

Abstract
The process of programmed cell death, or apoptosis, is generally characterized by distinct morphological characteristics and energy-dependent biochemical mechanisms. Apoptosis is considered a vital component of various processes including normal cell turnover, proper development and functioning of the immune system, hormone-dependent atrophy, embryonic development and chemical-induced cell death. Inappropriate apoptosis (either too little or too much) is a factor in many human conditions including neurodegenerative diseases, ischemic damage, autoimmune disorders and many types of cancer. The ability to modulate the life or death of a cell is recognized for its immense therapeutic potential. Therefore, research continues to focus on the elucidation and analysis of the cell cycle machinery and signaling pathways that control cell cycle arrest and apoptosis. To that end, the field of apoptosis research has been moving forward at an alarmingly rapid rate. Although many of the key apoptotic proteins have been identified, the molecular mechanisms of action or inaction of these proteins remain to be elucidated. The goal of this review is to provide a general overview of current knowledge on the process of apoptosis including morphology, biochemistry, the role of apoptosis in health and disease, detection methods, as well as a discussion of potential alternative forms of apoptosis.
Keywords: Apoptosis, programmed cell death, intrinsic/extrinsic pathway, granzyme A/B, perforin, autophagy
Introduction
The term apoptosis (a-po-toe-sis) was first used in a now-classic paper by Kerr, Wyllie, and Currie in 1972 to describe a morphologically distinct form of cell death, although certain components of the apoptosis concept had been explicitly described many years previously (Kerr et al., 1972; Paweletz, 2001; Kerr, 2002). Our understanding of the mechanisms involved in the process of apoptosis in mammalian cells transpired from the investigation of programmed cell death that occurs during the development of the nematode Caenorhabditis elegans (Horvitz, 1999). In this organism 1090 somatic cells are generated in the formation of the adult worm, of which 131 of these cells undergo apoptosis or “programmed cell death.” These 131 cells die at particular points during the development process, which is essentially invariant between worms, demonstrating the remarkable accuracy and control in this system. Apoptosis has since been recognized and accepted as a distinctive and important mode of “programmed” cell death, which involves the genetically determined elimination of cells. However, it is important to note that other forms of programmed cell death have been described and other forms of programmed cell death may yet be discovered (Formigli et al., 2000; Sperandio et al., 2000; Debnath et al., 2005).
Apoptosis occurs normally during development and aging and as a homeostatic mechanism to maintain cell populations in tissues. Apoptosis also occurs as a defense mechanism such as in immune reactions or when cells are damaged by disease or noxious agents (Norbury and Hickson, 2001). Although there are a wide variety of stimuli and conditions, both physiological and pathological, that can trigger apoptosis, not all cells will necessarily die in response to the same stimulus. Irradiation or drugs used for cancer chemotherapy results in DNA damage in some cells, which can lead to apoptotic death through a p53-dependent pathway. Some hormones, such as corticosteroids, may lead to apoptotic death in some cells (e.g., thymocytes) although other cells are unaffected or even stimulated.
Some cells express Fas or TNF receptors that can lead to apoptosis via ligand binding and protein cross-linking. Other cells have a default death pathway that must be blocked by a survival factor such as a hormone or growth factor. There is also the issue of distinguishing apoptosis from necrosis, two processes that can occur independently, sequentially, as well as simultaneously (Hirsch, 1997; Zeiss, 2003). In some cases it’s the type of stimuli and/or the degree of stimuli that determines if cells die by apoptosis or necrosis. At low doses, a variety of injurious stimuli such as heat, radiation, hypoxia and cytotoxic anticancer drugs can induce apoptosis but these same stimuli can result in necrosis at higher doses. Finally, apoptosis is a coordinated and often energy-dependent process that involves the activation of a group of cysteine proteases called “caspases” and a complex cascade of events that link the initiating stimuli to the final demise of the cell.
Morphology of Apoptosis
Light and electron microscopy have identified the various morphological changes that occur during apoptosis (Hacker, 2000). During the early process of apoptosis, cell shrinkage and pyknosis are visible by light microscopy (Kerr et al., 1972). With cell shrinkage, the cells are smaller in size, the cytoplasm is dense and the organelles are more tightly packed. Pyknosis is the result of chromatin condensation and this is the most characteristic feature of apoptosis. On histologic examination with hematoxylin and eosin stain, apoptosis involves single cells or small clusters of cells. The apoptotic cell appears as a round or oval mass with dark eosinophilic cytoplasm and dense purple nuclear chromatin fragments (Figure 1). Electron microscopy can better define the subcellular changes. Early during the chromatin condensation phase, the electron-dense nuclear material characteristically aggregates peripherally under the nuclear membrane although there can also be uniformly dense nuclei (Figures 2A, 2B).
Figure 1.
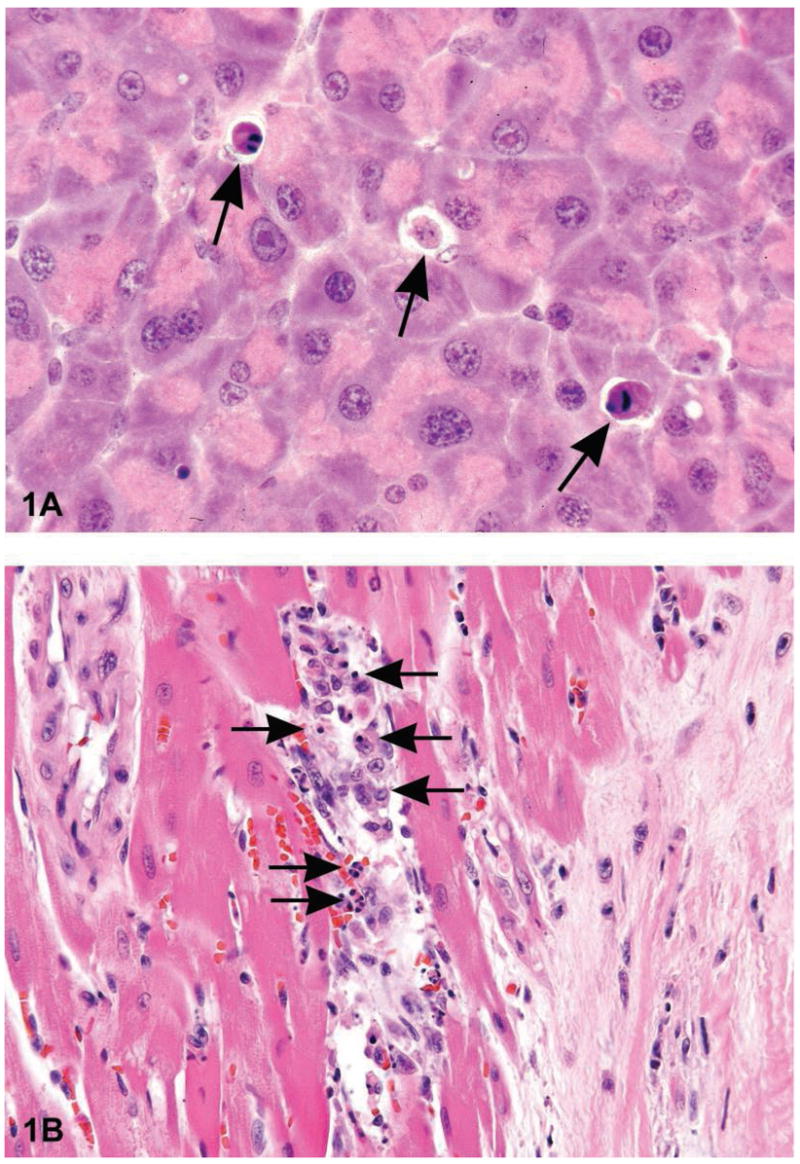
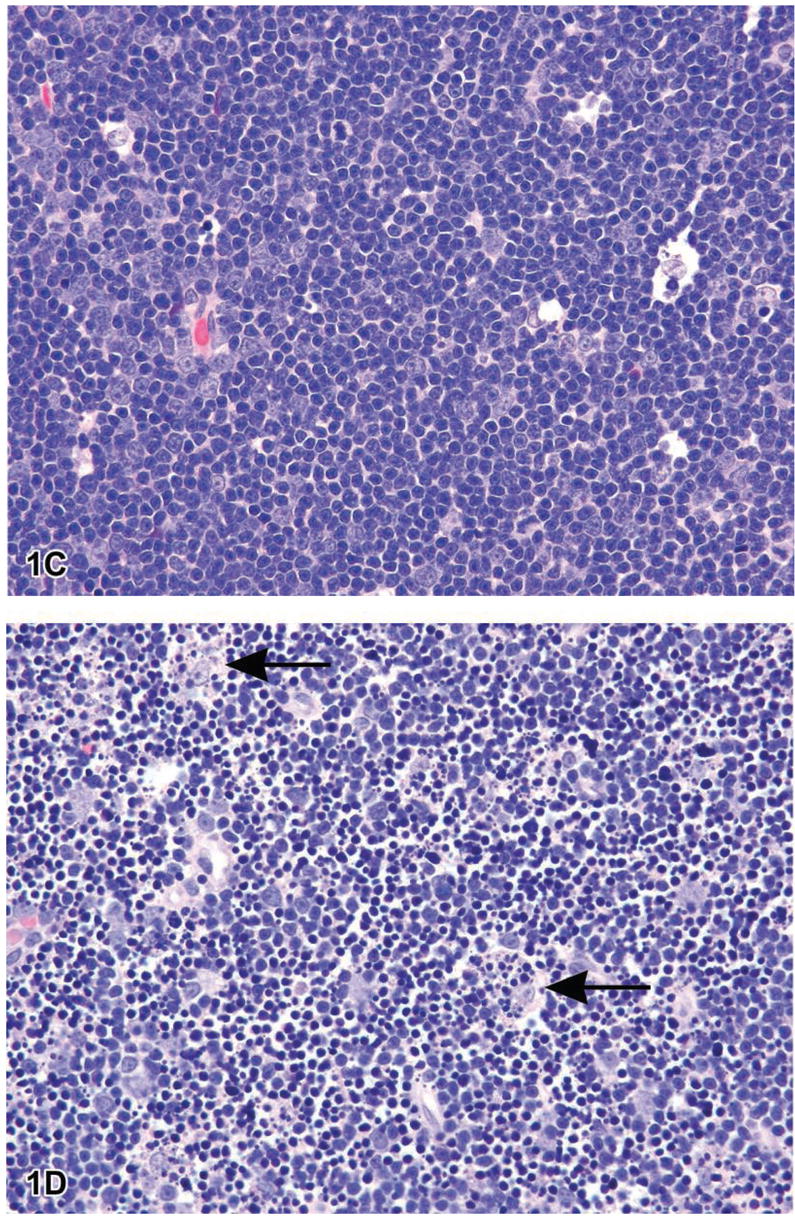
Figure 1A is a photomicrograph of a section of exocrine pancreas from a B6C3F1 mouse. The arrows indicate apoptotic cells that are shrunken with condensed cytoplasm. The nuclei are pyknotic and fragmented. Note the lack of inflammation. Figure 1B is an image of myocardium from a 14 week-old rat treated with ephedrine (25 mg/kg) and caffeine (30 mg/kg). Within the interstitial space there are apoptotic cells with condensed cytoplasm, condensed and hyperchromatic chromatin and fragmented nuclei (long arrows). Admixed with the apoptotic bodies are macrophages, some with engulfed apoptotic bodies (arrowheads) (Howden et al., 2004, by permission of the American Journal of Physiology). Figure 1C is a photomicrograph of normal thymus tissue from a control Sprague–Dawley rat for comparison. Figure ID illustrates sheets of apoptotic cells in the thymus from a rat that was treated with dexamethasone to induce lymphocyte apoptosis. Under physiological conditions, apoptosis typically affects single cells or small clusters of cells. However, the degree of apoptosis in this treated rat is more severe due to the amount of dexamethasone administered (1 mg/kg bodyweight) and the time posttreatment (12 hours). The majority of lymphocytes are apoptotic although there are a few interspersed cells that are morphologically normal and most likely represent lymphoblasts or macrophages. The apoptotic lymphocytes are small and deeply basophilic with pyknotic and often-fragmented nuclei. Macrophages are present with engulfed cytoplasmic apoptotic bodies (arrows). H&E.
Figure 2.
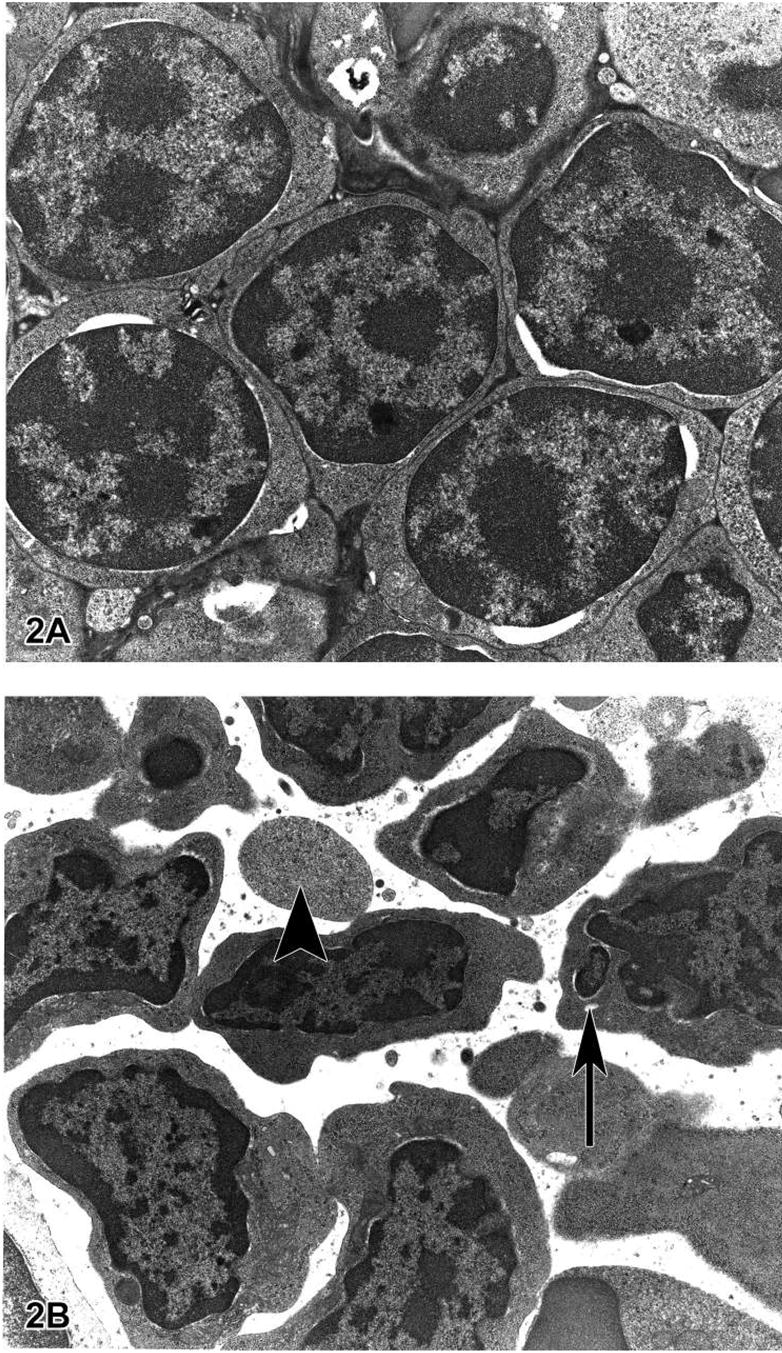
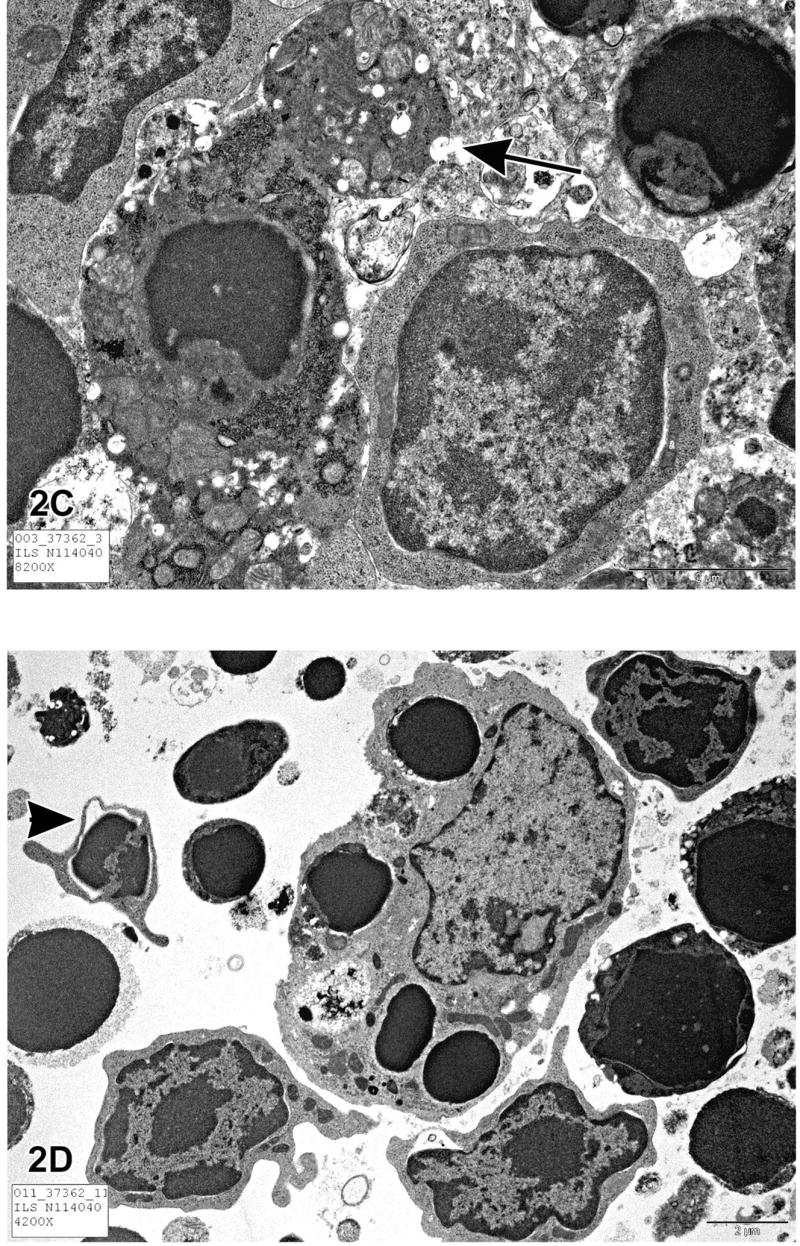
Figure 2A is a transmission electron micrograph (TEM) of the normal thymus tissue depicted in Figure 1C. The lymphocytes are closely packed, have large nuclei and scant cytoplasm. Figure 2B is a TEM of apoptotic thymic lymphocytes in an early phase of apoptosis with condensed and peripheralized chromatin. The cytoplasm is beginning to condense and the cell outlines are irregular. The arrow indicates a fragmented section of nucleus and the arrowhead most likely indicates an apoptotic body that seems to contain predominantly cytoplasm without organelles or nuclear material. Figure 2C illustrates an apoptotic lymphocyte in the process of “budding” or extrusion of membrane-bound cytoplasm containing organelles (arrow). Once the budding has occurred, this extruded fragment will be an “apoptotic body.” These apoptotic bodies are membrane-bound and thus do not release cytoplasmic contents into the interstitium. Macrophages or other adjacent healthy cells subsequently engulf the apoptotic bodies. For these reasons, apoptosis does not incite an inflammatory reaction. Figure 2D is a TEM of a section of thymus with lymphocytes in various stages of apoptosis. The large cell in the center of the photomicrograph is a macrophage with engulfed intracytoplasmic apoptotic bodies. This macrophage is also called a “tingible body macrophage.” The arrowhead indicates a lymphocyte in an advanced stage of apoptosis with nuclear fragmentation.
Extensive plasma membrane blebbing occurs followed by karyorrhexis and separation of cell fragments into apoptotic bodies during a process called “budding.” Apoptotic bodies consist of cytoplasm with tightly packed organelles with or without a nuclear fragment (Figure 2C). The organelle integrity is still maintained and all of this is enclosed within an intact plasma membrane. These bodies are subsequently phagocytosed by macrophages, parenchymal cells, or neoplastic cells and degraded within phagolysosomes (Figure 2D). Macrophages that engulf and digest apoptotic cells are called “tingible body macrophages” and are frequently found within the reactive germinal centers of lymphoid follicles or occasionally within the thymic cortex. The tingible bodies are the bits of nuclear debris from the apoptotic cells. There is essentially no inflammatory reaction associated with the process of apoptosis nor with the removal of apoptotic cells because: (1) apoptotic cells do not release their cellular constituents into the surrounding interstitial tissue; (2) they are quickly phagocytosed by surrounding cells thus likely preventing secondary necrosis; and, (3) the engulfing cells do not produce anti-inflammatory cytokines (Savill and Fadok, 2000; Kurosaka et al., 2003).
Distinguishing Apoptosis from Necrosis
The alternative to apoptotic cell death is necrosis, which is considered to be a toxic process where the cell is a passive victim and follows an energy-independent mode of death. But since necrosis refers to the degradative processes that occur after cell death, it is considered by some to be an inappropriate term to describe a mechanism of cell death. Oncosis is therefore used to describe a process that leads to necrosis with karyolysis and cell swelling whereas apoptosis leads to cell death with cell shrinkage, pyknosis, and karyorrhexis. Therefore the terms “oncotic cell death” and “oncotic necrosis” have been proposed as alternatives to describe cell death that is accompanied by cell swelling, but these terms are not widely used at this time (Majno and Joris, 1995; Levin et al., 1999).
Although the mechanisms and morphologies of apoptosis and necrosis differ, there is overlap between these two processes. Evidence indicates that necrosis and apoptosis represent morphologic expressions of a shared biochemical network described as the “apoptosis-necrosis continuum” (Zeiss, 2003). For example, two factors that will convert an ongoing apoptotic process into a necrotic process include a decrease in the availability of caspases and intracellular ATP (Leist et al., 1997; Denecker et al., 2001). Whether a cell dies by necrosis or apoptosis depends in part on the nature of the cell death signal, the tissue type, the developmental stage of the tissue and the physiologic milieu (Fiers et al., 1999; Zeiss, 2003).
Using conventional histology, it is not always easy to distinguish apoptosis from necrosis, and they can occur simultaneously depending on factors such as the intensity and duration of the stimulus, the extent of ATP depletion and the availability of caspases (Zeiss, 2003). Necrosis is an uncontrolled and passive process that usually affects large fields of cells whereas apoptosis is controlled and energy-dependent and can affect individual or clusters of cells. Necrotic cell injury is mediated by two main mechanisms; interference with the energy supply of the cell and direct damage to cell membranes.
Some of the major morphological changes that occur with necrosis include cell swelling; formation of cytoplasmic vacuoles; distended endoplasmic reticulum; formation of cytoplasmic blebs; condensed, swollen or ruptured mitochondria; disaggregation and detachment of ribosomes; disrupted organelle membranes; swollen and ruptured lysosomes; and eventually disruption of the cell membrane (Kerr et al., 1972; Majno and Joris, 1995; Trump et al., 1997). This loss of cell membrane integrity results in the release of the cytoplasmic contents into the surrounding tissue, sending chemotatic signals with eventual recruitment of inflammatory cells. Because apoptotic cells do not release their cellular constituents into the surrounding interstitial tissue and are quickly phagocytosed by macrophages or adjacent normal cells, there is essentially no inflammatory reaction (Savill and Fadok, 2000; Kurosaka et al., 2003). It is also important to note that pyknosis and karyorrhexis are not exclusive to apoptosis and can be a part of the spectrum of cytomorphological changes that occurs with necrosis (Cotran et al., 1999). Table 1 compares some of the major morphological features of apoptosis and necrosis.
Table 1.
Comparison of morphological features of apoptosis and necrosis.
| Apoptosis | Necrosis |
|---|---|
| Single cells or small clusters of cells | Often contiguous cells |
| Cell shrinkage and convolution | Cell swelling |
| Pyknosis and karyorrhexis | Karyolysis, pyknosis, and karyorrhexis |
| Intact cell membrane | Disrupted cell membrane |
| Cytoplasm retained in apoptotic bodies | Cytoplasm released |
| No inflammation | Inflammation usually present |
Is Apoptosis an Irreversible Process?
Many of the genes that control the killing and engulfment processes of programmed cell death have been identified, and the molecular mechanisms underlying these processes have proven to be evolutionarily conserved (Metzstein et al., 1998). Until recently, apoptosis has traditionally been considered an irreversible process with caspase activation committing a cell to death and the engulfment genes serving the purpose of dead cell removal. However, the uptake and clearance of apoptotic cells by macrophages may involve more than just the removal of cell debris. Hoeppner et al. have shown that blocking engulfment genes in C. elegans embryos enhances cell survival when cells are subjected to weak pro-apoptotic signals (Hoeppner et al., 2001).
Reddien et al. demonstrated that, in C. elegans, mutations that cause partial loss of function of killer genes allow the survival of some cells that are programmed to die via apoptosis, and mutations in engulfment genes enhance the frequency of this cell survival. Moreover, mutations in engulfment genes alone allowed the survival and differentiation of some cells that were otherwise destined to die via apoptosis (Reddien et al., 2001). These findings suggest that genes that mediate corpse removal can also function to actively kill cells. In other words, the engulfing cells may act to ensure that cells triggered to undergo apoptosis will die rather than recover after the initial stages of death.
In vertebrates, there is some evidence of a potential role for macrophages in promoting the death of cells in some tissues. Elimination of macrophages in the anterior chamber of the rat eye resulted in the survival of vascular endothelial cells that normally undergo apoptosis (Diez-Roux and Lang, 1997). Other studies have demonstrated that inhibition of macrophages can disrupt the remodeling of tissues in the mouse eye or in the tadpole tail during regression; two processes that involve apoptosis (Lang and Bishop, 1993; Little and Flores, 1993). Geske and coworkers (2001) demonstrated that early p53-induced apoptotic cells can be rescued from the apoptotic program if the apoptotic stimulus is removed. Their research suggests that DNA repair is activated early in the p53-induced apoptotic process and that this DNA repair may be involved in reversing the cell death pathway in some circumstances.
Mechanisms of Apoptosis
The mechanisms of apoptosis are highly complex and sophisticated, involving an energy-dependent cascade of molecular events (Figure 3). To date, research indicates that there are two main apoptotic pathways: the extrinsic or death receptor pathway and the intrinsic or mitochondrial pathway. However, there is now evidence that the two pathways are linked and that molecules in one pathway can influence the other (Igney and Krammer, 2002). There is an additional pathway that involves T-cell mediated cytotoxicity and perforin-granzyme-dependent killing of the cell. The perforin/granzyme pathway can induce apoptosis via either granzyme B or granzyme A. The extrinsic, intrinsic, and granzyme B pathways converge on the same terminal, or execution pathway. This pathway is initiated by the cleavage of caspase-3 and results in DNA fragmentation, degradation of cytoskeletal and nuclear proteins, cross-linking of proteins, formation of apoptotic bodies, expression of ligands for phagocytic cell receptors and finally uptake by phagocytic cells. The granzyme A pathway activates a parallel, caspase-independent cell death pathway via single stranded DNA damage (Martinvalet et al., 2005).
Figure 3.
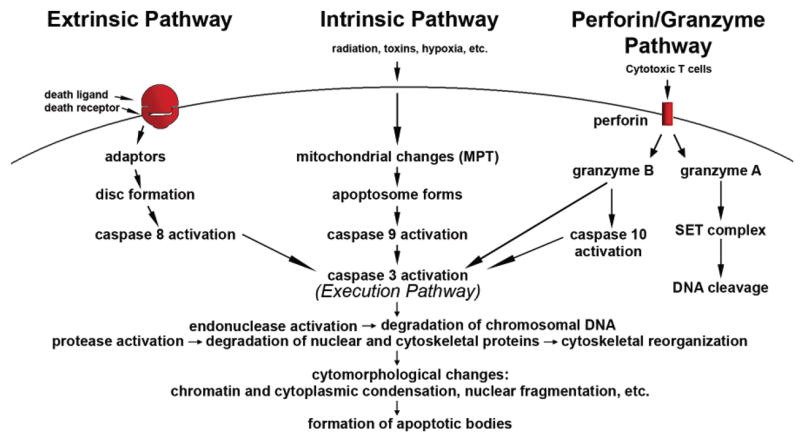
Schematic representation of apoptotic events. The two main pathways of apoptosis are extrinsic and intrinsic as well as a perforin/granzyme pathway. Each requires specific triggering signals to begin an energy-dependent cascade of molecular events. Each pathway activates its own initiator caspase (8, 9, 10) which in turn will activate the executioner caspase-3. However, granzyme A works in a caspase-independent fashion. The execution pathway results in characteristic cytomorphological features including cell shrinkage, chromatin condensation, formation of cytoplasmic blebs and apoptotic bodies and finally phagocytosis of the apoptotic bodies by adjacent parenchymal cells, neoplastic cells or macrophages.
Biochemical Features
Apoptotic cells exhibit several biochemical modifications such as protein cleavage, protein cross-linking, DNA breakdown, and phagocytic recognition that together result in the distinctive structural pathology described previously (Hengartner, 2000). Caspases are widely expressed in an inactive proenzyme form in most cells and once activated can often activate other procaspases, allowing initiation of a protease cascade. Some procaspases can also aggregate and autoactivate. This proteolytic cascade, in which one caspase can activate other caspases, amplifies the apoptotic signaling pathway and thus leads to rapid cell death.
Caspases have proteolytic activity and are able to cleave proteins at aspartic acid residues, although different caspases have different specificities involving recognition of neighboring amino acids. Once caspases are initially activated, there seems to be an irreversible commitment towards cell death. To date, ten major caspases have been identified and broadly categorized into initiators (caspase-2,-8,-9,-10), effectors or executioners (caspase-3,-6,-7) and inflammatory caspases (caspase-1,-4,-5) (Cohen, 1997; Rai et al., 2005). The other caspases that have been identified include caspase-11, which is reported to regulate apoptosis and cytokine maturation during septic shock, caspase-12, which mediates endoplasmic-specific apoptosis and cytotoxicity by amyloid-β, caspase-13, which is suggested to be a bovine gene, and caspase-14, which is highly expressed in embryonic tissues but not in adult tissues (Hu et al., 1998; Nakagawa et al., 2000, Koenig et al., 2001; Kang et al., 2002).
Extensive protein cross-linking is another characteristic of apoptotic cells and is achieved through the expression and activation of tissue transglutaminase (Nemes et al., 1996). DNA breakdown by Ca2+-and Mg2+-dependent endonucleases also occurs, resulting in DNA fragments of 180 to 200 base pairs (Bortner et al., 1995). A characteristic “DNA ladder” can be visualized by agarose gel electrophoresis with an ethidium bromide stain and ultraviolet illumination.
Another biochemical feature is the expression of cell surface markers that result in the early phagocytic recognition of apoptotic cells by adjacent cells, permitting quick phagocytosis with minimal compromise to the surrounding tissue. This is achieved by the movement of the normal inward-facing phosphatidylserine of the cell’s lipid bilayer to expression on the outer layers of the plasma membrane (Bratton et al., 1997). Although externalization of phosphatidylserine is a well-known recognition ligand for phagocytes on the surface of the apoptotic cell, recent studies have shown that other proteins are also be exposed on the cell surface during apoptotic cell clearance. These include Annexin I and calreticulin.
Annexin V is a recombinant phosphatidylserine-binding protein that interacts strongly and specifically with phosphatidylserine residues and can be used for the detection of apoptosis (Van Engeland et al., 1998; Arur et al., 2003). Calreticulin is a protein that binds to an LDL-receptor related protein on the engulfing cell and is suggested to cooperate with phosphatidylserine as a recognition signal (Gardai et al., 2005). The adhesive glycoprotein, thrombospondin-1, can be expressed on the outer surface of activated microvascular endothelial cells and, in conjunction with CD36, caspase-3-like proteases and other proteins, induce receptor-mediated apoptosis (Jimenez et al., 2000).
Extrinsic Pathway
The extrinsic signaling pathways that initiate apoptosis involve transmembrane receptor-mediated interactions. These involve death receptors that are members of the tumor necrosis factor (TNF) receptor gene superfamily (Locksley et al., 2001). Members of the TNF receptor family share similar cyteine-rich extracellular domains and have a cytoplasmic domain of about 80 amino acids called the “death domain” (Ashkenazi and Dixit, 1998). This death domain plays a critical role in transmitting the death signal from the cell surface to the intracellular signaling pathways. To date, the best-characterized ligands and corresponding death receptors include FasL/FasR, TNF-α/TNFR1, Apo3L/DR3, Apo2L/DR4 and Apo2L/DR5 (Chicheportiche et al., 1997; Ashkenazi et al., 1998; Peter and Kramer, 1998; Suliman et al., 2001; Rubio-Moscardo et al., 2005).
The sequence of events that define the extrinsic phase of apoptosis are best characterized with the FasL/FasR and TNF-α/TNFR1 models. In these models, there is clustering of receptors and binding with the homologous trimeric ligand. Upon ligand binding, cytplasmic adapter proteins are recruited which exhibit corresponding death domains that bind with the receptors. The binding of Fas ligand to Fas receptor results in the binding of the adapter protein FADD and the binding of TNF ligand to TNF receptor results in the binding of the adapter protein TRADD with recruitment of FADD and RIP (Hsu et al., 1995; Grimm et al., 1996; Wajant, 2002). FADD then associates with procaspase-8 via dimerization of the death effector domain. At this point, a death-inducing signaling complex (DISC) is formed, resulting in the auto-catalytic activation of procaspase-8 (Kischkel et al., 1995).
Once caspase-8 is activated, the execution phase of apoptosis is triggered. Death receptor-mediated apoptosis can be inhibited by a protein called c-FLIP which will bind to FADD and caspase-8, rendering them ineffective (Kataoka et al., 1998; Scaffidi, 1999). Another point of potential apoptosis regulation involves a protein called Toso, which has been shown to block Fas-induced apoptosis in T cells via inhibition of caspase-8 processing (Hitoshi et al., 1998). Table 2 lists the major extrinsic pathway proteins with common abbreviations and some of the alternate nomenclature used for each protein.
Table 2.
Extrinsic pathway proteins, abbreviations, and alternate nomenclature.
| Abbreviation | Protein Name | Select Alternate Nomenclature |
|---|---|---|
| TNF-α | Tumor necrosis factor alpha | TNF ligand, TNFA, cachectin |
| TNFR1 | Tumor necrosis factor receptor 1 | TNF receptor, TNFRSF1A, p55 TNFR, CD120a |
| FasL | Fatty acid synthetase ligand | Fas ligand, TNFSF6, Apo1, apoptosis antigen ligand 1, CD95L, CD178, APT1LG1 |
| FasR | Fatty acid synthetase receptor | Fas receptor, TNFRSF6, APT1, CD95 |
| Apo3L | Apo3 ligand | TNFSF12, Apo3 ligand, TWEAK, DR3LG |
| DR3 | Death receptor 3 | TNFRSF12, Apo3, WSL-1, TRAMP, LARD, DDR3 |
| Apo2L | Apo2 ligand | TNFSF10, TRAIL, TNF-related apoptosis inducing ligand |
| DR4 | Death receptor 4 | TNFRSF10A, TRAILR1, APO2 |
| DR5 | Death receptor 5 | TNFRS10B, TRAIL-R2, TRICK2, KILLER, ZTNFR9 |
| FADD | Fas-associated death domain | MORT1 |
| TRADD | TNF receptor-associated death domain | TNFRSF1A associated via death domain |
| RIP | Receptor-interacting protein | RIPK1 |
| DED | Death effector domain | Apoptosis antagonizing transcription factor, CHE1 |
| caspase-8 | Cysteinyl aspartic acid-protease 8 | FLICE, FADD-like Ice, Mach-1, Mch5 |
| c-FLIP | FLICE-inhibitory protein | Casper, I-FLICE, FLAME-1, CASH, CLARP, MRIT |
References:
Human Protein Reference Database 〈http://www.hprd.org/〉.
ExPASy Proteomics Server 〈http://ca.expasy.org/〉.
Perforin/granzyme Pathway
T-cell mediated cytotoxicity is a variant of type IV hypersensitivity where sensitized CD8+ cells kill antigen-bearing cells. These cytotoxic T lymphocytes (CTLs) are able to kill target cells via the extrinsic pathway and the FasL/FasR interaction is the predominant method of CTL-induced apoptosis (Brunner et al., 2003). However, they are also able to exert their cytotoxic effects on tumor cells and virus-infected cells via a novel pathway that involves secretion of the transmembrane pore-forming molecule perforin with a subsequent exophytic release of cytoplasmic granules through the pore and into the target cell (Trapani and Smyth, 2002). The serine proteases granzyme A and granzyme B are the most important component within the granules (Pardo et al., 2004).
Granzyme B will cleave proteins at aspartate residues and will therefore activate procaspase-10 and can cleave factors like ICAD (Inhibitor of Caspase Activated DNAse) (Sakahira et al., 1998). Reports have also shown that granzyme B can utilize the mitochondrial pathway for amplification of the death signal by specific cleavage of Bid and induction of cytochrome c release (Barry and Bleackley, 2002; Russell and Ley, 2002). However, granzyme B can also directly activate caspase-3. In this way, the upstream signaling pathways are bypassed and there is direct induction of the execution phase of apoptosis.
It is suggested that both the mitochondrial pathway and direct activation of caspase-3 are critical for granzyme B-induced killing (Goping et al., 2003). Recent findings indicate that this method of granzyme B cytotoxicity is critical as a control mechanism for T cell expansion of type 2 helper T (Th2) cells (Devadas et al., 2006). Moreover, findings indicate that neither death receptors nor caspases are involved with the T cell receptor-induced apoptosis of activated Th2 cells because blocking their ligands has no effect on apoptosis. On the other hand, Fas-Fas ligand interaction, adapter proteins with death domains and caspases are all involved in the apoptosis and regulation of cytotoxic Type 1 helper cells whereas granzyme B has no effect.
Granzyme A is also important in cytotoxic T cell induced apoptosis and activates caspase independent pathways. Once in the cell, granzyme A activates DNA nicking via DNAse NM23-H1, a tumor suppressor gene product (Fan et al., 2003). This DNAse has an important role in immune surveillance to prevent cancer through the induction of tumor cell apoptosis. The nucleosome assembly protein SET normally inhibits the NM23-H1 gene. Granzyme A protease cleaves the SET complex thus releasing inhibition of NM23-H1, resulting in apoptotic DNA degradation. In addition to inhibiting NM23-H1, the SET complex has important functions in chromatin structure and DNA repair. The proteins that make up this complex (SET, Ape1, pp32, and HMG2) seem to work together to protect chromatin and DNA structure (Lieberman and Fan, 2003). Therefore, inactivation of this complex by granzyme A most likely also contributes to apoptosis by blocking the maintenance of DNA and chromatin structure integrity.
Intrinsic Pathway
The intrinsic signaling pathways that initiate apoptosis involve a diverse array of non-receptor-mediated stimuli that produce intracellular signals that act directly on targets within the cell and are mitochondrial-initiated events. The stimuli that initiate the intrinsic pathway produce intracellular signals that may act in either a positive or negative fashion. Negative signals involve the absence of certain growth factors, hormones and cytokines that can lead to failure of suppression of death programs, thereby triggering apoptosis. In other words, there is the withdrawal of factors, loss of apoptotic suppression, and subsequent activation of apoptosis. Other stimuli that act in a positive fashion include, but are not limited to, radiation, toxins, hypoxia, hyperthermia, viral infections, and free radicals.
All of these stimuli cause changes in the inner mitochondrial membrane that results in an opening of the mitochondrial permeability transition (MPT) pore, loss of the mitochondrial transmembrane potential and release of two main groups of normally sequestered pro-apoptotic proteins from the intermembrane space into the cytosol (Saelens et al., 2004). The first group consists of cytochrome c, Smac/DIABLO, and the serine protease HtrA2/Omi (Cai et al., 1998; Du et al., 2000; Loo et al., 2002; Garrido et al., 2005). These proteins activate the caspase-dependent mitochondrial pathway. Cytochrome c binds and activates Apaf-1 as well as procaspase-9, forming an “apoptosome” (Chinnaiyan, 1999; Hill et al., 2004).
The clustering of procaspase-9 in this manner leads to caspase-9 activation. Smac/DIABLO and HtrA2/Omi are reported to promote apoptosis by inhibiting IAP (inhibitors of apoptosis proteins) activity (van Loo et al., 2002a; Schimmer, 2004). Additional mitochondrial proteins have also been identified that interact with and suppress the action of IAP however gene knockout experiments suggest that binding to IAP alone may not be enough evidence to label a mitochondrial protein as “pro-apoptotic” (Ekert and Vaux, 2005).
The second group of pro-apoptotic proteins, AIF, endonuclease G and CAD, are released from the mitochondria during apoptosis, but this is a late event that occurs after the cell has committed to die. AIF translocates to the nucleus and causes DNA fragmentation into ~50–300 kb pieces and condensation of peripheral nuclear chromatin (Joza et al., 2001). This early form of nuclear condensation is referred to as “stage I” condensation (Susin et al., 2000). Endonuclease G also translocates to the nucleus where it cleaves nuclear chromatin to produce oligonucleosomal DNA fragments (Li et al., 2001). AIF and endonuclease G both function in a caspase-independent manner. CAD is subsequently released from the mitochondria and translocates to the nucleus where, after cleavage by caspase-3, it leads to oligonucleosomal DNA fragmentation and a more pronounced and advanced chromatin condensation (Enari et al., 1998). This later and more pronounced chromatin condensation is referred to as “stage II” condensation (Susin et al., 2000).
The control and regulation of these apoptotic mitochondrial events occurs through members of the Bcl-2 family of proteins (Cory and Adams, 2002). The tumor suppressor protein p53 has a critical role in regulation of the Bcl-2 family of proteins, however the exact mechanisms have not yet been completely elucidated (Schuler and Green, 2001). The Bcl-2 family of proteins governs mitochondrial membrane permeability and can be either pro-apoptotic or anti-apoptotic. To date, a total of 25 genes have been identified in the Bcl-2 family. Some of the anti-apoptotic proteins include Bcl-2, Bcl-x, Bcl-XL, Bcl-XS, Bcl-w, BAG, and some of the pro-apoptotic proteins include Bcl-10, Bax, Bak, Bid, Bad, Bim, Bik, and Blk. These proteins have special significance since they can determine if the cell commits to apoptosis or aborts the process. It is thought that the main mechanism of action of the Bcl-2 family of proteins is the regulation of cytochrome c release from the mitochondria via alteration of mitochondrial membrane permeability.
A few possible mechanisms have been studied but none have been proven definitively. Mitochondrial damage in the Fas pathway of apoptosis is mediated by the caspase-8 cleavage of Bid (Li et al., 1998; Esposti, 2002). This is one example of the “cross-talk” between the death-receptor (extrinsic) pathway and the mitochondrial (intrinsic) pathway (Igney and Krammer, 2002). Serine phosphorylation of Bad is associated with 14-3-3, a member of a family of multifunctional phosphoserine binding molecules. When Bad is phosphorylated, it is trapped by 14-3-3 and sequestered in the cytosol but once Bad is unphosphorylated, it will translocate to the mitochondria to release cytochrome C (Zha, et al., 1996).
Bad can also heterodimerize with Bcl-Xl or Bcl-2, neutralizing their protective effect and promoting cell death (Yang et al., 1995). When not sequestered by Bad, both Bcl-2 and Bcl-Xl inhibit the release of cytochrome C from the mitochondria although the mechanism is not well understood. Reports indicate that Bcl-2 and Bcl-XL inhibit apoptotic death primarily by controlling the activation of caspase proteases (Newmeyer et al., 2000). An additional protein designated “Aven” appears to bind both Bcl-Xl and Apaf-1, thereby preventing activation of procaspase-9 (Chau et al., 2000). There is evidence that overexpression of either Bcl-2 or Bcl-Xl will down-regulate the other, indicating a reciprocal regulation between these two proteins.
Puma and Noxa are two members of the Bcl2 family that are also involved in pro-apoptosis. Puma plays an important role in p53-mediated apoptosis. It was shown that, in vitro, overexpression of Puma is accompanied by increased BAX expression, BAX conformational change, translocation to the mitochondria, cytochrome c release and reduction in the mitochondrial membrane potential (Liu et al., 2003). Noxa is also a candidate mediator of p53-induced apoptosis. Studies show that this protein can localize to the mitochondria and interact with anti-apoptotic Bcl-2 family members, resulting in the activation of caspase-9 (Oda et al., 2000). Since both Puma and Noxa are induced by p53, they might mediate the apoptosis that is elicited by geno-toxic damage or oncogene activation. The Myc oncoprotein has also been reported to potentiate apoptosis through both p53-dependent and -independent mechanisms (Meyer et al., 2006).
Further elucidation of these pathways should have important implications for tumorigenesis and therapy. Table 3 lists the major intrinsic pathway proteins with common abbreviations and some of the alternate nomenclature used for each protein.
Table 3.
Intrinsic pathway proteins, abbreviations, and alternate nomenclature.
| Abbreviation | Protein Name | Select Alternate Nomenclature |
|---|---|---|
| Smac/DIABLO | Second mitochondrial activator of caspases/direct IAP binding protein with low PI | None |
| HtrA2/Omi | High-temperature requirement | Omi stress regulated endoprotease, serine protease Omi protein A2 |
| IAP | Inhibitor of Apoptosis Proteins | XIAP, API3, ILP, HILP, HIAP2, cIAP1, API1, MIHB, NFR2-TRAF signaling complex protein |
| Apaf-1 | Apoptotic protease activating factor | APAF1 |
| Caspase-9 | Cysteinyl aspartic acid-protease-9 | ICE-LAP6, Mch6, Apaf-3 |
| AIF | Apoptosis Inducing Factor | Programmed cell death protein 8, mitochondrial |
| CAD | Caspase-Activated DNAse | CAD/CPAN/DFF40 |
| Bcl-2 | B-cell lymphoma protein 2 | Apoptosis regulator Bcl-2 |
| Bcl-x | BCL2 like 1 | BCL2 related protein |
| Bcl-XL | BCL2 related protein, long isoform | BCL2L protein, long form of Bcl-x |
| Bcl-XS | BCL2 related protein, short isoform | |
| Bcl-w | BCL2 like 2 protein | Apoptosis regulator BclW |
| BAG | BCL2 associated athanogene | BAG family molecular chaperone regulator |
| Bcl-10 | B-cell lymphoma protein 10 | mE10, CARMEN, CLAP, CIPER |
| BAX | BCL2 associated X protein | Apoptosis regulator BAX |
| BAK | BCL2 antagonist killer 1 | BCL2L7, cell death inhibitor 1 |
| BID | BH3 interacting domain death agonist | p22 BID |
| BAD | BCL2 antagonist of cell death | BCL2 binding protein, BCL2L8, BCL2 binding component 6, BBC6, Bcl-XL/Bcl-2 associated death promoter |
| BIM | BCL2 interacting protein BIM | BCL2 like 11 |
| BIK | BCL2 interacting killer | NBK, BP4, BIP1, apoptosis inducing NBK |
| Blk | Bik-like killer protein | B lymphoid tyrosine kinase, p55-BLK, MGC10442 |
| Puma | BCL2 binding component 3 | JFY1, PUMA/JFY1, p53 up-regulated modulator of apoptosis |
| Noxa | Phorbol-12-myristate-13-acetate-induced protein 1 | PMA induced protein 1, APR |
| 14-3-3 | Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein | 14-3-3 eta, theta, zeta, beta, epsilon, sigma, gamma |
| Aven | Cell death regulator Aven | None |
| Myc | Oncogene Myc | c-myc, Myc proto-oncogene protein |
References
Human Protein Reference Database 〈http://www.hprd.org/〉.
ExPASy Proteomics Server 〈http://ca.expasy.org/〉.
Execution Pathway
The extrinsic and intrinsic pathways both end at the point of the execution phase, considered the final pathway of apoptosis. It is the activation of the execution caspases that begins this phase of apoptosis. Execution caspases activate cytoplasmic endonuclease, which degrades nuclear material, and proteases that degrade the nuclear and cytoskeletal proteins. Caspase-3, caspase-6, and caspase-7 function as effector or “executioner” caspases, cleaving various substrates including cytokeratins, PARP, the plasma membrane cytoskeletal protein alpha fodrin, the nuclear protein NuMA and others, that ultimately cause the morphological and biochemical changes seen in apoptotic cells (Slee et al., 2001).
Caspase-3 is considered to be the most important of the executioner caspases and is activated by any of the initiator caspases (caspase-8, caspase-9, or caspase-10). Caspase-3 specifically activates the endonuclease CAD. In proliferating cells CAD is complexed with its inhibitor, ICAD. In apoptotic cells, activated caspase-3 cleaves ICAD to release CAD (Sakahira et al., 1998). CAD then degrades chromosomal DNA within the nuclei and causes chromatin condensation. Caspase-3 also induces cytoskeletal reorganization and disintegration of the cell into apoptotic bodies. Gelsolin, an actin binding protein, has been identified as one of the key substrates of activated caspase-3.
Gelsolin will typically act as a nucleus for actin polymerization and will also bind phosphatidylinositol biphosphate, linking actin organization and signal transduction. Caspase-3 will cleave gelsolin and the cleaved fragments of gelsolin, in turn, cleave actin filaments in a calcium independent manner. This results in disruption of the cytoskeleton, intracellular transport, cell division, and signal transduction (Kothakota et al., 1997).
Phagocytic uptake of apoptotic cells is the last component of apoptosis. Phospholipid asymmetry and externalization of phosphatidylserine on the surface of apoptotic cells and their fragments is the hallmark of this phase. Although the mechanism of phosphatidylserine translocation to the outer leaflet of the cell during apoptosis is not well understood, it has been associated with loss of aminophospholipid translocase activity and nonspecific flip-flop of phospholipids of various classes (Bratton et al., 1997). Research indicates that Fas, caspase-8, and caspase-3 are involved in the regulation of phosphatidylserine externalization on oxidatively stressed erythrocytes however caspase-independent phosphatidylserine exposure occurs during apoptosis of primary T lymphocytes (Ferraro-Peyret et al., 2002; Mandal et al., 2005).
The appearance of phosphotidylserine on the outer leaflet of apoptotic cells then facilitates noninflammatory phagocytic recognition, allowing for their early uptake and disposal (Fadok et al., 2001). This process of early and efficient uptake with no release of cellular constituents, results in essentially no inflammatory response. Table 4 lists the major proteins in the execution pathway with common abbreviations and some of the alternate nomenclature used for each protein.
Table 4.
Execution pathway proteins, abbreviations, and alternate nomenclature.
| Abbreviation | Protein name | Select alternate nomenclature |
|---|---|---|
| Caspase-3 | Cysteinyl aspartic acid-protease-3 | CPP32, Yama, Apopain, SCA-1, LICE |
| Caspase-6 | Cysteinyl aspartic acid-protease-6 | Mch-2 |
| Caspase-7 | Cysteinyl aspartic acid-protease-7 | Mch-3, ICE-LAP-3, CMH-1 |
| Caspase-10 | Cysteinyl aspartic acid-protease-10 | Mch4, FLICE-2 |
| PARP | Poly (ADP-ribose) polymerase | ADP ribosyl transferase, ADPRT1, PPOL |
| Alpha fodrin | Spectrin alpha chain | Alpha-II spectrin, fodrin alpha chain |
| NuMA | Nuclear mitotic apparatus protein | SP-H antigen |
| CAD | Caspase-activated DNAse | DNA fragmentation factor subunit beta, DFF-40, caspase-activated nuclease, CPAN |
| ICAD | Inhibitor of CAD | DNA fragmentation factor subunit alpha, DFF-45 |
References
Human Protein Reference Database 〈http://www.hprd.org/〉
ExPASy Proteomics Server 〈http://ca.expasy.org/〉
Physiologic Apoptosis
The role of apoptosis in normal physiology is as significant as that of its counterpart, mitosis. It demonstrates a complementary but opposite role to mitosis and cell proliferation in the regulation of various cell populations. It is estimated that to maintain homeostasis in the adult human body, around 10 billion cells are made each day just to balance those dying by apoptosis (Renehan et al., 2001). And that number can increase significantly when there is increased apoptosis during normal development and aging or during disease.
Apoptosis is critically important during various developmental processes. As examples, both the nervous system and the immune system arise through overproduction of cells. This initial overproduction is then followed by the death of those cells that fail to establish functional synaptic connections or productive antigen specificities, respectively (Nijhawan et al., 2000; Opferman and Korsmeyer, 2003).
Apoptosis is also necessary to rid the body of pathogen-invaded cells and is a vital component of wound healing in that it is involved in the removal of inflammatory cells and the evolution of granulation tissue into scar tissue (Greenhalgh, 1998). Dysregulation of apoptosis during wound healing can lead to pathologic forms of healing such as excessive scarring and fibrosis. Apoptosis is also needed to eliminate activated or auto-aggressive immune cells either during maturation in the central lymphoid organs (bone marrow and thymus) or in peripheral tissues (Osborne, 1996).
Additionally, apoptosis is central to remodeling in the adult, such as the follicular atresia of the postovulatory follicle and post-weaning mammary gland involution, to name a couple of examples (Tilly, 1991; Lund et al., 1996). Furthermore, as organisms grow older, some cells begin to deteriorate at a faster rate and are eliminated via apoptosis. One theory is that oxidative stress plays a primary role in the pathophysiology of age-induced apoptosis via accumulated free-radical damage to mitochondrial DNA (Harman, 1992; Ozawa, 1995). It is clear that apoptosis has to be tightly regulated since too little or too much cell death may lead to pathology, including developmental defects, autoimmune diseases, neurodegeneration, or cancer.
Pathologic Apoptosis
Abnormalities in cell death regulation can be a significant component of diseases such as cancer, autoimmune lymphoproliferative syndrome, AIDS, ischemia, and neurode-generative diseases such as Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, and Amyotrophic Lateral Sclerosis. Some conditions feature insufficient apoptosis whereas others feature excessive apoptosis.
Cancer is an example where the normal mechanisms of cell cycle regulation are dysfunctional, with either an overproliferation of cells and/or decreased removal of cells (King and Cidlowski, 1998). In fact, suppression of apoptosis during carcinogenesis is thought to play a central role in the development and progression of some cancers (Kerr et al., 1994). There are a variety of molecular mechanisms that tumor cells use to suppress apoptosis.
Tumor cells can acquire resistance to apoptosis by the expression of anti-apoptotic proteins such as Bcl-2 or by the down-regulation or mutation of pro-apoptotic proteins such as Bax. The expression of both Bcl-2 and Bax is regulated by the p53 tumor suppressor gene (Miyashita, 1994). Certain forms of human B cell lymphoma have overexpression of Bcl-2, and this is one of the first and strongest lines of evidence that failure of cell death contributes to cancer (Vaux et al., 1988). Another method of apoptosis suppression in cancer involves evasion of immune surveillance (Smyth et al., 2001).
Certain immune cells (T cells and natural killer cells) normally destroy tumor cells via the perforin/granzyme B pathway or the death-receptor pathway. In order to evade immune destruction, some tumor cells will diminish the response of the death receptor pathway to FasL produced by T cells. This has been shown to occur in a variety of ways including down-regulation of the Fas receptor on tumor cells. Other mechanisms include expression of nonfunctioning Fas receptor, secretion of high levels of a soluble form of the Fas receptor that will sequester the Fas ligand or expression of Fas ligand on the surface of tumor cells (Cheng et al., 1994; Cefai et al., 2001; Elnemr et al., 2001). In fact, some tumor cells are capable of a Fas ligand-mediated “counterattack” that results in apoptotic depletion of activated tumor infiltrating lymphocytes (Koyama et al., 2001).
Alterations of various cell signaling pathways can result in dysregulation of apoptosis and lead to cancer. The p53 tumor suppressor gene is a transcription factor that regulates the cell cycle and is the most widely mutated gene in human tumorigenesis (Wang and Harris, 1997). The critical role of p53 is evident by the fact that it is mutated in over 50% of all human cancers. p53 can activate DNA repair proteins when DNA has sustained damage, can hold the cell cycle at the G1/S regulation point on DNA damage recognition, and can initiate apoptosis if the DNA damage proves to be irreparable (Pientenpol and Stewart, 2002). Tumorigenesis can occur if this system goes awry. If the p53 gene is damaged, then tumor suppression is severely reduced. The p53 gene can be damaged by radiation, various chemicals, and viruses such as the Human papillomavirus (HPV). People who inherit only one functional copy of this gene will most likely develop Li–Fraumeni syndrome, which is characterized by the development of tumors in early adulthood (Varley et al., 1997; Gu et al., 2001).
The ataxia telangiectasia-mutated gene (ATM) has also been shown to be involved in tumorigenesis via the ATM/p53 signaling pathway (Kitagawa and Kastan, 2005). The ATM gene encodes a protein kinase that acts as a tumor suppressor. ATM activation, via ionizing radiation damage to DNA, stimulates DNA repair and blocks cell cycle progression. One mechanism through which this occurs is ATM dependent phosphorylation of p53 (Kurz and Lees-Miller, 2004).
As mentioned previously, p53 then signals growth arrest of the cell at a checkpoint to allow for DNA damage repair or can cause the cell to undergo apoptosis if the damage cannot be repaired. This system can also be inactivated by a number of mechanisms including somatic genetic/epigenetic alterations and expression of oncogenic viral proteins such as the HPV, leading to tumorigenesis (Bolt et al., 2005). Other cell signaling pathways can also be involved in tumor development. For example, upregulation of the phosphatidylinositol 3-kinase/AKT pathway in tumor cells renders them independent of survival signals. In addition to regulation of apoptosis, this pathway regulates other cellular processes, such as proliferation, growth, and cytoskeletal rearrangement (Vivanco and Sawyers, 2002).
In addition to cancer, too little apoptosis can also result in diseases such as autoimmune lymphoproliferative syndrome (ALPS) (Worth et al., 2006). This occurs when there is insufficient apoptosis of auto-aggressive T cells, resulting in multiple autoimmune diseases. An overproliferation of B cells occurs as well, resulting in excess immunoglobulin production, leading to autoimmunity. Some of the common diseases of ALPS include hemolytic anemia, immune-mediated thrombocytopenia, and autoimmune neutropenia. The different types of this condition are caused by different mutations. Type 1A results from a mutation in the death domain of the Fas receptor, Type 1B results from a mutation in Fas ligand and Type 2 results from a mutation in caspase-10, reducing its activity.
Excessive apoptosis may also be a feature of some conditions such as autoimmune diseases, neurodegenerative diseases, and ischemia-associated injury. Autoimmune deficiency syndrome (AIDS) is an example of an autoimmune disease that results from infection with the human immunodeficiency virus (HIV) (Li et al., 1995). This virus infects CD4+ T cells by binding to the CD4 receptor. The virus is subsequently internalized into the T cell where the HIV Tat protein is thought to increase the expression of the Fas receptor, resulting in excessive apoptosis of T cells.
Alzheimer’s disease is a neurodegenerative condition that is thought to be caused by mutations in certain proteins such as APP (amyloid precursor protein) and presenilins. Presenilins are thought to be involved in the processing of APP to amyloid β. This condition is associated with the deposition of amyloid β in extracellular deposits known as plaques and amyloid β is thought to be neurotoxic when found in aggregated plaque form. Amyloid β is thought to induce apoptosis by causing oxidative stress or by triggering increased Fas ligand expressions in neurons and glia. It may also activate microglia, which would result in TNFα secretion and activation of the TNF-R1, leading to apoptosis (Ethell and Buhler, 2003).
Excessive apoptosis is also thought to play an important role in various ischemia-associated injuries. One example is myocardial ischemia caused by an insufficient blood supply, leading to a decrease in oxygen delivery to, and subsequent death of, the cardiomyocytes. Although necrosis does occur, overexpression of BAX has been detected in ischemic myocardial tissue and therapy aimed at reducing apoptosis has shown some success in reducing the degree of tissue damage (Hochhauser et al., 2003). One hypothesis is that the damage produced by ischemia is capable of initiating apoptosis but if ischemia is prolonged, necrosis occurs. If energy production is restored, as with reperfusion, the apoptotic cascade that was initiated by ischemia may proceed (Freude et al., 2000). Although the extent to which apoptosis is involved in myocardial ischemia remains to be clarified, there is clear evidence that supports a role for this mode of cell death.
Inhibition of Apoptosis
There are many pathological conditions that feature excessive apoptosis (neurodegenerative diseases, AIDS, ischemia, etc.) and thus may benefit from artificially inhibiting apoptosis. As our understanding of the field evolves, the identification and exploitation of new targets remains a considerable focus of attention (Nicholson, 2000). A short list of potential methods of anti-apoptotic therapy includes stimulation of the IAP (inhibitors of apoptosis proteins) family of proteins, caspase inhibition, PARP (poly [ADP-ribose] polymerase) inhibition, stimulation of the PKB/Akt (protein kinase B) pathway, and inhibition of Bcl-2 proteins.
The IAP family of proteins is perhaps the most important regulators of apoptosis due to the fact that they regulate both the intrinsic and extrinsic pathways (Deveraux and Reed, 1999). Eight human IAP proteins have now been identified although XIAP (X-linked mammalian inhibitor of apoptosis protein) and survivin remain the better-known members (Silke et al., 2002; Colnaghi et al., 2006). So far, members of the IAP family have been investigated as therapeutic targets for the treatment of stroke, spinal cord injuries, multiple sclerosis as well as cancer. The synthetic nonspecific caspase inhibitor z-VAD-fmk was shown to reduce the severity of myocardial reperfusion injury in rat and mouse models of myocardial infarction (Mocanu et al., 2000). Specific inhibitors of caspase activity may also prove beneficial. ICE (Interleukin-1 beta-converting enzyme), also called caspase I, is a cysteine protease that appears to mediate intracellular protein degradation during apoptosis (Livingston, 1997). ICE inhibitors have been developed to treat rheumatoid arthritis and other inflammatory conditions via reduction of interleukin 1β (Le and Abbenante, 2005).
Due to the dual role of PARP-1 in both DNA repair and apoptosis, the pharmacological use of PARP-1 inhibitors may be able to attenuate ischemic and inflammatory cell and organ injury or may be able to enhance the cytotoxicity of antitumor agents (Graziani and Szabo, 2005). Recent research with PARP-1 knockout mice indicates that the use of PARP-1 inhibitors may be an effective therapy for the injury associated with myocardial ischemia and reperfusion injury (Zhou et al., 2006). Infusion of insulin-like growth factor-1 (IGF-1), which stimulates PKB/Akt signaling and promotes cell survival, was shown to be beneficial in animal models of myocardial ischemia (Fujio et al., 2000).
Other studies with transgenic models of cardiac ischemia and global brain ischemia indicate that inhibiting the expression and/or function of Bax can prevent cytochrome c release from mitochondria, inhibit the decrease in the mitochondrial membrane potential, and protect cells against apoptosis (Hochhauser et al., 2003; Hetz et al., 2005). The potential therapeutic modalities mentioned here represent just a few of the past and current research efforts in this field. As the molecular and biochemical complexities of apoptosis continue to be elucidated, new therapeutic strategies will continue to evolve.
Assays for Apoptosis
Since apoptosis occurs via a complex signaling cascade that is tightly regulated at multiple points, there are many opportunities to evaluate the activity of the proteins involved. As the activators, effectors and regulators of this cascade continue to be elucidated, a large number of apoptosis assays are devised to detect and count apoptotic cells. However, many features of apoptosis and necrosis can overlap, and it is therefore crucial to employ two or more distinct assays to confirm that cell death is occurring via apoptosis. One assay may detect early (initiation) apoptotic events and a different assay may target a later (execution) event. The second assay, used to confirm apoptosis, is generally based on a different principle. Multiplexing, which is the ability to gather more than one set of data from the same sample, is another methodology for apoptosis detection that is becoming increasingly popular. There are a large variety of assays available, but each assay has advantages and disadvantages which may make it acceptable to use for one application but inappropriate for another application (Watanabe et al., 2002; Otsuki et al., 2003). Therefore, when choosing methods of apoptosis detection in cells, tissues or organs, understanding the pros and cons of each assay is crucial.
Understanding the kinetics of cell death in each model system is also critical. Some proteins, such as caspases, are expressed only transiently. Cultured cells undergoing apoptosis in vitro will eventually undergo secondary necrosis. Apoptotic cells in any system can die and disappear relatively quickly. The time from initiation of apoptosis to completion can occur as quickly as 2–3 hours. Therefore a false negative can occur if the assay is done too early or too late. Moreover, apoptosis can occur at low frequency or in specific sites within organs, tissues and cultures. In such cases, the ability to rapidly survey large areas could be useful. In general, if detailed information on the mechanism of cell death is desired, the duration of toxin exposure, the concentration of the test compound and the choice of assay endpoint become critical.
A detailed description of all methodologies and assays for detecting apoptosis is beyond the scope of this article. However, some of the most commonly employed assays are mentioned and briefly described. Apoptosis assays, based on methodology, can be classified into six major groups and a subset of the available assays in each group is indicated and briefly discussed:
Cytomorphological alterations
DNA fragmentation
Detection of caspases, cleaved substrates, regulators and inhibitors
Membrane alterations
Detection of apoptosis in whole mounts 6. Mitochondrial assays.
Cytomorphological Alterations
The evaluation of hematoxylin and eosin-stained tissue sections with light microscopy does allow the visualization of apoptotic cells. Although a single apoptotic cell can be detected with this method, confirmation with other methods may be necessary. Because the morphological events of apoptosis are rapid and the fragments are quickly phagocytized, considerable apoptosis may occur in some tissues before it is histologically apparent. Additionally, this method detects the later events of apoptosis, so cells in the early phase of apoptosis will not be detected.
Semi-ultrathin sections from an epoxy-resin-embedded block can be stained with toluidine blue or methylene blue to reveal intensely stained apoptotic cells when evaluated by standard light microscopy. This methodology depends on the nuclear and cytoplasmic condensation that occurs during apoptosis. The tissue and cellular details are preserved with this technique and surveys of large tissue regions are distinct advantages. However, smaller apoptotic bodies will not be detected and healthy cells with large dense intracellular granules can be mistaken for apoptotic cells or debris. Additionally, there is loss of antigenicity during processing so that immunohistological or enzyme assays cannot be performed on the same tissue. However, this tissue may be used for transmission electron microscopy (TEM).
TEM is considered the gold standard to confirm apoptosis. This is because categorization of an apoptotic cell is irrefutable if the cell contains certain ultrastructural morphological characteristics (White and Cinti, 2004). These characteristics are: (1) electron-dense nucleus (marginalization in the early phase); (2) nuclear fragmentation; (3) intact cell membrane even late in the cell disintegration phase; (4) disorganized cytoplasmic organelles; (5) large clear vacuoles; and (6) blebs at the cell surface. As apoptosis progresses, these cells will lose the cell-to-cell adhesions and will separate from neighboring cells. During the later phase of apoptosis, the cell will fragment into apoptotic bodies with intact cell membranes and will contain cytoplasmic organelles with or without nuclear fragments. Phagocytosis of apoptotic bodies can also be appreciated with TEM. The main disadvantages of TEM are the cost, time expenditure, and the ability to only assay a small region at a time. Other disadvantages include the difficulty in detecting apoptotic cells due to their transient nature and the inability to detect apoptotic cells at the earliest stages.
DNA Fragmentation
The DNA laddering technique is used to visualize the endonuclease cleavage products of apoptosis (Wyllie, 1980). This assay involves extraction of DNA from a lysed cell homogenate followed by agarose gel electrophoresis. This results in a characteristic “DNA ladder” with each band in the ladder separated in size by approximately 180 base pairs. This methodology is easy to perform, has a sensitivity of 1 × 106 cells (i.e., level of detection is as few as 1,000,000 cells), and is useful for tissues and cell cultures with high numbers of apoptotic cells per tissue mass or volume, respectively. On the other hand, it is not recommended in cases with low numbers of apoptotic cells. There are other disadvantages to this assay. Since DNA fragmentation occurs in the later phase of apoptosis, the absence of a DNA ladder does not eliminate the potential that cells are undergoing early apoptosis. Additionally, DNA fragmentation can occur during preparation making it difficult to produce a nucleosome ladder and necrotic cells can also generate DNA fragments.
The TUNEL (Terminal dUTP Nick End-Labeling) method is used to assay the endonuclease cleavage products by enzymatically end-labeling the DNA strand breaks (Kressel and Groscurth, 1994; Ito and Otsuki, 1998). Terminal transferase is used to add labeled UTP to the 3′-end of the DNA fragments. The dUTP can then be labeled with a variety of probes to allow detection by light microscopy, fluorescence microscopy or flow cytometry (Figure 4). The assays are available as kits and can be acquired from a variety of companies. This assay is also very sensitive, allowing detection of a single cell via fluorescence microscopy or as few as ~100 cells via flow cytometry. It is also a fast technique and can be completed within 3 hours. The disadvantages are cost and the unknown parameter of how many DNA strand breaks are necessary for detection by this method. This method is also subject to false positives from necrotic cells and cells in the process of DNA repair and gene transcription. For these reasons, it should be paired with another assay.
Figure 4.
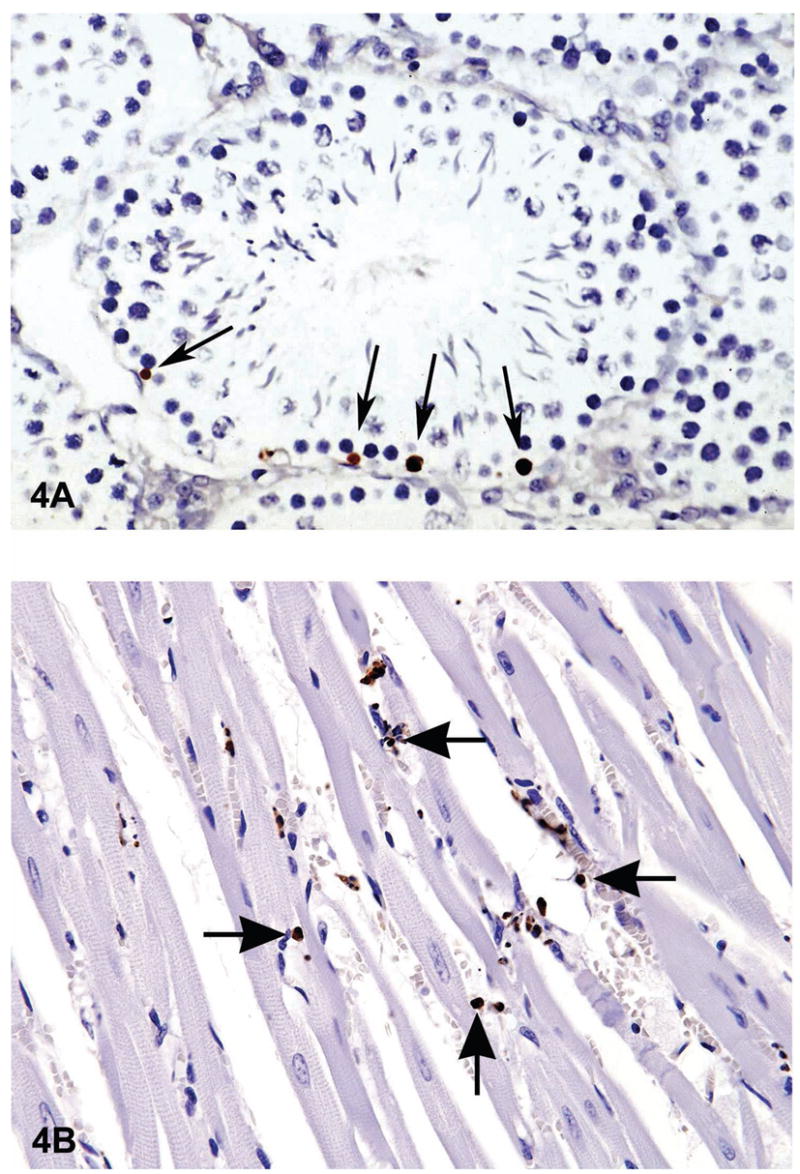
There are many ways of detecting apoptosis at different stages on histological sections. One commonly used method is called TUNEL (Terminal deoxynucleotidyl Transferase Biotin-dUTP Nick End Labeling). One of the major characteristics of apoptosis is the degradation of DNA after the activation of Ca/Mg dependent endonucleases. This DNA cleavage leads to strand breaks within the DNA. However, necrosis can also result in similar DNA cleavage. Therefore an additional method should be used to confirm apoptosis. The TUNEL method identifies cells with DNA strand breaks in situ by using terminal deoxynucleotidyl transferase (TdT) to transfer biotin-dUTP to the cleaved ends of the DNA. The biotin-labeled cleavage sites are then detected by reaction with HRP (horse radish peroxidase) conjugated streptavidin and visualized by DAB (diaminobenzidine), which gives a brown color. The tissue is typically counterstained with Toluidine blue to allow evaluation of tissue architecture. Figure 4A a photomicrograph of a section of testes from a B6C3F1 mouse that was in a National Toxicology Program bioassay. The TUNEL assay was used on this section of tissue to detect apoptotic cells. The seminiferous tubule is cut in cross section and has sperm, spermatogonia and spermatocytes in various stages of development. The arrows point to labeled (dark brown) apoptotic spermatogonia in the basal layer of the seminiferous epithelium. Figure 4B is an image of myocardium from 14 week-old rat treated with ephedrine (25 mg/kg) and caffeine (30 mg/kg). There are multiple apoptotic cells that have stained brown with the TUNEL method (arrows).
Detection of Caspases, Cleaved Substrates, Regulators, and Inhibitors
There are more than 13 known caspases (procaspases or active cysteine caspases) that can be detected using various types of caspase activity assays (Gurtu et al., 1997). There are also immunohistochemistry assays that can detect cleaved substrates such as PARP and known cell modifications such as phosphorylated histones (Figure 5A) (Love et al., 1999; Talasz et al., 2002). Fluorescently conjugated caspase inhibitors can also be used to label active caspases within cells (Grabarek et al., 2002). Caspase activation can be detected in a variety of ways including western blot, immunoprecipitation and immunohistochemistry (Figure 5B). Both polyclonal and monoclonal antibodies are available to both procaspases and active caspases.
Figure 5.
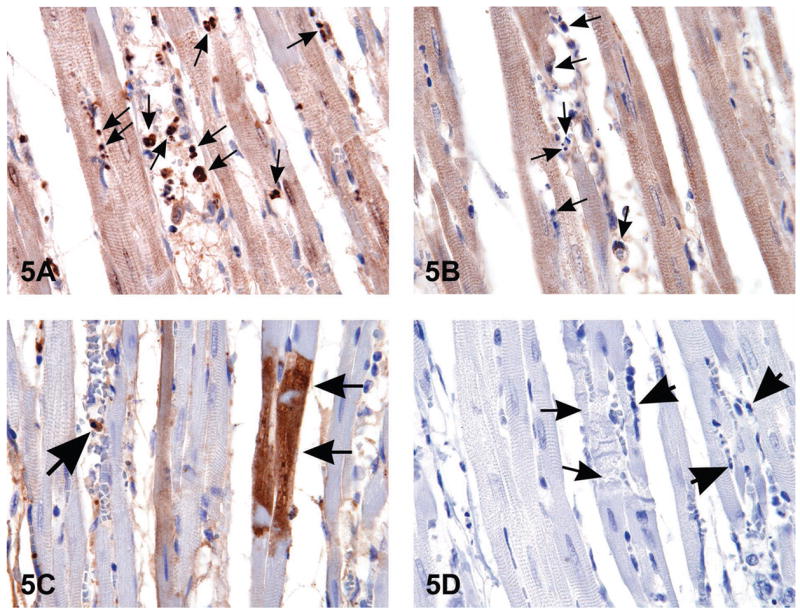
When histological changes suggestive of apoptosis are detected in H&E stained tissues (i.e., Figure 1B), confirmation of apoptosis can be obtained with a variety of additional stains. The images in Figure 5 are of myocardium from 14-week-old rats treated with ephedrine (25 mg/kg) and caffeine (30 mg/kg). Figure 5A is the interventricular septum stained by anti-phospho-H2A.X, a histone variant that becomes phosphorylated during apoptosis; note the strongly stained apoptotic bodies (arrows). Figure 5B demonstrates the negative control of the same section shown in (A); nonimmune rabbit IgG was used at equivalent conditions in place of the primary antibody. Note the unstained apoptotic bodies (arrows). Figure 5C illustrates cleaved caspase-3 staining for apoptosis revealing the presence of intracytoplasmic positive myofibers (small arrows) located in the interventricular septum; large arrow indicates an apoptotic body. Figure 5D is a negative control of the same heart demonstrated in 5C. The small arrows demonstrate degenerating myofibers; large arrows indicate apoptotic bodies. Nyska et al., 2005, by permission 2 of Oxford University Press.
One method of caspase detection requires cell lysis in order to release the enzymes into the solution, coating of microwells with anti-caspases; followed by detection with a fluorescent labeled substrate. Detection of caspase activity by this method usually requires 1 × 105 cells. This technique allows selection for individual initiator or execution caspases. It also allows for rapid and consistent quantification of apoptotic cells. The major disadvantage is that the integrity of the sample is destroyed thereby eliminating the possibility of localizing the apoptotic event within the tissue or determining the type of cell that is undergoing apoptosis. Another disadvantage is that caspase activation does not necessarily indicate that apoptosis will occur. Moreover, there is tremendous overlap in the substrate preferences of the members of the caspase family, affecting the specificity of the assay.
Apoptosis PCR microarray is a relatively new methodology that uses real-time PCR to profile the expression of at least 112 genes involved in apoptosis (Hofmann et al., 2001; Vallat et al., 2003). These PCR microarrays are designed to determine the expression profile of genes that encode key ligands, receptors, intracellular modulators, and transcription factors involved in the regulation of programmed cell death. Genes involved in anti-apoptosis can also be assessed with this methodology. Comparison of gene expression in cells or tissues can be performed between test samples and controls. This type of assay allows for the evaluation of the expression of a focused panel of genes related to apoptosis and several companies offer apoptosis pathway-specific gene panels.
Hierarchical cluster analysis of genes can reveal distinct temporal expression patterns of transcriptional activation and/or repression. However, interpretation of the results can be confounded by the large number of analyzed genes and by the methodological complexity. This methodology uses a 96-well plate and as little as 5 nanograms of total RNA. Every mRNA, or transcript, is labeled with a marker, such as a fluorescent dye. A real-time PCR instrument is used for expression profiling. The location and intensity of the resulting signals give an estimate of the quantity of each transcript in the sample. The microarray test should be combined with a different methodology to confirm apoptosis.
Membrane Alterations
Externalization of phosphatidylserine residues on the outer plasma membrane of apoptotic cells allows detection via An-nexin V in tissues, embryos or cultured cells (Bossy-Wetzel and Green, 2000). Once the apoptotic cells are bound with FITC-labeled Annexin V, they can be visualized with fluorescent microscopy. The advantages are sensitivity (can detect a single apoptotic cell) and the ability to confirm the activity of initiator caspases. The disadvantage is that the membranes of necrotic cells are labeled as well. Therefore a critical control is to demonstrate the membrane integrity of the phosphatidylserine-positive cells. Since loss of membrane integrity is a pathognomonic feature of necrotic cell death, necrotic cells will stain with specific membrane-impermeant nucleic acid dyes such as propidium iodide and trypan blue. Likewise, the membrane integrity of apoptotic cells can be demonstrated by the exclusion of these dyes. The transfer of phosphatidylserine to the outside of the cell membrane will also permit the transport of certain dyes into the cell in a unidirectional manner. As the cell accumulates dye and shrinks in volume, the cell dye content becomes more concentrated and can be visualized with light microscopy. This dye-uptake bioassay works on cell cultures, does not label necrotic cells, and has a high level of sensitivity (can detect a single apoptotic cell).
Detection of Apoptosis in Whole Mounts
Apoptosis can also be visualized in whole mounts of embryos or tissues using dyes such as acridine orange (AO), Nile blue sulfate (NBS), and neutral red (NR) (Zucker et al., 2000). Since these dyes are acidophilic, they are concentrated in areas of high lysosomal and phagocytotic activity. The results would need to be validated with other apoptosis assays because these dyes cannot distinguish between lysosomes degrading apoptotic debris from degradation of other debris such as microorganisms. Although all of these dyes are fast and inexpensive, they have certain disadvantages. AO is toxic and mutagenic and quenches rapidly under standard conditions whereas NBS and NR do not penetrate thick tissues and can be lost during preparation for sectioning. Lyso-Tracker Red is another dye that acts in a similar way; however this dye can be used with laser confocal microscopy to provide 3-dimensional imaging of apoptotic cells. This dye is stable during processing, penetrates thick tissues and is resistant to quenching. This dye can be used for cell culture as well as whole mounts of embryos, tissues, or organs.
Mitochondrial Assays
Mitochondrial assays and cytochrome c release allow the detection of changes in the early phase of the intrinsic pathway. Laser scanning confocal microscopy (LSCM) creates submicron thin optical slices through living cells that can be used to monitor several mitochondrial events in intact single cells over time (Bedner et al., 1999; Darzynkiewicz et al., 1999). Mitochondrial permeability transition (MPT), depolarization of the inner mitochondrial membrane, Ca2+ fluxes, mitochondrial redox status, and reactive oxygen species can all be monitored with this methodology. The main disadvantage is that the mitochondrial parameters that this methodology monitors can also occur during necrosis. The electrochemical gradient across the mitochondrial outer membrane (MOM) collapses during apoptosis, allowing detection with a fluorescent cationic dye (Poot and Pierce, 1999). In healthy cells this lipophilic dye accumulates in the mitochondria, forming aggregates that emit a specific fluorescence. In apoptotic cells the MOM does not maintain the electrochemical gradient and the cationic dye diffuses into the cytoplasm where it emits a fluorescence that is different from the aggregated form.
Other mitochondrial dyes can be used that measure the redox potential or metabolic activity of the mitochondria in cells. However, these dyes do not address the mechanism of cell death and should be used in conjunction with other apoptosis detection methods such as a caspase assay.
Cytochrome c release from the mitochondria can also be assayed using fluorescence and electron microscopy in living or fixed cells (Scorrano et al., 2002). However, cytochrome c becomes unstable once it is released into the cytoplasm (Goldstein et al., 2000). Therefore a non-apoptotic control should be used to ensure that the staining conditions used are able to detect any available cytochrome c.
Apoptotic or anti-apoptotic regulator proteins such as Bax, Bid, and Bcl-2 can also be detected using fluorescence and confocal microscopy (Tsien, 1998; Zhang et al., 2002). However, the fluorescent protein tag may alter the interaction of the native protein with other proteins. Therefore, other apoptosis assays should be used to confirm the results.
Other Forms of Programmed Cell Death
There is evidence of other forms of non-apoptotic programmed cell death that should also be considered since they may lead to new insights into cell death programs and reveal their potentially unique roles in development, homeostasis, neoplasia and degeneration. It has become increasingly apparent that cell death mechanisms include a highly diverse array of phenotypes and molecular mechanisms. And there is evidence that modulation of one form of cell death may lead to another. Because other types of cell death may require gene activation and function in an energy dependent manner, they are also considered to be forms of “programmed cell death.” Therefore, there is some resistance to the exclusive use of the term “programmed cell death” to specifically describe apoptosis.
There are necrotic-like phenotypes that require gene activation and protein synthesis so they are, strictly speaking, forms of programmed cell death (Proskuryakov et al., 2003). These forms of cell death that have certain morphological features of both necrosis and apoptosis have been given the term “aponecrosis” (Formigli et al., 2000). By affecting the mitochondrial respiratory chain with antimycin A, Formigli and coworkers induced a type of cell death that shared dynamic, molecular, and morphological features with both apoptosis and necrosis.
Caspase-independent mechanisms of neuronal cell death have also been identified. This specific type of programmed cell death may involve specific mitochondrial factors. In experimental models, apoptosis-inducing factor (AIF) and endonuclease G promote this type of cell death; however, Smac/DIABLO and HtrA2/Omi may also contribute (Ravagnan et al., 2002; van Loo et al., 2002b). Oppenheim and coworkers (2001) have shown that programmed cell death occurs in developing mammalian neurons, even after the genetic deletion of caspases. Other research has shown that inhibition of the caspase execution machinery may only temporarily rescue damaged neurons and that classical apoptotic features can still appear in caspase-inhibited neurons (Volbracht et al., 2001). It appears that caspase-dependent and caspase-independent mechanisms of neuronal cell death may depend on brain region, cell type, and age.
Sperandio and coworkers (2000) have described a form of programmed cell death that is morphologically and biochemically distinct from apoptosis, dubbed “paraptosis.” Although this form of cell death does not respond to caspase inhibitors or BCL-XL, it is driven by an alternative form of caspase-9 activity that is Apaf-1 independent. This alternative form of programmed cell death is reported to occur during development and in transgenic models of Huntington’s disease and human amyotrophic lateral sclerosis (Dal Canto and Gurney, 1994; Turmaine et al., 2000).
There is speculation that “autophagy” represents another mechanism for programmed cell death and, similar to apoptosis, has important roles in developmental processes, human diseases and cellular responses to nutrient deprivation (Schwartz et al., 1993; Gozuacik and Kimchi, 2004; Debnath et al., 2005). Other terms used synonymously are “macroautophagy” and “autophagic type II cell death” (Klionsky and Emr, 2000). Autophagic cell death is characterized by the sequestration of cytoplasm and organelles in double or multimembrane vesicles and delivery to the cells own lysosomes for subsequent degradation (Noda et al., 2002). In a sense, the cell “cannibalizes” itself. The mechanisms and morphology of autophagy are evolutionarily conserved with strong similarities between organisms as diverse as animals, plants and yeast. The process of autophagy depends on both continuous protein synthesis and the continuous presence of ATP. The molecular mechanisms have been extensively studied in yeast and mammalian orthologues continue to be elucidated (Ohsumi, 2001; Huang and Klionsky, 2002).
This distinction of a autophagic programmed cell death was made because it was determined that some cells would undergo caspase-independent gene-activated cell death but would display few of the ultrastructural features characteristic of apoptosis (Table 1) and would not exhibit DNA laddering (Cohen, 1991). However these cells do require de novo gene expression with an increase in expression of the polyubiquitin gene, similar to apoptosis (Ohsumi, 2001; Gozuacik and Kimchi, 2004). Specifically, autophagy occurs in all eukaryotic cells and involves the dynamic rearrangement of subcellular membranes to sequester cytoplasm and organelles for delivery to the lysosome or vacuole where degradation occurs. This is considered to be the major inducible pathway for the general turnover of cytoplasmic components.
A unique ubiquitin-like protein conjugation system and a protein complex that directs membrane docking and fusion at the lysosome or vacuole are important components of autophagy. In general, the process of autophagy can be divided into 4 steps: (1) induction, (2) formation of the autophagosome, (3) fusion with the lysosome or vacuole, and (4) autophagic body breakdown and recycling. Although the molecular details are still being elucidated, the regulation of this process occurs through various kinases, phosphatases and guanosine triphosphatases (GTPases).
There are some settings where autophagy and apoptosis seem to be interconnected and the idea of “molecular switches” between the two processes has been introduced (Piacentini et al., 2003). This is similar to the concept of the necrosis-apoptosis “continuum” or “paradox” which suggests that both apoptosis and necrosis represent morphologic expressions of a shared biochemical network in which the route of cell death depends on a variety of factors such as the physiologic milieu, developmental stage, tissue type, and the nature of the cell death signal (Hirsch et al., 1997; Zeiss, 2003). However, the core apoptotic pathway can be diverted to induce a necrotic phenotype by alteration of the availability of intracellular ATP and the availability of caspases.
A similar relationship may occur between apoptosis and autophagy. It has been suggested that mitochondria may be central organelles that integrate both apoptosis and autophagy (Elmore et al., 2001). Moreover, some of the same signals that are involved in apoptosis may also be involved in autophagy. For example, in both apoptosis and autophagy, there is the coordinated regulation of Akt (protein kinase B) and p70S6 kinase. Other proteins that may be part of the network connecting the two types of cell death include DAPk, Beclin 1, BNIP3, HSpin1, or protymosin-α (Klionsky and Emr, 2000). Malignant transformation is another link between autophagy and apoptosis (Gozuacik and Kimchi, 2004).
The role of genetic alterations in the pathways that control cellular proliferation/apoptosis in cancer development has been well established. Similarly, a correlation between reduced autophagy and cancer has also been documented. Studies have indicated that during malignant transformation several proteins and pathways that are related to autophagy signaling are deregulated resulting in reduced autophagocytic activity. This suggests that, in some circumstances, autophagy may function as a safeguard mechanism that restricts uncontrolled cell growth. Autophagy has also been considered a protective mechanism against apoptosis. Lemasters and coworkers (1998) observed that depolarized mitochondria, a feature of apoptosis, are rapidly eliminated by autophagy in primary hepatocytes. Eliminating damaged mitochondria prevents the release of pro-apoptotic substances from the mitochondria, thus preventing apoptosis.
Autophagy is considered the major cellular mechanism for disposing of long-lived proteins and cytoplasmic organelles; however, the concept of autophagic cell death has been a matter of debate within the scientific community. Since there is a distinct advantage of increased autophagy in various physiological and stress conditions, it has been suggested that autophagy represents an important adaptive mechanism that attempts to rescue cells from death. In other words, the presence of autophagic vesicles in dying cells may reflect an adaptive response to maintain cell survival under stress conditions rather than a reflection of “autophagic cell death.” Alternatively, are apoptosis and autophagic cell death mutually exclusive or can both apply in a situation similar to the apoptosis-necrosis continuum? It may be that the type of cell death depends on the severity of the response, the influences of cellular constituents and/or the influences of other signaling pathways.
There are reports that apoptosis and autophagic programmed cell death are not mutually exclusive and the diverse morphologies are attributed, in part, to distinct biochemical and molecular events (Gozuacik and Kimchi, 2004). In certain cells and under certain conditions the morphological features of autophagy may occur prior to apoptotic cell death, representing an early phase of apoptosis. The controversy still remains, however interest in the field of autophagic cell death is constantly increasing with the emergence of new assays and markers for elucidating the molecular basis of autophagy and its possible implications in programmed cell death and malignant cell transformation.
Conclusions
Apoptosis is regarded as a carefully regulated energy-dependent process, characterized by specific morphological and biochemical features in which caspase activation plays a central role. Although many of the key apoptotic proteins that are activated or inactivated in the apoptotic pathways have been identified, the molecular mechanisms of action or activation of these proteins are not fully understood and are the focus of continued research. The importance of understanding the mechanistic machinery of apoptosis is vital because programmed cell death is a component of both health and disease, being initiated by various physiologic and pathologic stimuli. Moreover, the widespread involvement of apoptosis in the pathophysiology of disease lends itself to therapeutic intervention at many different checkpoints. Understanding the mechanisms of apoptosis, and other variants of programmed cell death, at the molecular level provides deeper insight into various disease processes and may thus influence therapeutic strategy.
Acknowledgments
The authors wish to acknowledge Elizabeth Ney of the National Institute of Environmental Health Sciences for graphic design of Figure 3.
References
- Arur S, Uche UE, Rezaul K, Fong M, Scranton V, Cowan AE, Mohler W, Han DK. Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Dev Cell. 2003;4:587–98. doi: 10.1016/s1534-5807(03)00090-x. [DOI] [PubMed] [Google Scholar]
- Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–8. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- Barry M, Bleackley RC. Cytotoxic T lymphocytes: all roads lead to death. Nat Rev Immunol. 2002;2:401–9. doi: 10.1038/nri819. [DOI] [PubMed] [Google Scholar]
- Bedner E, Li X, Gorczyca W, Melamed MR, Darzynkiewicz Z. Analysis of apoptosis by laser scanning cytometry. Cytometry. 1999;35:181–95. doi: 10.1002/(sici)1097-0320(19990301)35:3<181::aid-cyto1>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Bortner CD, Oldenburg NB, Cidlowski JA. The role of DNA fragmentation in apoptosis. Trends Cell Biol. 1995;5:21–6. doi: 10.1016/s0962-8924(00)88932-1. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Green DR. Detection of apoptosis by Annexin V labeling. Methods Enzymol. 2000;322:15–18. doi: 10.1016/s0076-6879(00)22004-1. [DOI] [PubMed] [Google Scholar]
- Bratton DL, Fadok VA, Richter DA, Kailey JM, Guthrie LA, Henson PM. Appearance of phosphatidylserine on apoptotic cells requires calcium-mediated nonspecific flip-flop and is enhanced by loss of the aminophospholipid translocase. J Biol Chem. 1997;272:26159–65. doi: 10.1074/jbc.272.42.26159. [DOI] [PubMed] [Google Scholar]
- Brunner T, Wasem C, Torgler R, Cima I, Jakob S, Corazza N. Fas (CD95/Apo-1) ligand regulation in T cell homeostasis, cell-mediated cytotoxicity and immune pathology. Semin Immunol. 2003;15:167–76. doi: 10.1016/s1044-5323(03)00035-6. [DOI] [PubMed] [Google Scholar]
- Chau BN, Cheng EH, Kerr DA, Hardwick JM. Aven, a novel inhibitor of caspase activation, binds Bcl-xL and Apaf-1. Mol Cell. 2000;6:31–40. [PubMed] [Google Scholar]
- Cheng J, Zhou T, Liu C, Shapiro JP, Brauer MJ, Kiefer MC, Barr PJ, Mountz JD. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science. 1994;263:1759–62. doi: 10.1126/science.7510905. [DOI] [PubMed] [Google Scholar]
- Chicheportiche Y, Bourdon PR, Xu H, Hsu YM, Scott H, Hession C, Garcia I, Browning JL. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J Biol Chem. 1997;272:32401–10. doi: 10.1074/jbc.272.51.32401. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM. The apoptosome: heart and soul of the cell death machine. Neoplasia. 1999;1:5–15. doi: 10.1038/sj.neo.7900003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326(Pt 1):1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JJ. Programmed cell death in the immune system. Adv Immunol. 1991;50:55–85. doi: 10.1016/s0065-2776(08)60822-6. [DOI] [PubMed] [Google Scholar]
- Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–56. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- Cotran RS, Kumar V, Collins T. Cellular pathology I: cell injury and cell death. In: Cortan RS, Kumar V, Collins T, editors. Robbins Pathologic Basis of Disease. 6. W.B. Saunders Co; Philadelphia, PA: 1999. pp. 1–29. [Google Scholar]
- Dal Canto MC, Gurney ME. Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am J Pathol. 1994;145:1271–9. [PMC free article] [PubMed] [Google Scholar]
- Darzynkiewicz Z, Bedner E, Li X, Gorczyca W, Melamed MR. Laser-scanning cytometry: A new instrumentation with many applications. Exp Cell Res. 1999;249:1–12. doi: 10.1006/excr.1999.4477. [DOI] [PubMed] [Google Scholar]
- Debnath J, Baehrecke EH, Kroemer G. Does autophagy contribute to cell death? Autophagy. 2005;1:66–74. doi: 10.4161/auto.1.2.1738. [DOI] [PubMed] [Google Scholar]
- Denecker G, Vercammen D, Declercq W, Vandenabeele P. Apoptotic and necrotic cell death induced by death domain receptors. Cell Mol Life Sci. 2001;58:356–70. doi: 10.1007/PL00000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devadas S, Das J, Liu C, Zhang L, Roberts AI, Pan Z, Moore PA, Das G, Shi Y. Granzyme B is critical for T cell receptor-induced cell death of type 2 helper T cells. Immunity. 2006;25:237–47. doi: 10.1016/j.immuni.2006.06.011. [DOI] [PubMed] [Google Scholar]
- Deveraux QL, Reed JC. IAP family proteins—suppressors of apoptosis. Genes Dev. 1999;13:239–52. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- Diez-Roux G, Lang RA. Macrophages induce apoptosis in normal cells in vivo. Development. 1997;124:3633–8. doi: 10.1242/dev.124.18.3633. [DOI] [PubMed] [Google Scholar]
- Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Ekert PG, Vaux DL. The mitochondrial death squad: hard- ened killers or innocent bystanders? Curr Opin Cell Biol. 2005;17:626–30. doi: 10.1016/j.ceb.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001;15:2286–7. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- Elnemr A, Ohta T, Yachie A, Kayahara M, Kitagawa H, Ninomiya I, Fushida S, Fujimura T, Nishimura G, Shimizu K, Miwa K. Human pancreatic cancer cells express non-functional Fas receptors and counterattack lymphocytes by expressing Fas ligand; a potential mechanism for immune escape. Int J Oncol. 2001;18:33–9. [PubMed] [Google Scholar]
- Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- Esposti MD. The roles of Bid. Apoptosis. 2002;7:433–40. doi: 10.1023/a:1020035124855. [DOI] [PubMed] [Google Scholar]
- Ethell DW, Buhler LA. Fas ligand-mediated apoptosis in degenerative disorders of the brain. J Clin Immunol. 2003;23:439–46. doi: 10.1023/b:joci.0000010420.96419.a8. [DOI] [PubMed] [Google Scholar]
- Fadok VA, de Cathelineau A, Daleke DL, Henson PM, Bratton DL. Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J Biol Chem. 2001;276:1071–7. doi: 10.1074/jbc.M003649200. [DOI] [PubMed] [Google Scholar]
- Fan Z, Beresford PJ, Oh DY, Zhang D, Lieberman J. Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL- mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell. 2003;112:659–72. doi: 10.1016/s0092-8674(03)00150-8. [DOI] [PubMed] [Google Scholar]
- Ferraro-Peyret C, Quemeneur L, Flacher M, Revillard JP, Genestier L. Caspase-independent phosphatidylserine exposure during apoptosis of primary T lymphocytes. J Immunol. 2002;169:4805–10. doi: 10.4049/jimmunol.169.9.4805. [DOI] [PubMed] [Google Scholar]
- Fiers W, Beyaert R, Declercq W, Vandenabeele P. More than one way to die: apoptosis, necrosis and reactive oxygen damage. Oncogene. 1999;18:7719–30. doi: 10.1038/sj.onc.1203249. [DOI] [PubMed] [Google Scholar]
- Formigli L, Papucci L, Tani A, Schiavone N, Tempestini A, Orlandini GE, Capaccioli S, Orlandini SZ. Aponecrosis: morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J Cell Physiol. 2000;182:41–9. doi: 10.1002/(SICI)1097-4652(200001)182:1<41::AID-JCP5>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Freude B, Masters TN, Robicsek F, Fokin A, Kostin S, Zimmermann R, Ullmann C, Lorenz-Meyer S, Schaper J. Apoptosis is initiated by myocardial ischemia and executed during reperfusion. J Mol Cell Cardiol. 2000;32:197–208. doi: 10.1006/jmcc.1999.1066. [DOI] [PubMed] [Google Scholar]
- Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia- reperfusion injury in mouse heart. Circulation. 2000;101:660–7. doi: 10.1161/01.cir.101.6.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–34. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, Kroemer G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006;13:1423–33. doi: 10.1038/sj.cdd.4401950. [DOI] [PubMed] [Google Scholar]
- Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol. 2000;2:156–62. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- Goping IS, Barry M, Liston P, Sawchuk T, Constantinescu G, Michalak KM, Shostak I, Roberts DL, Hunter AM, Korneluk R, Bleackley RC. Granzyme B-induced apoptosis requires both direct caspase activation and relief of caspase inhibition. Immunity. 2003;18:355–65. doi: 10.1016/s1074-7613(03)00032-3. [DOI] [PubMed] [Google Scholar]
- Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- Grabarek J, Amstad P, Darzynkiewicz Z. Use of fluorescently labeled caspase inhibitors as affinity labels to detect activated caspases. Hum Cell. 2002;15:1–12. doi: 10.1111/j.1749-0774.2002.tb00094.x. [DOI] [PubMed] [Google Scholar]
- Graziani G, Szabo C. Clinical perspectives of PARP inhibitors. Pharmacol Res. 2005;52:109–18. doi: 10.1016/j.phrs.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Greenhalgh DG. The role of apoptosis inwound healing. Int J Biochem Cell Biol. 1998;30:1019–30. doi: 10.1016/s1357-2725(98)00058-2. [DOI] [PubMed] [Google Scholar]
- Gu J, Kawai H, Wiederschain D, Yuan ZM. Mechanism of functional inactivation of a Li-Fraumeni syndrome p53 that has a mutation outside of the DNA-binding domain. Cancer Res. 2001;61:1741–6. [PubMed] [Google Scholar]
- Gurtu V, Kain SR, Zhang G. Fluorometric and colorimetric detection of caspase activity associated with apoptosis. Anal Biochem. 1997;251:98–102. doi: 10.1006/abio.1997.2220. [DOI] [PubMed] [Google Scholar]
- Hacker G. The morphology of apoptosis. Cell Tissue Res. 2000;301:5–17. doi: 10.1007/s004410000193. [DOI] [PubMed] [Google Scholar]
- Harman D. Role of free radicals in aging and disease. Ann N Y Acad Sci. 1992;673:126–41. doi: 10.1111/j.1749-6632.1992.tb27444.x. [DOI] [PubMed] [Google Scholar]
- Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–6. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- Hetz C, Vitte PA, Bombrun A, Rostovtseva TK, Montessuit S, Hiver A, Schwarz MK, Church DJ, Korsmeyer SJ, Martinou JC, Antonsson B. Bax channel inhibitors prevent mitochondrion- mediated apoptosis and protect neurons in a model of global brain ischemia. J Biol Chem. 2005;280:42960–70. doi: 10.1074/jbc.M505843200. [DOI] [PubMed] [Google Scholar]
- Hill MM, Adrain C, Duriez PJ, Creagh EM, Martin SJ. Analysis of the composition, assembly kinetics and activity of native Apaf-1 apoptosomes. Embo J. 2004;23:2134–45. doi: 10.1038/sj.emboj.7600210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch T, Marchetti P, Susin SA, Dallaporta B, Zamzami N, Marzo I, Geuskens M, Kroemer G. The apoptosis-necrosis paradox. Apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene. 1997;15:1573–81. doi: 10.1038/sj.onc.1201324. [DOI] [PubMed] [Google Scholar]
- Hitoshi Y, Lorens J, Kitada SI, Fisher J, LaBarge M, Ring HZ, Francke U, Reed JC, Kinoshita S, Nolan GP. Toso, a cell surface, specific regulator of Fas-induced apoptosis in T cells. Immunity. 1998;8:461–71. doi: 10.1016/s1074-7613(00)80551-8. [DOI] [PubMed] [Google Scholar]
- Hochhauser E, Kivity S, Offen D, Maulik N, Otani H, Barhum Y, Pannet H, Shneyvays V, Shainberg A, Goldshtaub V, Tobar A, Vidne BA. Bax ablation protects against myocardial ischemia-reperfusion injury in transgenic mice. Am J Physiol Heart Circ Physiol. 2003;284:H2351–9. doi: 10.1152/ajpheart.00783.2002. [DOI] [PubMed] [Google Scholar]
- Hoeppner DJ, Hengartner MO, Schnabel R. Engulfment genes cooperate with ced-3 to promote cell death in Caenorhabditis elegans. Nature. 2001;412:202–6. doi: 10.1038/35084103. [DOI] [PubMed] [Google Scholar]
- Hofmann WK, de Vos S, Tsukasaki K, Wachsman W, Pinkus GS, Said JW, Koeffler HP. Altered apoptosis pathways in mantle cell lymphoma detected by oligonucleotide microarray. Blood. 2001;98:787–94. doi: 10.1182/blood.v98.3.787. [DOI] [PubMed] [Google Scholar]
- Horvitz HR. Genetic control of programmed cell death in the nematode Caenorhabditis elegans. Cancer Res. 1999;59:1701s–1706s. [PubMed] [Google Scholar]
- Howden R, Hanlon PR, Petranka JG, Kleeberger S, Bucher J, Dunnick J, Nyska A, Murphy E. Ephedrine plus caffeine causes age-dependent cardiovascular responses in Fischer 344 rats. Am J Physiol Heart Circ Physiol. 2005;288:2219–24. doi: 10.1152/ajpheart.01164.2004. [DOI] [PubMed] [Google Scholar]
- Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- Hu S, Snipas SJ, Vincenz C, Salvesen G, Dixit VM. Caspase- 14 is a novel developmentally regulated protease. J Biol Chem. 1998;273:29648–53. doi: 10.1074/jbc.273.45.29648. [DOI] [PubMed] [Google Scholar]
- Huang WP, Klionsky DJ. Autophagy in yeast: a review of the molecular machinery. Cell Struct Funct. 2002;27:409–20. doi: 10.1247/csf.27.409. [DOI] [PubMed] [Google Scholar]
- Igney FH, Krammer PH. Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer. 2002;2:277–88. doi: 10.1038/nrc776. [DOI] [PubMed] [Google Scholar]
- Jimenez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med. 2000;6:41–8. doi: 10.1038/71517. [DOI] [PubMed] [Google Scholar]
- Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CY, Sasaki T, Elia AJ, Cheng HY, Ravagnan L, Ferri KF, Zamzami N, Wakeham A, Hakem R, Yoshida H, Kong YY, Mak TW, Zuniga- Pflucker JC, Kroemer G, Penninger JM. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;410:549–54. doi: 10.1038/35069004. [DOI] [PubMed] [Google Scholar]
- Kang SJ, Wang S, Kuida K, Yuan J. Distinct downstream pathways of caspase-11 in regulating apoptosis and cytokine maturation during septic shock response. Cell Death Differ. 2002;9:1115–25. doi: 10.1038/sj.cdd.4401087. [DOI] [PubMed] [Google Scholar]
- Kataoka T, Schroter M, Hahne M, Schneider P, Irmler M, Thome M, Froelich CJ, Tschopp J. FLIP prevents apoptosis induced by death receptors but not by perforin/granzyme B, chemotherapeutic drugs, and gamma irradiation. J Immunol. 1998;161:3936–42. [PubMed] [Google Scholar]
- Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8:297–303. doi: 10.1016/s1074-7613(00)80535-x. [DOI] [PubMed] [Google Scholar]
- Kerr JF. History of the events leading to the formulation of the apoptosis concept. Toxicology. 2002;181–182:471–4. doi: 10.1016/s0300-483x(02)00457-2. [DOI] [PubMed] [Google Scholar]
- Kerr JF, Winterford CM, Harmon BV. Apoptosis. Its significance in cancer and cancer therapy. Cancer. 1994;73:2013–26. doi: 10.1002/1097-0142(19940415)73:8<2013::aid-cncr2820730802>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King KL, Cidlowski JA. Cell cycle regulation and apoptosis. Annu Rev Physiol. 1998;60:601–17. doi: 10.1146/annurev.physiol.60.1.601. [DOI] [PubMed] [Google Scholar]
- Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME. Cytotoxicity-dependent APO-1 (Fas/CD95)- associated proteins form a death-inducing signaling complex (DISC) with the receptor. Embo J. 1995;14:5579–88. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–21. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig U, Eckhart L, Tschachler E. Evidence that caspase-13 is not a human but a bovine gene. Biochem Biophys Res Commun. 2001;285:1150–4. doi: 10.1006/bbrc.2001.5315. [DOI] [PubMed] [Google Scholar]
- Kothakota S, Azuma T, Reinhard C, Klippel A, Tang J, Chu K, McGarry TJ, Kirschner MW, Koths K, Kwiatkowski DJ, Williams LT. Caspase-3-generated fragment of gelsolin: effector of morphological change in apoptosis. Science. 1997;278:294–8. doi: 10.1126/science.278.5336.294. [DOI] [PubMed] [Google Scholar]
- Koyama S, Koike N, Adachi S. Fas receptor counterattack against tumor-infiltrating lymphocytes in vivo as a mechanism of immune escape in gastric carcinoma. J Cancer Res Clin Oncol. 2001;127:20–6. doi: 10.1007/s004320000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kressel M, Groscurth P. Distinction of apoptotic and necrotic cell death by in situ labelling of fragmented DNA. Cell Tissue Res. 1994;278:549–56. doi: 10.1007/BF00331373. [DOI] [PubMed] [Google Scholar]
- Kurosaka K, Takahashi M, Watanabe N, Kobayashi Y. Silent cleanup of very early apoptotic cells by macrophages. J Immunol. 2003;171:4672–9. doi: 10.4049/jimmunol.171.9.4672. [DOI] [PubMed] [Google Scholar]
- Kurz EU, Lees-Miller SP. DNA damage-induced activation of ATM and ATM-dependent signaling pathways. DNA Repair (Amst) 2004;3:889–900. doi: 10.1016/j.dnarep.2004.03.029. [DOI] [PubMed] [Google Scholar]
- Lang RA, Bishop JM. Macrophages are required for cell death and tissue remodeling in the developing mouse eye. Cell. 1993;74:453–62. doi: 10.1016/0092-8674(93)80047-i. [DOI] [PubMed] [Google Scholar]
- Le GT, Abbenante G. Inhibitors of TACE and Caspase-1 as anti-inflammatory drugs. Curr Med Chem. 2005;12:2963–77. doi: 10.2174/092986705774462851. [DOI] [PubMed] [Google Scholar]
- Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med. 1997;185:1481–6. doi: 10.1084/jem.185.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE, Bradham CA, Brenner DA, Herman B. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta. 1998;1366:177–96. doi: 10.1016/s0005-2728(98)00112-1. [DOI] [PubMed] [Google Scholar]
- Levin S, Bucci TJ, Cohen SM, Fix AS, Hardisty JF, LeGrand EK, Maronpot RR, Trump BF. The nomenclature of cell death: recommendations of an ad hoc Committee of the Society of Toxicologic Pathologists. Toxicol Pathol. 1999;27:484–90. doi: 10.1177/019262339902700419. [DOI] [PubMed] [Google Scholar]
- Li CJ, Friedman DJ, Wang C, Metelev V, Pardee AB. Induction of apoptosis in uninfected lymphocytes by HIV-1 Tat protein. Science. 1995;268:429–31. doi: 10.1126/science.7716549. [DOI] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase-8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–9. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- Lieberman J, Fan Z. Nuclear war: the granzyme A-bomb. Curr Opin Immunol. 2003;15:553–9. doi: 10.1016/s0952-7915(03)00108-0. [DOI] [PubMed] [Google Scholar]
- Little GH, Flores A. Inhibition of programmed cell death by catalase and phenylalanine methyl ester. Comp Biochem Physiol Comp Physiol. 1993;105:79–83. doi: 10.1016/0300-9629(93)90176-5. [DOI] [PubMed] [Google Scholar]
- Liu FT, Newland AC, Jia L. Bax conformational change is a crucial step for PUMA-mediated apoptosis in human leukemia. Biochem Biophys Res Commun. 2003;310:956–62. doi: 10.1016/j.bbrc.2003.09.109. [DOI] [PubMed] [Google Scholar]
- Livingston DJ. In vitro and in vivo studies of ICE inhibitors. J Cell Biochem. 1997;64:19–26. [PubMed] [Google Scholar]
- Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- Love S, Barber R, Wilcock GK. Increased poly(ADP- ribosyl)ation of nuclear proteins in Alzheimer’s disease. Brain. 1999;122(Pt 2):247–53. doi: 10.1093/brain/122.2.247. [DOI] [PubMed] [Google Scholar]
- Lund LR, Romer J, Thomasset N, Solberg H, Pyke C, Bissell MJ, Dano K, Werb Z. Two distinct phases of apoptosis in mammary gland involution: proteinase-independent and -dependent pathways. Development. 1996;122:181–93. doi: 10.1242/dev.122.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- Mandal D, Mazumder A, Das P, Kundu M, Basu J. Fas-, caspase-8-, and caspase-3-dependent signaling regulates the activity of the aminophospholipid translocase and phosphatidylserine externalization in human erythrocytes. J Biol Chem. 2005;280:39460–7. doi: 10.1074/jbc.M506928200. [DOI] [PubMed] [Google Scholar]
- Martinvalet D, Zhu P, Lieberman J. Granzyme A induces caspase- independent mitochondrial damage, a required first step for apoptosis. Immunity. 2005;22:355–70. doi: 10.1016/j.immuni.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Metzstein MM, Stanfield GM, Horvitz HR. Genetics of programmed cell death in C. elegans: past, present and future. Trends Genet. 1998;14:410–6. doi: 10.1016/s0168-9525(98)01573-x. [DOI] [PubMed] [Google Scholar]
- Meyer N, Kim SS, Penn LZ. The Oscar-worthy role of Myc in apoptosis. Semin Cancer Biol. 2006;16:275–87. doi: 10.1016/j.semcancer.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Liebermann DA, Hoffman B, Reed JC. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 1994;9:1799–805. [PubMed] [Google Scholar]
- Mocanu MM, Baxter GF, Yellon DM. Caspase inhibition and limitation of myocardial infarct size: protection against lethal reperfusion injury. Br J Pharmacol. 2000;130:197–200. doi: 10.1038/sj.bjp.0703336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- Nemes Z, Jr, Friis RR, Aeschlimann D, Saurer S, Paulsson M, Fesus L. Expression and activation of tissue transglutaminase in apoptotic cells of involuting rodent mammary tissue. Eur J Cell Biol. 1996;70:125–33. [PubMed] [Google Scholar]
- Newmeyer DD, Bossy-Wetzel E, Kluck RM, Wolf BB, Beere HM, Green DR. Bcl-xL does not inhibit the function of Apaf-1. Cell Death Differ. 2000;7:402–7. doi: 10.1038/sj.cdd.4400665. [DOI] [PubMed] [Google Scholar]
- Nicholson DW. From bench to clinic with apoptosis-based therapeutic agents. Nature. 2000;407:810–6. doi: 10.1038/35037747. [DOI] [PubMed] [Google Scholar]
- Nijhawan D, Honarpour N, Wang X. Apoptosis in neural development and disease. Annu Rev Neurosci. 2000;23:73–87. doi: 10.1146/annurev.neuro.23.1.73. [DOI] [PubMed] [Google Scholar]
- Noda T, Suzuki K, Ohsumi Y. Yeast autophagosomes: de novo formation of a membrane structure. Trends Cell Biol. 2002;12:231–5. doi: 10.1016/s0962-8924(02)02278-x. [DOI] [PubMed] [Google Scholar]
- Norbury CJ, Hickson ID. Cellular responses to DNA damage. Annu Rev Pharmacol Toxicol. 2001;41:367–401. doi: 10.1146/annurev.pharmtox.41.1.367. [DOI] [PubMed] [Google Scholar]
- Nyska A, Murphy E, Foley JF, Collins BJ, Petranka J, Howden R, Hanlon P, Dunnick JK. Acute hemorrhagic myocardial necrosis and sudden death of rats exposed to a combination of ephedrine and caffeine. Toxicol Sci. 2005;83:388–96. doi: 10.1093/toxsci/kfi034. [DOI] [PubMed] [Google Scholar]
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–8. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2:211–6. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- Opferman JT, Korsmeyer SJ. Apoptosis in the development and maintenance of the immune system. Nat Immunol. 2003;4:410–5. doi: 10.1038/ni0503-410. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Flavell RA, Vinsant S, Prevette D, Kuan CY, Rakic P. Programmed cell death of developing mammalian neurons after genetic deletion of caspases. J Neurosci. 2001;21:4752–60. doi: 10.1523/JNEUROSCI.21-13-04752.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne BA. Apoptosis and the maintenance of homoeostasis in the immune system. Curr Opin Immunol. 1996;8:245–54. doi: 10.1016/s0952-7915(96)80063-x. [DOI] [PubMed] [Google Scholar]
- Otsuki Y, Li Z, Shibata MA. Apoptotic detection methods–from morphology to gene. Prog Histochem Cytochem. 2003;38:275–339. doi: 10.1016/s0079-6336(03)80002-5. [DOI] [PubMed] [Google Scholar]
- Ozawa T. Mechanism of somatic mitochondrial DNA mutations associated with age and diseases. Biochim Biophys Acta. 1995;1271:177–89. doi: 10.1016/0925-4439(95)00026-z. [DOI] [PubMed] [Google Scholar]
- Paweletz N. Walther Flemming: pioneer of mitosis research. Nat Rev Mol Cell Biol. 2001;2:72–5. doi: 10.1038/35048077. [DOI] [PubMed] [Google Scholar]
- Peter ME, Krammer PH. Mechanisms of CD95 (APO-1/Fas)- mediated apoptosis. Curr Opin Immunol. 1998;10:545–51. doi: 10.1016/s0952-7915(98)80222-7. [DOI] [PubMed] [Google Scholar]
- Piacentini M, Evangelisti C, Mastroberardino PG, Nardacci R, Kroemer G. Does prothymosin-alpha act as molecular switch between apoptosis and autophagy? Cell Death Differ. 2003;10:937–9. doi: 10.1038/sj.cdd.4401282. [DOI] [PubMed] [Google Scholar]
- Pietenpol JA, Stewart ZA. Cell cycle checkpoint signaling: cell cycle arrest versus apoptosis. Toxicology. 2002;181–182:475–81. doi: 10.1016/s0300-483x(02)00460-2. [DOI] [PubMed] [Google Scholar]
- Poot M, Pierce RH. Detection of changes in mitochondrial function during apoptosis by simultaneous staining with multiple fluorescent dyes and correlated multiparameter flow cytometry. Cytometry. 1999;35:311–7. doi: 10.1002/(sici)1097-0320(19990401)35:4<311::aid-cyto3>3.3.co;2-5. [DOI] [PubMed] [Google Scholar]
- Proskuryakov SY, Konoplyannikov AG, Gabai VL. Necrosis: a specific form of programmed cell death? Exp Cell Res. 2003;283:1–16. doi: 10.1016/s0014-4827(02)00027-7. [DOI] [PubMed] [Google Scholar]
- Rai NK, Tripathi K, Sharma D, Shukla VK. Apoptosis: a basic physiologic process in wound healing. Int J Low Extrem Wounds. 2005;4:138–44. doi: 10.1177/1534734605280018. [DOI] [PubMed] [Google Scholar]
- Ravagnan L, Roumier T, Kroemer G. Mitochondria, the killer organelles and their weapons. J Cell Physiol. 2002;192:131–7. doi: 10.1002/jcp.10111. [DOI] [PubMed] [Google Scholar]
- Reddien PW, Cameron S, Horvitz HR. Phagocytosis promotes programmed cell death in C. elegans. Nature. 2001;412:198–202. doi: 10.1038/35084096. [DOI] [PubMed] [Google Scholar]
- Renehan AG, Booth C, Potten CS. What is apoptosis, and why is it important? Bmj. 2001;322:1536–8. doi: 10.1136/bmj.322.7301.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Moscardo F, Blesa D, Mestre C, Siebert R, Balasas T, Benito A, Rosenwald A, Climent J, Martinez JI, Schilhabel M, Karran EL, Gesk S, Esteller M, deLeeuw R, Staudt LM, Fernandez-Luna JL, Pinkel D, Dyer MJ, Martinez-Climent JA. Characterization of 8p21.3 chromosomal deletions in B-cell lymphoma: TRAIL-R1 and TRAIL-R2 as candidate dosage-dependent tumor suppressor genes. Blood. 2005;106:3214–22. doi: 10.1182/blood-2005-05-2013. [DOI] [PubMed] [Google Scholar]
- Russell JH, Ley TJ. Lymphocyte-mediated cytotoxicity. Annu Rev Immunol. 2002;20:323–70. doi: 10.1146/annurev.immunol.20.100201.131730. [DOI] [PubMed] [Google Scholar]
- Saelens X, Festjens N, Vande Walle L, van Gurp M, van Loo G, Vandenabeele P. Toxic proteins released from mitochondria in cell death. Oncogene. 2004;23:2861–74. doi: 10.1038/sj.onc.1207523. [DOI] [PubMed] [Google Scholar]
- Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–9. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature. 2000;407:784–8. doi: 10.1038/35037722. [DOI] [PubMed] [Google Scholar]
- Scaffidi C, Schmitz I, Krammer PH, Peter ME. The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem. 1999;274:1541–8. doi: 10.1074/jbc.274.3.1541. [DOI] [PubMed] [Google Scholar]
- Schimmer AD. Inhibitor of apoptosis proteins: translating basic knowledge into clinical practice. Cancer Res. 2004;64:7183–90. doi: 10.1158/0008-5472.CAN-04-1918. [DOI] [PubMed] [Google Scholar]
- Schuler M, Green DR. Mechanisms of p53-dependent apoptosis. Biochem Soc Trans. 2001;29:684–8. doi: 10.1042/0300-5127:0290684. [DOI] [PubMed] [Google Scholar]
- Schwartz LM, Smith SW, Jones ME, Osborne BA. Do all programmed cell deaths occur via apoptosis? Proc Natl Acad Sci USA. 1993;90:980–4. doi: 10.1073/pnas.90.3.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- Silke J, Hawkins CJ, Ekert PG, Chew J, Day CL, Pakusch M, Verhagen AM, Vaux DL. The anti-apoptotic activity of XIAP is retained upon mutation of both the caspase-3- and caspase-9-interacting sites. J Cell Biol. 2002;157:115–24. doi: 10.1083/jcb.200108085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slee EA, Adrain C, Martin SJ. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J Biol Chem. 2001;276:7320–6. doi: 10.1074/jbc.M008363200. [DOI] [PubMed] [Google Scholar]
- Smyth MJ, Godfrey DI, Trapani JA. A fresh look at tumor immunosurveillance and immunotherapy. Nat Immunol. 2001;2:293–9. doi: 10.1038/86297. [DOI] [PubMed] [Google Scholar]
- Sperandio S, de Belle I, Bredesen DE. An alternative, non- apoptotic form of programmed cell death. Proc Natl Acad Sci USA. 2000;97:14376–81. doi: 10.1073/pnas.97.26.14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suliman A, Lam A, Datta R, Srivastava RK. Intracellular mechanisms of TRAIL: apoptosis through mitochondrial-dependent and -independent pathways. Oncogene. 2001;20:2122–33. doi: 10.1038/sj.onc.1204282. [DOI] [PubMed] [Google Scholar]
- Susin SA, Daugas E, Ravagnan L, Samejima K, Zamzami N, Loeffler M, Costantini P, Ferri KF, Irinopoulou T, Prevost MC, Brothers G, Mak TW, Penninger J, Earnshaw WC, Kroemer G. Two distinct pathways leading to nuclear apoptosis. J Exp Med. 2000;192:571–80. doi: 10.1084/jem.192.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talasz H, Helliger W, Sarg B, Debbage PL, Puschendorf B, Lindner H. Hyperphosphorylation of histone H2A.X and dephosphorylation of histone H1 subtypes in the course of apoptosis. Cell Death Differ. 2002;9:27–39. doi: 10.1038/sj.cdd.4400925. [DOI] [PubMed] [Google Scholar]
- Tilly JL, Kowalski KI, Johnson AL, Hsueh AJ. Involvement of apoptosis in ovarian follicular atresia and postovulatory regression. Endocrinology. 1991;129:2799–801. doi: 10.1210/endo-129-5-2799. [DOI] [PubMed] [Google Scholar]
- Trapani JA, Smyth MJ. Functional significance of the perforin/granzyme cell death pathway. Nat Rev Immunol. 2002;2:735–47. doi: 10.1038/nri911. [DOI] [PubMed] [Google Scholar]
- Trump BF, Berezesky IK, Chang SH, Phelps PC. The pathways of cell death: oncosis, apoptosis, and necrosis. Toxicol Pathol. 1997;25:82–8. doi: 10.1177/019262339702500116. [DOI] [PubMed] [Google Scholar]
- Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–44. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington’s disease. Proc Natl Acad Sci USA. 2000;97:8093–7. doi: 10.1073/pnas.110078997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallat L, Magdelenat H, Merle-Beral H, Masdehors P, Potocki de Montalk G, Davi F, Kruhoffer M, Sabatier L, Orntoft TF, Delic J. The resistance of B-CLL cells to DNA damage-induced apoptosis defined by DNA microarrays. Blood. 2003;101:4598–606. doi: 10.1182/blood-2002-06-1743. [DOI] [PubMed] [Google Scholar]
- van Loo G, van Gurp M, Depuydt B, Srinivasula SM, Rodriguez I, Alnemri ES, Gevaert K, Vandekerckhove J, Declercq W, Vandenabeele P. The serine protease Omi/HtrA2 is released from mitochondria during apoptosis. Omi interacts with caspase-inhibitor XIAP and induces enhanced caspase activity. Cell Death Differ. 2002a;9:20–6. doi: 10.1038/sj.cdd.4400970. [DOI] [PubMed] [Google Scholar]
- van Loo G, Saelens X, van Gurp M, MacFarlane M, Martin SJ, Vandenabeele P. The role of mitochondrial factors in apoptosis: a Russian roulette with more than one bullet. Cell Death Differ. 2002b;9:1031–42. doi: 10.1038/sj.cdd.4401088. [DOI] [PubMed] [Google Scholar]
- Varley JM, Evans DG, Birch JM. Li-Fraumeni syndrome—a molecular and clinical review. Br J Cancer. 1997;76:1–14. doi: 10.1038/bjc.1997.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux DL. Apoptosis and toxicology—what relevance? Toxicology. 2002;181–182:3–7. doi: 10.1016/s0300-483x(02)00248-2. [DOI] [PubMed] [Google Scholar]
- Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–2. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Volbracht C, Leist M, Kolb SA, Nicotera P. Apoptosis in caspase-inhibited neurons. Mol Med. 2001;7:36–48. [PMC free article] [PubMed] [Google Scholar]
- Wajant H. The Fas signaling pathway: more than a paradigm. Science. 2002;296:1635–6. doi: 10.1126/science.1071553. [DOI] [PubMed] [Google Scholar]
- Wang XW, Harris CC. p53 tumor-suppressor gene: clues to molecular carcinogenesis. J Cell Physiol. 1997;173:247–55. doi: 10.1002/(SICI)1097-4652(199711)173:2<247::AID-JCP30>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Hitomi M, van der Wee K, Rothenberg F, Fisher SA, Zucker R, Svoboda KK, Goldsmith EC, Heiskanen KM, Nieminen AL. The pros and cons of apoptosis assays for use in the study of cells, tissues, and organs. Microsc Microanal. 2002;8:375–91. doi: 10.1017/S1431927602010346. [DOI] [PubMed] [Google Scholar]
- White MK, Cinti C. A morphologic approach to detect apoptosis based on electron microscopy. Methods Mol Biol. 2004;285:105–11. doi: 10.1385/1-59259-822-6:105. [DOI] [PubMed] [Google Scholar]
- Worth A, Thrasher AJ, Gaspar HB. Autoimmune lymphoproliferative syndrome: molecular basis of disease and clinical phenotype. Br J Haematol. 2006;133:124–40. doi: 10.1111/j.1365-2141.2006.05993.x. [DOI] [PubMed] [Google Scholar]
- Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284:555–6. doi: 10.1038/284555a0. [DOI] [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–91. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- Zeiss CJ. The apoptosis-necrosis continuum: insights from genetically altered mice. Vet Pathol. 2003;40:481–95. doi: 10.1354/vp.40-5-481. [DOI] [PubMed] [Google Scholar]
- Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87:619–28. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- Zhang J, Campbell RE, Ting AY, Tsien RY. Creating new fluorescent probes for cell biology. Nat Rev Mol Cell Biol. 2002;3:906–18. doi: 10.1038/nrm976. [DOI] [PubMed] [Google Scholar]
- Zhou HZ, Swanson RA, Simonis U, Ma X, Cecchini G, Gray MO. Poly(ADP-ribose) polymerase-1 hyperactivation and impairment of mitochondrial respiratory chain complex I function in reperfused mouse hearts. Am J Physiol Heart Circ Physiol. 2006;291:H714–23. doi: 10.1152/ajpheart.00823.2005. [DOI] [PubMed] [Google Scholar]
- Zhu P, Zhang D, Chowdhury D, Martinvalet D, Keefe D, Shi L, Lieberman J. Granzyme A, which causes single-stranded DNA damage, targets the double-strand break repair protein Ku70. EMBO Rep. 2006;7:431–7. doi: 10.1038/sj.embor.7400622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RM, Hunter ES, 3rd, Rogers JM. Confocal laser scanning microscopy of morphology and apoptosis in organogenesis-stage mouse embryos. Methods Mol Biol. 2000;135:191–202. doi: 10.1385/1-59259-685-1:191. [DOI] [PubMed] [Google Scholar]