in the DNase I cleavage pattern of this DNA on the surface of the histone octamer (data not shown). The data also indicate a trend toward higher amplitudes with an increasing extent of substitution.
Figure 5.
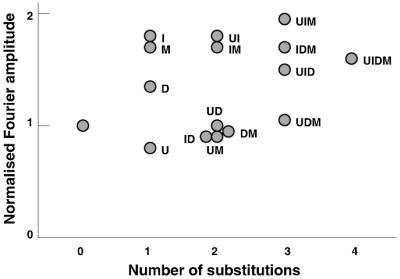
Normalized Fourier amplitudes of hydroxyl-radical cleavage profiles for different substituted DNAs. The Fourier transform was calculated from densitometric profiles of the hydroxyl-radical cleavage patterns. Only the normalized amplitudes of the main harmonic are shown. The number of substitutions is specified on the abscissa as in Fig. 3. The values shown are the average of two determinations.
DISCUSSION
The wrapping of DNA by the histone octamer essentially is a mechanical deformation that is modulated by base-stacking interactions. The exocyclic groups of DNA would be expected to affect the mechanics of bending in two ways. First, their presence could exercise constraints on the compressibility of both the major and the minor grooves (27), more especially at extreme, protein-induced deformations. Second, addition or loss of the purine 2-amino group, by altering the number of hydrogen bonds in a base pair, also will affect the rotational freedom of the substituted bases about the long axis of the base pair and consequently alter the range of available base-stacking interactions (28).
The first studies to address the effects of exocyclic groups on nucleosome formation showed that structurally intact core particles could be reconstituted with phage T4 DNA in which all the cytosine residues are substituted with glucosylated 5-hydroxymethyl cytosine (29). We observe experimentally that both the presence and location of exocyclic groups have a substantial influence on the affinity for the histone octamer. Addition of exocyclic groups decreases and their removal increases affinity. It must be remembered that the changes in free energy of histone octamer–DNA association that we have observed are global values averaging the small changes at many individual positions, some of which even could be of opposite sign.
Previous work has shown that removal or addition of these exocyclic groups influenced the binding of two other DNA-bending proteins: FIS, which binds in the major groove (17), and HMG-D, which binds in the minor groove. Notably, substitutions in the noncontacted groove of DNA had substantial effects on DNA-binding affinity for both these proteins. For HMG-D, the greatest effects of exocyclic group removal in the noncontacted groove occurred at the inferred position of maximum curvature (18). In the nucleosome core particle the majority of protein–DNA contacts occur between charged amino acid side chains and the sugar–phosphate backbone (9). Arginine residues penetrate at certain locations into the minor groove, and there is a sole example of a side-chain–base contact in the major groove. Because of the thermodynamic equivalence of major- and minor-groove substitutions and the disparity between histone contacts in the major and minor grooves, we think it unlikely that direct protein–DNA contacts are significantly influenced by changing the exocyclic groups.
The relative changes in FIS and HMG-D affinity resulting from the substitutions are qualitatively similar to those found here for the histone octamer. This implies that the effects on affinity reflect changes in a general physicochemical property of the DNA rather than some specific effect on nucleosome formation. We suggest that the simplest explanation of the data is that addition of exocyclic groups decreases and their removal increases the local deformability of DNA by maximizing and minimizing, respectively, the steric resistance to bending. Such resistance would occur when the DNA bend compressed a groove so that adjacent exocyclic groups could not be accommodated in their normal conformation. Our observation that DAP-substituted tyrT DNA circularizes at a significantly slower rate than unsubstituted DNA (A.M. and J. Morgan, unpublished observations) is fully in accord with this notion.
The notion that DNA deformability is a major determinant of histone octamer affinity is supported by the observation that when present in multiple repeats, the most deformable of the DNA trinucleotides, CAG/CTG (11, 30), forms hyperstable core particles (31). Similarly natural DNA sequences enriched in other deformable trinucleotides have a high affinity for the histone octamer relative to bulk chromosomal DNA (23).
We have assessed the positioning of the reconstituted core particle by two criteria: the positions of maximum cleavage by hydroxyl radical and the amplitude of periodic modulations in cleavage frequency. The former indicates the preferred rotational setting and the latter is influenced both by the precision of positioning, i.e., the preference for occupation of a unique rotational setting, and by the facility of DNA wrapping. A decrease in the precision of positioning should increase the number and variety of sites available to the histone octamer, possibly leading to an increase in affinity. We would also expect those substitutions that decrease affinity but increase amplitude to restrict the available rotational settings and those substitutions with the opposite effects to increase the choice of rotational settings. Conversely, substitutions that increase a preexisting global anisotropic flexibility would be expected to facilitate wrapping by enabling close protein–DNA contacts and increase both affinity and amplitude without changing the rotational setting.
A major conclusion from these experiments is that the rotational setting of the histone octamer on tyrT DNA is highly sensitive to the nature and number of exocyclic groups in the molecule. Addition of either a pyrimidine 5-methyl group (M) in the major groove or a purine 2-amino group (D) in the minor groove changes the rotational setting of DNA on nucleosome core particles and also increases the amplitude of cleavage modulations. Conversely, removal of exocyclic groups from either the major or the minor grooves or from both does not substantially change the average preferred rotational setting. In general, for single substitutions the greatest shifts of rotational setting are observed when the exocyclic “load” is increased, i.e., when the occupation of the major and minor grooves together by exocyclic groups is greater than that for normal DNA. However, whereas total removal of the 5-methyl group (U) results in loss of precision of positioning, total removal of the 2-amino group (I) increases precision. One explanation for the observed changes in rotational setting on addition of exocyclic groups is that steric factors constrain the available settings. The potential for local steric clashes would be most pronounced at positions where groove compression was greatest. The rotational position adopted then would be that which minimized steric clash between adjacent exocyclic groups and thus would be strongly sequence-dependent. The poor precision of positioning of U-substituted DNA is also fully consistent with this explanation. Here, loss of the pyrimidine 5-methyl group would eliminate the potential for steric clashes in the major groove and consequently reduce the sequence dependence of positioning. In other words, increased local flexibility would be accompanied by a loss of selectivity.
Steric considerations alone do not explain the effect of certain of the I-substitutions on the cleavage amplitude. We observe that nucleosomal particles assembled with I-, UI-, and IM-substituted DNAs all possess high amplitudes of hydroxyl-radical cleavage modulation. In addition, these same DNA species are characterized by an increased apparent intrinsic curvature, possibly because of the stacking of an I-C or I-M base pair on adjacent A-T pairs, thus creating the structural equivalent of a short dA:dT tract (27, 32–34). Indeed, recent crystal structures have shown that single I-C base pairs sandwiched between A-T pairs in the sequence AIA exhibit the high propeller twist normally characteristic of A-T but not G-C base pairs (32). The resultant conformations of the AI and IA base steps thus are very similar to that of an AA step. Consequently, the increased amplitude observed with the I-, IM-, and UI-substituted DNAs is most likely to be a result of an increased global anisotropic flexibility facilitating DNA wrapping. Strikingly, when the potential for this stacking interaction is abolished by the substitution of 2,6-diaminopurine for adenine, inosine substitution decreases rather than increases the cleavage amplitude (Fig. 5, ID DNA).
The conservation of rotational setting in the tetrasubstituted DNA, in which the patterns of the base pair H bonding and attachment of exocyclic groups are switched relative to unsubstituted DNA, shows that the directionality of the anisotropic flexibility is relatively insensitive to the number of hydrogen bonds per base pair and to the nature of the exocyclic groups. Although the nature of the spine of hydration in the minor groove is known to be affected by the presence of the purine 2-amino group (1), the thermodynamic equivalence of major and minor groove substitutions suggests that any effects on flexibility attributable to changes in the water spines are unlikely to be a major factor. In free DNA the preferred conformations of base steps are determined by van der Waals and electrostatic interactions between adjacent base pairs (1–7). The electrostatic interactions are dominated by a large dipole on G-C pairs contrasted with a diffuse distribution of charge on A-T base pairs. The analogues used for the substitution of G-C pairs in this study conserve the patch of negative charge formed by N1 and O6 of guanine and thus are likely to conserve the direction of the dipole in G-C pairs (2). This would result, in turn, in the conservation of the induced slide at GG and GC steps, and hence the bendability (2, 35, 36). However, because the methyl group acts as an electron donor we would expect the addition of a pyrimidine 5-methyl group to decrease the magnitude of the dipole on a G-C base pair and the consequent contribution of this dipole to the slide component of base-step conformation (2, 35, 36). By the same argument, the T → U substitution involving the loss of a 5-methyl group should increase the dipole on an A-T base pair. Similarly, the loss of a purine 2-amino group from a G-C base pair and its addition to an A-T base pair would be expected to reduce and increase the respective dipoles (35, 36). These changes in the magnitude of the dipole and the stereochemistry will tend, on average, to favor a narrower minor groove for altered “G-C” base pairs and a wider minor groove for altered “A-T” base pairs. For the singly and doubly substituted DNAs we cannot discern any straightforward correlation with such a projected change in dipole moment. However, the general trends toward higher affinity and greater precision in positioning for the tri- and tetrasubstituted DNA species would be consistent with such an effect. Overall, we conclude that the rotational setting adopted by the substituted DNA species on the histone octamer depends both on simple steric effects and also on base-stacking interactions. It seems likely that the steric effects are sequence-dependent.
Of the base analogues that we have studied only 5-methyl cytosine occurs naturally. Methylation of CpG dinucleotides normally is associated with repression of transcription (37, 38). Although previous in vitro studies (39, 40) found little, if any, effect of CpG methylation on nucleosome positioning, Davey et al. (41) demonstrated recently that methylation of a set of contiguous CpG dinucleotides, normally located close to the nucleosome dyad, excludes reconstitution at this position. This result is consistent with the observations and interpretation reported here. Our data also suggest that 5-methylation of an appropriately located cytosine(s) may have the potential to alter the rotational (and necessarily the translational) setting of a core particle and therefore change the accessibility of protein-binding sequences to their ligands.
We conclude that with increasing protein-induced DNA bending the steric constraints imposed by the exocyclic groups in both the major and minor grooves become more energetically dominant and affect both the formation and positioning of the nucleosome core particle. In addition, these effects may be modulated by changes in base-stacking interactions. Natural DNA contains A-T and G-C base pairs yet our experiments show that DNA with I-M and D-U base pairs is wrapped efficiently by the histone octamer. Why then should A-T and G-C base pairs be apparently preferred by natural selection? We note that for A-T base pairs both the base-stacking interactions and the distribution of exocyclic groups favor a narrow minor groove whereas for G-C base pairs both favor a wide minor groove. Together these characteristics would allow bendability to be combined with specificity.
Acknowledgments
We thank Gianluca Fulgenzi and Judy Smith for computer help in the hydroxyl-radical pattern analysis, the Cancer Research Campaign and the International Association for Cancer Research for support to M.W., and the European Commission for grants to both M.W. (Grant BMH4-CT96–0848) and A.T. (Grant ERBCHRXT940447).
ABBREVIATIONS
- np
nucleosome particle
- ncp
nucleosome core particle
- U
2′-deoxyuridine
- I
2′-deoxyinosine
- D
2,6-diaminopurine2′-deoxyriboside
- M
2′-deoxy-5-methylcytidine
References
- 1.Yanagi K, Privé G G, Dickerson R E. J Mol Biol. 1991;217:201–214. doi: 10.1016/0022-2836(91)90620-l. [DOI] [PubMed] [Google Scholar]
- 2.Hunter C A. J Mol Biol. 1993;230:1025–1054. doi: 10.1006/jmbi.1993.1217. [DOI] [PubMed] [Google Scholar]
- 3.Young M A, Ravishanker G, Beveridge D L, Berman H M. Biophys J. 1995;68:2454–2468. doi: 10.1016/S0006-3495(95)80427-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gorin A A, Zhurkin V B, Olson W K. J Mol Biol. 1995;247:34–48. doi: 10.1006/jmbi.1994.0120. [DOI] [PubMed] [Google Scholar]
- 5.El Hassan M A, Calladine C R. J Mol Biol. 1996;259:95–103. doi: 10.1006/jmbi.1996.0304. [DOI] [PubMed] [Google Scholar]
- 6.Hagerman K R, Hagerman P J. J Mol Biol. 1996;260:207–223. doi: 10.1006/jmbi.1996.0393. [DOI] [PubMed] [Google Scholar]
- 7.Hunter C A, Lu X-J. J Mol Biol. 1997;265:603–619. doi: 10.1006/jmbi.1996.0755. [DOI] [PubMed] [Google Scholar]
- 8.Richmond T J, Finch J T, Rushton B, Rhodes D, Klug A. Nature (London) 1984;311:532–537. doi: 10.1038/311532a0. [DOI] [PubMed] [Google Scholar]
- 9.Luger K, Mäder A W, Richmond R K, Sargent D L, Richmond T J. Nature (London) 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 10.Drew H R, Travers A A. J Mol Biol. 1985;186:773–790. doi: 10.1016/0022-2836(85)90396-1. [DOI] [PubMed] [Google Scholar]
- 11.Satchwell S C, Drew H R, Travers A A. J Mol Biol. 1986;191:659–675. doi: 10.1016/0022-2836(86)90452-3. [DOI] [PubMed] [Google Scholar]
- 12.Travers A A, Klug A. Philos Trans R Soc London B. 1987;317:537–561. doi: 10.1098/rstb.1987.0080. [DOI] [PubMed] [Google Scholar]
- 13.Drew H R, McCall M J, Calladine C R. In: DNA Topology and Its Biological Effects. Cozzarelli N R, Wang J C, editors. Cold Spring Harbor, NY: Cold Spring Harbor Lab. Press; 1990. pp. 1–56. [Google Scholar]
- 14.Shrader T E, Crothers D M. Proc Natl Acad Sci USA. 1989;86:7418–7422. doi: 10.1073/pnas.86.19.7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shrader T E, Crothers D M. J Mol Biol. 1990;216:69–84. doi: 10.1016/S0022-2836(05)80061-0. [DOI] [PubMed] [Google Scholar]
- 16.Lowary P T, Widom J. Proc Natl Acad Sci USA. 1997;94:1183–1188. doi: 10.1073/pnas.94.4.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bailly C, Waring M J, Travers A A. J Mol Biol. 1995;253:1–7. doi: 10.1006/jmbi.1995.0530. [DOI] [PubMed] [Google Scholar]
- 18.Bailly C, Payet D, Travers A A, Waring M J. Proc Natl Acad Sci USA. 1996;93:13623–13628. doi: 10.1073/pnas.93.24.13623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grove A, Galeone A, Mayol L, Geiduschek E P. J Mol Biol. 1996;260:120–125. doi: 10.1006/jmbi.1996.0386. [DOI] [PubMed] [Google Scholar]
- 20.Grove A, Figueiredo M L, Galeone A, Mayol L, Geiduschek E P. J Biol Chem. 1997;272:13084–13087. doi: 10.1074/jbc.272.20.13084. [DOI] [PubMed] [Google Scholar]
- 21.Lutter L C. J Mol Biol. 1978;124:391–420. doi: 10.1016/0022-2836(78)90306-6. [DOI] [PubMed] [Google Scholar]
- 22.Drew H R, Calladine C R. J Mol Biol. 1987;206:451–463. doi: 10.1016/0022-2836(87)90333-0. [DOI] [PubMed] [Google Scholar]
- 23.Widlund, H. R., Cao, H., Simonsson, S., Magnusson, E., Simonsson, T., Nielsen, P. E., Kahn, J. D., Crothers, D. M. & Kubista, M. J. Mol. Biol. 267, 807–817. [DOI] [PubMed]
- 24.Tullius T D, Dombroski B A. Science. 1985;230:679–681. doi: 10.1126/science.2996145. [DOI] [PubMed] [Google Scholar]
- 25.Price M A, Tullius T D. Methods Enzymol. 1992;212:194–219. doi: 10.1016/0076-6879(92)12013-g. [DOI] [PubMed] [Google Scholar]
- 26.Smith J, Singh M. BioTechniques. 1996;20:1082–1087. doi: 10.2144/96206bc01. [DOI] [PubMed] [Google Scholar]
- 27.Diekmann S, von Kitzing E, McLaughlin L W, Ott J, Eckstein F. Proc Natl Acad Sci USA. 1987;84:8257–8261. doi: 10.1073/pnas.84.23.8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calladine C R, Drew H R. Understanding DNA. 2nd Ed. London: Academic; 1997. [Google Scholar]
- 29.McGhee J D, Felsenfeld G. J Mol Biol. 1982;158:685–698. doi: 10.1016/0022-2836(82)90254-6. [DOI] [PubMed] [Google Scholar]
- 30.Brukner I, Sanchez R, Suck D, Pongor S. EMBO J. 1995;14:1812–1818. doi: 10.1002/j.1460-2075.1995.tb07169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J H, Griffith J. Genomics. 1995;25:570–573. doi: 10.1016/0888-7543(95)80061-p. [DOI] [PubMed] [Google Scholar]
- 32.Shatzky-Schwartz M, Arbuckle N D, Eisenstein M, Rabinovich D, Bareket-Somish A, Luis B F, Shakked Z. J Mol Biol. 1997;267:595–623. doi: 10.1006/jmbi.1996.0878. [DOI] [PubMed] [Google Scholar]
- 33.Koo H-S, Crothers D M. Biochemistry. 1987;26:3745–3748. doi: 10.1021/bi00386a070. [DOI] [PubMed] [Google Scholar]
- 34.Hagerman P J. Biochemistry. 1990;29:1980–1983. doi: 10.1021/bi00460a003. [DOI] [PubMed] [Google Scholar]
- 35.Gallego J, Luque F J, Orozco M, Burgos C, Alvarez-Builla J, Rodrigo M M, Gago F. J Med Chem. 1994;37:1602–1609. doi: 10.1021/jm00037a010. [DOI] [PubMed] [Google Scholar]
- 36.Gallego J, Pascual-Teresa A R, Pisabarro M T, Gago F. In: QSAR and Molecular Modelling: Concepts, Computational Tools and Applications. Sanz F, Giraldo J, Manaut F, editors. Barcelona: Prous Science Publishers; 1995. pp. 274–281. [Google Scholar]
- 37.Bird A P. Nature (London) 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 38.Siegfried Z, Cedar H. Curr Biol. 1997;7:305–307. doi: 10.1016/s0960-9822(06)00144-8. [DOI] [PubMed] [Google Scholar]
- 39.Englander E W, Wolffe A P, Howard B H. J Biol Chem. 1993;268:19565–19573. [PubMed] [Google Scholar]
- 40.Nightingale K, Wolffe A P. J Biol Chem. 1995;270:4197–4200. doi: 10.1074/jbc.270.9.4197. [DOI] [PubMed] [Google Scholar]
- 41.Davey C, Pennings S, Allan J. J Mol Biol. 1997;267:276–288. doi: 10.1006/jmbi.1997.0899. [DOI] [PubMed] [Google Scholar]