

Abstract
The past decade has seen a significant increase in the number of potentially tolerogenic therapies for treatment of new-onset diabetes. However, most treatments are antigen nonspecific, and the mechanism for the maintenance of long-term tolerance remains unclear. In this study, we developed an antigen-specific therapy, insulin-coupled antigen-presenting cells, to treat diabetes in nonobese diabetic mice after disease onset. Using this approach, we demonstrate disease remission, inhibition of pathogenic T cell proliferation, decreased cytokine production, and induction of anergy. Moreover, we show that robust long-term tolerance depends on the programmed death 1 (PD-1)–programmed death ligand (PD-L)1 pathway, not the distinct cytotoxic T lymphocyte–associated antigen 4 pathway. Anti–PD-1 and anti–PD-L1, but not anti–PD-L2, reversed tolerance weeks after tolerogenic therapy by promoting antigen-specific T cell proliferation and inflammatory cytokine production directly in infiltrated tissues. PD-1–PD-L1 blockade did not limit T regulatory cell activity, suggesting direct effects on pathogenic T cells. Finally, we describe a critical role for PD-1–PD-L1 in another powerful immunotherapy model using anti-CD3, suggesting that PD-1–PD-L1 interactions form part of a common pathway to selectively maintain tolerance within the target tissues.
Autoimmune diabetes results from a systemic breakdown in the central and peripheral mechanisms of tolerance (1). Autoreactive T cells respond to autoantigens in conjunction with costimulatory signals promoting initial T cell activation resulting in selective expansion, differentiation, tissue invasion, and ultimately destruction. Thus, many attempts to regulate autoimmunity have focused on therapies targeting early activation. However, these therapies, often effective for preventing disease, have provided little success in abrogating or reversing ongoing disease (2). In sharp contrast, targeting the TCR complex has been successful in abrogating autoimmunity and inducing tolerance after disease onset has occurred (3). To this end, FcR-nonbinding anti-CD3 mAb and anti-thymocyte globulin have been among the most effective therapies for reversing diabetes in nonobese diabetic (NOD) mice, with initial efficacy in patients with type 1 diabetes (T1D) as well (3, 4). However, these broad-based immunosuppressive treatments have potentially significant side effects, including increased development of infections and cancer. In contrast, antigen-specific therapies for treatment of autoimmune diabetes “after” disease onset, although potentially safer, have been less successful. Administration of antigen-pulsed ethylene carbodiimide–fixed APCs has been used efficiently to induce specific tolerance and ameliorate ongoing experimental autoimmune encephalitis (EAE) disease in mice (5). Interestingly, like anti-CD3 therapy, the efficacy of the antigen-coupled APC therapy depends on a direct effect on pathogenic T cells within the inflamed tissue (6, 7), implying that a cell-intrinsic mechanism induces and maintains tolerance in the setting of continual autoantigen exposure. Finally, recent studies have positioned insulin as the primary autoantigen in T1D in mice and man (8).
Several cell surface molecules have been implicated in the control of immune responses in various tolerance settings. Many studies by our group and others have demonstrated the importance of cytotoxic T lymphocyte–associated antigen 4 (CTLA-4) in immune tolerance. Engagement of CTLA-4 down-regulates T cell proliferation and cytokine production by altering TCR complex phosphorylation. In a variety of autoimmune diseases, CTLA-4 engagement is critical to initiate a tolerogenic response and in some settings regulates T regulatory (T reg) cell activity. Less is known about a related CD28/CTLA-4 family member, programmed death 1 (PD-1; CD279). Recent studies have shown that PD-1 engagement on activated T cells limits proliferation and IFN-γ production, and increases apoptosis (9). PD-1 interacts with two B7 family ligands, programmed death ligand (PD-L)1 (B7-H1 and CD274) and PD-L2 (B7-DC and CD273), whose expression patterns are distinct (9). PD-L1 is widely distributed on leukocytes, nonhematopoietic cells, and in nonlymphoid tissues including islets, whereas PD-L2 is expressed exclusively on dendritic cells and monocytes (for review see reference 9). Disruption of PD-1 results in autoimmune cardiomyopathy (10), progressive arthritis (11), lupus-like glomerulonephritis (12), and exacerbated EAE in mice (13). Most importantly, blocking the PD-1–PD-L1 pathway in NOD mice results in diabetes (14), with PD-1–PD-L1 interactions critical within the pancreas for limiting T cell function (15). Although these findings could not distinguish a role for PD-1 in the initiation of immunity versus tolerance maintenance, this pathway was positioned as a critical regulator of immunity and may play a significant role in tolerance in normal and therapeutic settings.
Thus, in this study, we took advantage of the robust tolerance-inducing treatments, FcR-nonbinding anti-CD3 and insulin-coupled fixed APCs, to examine the role of PD-1 in active tolerance in NOD mice. We show that, unlike CTLA-4, PD-1–PD-L1 is involved in both tolerance induction and maintenance after antigen-coupled splenocytes (antigen-SP) or anti-CD3 treatments. PD-L1 blockade did not affect T reg cell function but directly effected diabetogenic T cells, reversing T cell anergy and promoting tissue destruction and the rapid development of autoimmune diabetes. These results suggest that the PD-1–PD-L1 pathway controls autoimmunity by directly controlling pathogenic T cells at the site of tissue attack, making this pathway an ideal target for novel therapeutics to induce immune tolerance.
RESULTS
Antigen-specific tolerance regulates autoimmune diabetes
Autoimmune diabetes is a chronic disease exemplified by the development of progressive autoreactive antibodies and T cells. Although multiple approaches have been used to prevent diabetes in the spontaneous NOD mouse model, there has been less success in reversing disease after onset. One of the approaches successfully used in other autoimmune diseases in mice is the i.v. injection of antigen-coupled APCs. It has recently been reported that insulin is a key target during the pathogenesis of T1D in the NOD mouse (8). Therefore, we tested the use of insulin-coupled fixed splenocytes (INS-SP) as a therapy to reverse autoimmune diabetes. New-onset diabetic NOD mice were treated with INS-SP–coupled cells and monitored for disease remission. As seen in Fig. 1 A, INS-SP treatment reverses diabetes in ∼50% of the mice. This effect is durable, lasting >25 wk after therapy (Fig. 1 A). Similar studies using other islet proteins implicated in diabetes, including glutamic acid decarboxylase (GAD), the dominant peptides GAD 206–220, GAD 217–236, and GAD 524–536, and islet-specific glucose- 6-phosphatase catalytic subunit–related protein peptide NRP-V7, had no effect in reversing diabetes (not depicted; n ≥ 9/group), even though immunological responses to these proteins have been detected in disease (1). These results indicate that insulin is a critical autoantigen during the pathogenesis of spontaneous diabetes. Moreover, this result demonstrates an antigen-specific therapy that can reverse diabetes in the NOD mouse after clinical disease onset (2).
Figure 1.

Antigen-specific coupled cell tolerance induces diabetes remission of ongoing spontaneous autoimmune diabetes by the induction of T cell anergy. (A) New-onset spontaneously diabetic NOD mice were treated with INS-SP or control SHAM-SP. The percent of mice that entered disease remission after INS-SP treatment (•; n = 19) are shown compared with SHAM-SP controls (◯; n = 9). (B) Adoptive transfer diabetes tolerance model. Diabetes was induced by the transfer of activated BDC2.5 T cells to NOD recipients. The next day, mice received p31-SP or SHAM-SP. Individual glucose readings for p31-SP–tolerized (•; n = 8) and diabetic SHAM-SP mice (◯; n = 8) are shown. (C–E) p31-SP–tolerized mice have decreased antigen-specific proliferation and cytokine production. 7 d after BDC2.5 transfer and p31-SP tolerance, pLN cells were isolated and in vitro cultured with p31. (C) Antigen-specific proliferation was determined from p31-SP– (•) or SHAM-SP– (◯) treated mice. (D) Antigen-specific IFN-γ (ng/ml) and (E) IL-2 (pg/ml) production from p31-SP– and SHAM-SP–tolerized mice. Gray bars, 0.1 μM p31 peptide; black bars, media control. These data are representative of three independent experiments.
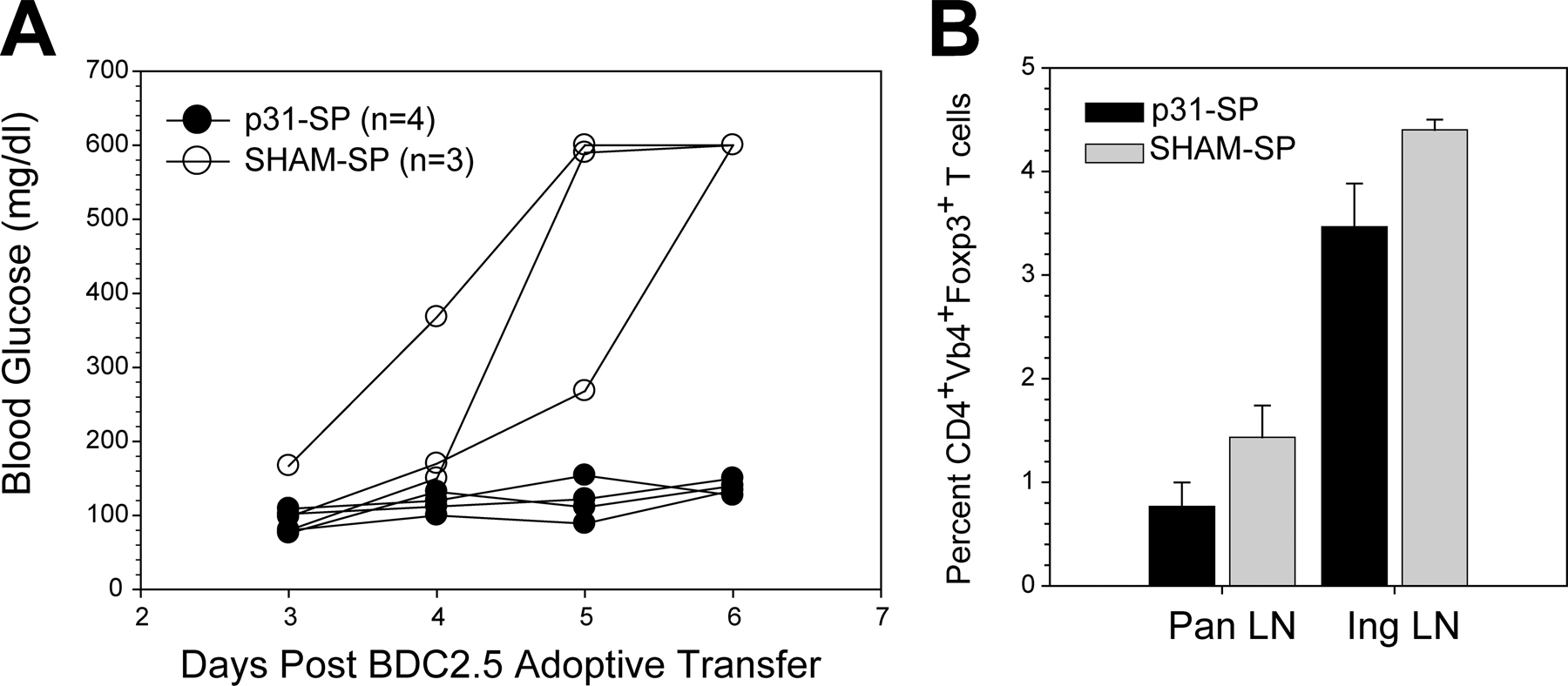
To determine the basis for tolerance in the insulin-SP setting, we used traceable T cells from islet antigen–specific TCR transgenic (Tg)+ mice. In this model, diabetes was induced in NOD mice by the adoptive transfer of activated CD4+ BDC2.5 Tg+ T cells, which recognize an islet antigen that can be mimicked by a peptide, termed 1040-p31 (16, 17). The next day, mice were treated with 1040-p31 peptide–coupled ethylene carbodiimide (ECDI)-fixed splenocytes (p31-SP) or irrelevant peptide–coupled ECDI-fixed splenocytes (SHAM-SP) as control. SHAM-SP control mice develop severe diabetes within 7 d of transfer, with 100% disease incidence (Fig. 1 B). p31-SP treatment, however, provided complete and long-lasting protection (>100 d; Fig. 1 B). This model replicated the results observed with insulin-SP in spontaneously diabetic NOD mice and therefore provided a useful tool to dissect the basis for tolerance.
Multiple peripheral pathways have been implicated in the development and maintenance of immune tolerance, including clonal deletion, anergy induction, and active suppression by T reg cells. We next examined these parameters in this antigen-fixed APC-induced tolerance model. First, BDC2.5 T cell numbers were monitored in control and p31-SP–treated mice. After antigen-SP treatment, there was an initial increase in BDC2.5 T cells in both p31-SP and SHAM-SP groups followed by a retraction phase (not depicted) similar to that previously reported in the EAE model (5). BDC2.5 cells were enumerated using congenic markers 4 wk after receiving antigen-SP therapy (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20061557/DC1). There was a reduction in the percentage of cells isolated from the pancreatic LN (pLN), but because of the low cell numbers in the pLN, the total number of BDC2.5 T cells was not different in p31-SP compared with SHAM-SP when all other peripheral lymphoid compartments were compared. These results indicate that BDC2.5 cells persist in p31-SP–treated mice. Next, we examined the responses of the BDC2.5 T cells exposed to p31-SP tolerogenic therapy 7 d after treatment. Proliferation to p31 peptide was significantly reduced in the pLN (Fig. 1 C), peripheral LN, and spleen (not depicted). Additionally, antigen-specific inflammatory cytokines including IFN-γ and IL-2 were significantly reduced after p31-SP treatment (Fig. 1, D and E). This decrease in cytokine production did not reflect a switch toward a Th2 phenotype as both IL-4 and IL-10 were not produced after T cell activation from either p31-SP– or SHAM-SP–treated mice (not depicted).
Lastly, we determined the role of T reg cells during antigen-SP tolerance induction. To assess this, TCR-α KO recipients, deficient of T cells including T reg cells, were given CD4+CD25− BDC2.5 TCR Tg+ T cells that had been depleted of CD4+CD25+ T reg cells followed by CD25-depleted p31-SP antigen–coupled splenocytes. These recipient mice were protected from developing diabetes (Fig. S2 A, available at http://www.jem.org/cgi/content/full/jem.20061577/DC1) and had decreased antigen-specific proliferation and cytokines (not depicted), indicating that antigen-SP is effective in the absence of T reg cells. Furthermore, we did not detect an increase of FoxP3+BDC2.5 TCR Tg+ T cells, indicating that antigen-SP tolerance did not induce adaptive T reg cells (Fig. S2 B). Collectively, these results indicate that antigen-SP therapy results in the induction of antigen-specific T cell anergy through an intrinsic mechanism.
p31-SP–coupled cells limit, but do not eliminate, pancreatic infiltration of antigen-specific T cells
We next investigated whether p31-SP treatment affected pancreatic insulitis. Diabetogenic BDC2.5 T cells were activated and transferred to NOD mice. The next day, mice were treated with p31-SP or SHAM-SP. 7 d after treatment, pancreatic islets were examined for the presence of mononuclear cell infiltrates. p31-SP treatment prevented severe insulitis; however, mild insulitis and perivascular infiltrates developed throughout the pancreas based on histological examination (magnification, 200×; Fig. 2 A) compared with SHAM-SP–injected mice that developed severe insulitis (Fig. 2 B). Quantification confirmed that there was more severe insulitis in SHAM-SP compared with p31-SP–protected mice (Fig. 2 C). Thus, p31-SP therapy prevents clinical disease but did not fully prevent cellular accumulation within the target organ, suggesting that there may be active tissue-specific regulation of these potentially pathogenic T cells.
Figure 2.

p31-SP–coupled cells prevent adoptive transfer of diabetes, but not peri-insulitis. Pancreata from p31-SP–tolerized and SHAM-SP control mice were stained with hematoxylin and eosin to determine clinical severity of insulitis. 100 pancreatic islets were scored for the presence of mononuclear infiltration. Representative pictures from NOD mice 7 d after receiving activated BDC2.5 T cells and either (A) p31-SP– or (B) SHAM-SP–coupled cells are shown. (C) Quantification of >100 pancreatic islets as a percentage for each histological score demonstrating that SHAM-SP–treated mice have more severe insulitis compared with p31-SP–protected mice (n = 5 mice/group; P = 0.0004).
Both PD-1 and CTLA-4 blockade inhibits the induction of peripheral CD4+ T cell tolerance
We next turned our attention toward cell surface inhibitory molecules as a potential mechanism for maintaining antigen-SP anergy. Several regulatory pathways have been implicated in controlling intrinsic T cell function. In this study, we sought to examine two such pathways, PD-1 and CTLA-4. We have previously shown that CTLA-4 is important for T cell tolerance during an immunogenic encounter with a model antigen, ovalbumin (5). These results raised the possibility that CTLA-4 would regulate tolerance induced in this system. Similarly, PD-1 has been shown to be involved in the control of tissue-specific autoimmunity (15, 18). PD-1 and CTLA-4 expression is rapidly up-regulated after T cell activation (for review see reference 9). We measured the expression of PD-1 and CTLA-4 on activated diabetogenic BDC2.5 CD4+ T cells to ensure that CTLA-4 and PD-1 were expressed both at the time of transfer and after antigen-specific treatment. Results shown in Fig. 3 A demonstrate significant expression of PD-1 and CTLA-4 from the transferred BDC2.5 CD4+ T cells. Therefore, we examined the role of CTLA-4 and PD-1 inhibitory pathways in peripheral tolerance using the antigen-coupled cell tolerance model. Activated BDC2.5 T cells were transferred to NOD mice followed 1 d later with p31-SP tolerogen or SHAM-SP. Each group was concurrently treated with control Ig, anti–CTLA-4, or anti–PD-1 and monitored for diabetes. SHAM-SP–treated mice developed diabetes by day 6, whereas p31-SP–treated mice were completely protected (Fig. 3, B and D). The protection correlated with reduced antigen-specific proliferation (Fig. 3, C and E). Anti–CTLA-4 and anti–PD-1, but not control Ig, surrounding tolerance induction resulted in rapid diabetes within 6 d of CTLA-4 or PD-1 blockade (Fig. 3, B and D). Anti–CTLA-4 and anti–PD-1 treatment not only prevented anergy induction, but also enhanced antigen-specific proliferation (Fig. 3, C and E, respectively).
Figure 3.

CTLA-4 and PD-1 blockade prevents the induction of antigen-SP tolerance and restores autoreactive T cell function. (A) Up-regulation of PD-1 and CTLA-4 expression on diabetogenic BDC2.5 T cells after p31 antigen activation in vitro (day 4; top) and 30 h after p31-SP transfer (bottom). Flow cytometric analysis was performed on congenic BDC2.5 CD90.1+ CD4+ T cells for PD-1 and CTLA-4 expression. CD4+ gated events are shown illustrating increased PD-1 and CTLA-4 expression on the majority of the CD90.1 cells compared with isotype hamster control staining. Numbers illustrate the percentage of cells in each quadrant. (B–E) Anti–CTLA-4, anti–PD-1, or IgG control was administered surrounding BDC transfer and coupled cell tolerance to determine the role of these inhibitory molecules during tolerance induction. (B) Diabetes incidence and (C) day 3 p31-specific proliferation is shown for p31-SP plus IgG-tolerized mice (•; n = 7), SHAM-SP plus IgG control mice (◯; n = 7), anti–CTLA-4 p31-SP mice (▴; n = 7), and anti–CTLA-4 SHAM-SP control mice (▿; n = 7). (D) Diabetes incidence and (E) day 3 p31-specific proliferation is shown for p31-SP plus IgG-tolerized mice (•; n = 5), SHAM-SP plus IgG control mice (◯; n = 5), anti–PD-1 p31-SP mice (▴; n = 5), and anti–PD-1 SHAM-SP control mice (▿; n = 5) illustrating PD-1 reversal of antigen- SP tolerance. (F) p31-specific IFN-γ production from p31-SP and SHAM-SP mice treated with IgG or anti–PD-1 (n = 3) demonstrating significant IFN-γ increase after anti–PD-1 treatment (P = 0.027). Results shown are representative from two independent experiments.
Anti–PD-1 treatment of p31-SP–tolerized mice resulted in enhanced cytokine production including IFN-γ and IL-2 compared with control Ig–treated mice (Fig. 3 F and not depicted). IFN-γ was significantly increased in anti–PD-1–treated mice (15.7 ± 0.8 ng/ml) compared with control IgG–treated tolerized animals (0.35 ± 0.06 ng/ml; Fig. 3 F; P = 0.027). The increased proliferation and effector cytokines after PD-1 blockade are consistent with a direct effect on the tolerized T cells and the induction of diabetes.
PD-L1, but not PD-L2, regulates the induction of CD4+ T cell tolerance
Experiments were performed using blocking antibodies to PD-L1 and PD-L2 to determine the relative importance of each ligand in tolerance induction. BDC2.5 T cells were activated and transferred to NOD mice. The next day, mice were treated with p31-SP or SHAM-SP together with anti– PD-L1, anti–PD-L2, or control Ig. p31-SP, but not SHAM-SP, treatment protected mice from developing diabetes (0/15; Table I). Anti–PD-L1, but not anti–PD-L2 or control Ig, administration prevented tolerance induction (18/18; Table I). Finally, we tested whether the need for PD-L1 expression on the p31 peptide ECDI-fixed APC was critical for tolerance induction. Antigen-coupled cells (antigen-SP) from WT or PD-L1–deficient NOD mice were used to induce tolerance in BDC2.5 T cell–transferred NOD mice. The NOD WT and NOD.PD-L1KO p31- coupled APCs were equally effective at inducing antigen-specific T cell tolerance and preventing diabetes (not depicted), suggesting that tolerance induction in this setting does not require PD-1–PD-L1 interaction on the fixed APCs. Thus, PD-L1 expression on host tissue is critical for the induction of tolerance in this model.
Table I.
Anti–PD-1 and anti–PD-L1, but not anti–PD-L2, therapy inhibits tolerance induction
| Couple cells | Treatment | No. diabetic/n | % Diabetes |
|---|---|---|---|
| p31-SP | Control | 0/15 | 0 |
| SHAM-SP | Control | 15/15 | 100 |
| p31-SP | Anti–PD-1 | 21/21 | 100 |
| SHAM-SP | Anti–PD-1 | 20/20 | 100 |
| p31-SP | Anti–PD-L1 | 18/18 | 100 |
| SHAM-SP | Anti–PD-L1 | 18/18 | 100 |
| p31-SP | Anti–PD-L2 | 0/9 | 0 |
| SHAM-SP | Anti–PD-L2 | 8/8 | 100 |
Disease was induced by BDC2.5 cell transfer to NOD mice. 24 h later, recipients received either p31-SP or SHAM-SP. Anti–PD-1, anti–PD-L1, anti–PD-L2, or IgG was administered on days (−1, 0, and +1) surrounding coupled cell transfer. Mice were monitored for diabetes for 6 wk. Disease incidence (No. diabetic/n) and percent diabetic for each group is shown.
PD-1–PD-L1, but not CTLA-4, plays a critical role in the maintenance of CD4+ T cell tolerance
The previous results suggested that antigen-SP injection induced a state of T cell anergy that is dependent on PD-1– PD-L1 engagement on host cells. We next investigated whether PD-1–PD-L1 was involved in the tissue- specific tolerance given the peri-insulitis observed after p31-SP tolerance. Activated BDC2.5 T cells were transferred to NOD mice, tolerized on day +1 with p31-SP cells, and treated with mAbs 2 wk after tolerance induction, a time point with nondestructive peri-insulitis. Injection of anti–PD-1 or anti–PD-L1, but not PD-L2, resulted in the reversal of tolerance and rapid development of diabetes (Fig. 4, A and B). Anti–PD-L1 led to an increase of antigen-specific BDC2.5 T cell accumulation, enhanced T cell expansion (Fig. 4 C), and inflammatory cytokines in the pancreas and pLN (not depicted), suggesting that the effects were limited to the local sites of autoantigen. However, in sharp contrast, unlike PD-1, CTLA-4 blockade during the maintenance phase of peripheral tolerance induced by antigen-SP therapy had no effect (Fig. 4 A). Therefore, CTLA-4 blockade was not sufficient to break tolerance. Thus, PD-1 is distinct as a negative regulator of tolerance in that it appears to play a critical role in the maintenance of already established tolerance. These results suggest that PD-1–PD-L1 plays a dual role in tolerance by antigen-SP, which is required for the induction and maintenance of T cell anergy in this model.
Figure 4.

PD-1 and PD-L1, but not CTLA-4, maintain peripheral tolerance. (A) p31-SP–tolerized NOD mice that received BDC T cells were treated with anti–PD-1, anti–CTLA-4, or IgG control (days 14, 16, 18, 20, 22, and 24; gray shaded area) to determine the role of these inhibitory molecules during tolerance maintenance. Diabetes incidence from p31-SP anti–PD-1 (100%; n = 10; ▾), p31-SP anti–CTLA-4 (0%; n = 10; ◯), and p31-SP IgG (0%; n = 10; •) is shown. (B) p31-SP–tolerized mice were injected with anti–PD-L1 or anti–PD-L2 on days +14, 16, 18, 20, 22, and 24 (gray shaded area) to determine which PD-1 ligands were important for tolerance. Diabetes incidence from p31-SP anti–PD-L1 (87.5%; n = 8; ◯) and complete protection in p31-SP anti–PD-L2 (0%; n = 8; •) is shown. (C) Diabetogenic CD4+CD90.1+ BDC2.5 T cell percentages from the pancreas and pLN were determined 7 d after anti–PD-1 treatment, illustrating an increase after anti–PD-1 injection. Results shown are representative from at least two independent experiments.
The previous results could not rule out that endogenous T cells, not the transferred BDC2.5 T cells, were responsible for the antigen-specific proliferation, cytokine production, and rapid induction of diabetes after anti–PD-1–anti–PD-L1. Thus, we took advantage of the CD28 costimulation–deficient NOD B7-2 KO mouse strain, which does not develop insulitis or progress and develop autoimmune diabetes due to limited activation of the potentially pathogenic T cells (19, 20). NOD B7-2 KO mice were treated with blocking anti–PD-L1 antibodies and followed for diabetes. B7-2KO mice were completely protected from developing diabetes compared with NOD controls (83.6% diabetic; Fig. 5 A). This diabetes-resistant model was exploited to directly study anti–PD-L1 treatment of BDC2.5 T cells and the development of diabetes without the confounding effects from polyclonal host T cells. Activated BDC2.5 T cells were transferred to NOD B7-2KO recipients. The next day, mice received either tolerogenic p31-SP cells or SHAM-SP. SHAM- SP–treated NOD B7-2 KO mice rapidly developed diabetes (Fig. 5 B). p31-SP cells induced complete tolerance and disease protection (Fig. 5 B). These results suggest that the previously activated BDC2.5 T cells did not need CD28/ B7-2–mediated costimulation to promote pathogenesis. Moreover, tolerance induction by antigen-coupled APCs did not require B7-2 expression on host cells. This result was in sharp contrast to the PD-1–PD-L1 blockade results and demonstrates the distinct role of these pathways in controlling tolerance at different stages of immune activation.
Figure 5.

PD-L1 blockade releases the antigen-specific BDC2.5 CD4+ T cells from anergy and allows the development of diabetes. (A) Anti–PD-L1–treated NOD B7-2 KO mice (n = 8; ◯) are protected from diabetes compared with WT NOD mice (n = 6; •). (B) Anti–PD-L1 precipitates clinical diabetes in B7-2 KO mice receiving BDC2.5 T cells. Disease was induced by BDC2.5 T cell transfer. 24 h later, recipient mice were treated with p31-SP to induce tolerance. 10 d after tolerance, B7-2 KO recipients were injected with anti–PD-L1 or IgG control (days +10, 12, 14, 16, 18, and 20; gray shaded area) and monitored for diabetes. Diabetes incidence for NOD B7-2KO SHAM-SP (▾; n = 3), NOD B7-2KO p31-SP plus anti–PD-L1 (•; n = 4), and NOD B7-2 KO p31-SP plus IgG (◯; n = 4) is shown. (C) BDC2.5 T cell antigen-specific cytokine production. pLN cells were isolated and activated in vitro for 6 h with p31 antigen at day 20 after receiving anti–PD-L1 or IgG and p31-SP treatment. Cells were stained with immune markers for transferred CD90.1 BDC2.5 CD4 T cells, IL-2, and IFN-γ. IFN-γ+ cells are shown as a percent of the total BDC2.5 T cells from three to five mice per group. Results demonstrate a significant increase in BDC2.5 IFN-γ+ cells from anti–PD-L1 compared with IgG controls (P = 0.007). These data are representative of two independent experiments.
Next, p31-SP–tolerized NOD B7-2 KO mice were treated with anti–PD-L1 or control IgG antibody 10 d after p31-SP tolerance was established. The administration of anti–PD-L1 resulted in tolerance reversal and rapid development of diabetes (Fig. 5 B). Anti–PD-L1 treatment not only abrogated tolerance, but also enhanced antigen-specific cytokines. To investigate if BDC2.5 T cells were truly functional after PD-L1 blockade, pLN cells were isolated from NOD mice seeded with activated BDC2.5 T cells, treated with either p31-SP or SHAM-SP, and injected with either anti–PD-L1 or IgG control. As demonstrated above, BDC2.5 T cells are completely anergic after p31-SP tolerance, as these cells do not make IFN-γ (Fig. 5 C) or IL-2 (not depicted). However, anti–PD-L1 abrogates tolerance and restores the ability of BDC2.5 cells to respond to antigen challenge with between 3 and 14% of the total BDC2.5 T cells producing IFN-γ, similar to SHAM-SP treatment (Fig. 5 C; P = 0.007). Anti–PD-L1 did not result in a significant increase in IFN-γ production by host CD4+ T cells. Less than 1% of polyclonal host CD4+ T cells made IFN-γ when stimulated with PMA and ionomycin (not depicted). Therefore, PD-L1 blockade precipitates diabetes in resistant NOD B7-2 KO animals by directly affecting previously tolerized BDC2.5 T cells. Moreover, PD-L1 blockade reverses anergy in BDC2.5 T cells weeks after tolerance induction resulting in diabetes and demonstrating a critical role for the PD-1–PD-L1 pathway for the maintenance of T cell tolerance.
PD-1–PD-L1 regulates the maintenance of peripheral tolerance in spontaneous polyclonal autoimmune diabetes
The above mechanistic studies were limited to the use of monoclonal islet antigen–specific TCR Tg+ T cells. Thus, it was critical to determine whether the PD-1 pathway was operational in tolerance-inducing therapies using intact NOD mice, which involves polyclonal T cell responses with a wide range of TCR specificity and antigen affinity (Fig. 1 A). New-onset NOD mice, tolerized with insulin-SP, were treated with anti–PD-L1 mAb 25 wk after tolerogenic therapy and disease remission. Tolerance was abrogated in all mice resulting in the rapid development of diabetes within 5 ± 2 d of anti–PD-L1 (Fig. 6, A and B). These results indicate that tolerance induced by INS-SP therapy is also dependent on PD-1–PD-L1 interactions long after tolerance induction.
Figure 6.

PD-L1 blockade breaks tolerance induced by insulin-SP in spontaneous autoimmune diabetes. New-onset spontaneously diabetic NOD mice were treated with insulin-SP or SHAM-SP and evaluated for disease remission. (A) Five mice that were treated with INS-SP and became euglycemic compared with five SHAM-SP diabetic control mice are shown. The five INS-SP–treated mice shown remained disease free for 25 wk after INS-SP therapy. At this time, mice were injected with anti–PD-L1 and followed for diabetes. After PD-L1 blockade, all five mice became diabetic within 5 ± 2 d. Diabetes incidence for INS-SP plus anti–PD-L1 (•; n = 5) and SHAM-SP control (◯; n = 5) is shown. (B) Individual mice and their blood glucose levels after INS-SP therapy and PD-L1 blockade. Each circle represents an individual mouse (n = 5).
Finally, we examined whether the role of the PD-1–PD-L1 pathway could be generalized to other TCR signaling–mediated tolerogenic therapies. We and others have previously reported that anti-CD3 prevents and reverses diabetes in NOD mice (4, 21, 22). This tolerance is robust and durable, with the majority of mice remaining normal glycemic for the rest of their lives. In the first set of experiments, we treated 5-wk-old prediabetic NOD mice with anti-CD3 to induce tolerance (Fig. 7 A). Mice were aged for an additional 12 wk (a time point when untreated mice start to become diabetic in our colony) and monitored for diabetes. None of the anti-CD3–treated animals became diabetic for the duration of the study (43 wk; Fig. 7 A), whereas all those receiving anti–PD-L1 mAb at 12 wk became diabetic within the subsequent 3-wk period (Fig. 7 A). In fact, anti–PD-L1 appeared to accelerate disease because only 75% of control (hamster Ig–treated) NOD mice were diabetic by 21 wk of age. In this regard, anti–PD-L1 precipitated rapid disease progression and complete (100%) penetration (Fig. 7 A; reference 14). These results demonstrate that anti-CD3–induced tolerance is abrogated by PD-L1 blockade even weeks after tolerance induction (Fig. 7 A). As another demonstration of the importance of the PD- 1–PD-L1 pathway in maintaining tolerance, we compared the ability of anti-CD3 therapy to induce durable tolerance after disease onset (21, 22). Newly diabetic NOD mice treated with anti-CD3 became normoglycemic and remained disease-free for the duration of the study (Fig. 7 B). Anti–PD-L1 administered to mice 12 wk after tolerance induction resulted in the rapid development of diabetes within 2 ± 1 d of mAb injection (Fig. 7 C). In addition, although anti-CD3 therapy effectively reversed diabetes in NOD mice after disease onset, anti-CD3 therapy was ineffective at reversing disease in NOD.PD-L1 KO mice (Fig. 7 B). Collectively, these data indicate that both antigen-specific and anti-CD3–mediated tolerance is dependent on PD- 1–PD-L1 signaling.
Figure 7.

PD-L1 blockade abrogates anti-CD3–induced tolerance precipitating autoimmune diabetes. (A) 5-wk-old NOD mice were treated with anti-CD3 or IgG (days 0, 2, 4, 5, and 7). 12 wk after anti-CD3 tolerance induction, mice were injected with anti–PD-L1 mAb (days 84, 86, 88, 90, 92, and 94; gray shaded area) and monitored for diabetes. Disease incidence is shown for anti-CD3–treated NOD mice (◯; n = 7), diabetic IgG-treated NOD mice (•; n = 7), 17-wk-old NOD mice plus anti–PD-L1 (□; n = 4), NOD mice treated with anti-CD3 plus anti–PD-L1 12 wk after tolerance induction (▵; n = 8), and diabetic control IgG plus anti–PD-L1–treated NOD mice (▾; n = 5). (B) Anti-CD3 reverses spontaneous diabetes in WT NOD, but not NOD PDL1 KO, mice. New-onset NOD and NOD PD-L1 KO mice were treated with anti-CD3 or IgG and monitored for diabetes remission. 8 out of 10 NOD mice treated with anti-CD3 went into disease remission (•; n = 8). None of the NOD mice treated with IgG control went into disease remission (◯; n = 8). Similarly, none of the NOD.PD-L1 KO mice treated with anti-CD3 (▪; n = 6) or IgG control (□; n = 3) went into disease remission. (C) Anti–PD-L1 reverses anti-CD3–induced tolerance. New-onset spontaneously diabetic mice were treated with five injections of anti-CD3. After 12 wk, anti–PD-L1 was given to mice that had gone into disease remission. Blood glucose from four individual mice demonstrating rapid diabetes progression by day 3 after PD-L1 blockade is shown. (D) PD-L1 blockade does not inhibit T reg cell function in vivo. Disease was induced by the transfer of 106 BDC2.5 CD4+CD62L+CD25− (T eff cells) to NOD RAG KO recipients. Mice were divided into two groups and half received 106 CD4+CD62L+CD25+ (T reg cells) to control diabetes. At the time of disease transfer, mice received either IgG control or anti–PD-L1 mAb to determine PD-L1 effects on T reg cell function. Mice receiving T eff cells plus control IgG or anti–PD-L1 developed diabetes within 15 d (Teff + IgG; •; n = 5) and (Teff + anti–PD-L1; ◯; n = 5). Mice receiving T reg cells were protected from disease even in the presence of anti–PD-L1 administration. Shown are mice from (Teff+Treg+IgG; ▾; n = 5) and (Teff+Treg+anti–PD-L1; ▿; n = 5). These data are representative of two independent experiments.
Anti-CD3 therapy has been shown to induce tolerance by two distinct mechanisms: the induction of pathogenic T cell apoptosis and through the induction of T reg cells (4). We next tested whether PD-1–PD-L1 blockade was acting on effector cell function alone or T reg cell function as well. BDC2.5 T cells were sorted into T reg cells (CD4+CD25+CD62L+) and effector T (T eff) cells (CD4+CD25-CD62L+). NOD.RAG KO recipient mice received T eff cells alone or together with T reg cells. The addition of T reg cells with T eff cells suppressed the development of diabetes. Mice receiving BDC2.5 T eff cells with either IgG control or anti–PD-L1 developed diabetes within 11 d after transfer (Fig. 7 D); however, coinjection of T reg cells with T eff cells suppressed the induction of diabetes. These results indicated that T reg cells did not depend on PD-1–PD-L1 interactions for tolerance induction and prevention of autoimmune diabetes (Fig. 7 D). Thus, in this model, the PD-1–PD-L1 pathway reversed anergy in islet antigen–specific T cells by a direct effect on the pathogenic T cells.
DISCUSSION
The goal of this study was to examine the role of PD-1 in the induction and maintenance of tolerance in NOD mice that are prone to develop T1D. We demonstrate that insulin is a critical antigen involved in T1D pathogenesis, and that selectively silencing insulin-specific T cells can reverse disease progression and lead to long-term tolerance. This result demonstrates one of the first antigen-specific therapies for diabetes reversal after clinical onset (2). The mechanism for antigen-specific tolerance is the induction of T cell anergy maintained through the interactions of PD-1–PD-L1. Previous studies have documented that PD-1–PD-L1 are important for regulating T cell responses in vivo. This is most evident in PD-1 KO mice, which exhibit several autoimmune syndromes, and in humans where polymorphisms in PD-1 are associated with susceptibility to systemic lupus erythematosus, rheumatoid arthritis, and diabetes (23, 24). Furthermore, treatment of autoimmune-prone NOD mice with PD-1 antagonists precipitates disease. However, none of these studies has directly addressed the role of PD-1 in a tolerogenic setting. In this study, we demonstrate that PD-1– PD-L1 interactions control tolerance weeks to months after induction by suppressing immunity within target tissues. These studies took advantage of a newly developed model of tolerance in NOD mice where ECDI-fixed insulin-coupled spleen cells induced robust tolerance even after disease onset. Similarly, the PD-1–PD-L1 pathway, but not the PD-1–PD-L2 pathway, was critical for maintenance of tolerance in anti-CD3–treated mice. Collectively, these findings support a central role in the maintenance of tolerance in well-defined and clinically applicable tolerogenic treatments currently being developed for humans with T1D.
The observation that insulin-coupled fixed spleen cells induced robust tolerance even after disease onset supports recent suggestions that insulin is the dominant autoantigen in T1D (8). Tolerance induced by antigen-coupled spleen cells led to a rapid block in proliferation, decreased cytokine production, and protection from diabetes. This therapy has many of the attributes of anti-CD3 treatment, which has been shown to be effective for treatment of T1D in NOD mice and humans (3, 21). In those studies, the FcR-nonbinding anti-CD3 induced significant pathogenic T cell death or anergy in residual T cells that infiltrated the pancreatic tissue (25–28). Importantly, combination therapies using both anti-CD3 and insulin have recently been shown to synergize to induce robust antigen-specific tolerance (29).
In this model of tolerance, the fate of the antigen-SP cells is not completely understood. CFSE tracking studies have revealed that antigen-SP cells are present in peripheral tissues, including the lung, liver, pancreas, spleen, and peripheral LNs among other locations, within 6 h after transfer (unpublished data). Functionally, these cells mediate their effects within the first 48 h after transfer. Antigen-SP pretreatment followed by adoptive transfer of TCR Tg effector cells 48 h later did not result in tolerance, suggesting that the effects of the antigen-SP cells occur before this time point (unpublished data). The effects of this therapy are, however, antigen-specific. p31 peptide, but not whole insulin–coupled APCs, tolerized the BDC2.5 T cells and prevented diabetes (unpublished data). Similarly, p31 peptide–coupled cells did not reverse spontaneous diabetes, further demonstrating the antigen-specific nature of this therapy and a critical role for insulin in the pathogenesis of autoimmune diabetes.
Previous studies have shown that the PD-1–PD-L1 blockade resulted in exacerbated autoimmunity (13, 14). These effects suggest a role for PD-1–PD-L1 in immune tolerance; however, studies by Barber et al. (30) suggest that PD-1 may keep weakly pathogenic T cells in check or reverse the functional “exhaustion” of antigen-overstimulated T eff cells. Thus, these studies could not distinguish whether PD-1–PD-L1 promoted tolerance by increasing the threshold of previously exposed T eff cells or an active down-regulation of T cells involved in ongoing immunity. Distinguishing the precise role of PD-1 for tolerance reversal or bypassing the normal tolerogenic processes in these settings has important implications for PD-1 inhibition and the interpretation of previous studies. In the NOD mouse for instance, exacerbation of disease after PD-1–PD-L1 blockade could reflect abrogation of tolerance in the normal setting or might promote a more vigorous effector cell response from low affinity or weakly activated T cells. By studying tolerance models, direct assessment of PD-1 blockade on the antigen-specific T cells could be determined. The findings reported herein demonstrate a critical role for PD-1–PD-L1 interaction during the induction and maintenance of peripheral T cell tolerance in autoimmune diabetes. Blockade of the PD-1–PD-L1 pathway resulted in the proliferation and accumulation of autoreactive T cells and rapid progression of diabetes. Most importantly, the PD-1–PD-L1 blockade reversed anergy in islet antigen–specific T cells, suggesting a direct effect of PD-1 on the pathogenic T cells in this setting.
This last point is supported by the observation that the PD-1–PD-L1 blockade was effective even in the absence of T reg cells, as we have performed transfers with CD4+CD25− (T reg cell–depleted) pathogenic T cells into T reg cell–deficient hosts and still observed effective tolerance (Fig. S2 A). Moreover, we did not detect an increase of FoxP3+ TCR Tg+ T cells, indicating that antigen-SP tolerance did not induce adaptive T reg cells (Fig. S2 B). These results suggest that the PD-1–PD-L1 pathway functions in an T eff cell–intrinsic manner to modulate IFN-γ production and direct T cell function. In addition, these studies demonstrate that T reg cells function normally and prevent diabetes in the presence of PD-1 blockade, further suggesting a direct effect on the diabetogenic T cells.
It was surprising that the PD-1–PD-L1 interaction was not critical in the direct engagement of the activated T eff cells and p31-SP ECDI-treated APCs given the integral role of this pathway in controlling T cell activation in several systems (9). Rather, the PD-1–PD-L1 pathway acts downstream of initial antigen encounter. Thus, the PD-1–PD-L1 pathway is not critical for induction but rather the maintenance of tolerance in the target tissue in both the antigen-coupled fixed APCs and anti-CD3 models where antigen-specific T cells continue to reside in the target organ (although they are not pathogenic; reference 31). Therefore, we speculate that the PD-1–PD-L1 interactions maintain tolerance in the local infiltrated tissues. In this regard, we have observed that anti–PD-L1 treatment did not precipitate diabetes in models such as the NOD B7-2KO, where there is no infiltrate at the time of treatment (20) and, thus, the endogenous host CD4+ T cells were not able to induce disease when PD-1 was blocked. In contrast, blockade of PD-1 on the tolerized BDC2.5 T cells resulted in rapid reversal of anergy, including a dramatic increase in inflammatory cytokines, indicating that induction of tolerance through TCR-mediated pathways induces a state of T cell anergy that is maintained by PD-1–PD-L1 engagement.
CTLA-4 and PD-1 both inhibit T cell proliferation, cytokine production, and proximal TCR signaling (for review see reference 9). Thus, it is critical to understand the apparent paradox that each molecule is both necessary yet not sufficient for tolerance. The data suggest this paradox can be best reconciled by proposing that the two negative regulatory pathways function at different stages and sites during autoimmunity. For instance, unlike PD-L1, which is highly expressed in islets and other stromal cells (32), expression of the CTLA-4 ligands B7-1 and B7-2 is largely restricted to lymphoid tissues. Therefore, CTLA-4 may act proximal to initial T cell activation in the draining LNs after B7-1 and B7-2 engagement, whereas the PD-1–PD-L1 pathway may function within tissue sites distal to initial activation. Thus, PD-1 provides an additional opportunity to limit T cell function and prevent tissue damage downstream of CTLA-4. The data presented here support this model. Anti–CTLA-4 prevented the development of peripheral tolerance but did not abrogate established peripheral tolerance. PD-1 blockade, however, reversed the maintenance of tolerance, thus supporting a linear model of T eff cell inhibition with CTLA-4 acting early and PD-1 preventing diabetogenic T eff cells late, perhaps directly within the target organ. Furthermore, CTLA-4 has been shown to inhibit T reg cell suppression (33), but PD-1 blockade did not. These distinct differences create a scenario in which both T eff cells and T reg cells are inhibited by CTLA-4 signaling, perhaps early during the initiation of immunity, but PD-1 signaling may selectively inhibit T eff cells once they enter the target tissues. Thus, the use of PD-1 agonists locally in inflamed tissue may be effective for maintaining tolerance when used in conjunction with a tolerogen such as anti-CD3 or antigen-coupled fixed APCs. Moreover, local administration of anti–PD-1–PD-L1 antagonists in tumor settings where anergic T cells have been found in the infiltrate (e.g., renal cancer) may be effective for promoting tumor immunity (34). The distinct temporal and spatial expression of the PD-1–PD-L1 inhibitory pathway offers an attractive target for therapeutic intervention in transplantation, cancer therapy, and autoimmune disease settings.
MATERIALS AND METHODS
Mice.
Female NOD mice were purchased from Taconic. NOD-BDC2.5 TCR Tg+ mice (16) were provided by C. Benoist and D. Mathis (Harvard Medical School, Boston, MA). NOD.BDC2.5 TCR Tg+ mice were crossed to NOD.Thy1.1 mice to generate NOD.BDC2.5.Thy1.1 TCR Tg+ mice. C57BL/6 PD-L1 KO mice (15) were backcrossed 10 generations to NOD. NOD B7-2 KO mice were generated as described previously (19). NOD TCR-α KO and NOD RAG KO mice were purchased from The Jackson Laboratory. Mice were 3–10 wk old at the initiation of the experiments. All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco.
Antibodies.
mAbs to murine CD4 FITC, CD4 APC-Cy7 (RM4-5), CD8a PerCP (Ly-2), CD25 PE, CD25 APC (7D4, PC61), CD62L FITC, CD62L APC (MEL14), CD69 PE (H1.2F3), CD90.1 APC, CD90.1 FITC (HIS51), Vβ4 PE (KT4), IFN-γ APC (XMG1.2), IL-2 PE (JES6-5H4), IL-4 PE (11B11), IL-10 APC (JES5-16E3), Armenian hamster IgG1 PE, Armenian hamster control IgG2 PE, CD152 PE (UC10-4F10), CD279 PE (J43), and isotype controls were purchased from BD Biosciences. Anti-FoxP3 PE (FJK-16s) was purchased from eBioscience. Anti–PD-1 (RMP1-14), anti–PD-L1 (MIH5, MIH6), and anti–PD-L2 (TY25) were made as described previously (14, 35, 36). Anti-CD3 (145-2C11 or 2C11-IgG3) was made as described previously (37).
Antibody treatment.
Mice were treated intraperitoneally with 500 μg anti–PD-1, anti–PD-L1, anti–PD-L2, or IgG on day 0 and with 250 μg on days 2, 4, 6, 8, and 10 unless otherwise noted. 250 μg anti-CD3 or hamster IgG was injected intraperitoneally at a low dose (days 0 and 2) or high dose (days 0, 2, 4, 5, and 7) as indicated.
Antigens.
1040-p31 peptide (YVRPLWVRME) was purchased from Genemed Synthesis Inc. The amino acid composition was verified by mass spectrometry, and purity (>98%) was assessed by HPLC. Insulin was purchased from Novo Nordisk Pharmaceuticals Inc.
Activation of donor lymphocytes, cell culture, transfer, and induction of T cell tolerance.
NOD.BDC2.5.Thy1.1 TCR Tg+ lymphocytes were harvested and pooled from brachial, axillary, peri-aortic and pLNs, and the spleen. Cells were activated in vitro in the presence of 0.5 μM 1040-p31 peptide in complete DMEM containing 5 × 10−5 M 2-ME, 2 mM L-glutamine, 100 U/ml penicillin/streptomycin, 0.1 M nonessential amino acids (Invitrogen), and 10% FCS (Hy-clone). Cells were incubated at 37°C in a humidified atmosphere containing 5% CO2. The cells were harvested after 96 h and washed, and 5 × 106 T cells were transferred i.v. to naive prediabetic NOD or NOD.B7-2KO recipients. Tolerance was induced using i.v. injections of 50 × 106 chemically treated antigen-coupled syngeneic splenocytes (p31 or SHAM control), as described previously (5). To achieve 100% tolerance in the BDC2.5 T cell transfer experiments, both the antigen and the number of antigen-coupled splenocytes were titered to identify the lowest concentration of antigen and splenocytes required. Complete protection required 0.5 mg/ml antigen and 50 × 106 antigen-coupled splenocytes.
Assessment of diabetes and insulitis.
Blood glucose levels were measured from female NOD mice with an Accu-Chek glucose meter (Roche). Mice were determined diabetic with two consecutive readings of >250 mg/dL. For histological analysis, the pancreas was formalin fixed in 10% buffered formalin. Multiple 5-μm sections were stained with hematoxylin and eosin and scored blindly for severity of insulitis (Score: 0, no infiltrate; 1, peri-insulitis present; 2, 25>50%; 3, >50% of the islet is infiltrated; reference 20). The average insulitis percentages shown were determined from at least 100 islets from at least five mice per group.
In vitro T cell proliferation assays.
Cells were cultured in 96-well microtiter plates (Corning) at 5 × 105 cells/well in complete DMEM. Cells were pulsed with 1 μCi of 3H-TdR (MP Radiochemicals) during the last 8 h of a 96-h culture, and 3H-TdR uptake was detected using a Packard Topcount microplate scintillation counter (Packard Instrument Co.). The average proliferation in triplicate ± SEM is shown.
Flow cytometry.
For assessment of surface molecule and intracellular protein expression, cells were labeled with predetermined optimal antibody concentrations according to the manufacturer's staining protocol and 0.5 × 106 cells in the CD4 gate were acquired, as described previously (38, 39). Data acquisition was performed on an LSRII flow cytometer and analyzed using FACSDiva software (Becton Dickinson).
ELISA.
Assessment of cytokine production was tested for IL-2, IL-4, IL-10, and IFN-γ by commercial ELISA kits according to the manufacturer's recommended protocol (Endogen). Plates were developed using strepavidin-peroxidase (Zymed Laboratories) and OPD substrate (Sigma-Aldrich), and absorbance was read at 405 nm using a Vmax kinetic microplate reader (Molecular Devices).
Cell sorting.
NOD.BDC2.5 LN and splenic cells were stained with anti-CD4 FITC, anti-CD25 PE, and anti-CD62L APC. CD4+CD62L+CD25− (T eff cells) and CD4+CD62L+CD25+ (T reg cells) T cells were sorted using a MoFlo cytometer high speed cell sorter (DakoCytomation). All sorted populations had ≥98% cell purity.
Statistical analysis.
The statistical significance of cytokine levels, thymidine incorporation, and disease incidence was analyzed using the two-tailed Student's t test for comparisons of two means. Diabetes incidence statistical significance was analyzed using a nonparametric Wilcoxon signed rank test. Values of P ≤ 0.05 were considered significant.
Online supplemental material.
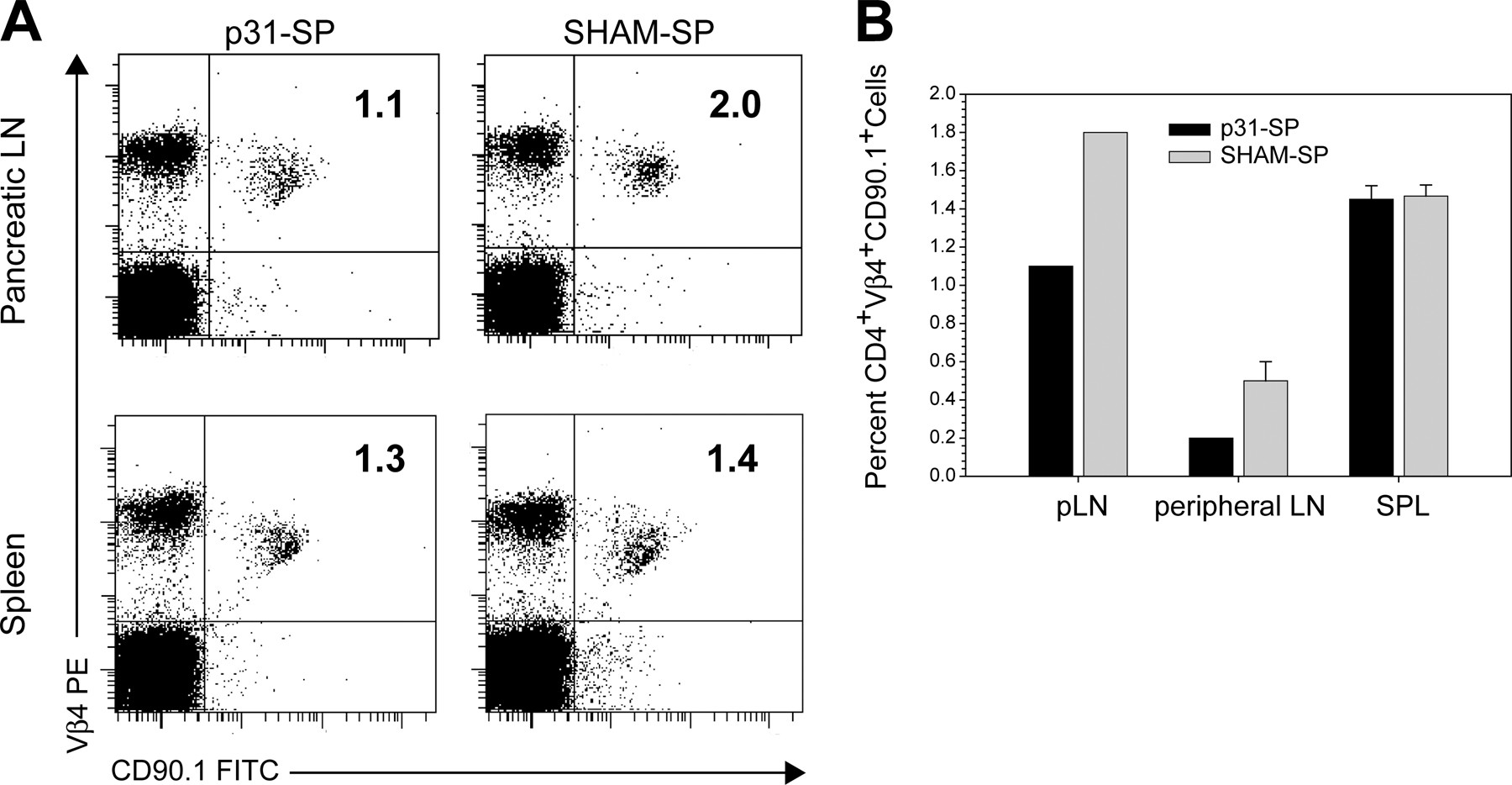
Fig. S1 shows (A) changes in the percentages of CD4+CD90.1+ BDC2.5 T cells 4 wk after Ag-SP or SHAM-SP therapy and (B) the average percentage of BDC2.5 cells ± SEM from pLNs, peripheral LNs, and spleens from p31-SP (black bars) or SHAM-SP (grey bars; three mice per group). Fig. S2 shows (A) that p31-SP therapy induced tolerance and prevented diabetes (•; n = 4) compared with SHAM-SP control (◯; n = 3) when NOD BDC2.5 CD4+CD25− (T reg cell–depleted) pathogenic T cells were transferred to NOD.TCRα KO T reg cell–deficient hosts in the presence of NOD.CD4+CD25− (T reg cell–depleted) polyclonal T cells to prevent lymphopenic-driven proliferation, and (B) the lack of induced adaptive T reg cells after p31-SP therapy based on staining for FoxP3, CD4, and Vβ4 to enumerate T reg cells. Figs. S1 and S2 are available at http://www.jem.org/cgi/content/full/jem.20061577/DC1.
Supplemental Material
Acknowledgments
The authors would like to thank P. Wegfahrt, P. Koudria, and J. Lang for technical assistance. We thank members of the Bluestone laboratory, Drs. A. Abbas, A. DeFranco, and M. Anderson for critical review of this manuscript and helpful discussions.
This work was supported by grants from the National Institutes of Health (P01 AI035297 and P30 DK63720 to J.A. Bluestone and AI56299 to M.H. Sayegh), the Juvenile Diabetes Research Foundation (JDRF) (10-2006-799 to B.T. Fife), and the JDRF Center Grant on Immunological Tolerance in Type 1 Diabetes (to M.H. Sayegh).
The authors have no conflicting financial interests.
Abbreviations used: antigen-SP, antigen-coupled splenocyte(s); CTLA-4, cytotoxic T lymphocyte–associated antigen 4; EAE, experimental autoimmune encephalitis; ECDI, ethylene carbodiimide; INS-SP, insulin-coupled fixed splenocyte(s); NOD, nonobese diabetic; p31-SP, 1040-p31 peptide–coupled ECDI-fixed splenocyte(s); PD-1, programmed death 1; PD-L, programmed death ligand; pLN, pancreatic LN; SHAM-SP, irrelevant peptide–coupled ECDI-fixed splenocyte(s); T1D, type 1 diabetes; T eff, effector T; Tg, transgenic; T reg, T regulatory.
M. Gubbels Bupp's present address is Roche Pharmaceuticals, Palo Alto, CA 94304.
References
- 1.Anderson, M.S., and J.A. Bluestone. 2005. The NOD mouse: a model of immune dysregulation. Annu. Rev. Immunol. 23:447–485. [DOI] [PubMed] [Google Scholar]
- 2.Shoda, L.K., D.L. Young, S. Ramanujan, C.C. Whiting, M.A. Atkinson, J.A. Bluestone, G.S. Eisenbarth, D. Mathis, A.A. Rossini, S.E. Campbell, et al. 2005. A comprehensive review of interventions in the NOD mouse and implications for translation. Immunity. 23:115–126. [DOI] [PubMed] [Google Scholar]
- 3.Herold, K.C., W. Hagopian, J.A. Auger, E. Poumian-Ruiz, L. Taylor, D. Donaldson, S.E. Gitelman, D.M. Harlan, D. Xu, R.A. Zivin, and J.A. Bluestone. 2002. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N. Engl. J. Med. 346:1692–1698. [DOI] [PubMed] [Google Scholar]
- 4.Belghith, M., J.A. Bluestone, S. Barriot, J. Megret, J.F. Bach, and L. Chatenoud. 2003. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat. Med. 9:1202–1208. [DOI] [PubMed] [Google Scholar]
- 5.Eagar, T.N., N.J. Karandikar, J.A. Bluestone, and S.D. Miller. 2002. The role of CTLA-4 in induction and maintenance of peripheral T cell tolerance. Eur. J. Immunol. 32:972–981. [DOI] [PubMed] [Google Scholar]
- 6.Masteller, E.L., and J.A. Bluestone. 2002. Immunotherapy of insulin-dependent diabetes mellitus. Curr. Opin. Immunol. 14:652–659. [DOI] [PubMed] [Google Scholar]
- 7.Bluestone, J.A., and Q. Tang. 2005. How do CD4+CD25+ regulatory T cells control autoimmunity? Curr. Opin. Immunol. 17:638–642. [DOI] [PubMed] [Google Scholar]
- 8.Nakayama, M., N. Abiru, H. Moriyama, N. Babaya, E. Liu, D. Miao, L. Yu, D.R. Wegmann, J.C. Hutton, J.F. Elliott, and G.S. Eisenbarth. 2005. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature. 435:220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greenwald, R.J., G.J. Freeman, and A.H. Sharpe. 2005. The B7 family revisited. Annu. Rev. Immunol. 23:515–548. [DOI] [PubMed] [Google Scholar]
- 10.Nishimura, H., T. Okazaki, Y. Tanaka, K. Nakatani, M. Hara, A. Matsumori, S. Sasayama, A. Mizoguchi, H. Hiai, N. Minato, and T. Honjo. 2001. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 291:319–322. [DOI] [PubMed] [Google Scholar]
- 11.Lin, S.C., J.H. Yen, J.J. Tsai, W.C. Tsai, T.T. Ou, H.W. Liu, and C.J. Chen. 2004. Association of a programmed death 1 gene polymorphism with the development of rheumatoid arthritis, but not systemic lupus erythematosus. Arthritis Rheum. 50:770–775. [DOI] [PubMed] [Google Scholar]
- 12.Nishimura, H., M. Nose, H. Hiai, N. Minato, and T. Honjo. 1999. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 11:141–151. [DOI] [PubMed] [Google Scholar]
- 13.Salama, A.D., T. Chitnis, J. Imitola, M.J. Ansari, H. Akiba, F. Tushima, M. Azuma, H. Yagita, M.H. Sayegh, and S.J. Khoury. 2003. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J. Exp. Med. 198:71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ansari, M.J., A.D. Salama, T. Chitnis, R.N. Smith, H. Yagita, H. Akiba, T. Yamazaki, M. Azuma, H. Iwai, S.J. Khoury, et al. 2003. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J. Exp. Med. 198:63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keir, M.E., S.C. Liang, I. Guleria, Y.E. Latchman, A. Qipo, L.A. Albacker, M. Koulmanda, G.J. Freeman, M.H. Sayegh, and A.H. Sharpe. 2006. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 203:883–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Katz, J.D., B. Wang, K. Haskins, C. Benoist, and D. Mathis. 1993. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 74:1089–1100. [DOI] [PubMed] [Google Scholar]
- 17.Judkowski, V., C. Pinilla, K. Schroder, L. Tucker, N. Sarvetnick, and D.B. Wilson. 2001. Identification of MHC class II-restricted peptide ligands, including a glutamic acid decarboxylase 65 sequence, that stimulate diabetogenic T cells from transgenic BDC2.5 nonobese diabetic mice. J. Immunol. 166:908–917. [DOI] [PubMed] [Google Scholar]
- 18.Wang, J., T. Yoshida, F. Nakaki, H. Hiai, T. Okazaki, and T. Honjo. 2005. Establishment of NOD-Pdcd1−/− mice as an efficient animal model of type I diabetes. Proc. Natl. Acad. Sci. USA. 102:11823–11828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salomon, B., L. Rhee, H. Bour-Jordan, H. Hsin, A. Montag, B. Soliven, J. Arcella, A.M. Girvin, J. Padilla, S.D. Miller, and J.A. Bluestone. 2001. Development of spontaneous autoimmune peripheral polyneuropathy in B7-2–deficient NOD mice. J. Exp. Med. 194:677–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bour-Jordan, H., B.L. Salomon, H.L. Thompson, G.L. Szot, M.R. Bernhard, and J.A. Bluestone. 2004. Costimulation controls diabetes by altering the balance of pathogenic and regulatory T cells. J. Clin. Invest. 114:979–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chatenoud, L., E. Thervet, J. Primo, and J.F. Bach. 1994. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc. Natl. Acad. Sci. USA. 91:123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chatenoud, L., J. Primo, and J.F. Bach. 1997. CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J. Immunol. 158:2947–2954. [PubMed] [Google Scholar]
- 23.Nielsen, C., D. Hansen, S. Husby, B.B. Jacobsen, and S.T. Lillevang. 2003. Association of a putative regulatory polymorphism in the PD-1 gene with susceptibility to type 1 diabetes. Tissue Antigens. 62:492–497. [DOI] [PubMed] [Google Scholar]
- 24.Prokunina, L., L. Padyukov, A. Bennet, U. de Faire, B. Wiman, J. Prince, L. Alfredsson, L. Klareskog, and M. Alarcon-Riquelme. 2004. Association of the PD-1.3A allele of the PDCD1 gene in patients with rheumatoid arthritis negative for rheumatoid factor and the shared epitope. Arthritis Rheum. 50:1770–1773. [DOI] [PubMed] [Google Scholar]
- 25.Smith, J.A., J.Y. Tso, M.R. Clark, M.S. Cole, and J.A. Bluestone. 1997. Nonmitogenic anti-CD3 monoclonal antibodies deliver a partial T cell receptor signal and induce clonal anergy. J. Exp. Med. 185:1413–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang, Q., J.A. Smith, G.L. Szot, P. Zhou, M.L. Alegre, K.J. Henriksen, C.B. Thompson, and J.A. Bluestone. 2003. CD28/B7 regulation of anti-CD3-mediated immunosuppression in vivo. J. Immunol. 170:1510–1516. [DOI] [PubMed] [Google Scholar]
- 27.Chatenoud, L. 2005. CD3-specific antibodies restore self-tolerance: mechanisms and clinical applications. Curr. Opin. Immunol. 17:632–637. [DOI] [PubMed] [Google Scholar]
- 28.Chen, W., J.A. Bluestone, and K.C. Herold. 2005. Achieving antigen-specific tolerance in diabetes: regulating specifically. Int. Rev. Immunol. 24:287–305. [DOI] [PubMed] [Google Scholar]
- 29.Bresson, D., L. Togher, E. Rodrigo, Y. Chen, J.A. Bluestone, K.C. Herold, and M. von Herrath. 2006. Anti-CD3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing Tregs. J. Clin. Invest. 116:1371–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barber, D.L., E.J. Wherry, D. Masopust, B. Zhu, J.P. Allison, A.H. Sharpe, G.J. Freeman, and R. Ahmed. 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 439:682–687. [DOI] [PubMed] [Google Scholar]
- 31.Chatenoud, L. 2003. CD3-specific antibody-induced active tolerance: from bench to bedside. Nat. Rev. Immunol. 3:123–132. [DOI] [PubMed] [Google Scholar]
- 32.Liang, S.C., Y.E. Latchman, J.E. Buhlmann, M.F. Tomczak, B.H. Horwitz, G.J. Freeman, and A.H. Sharpe. 2003. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur. J. Immunol. 33:2706–2716. [DOI] [PubMed] [Google Scholar]
- 33.Tang, Q., E.K. Boden, K.J. Henriksen, H. Bour-Jordan, M. Bi, and J.A. Bluestone. 2004. Distinct roles of CTLA-4 and TGF-beta in CD4+CD25+ regulatory T cell function. Eur. J. Immunol. 34:2996–3005. [DOI] [PubMed] [Google Scholar]
- 34.Blank, C., J. Kuball, S. Voelkl, H. Wiendl, B. Becker, B. Walter, O. Majdic, T.F. Gajewski, M. Theobald, R. Andreesen, and A. Mackensen. 2006. Blockade of PD-L1 (B7-H1) augments human tumor-specific T cell responses in vitro. Int. J. Cancer. 119:317–327. [DOI] [PubMed] [Google Scholar]
- 35.Yamazaki, T., H. Akiba, H. Iwai, H. Matsuda, M. Aoki, Y. Tanno, T. Shin, H. Tsuchiya, D.M. Pardoll, K. Okumura, et al. 2002. Expression of programmed death 1 ligands by murine T cells and APC. J. Immunol. 169:5538–5545. [DOI] [PubMed] [Google Scholar]
- 36.Tsushima, F., H. Iwai, N. Otsuki, M. Abe, S. Hirose, T. Yamazaki, H. Akiba, H. Yagita, Y. Takahashi, K. Omura, et al. 2003. Preferential contribution of B7-H1 to programmed death-1-mediated regulation of hapten-specific allergic inflammatory responses. Eur. J. Immunol. 33:2773–2782. [DOI] [PubMed] [Google Scholar]
- 37.Alegre, M.L., J.Y. Tso, H.A. Sattar, J. Smith, F. Desalle, M. Cole, and J.A. Bluestone. 1995. An anti-murine CD3 monoclonal antibody with a low affinity for Fc gamma receptors suppresses transplantation responses while minimizing acute toxicity and immunogenicity. J. Immunol. 155:1544–1555. [PubMed] [Google Scholar]
- 38.Fife, B.T., M.D. Griffin, A.K. Abbas, R.M. Locksley, and J.A. Bluestone. 2006. Inhibition of T cell activation and autoimmune diabetes using a B cell surface-linked CTLA-4 agonist. J. Clin. Invest. 116:2252–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang, Q., J.Y. Adams, A.J. Tooley, M. Bi, B.T. Fife, P. Serra, P. Santamaria, R.M. Locksley, M.F. Krummel, and J.A. Bluestone. 2006. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat. Immunol. 7:83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}