

Abstract
The physiological role of B cell lymphoma 2 (Bcl-2) homology 3–only proteins has been investigated in mice lacking the individual genes identifying rate-limiting roles for Bim (Bcl-2–interacting mediator of cell death) and Puma (p53–up-regulated modulator of apoptosis) in apoptosis induction. The loss of Bim protects lymphocytes from apoptosis induced by cytokine deprivation and deregulated Ca++ flux and interferes with the deletion of autoreactive lymphocytes and the shutdown of immune responses. In contrast, Puma is considered the key mediator of p53-induced apoptosis. To investigate the hypothesis that Bim and Puma have overlapping functions, we generated mice lacking both genes and found that bim−/−/puma−/− animals develop multiple postnatal defects that are not observed in the single knockout mice. Most strikingly, hyperplasia of lymphatic organs is comparable with that observed in mice overexpressing Bcl-2 in all hemopoietic cells exceeding the hyperplasia observed in bim−/− mice. Bim and Puma also have clearly overlapping functions in p53-dependent and -independent apoptosis. Their combined loss promotes spontaneous tumorigenesis, causing the malignancies observed in Bcl-2 transgenic mice, but does not exacerbate the autoimmunity observed in the absence of Bim.
Members of the B cell lymphoma 2 (Bcl-2) family regulate cell death in response to a wide range of stimuli, including growth factor or cytokine deprivation, DNA damage caused by UV or γ irradiation, and certain anticancer drugs. Members of the Bcl-2 family are characterized by structural motives called Bcl-2 homology (BH) domains. The prosurvival family members Bcl-2, Bcl-xL, Bcl-w, A1/Bfl-1, and Mcl-1 contain up to four such homology domains (BH1–4), whereas proapoptotic members of the same family either possess three out of the four BH domains (e.g., Bax [Bcl-2–associated protein X], Bak [Bcl-2 antagonist/killer], and Bok [Bcl-2–related ovarian killer]) or only the BH3 domain (1).
The BH3-only proteins Blk (Bik-like killer)/Bik (Bcl-2–like killer)/Nbk, Bid (Bcl-2–interacting domain death agonist), Bad (Bcl-2 antagonist of cell death), Harakiri/death protein 5, Noxa/Apr, Bmf (Bcl-2 modifying factor), Puma (p53–up-regulated modulator of apoptosis)/bbc3, and Bim (Bcl-2–interacting mediator of cell death)/Bod (Bcl-2–related ovarian death gene) can all induce apoptosis when overexpressed in cultured cells (1). This killing requires Bax or Bak (2), but how BH3-only proteins are activated by physiological stimuli or in response to genotoxins remains only partly understood. According to a current model (3, 4), Bcl-2–like prosurvival molecules can act as direct activators (Bid and Bim) or as derepressors (all others). In this model, the active form of Bid (caspase-truncated (t)Bid) or Bim are thought to bind to Bcl-2 prosurvival homologues in response to certain stress signals such as growth factor deprivation and prime mitochondria for the induction of apoptosis. Derepressor proteins are thought to free (t)Bid or Bim from sequestration by competitive binding to Bcl-2–like molecules. Once freed, the direct activators are proposed to interact physically with Bax and/or Bak, triggering their activation and subsequent apoptosis (3, 4). An alternative model favors the idea that BH3-only proteins have different, only partially overlapping binding preferences for their prosurvival Bcl-2–like relatives, and individual BH3-only proteins antagonize a specific subset of Bcl-2–like prosurvival molecules (5). According to this model, Bax and/or Bak are normally kept in check by binding to their prosurvival relatives and are activated when released as a result of BH3-only protein binding to the Bcl-2–like proteins (5).
The physiological role of BH3-only proteins has been addressed by analyzing mice lacking individual members of the family. The absence of single BH3-only proteins is mostly compatible with embryogenesis, with the exception of the partial lethality of bim−/− embryos (6), suggesting a high degree of redundancy among this class of proteins in early embryonic development. In the adult organism, however, tissue and cell type–specific defects have been observed in some but not all knockout mouse models (1). Lymphocytes from bim−/− mice were shown to be highly resistant to the effects of cytokine deprivation or Ca2+ flux and, to a lesser extend, also to glucocorticoid (GC) treatment (6). The loss of Bim causes lymphadenopathy and autoimmunity as a result of the inefficient deletion of autoreactive thymocytes and immature B cells (7–9). In addition, Bim is also critical for the deletion of antigen-activated T cells during the shutdown of an immune response (10, 11).
The BH3-only genes puma and noxa are direct transcriptional targets of the tumor suppressor p53, and gene targeting in mice confirmed crucial cell type–dependent functions in p53-induced apoptosis for both proteins (12–14). Puma-deficient mouse embryonic fibroblasts and lymphocytes are highly refractory to treatment with γ irradiation or treatment with DNA-damaging drugs such as etoposide. Lymphocytes lacking puma also show decreased sensitivity to certain p53-independent cell death stimuli such as cytokine deprivation, treatment with phorbol esters, or dexamethasone (14). Experiments with mice exposed to whole body γ irradiation or injected with GCs indicated that Puma and Bim might have overlapping functions in the killing of thymocytes and other lymphocyte populations in vivo (15).
We have generated mice lacking both Puma and Bim and demonstrate that these two proteins efficiently cooperate in mediating apoptosis in response to a broad range of p53-dependent and -independent apoptosis stimuli. Stress-induced cell death, spontaneous cell death, and death of activated T cells depend largely on these two BH3-only proteins. Autoimmunity caused by Bim deficiency was not exacerbated by the additional loss of Puma despite pronounced lymphadenopathy and thymic hyperplasia. Interestingly, the loss of Puma over Bim elevates the rate of spontaneous tumor development observed in aged bim−/− mice, confirming the intrinsic tumor suppressor potential of these proteins.
RESULTS
Generation of bim−/−/puma−/− double-deficient mice
Mice lacking both Bim and Puma were generated by intercrossing double heterozygotes (bim +/−/puma +/−) and subsequently by intercrossing bim +/−/puma−/− males with double heterozygous females. Consistent with the previously reported partial embryonic lethality of bim−/− mice in intercrosses of bim +/− heterozygotes on the mixed 129SV×C57BL/6 background (6), only ∼44% of bim−/− and ∼40% of the expected numbers of bim−/−/puma−/− mice were born alive on the inbred C57BL/6 background. This indicates that a substantial percentage of bim−/− and bim−/−/puma−/− embryos die in utero and suggests that Bim and Puma do not have overlapping functions during embryogenesis. Additional bim−/−/puma−/− mice were generated by crossing bim−/−/puma−/− males with bim +/−/puma−/− females.
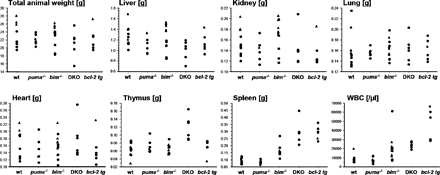
Histological examination of 8–12-wk-old bim−/−/puma−/− mice revealed the normal development of most organs, including the lung, liver, heart, and kidneys (unpublished data). In contrast, spleens and lymph nodes were enlarged and comparable in sizes with the ones observed in age-matched vav-bcl-2 transgenic (tg) mice but were clearly larger than those seen in bim−/− animals (P = 0.0001; Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20061552/DC1). Although neither the loss of Bim or Puma nor Bcl-2 overexpression caused increased thymic size or cellularity (6, 14, 16), animals that lacked both Bim and Puma displayed a significant increase in thymic organ weight (P < 0.0021; Fig. S1).
Although increased in size, the thymus displayed an architecture containing clearly demarcated cortical and medullar regions but showed an increase in the number of medullar lymphocytes, as reflected in the reduced contrast between cortex and medulla in hematoxylin and eosin–stained sections (Fig. S2 a, available at http://www.jem.org/cgi/content/full/jem.20061552/DC1). The architectures of spleens (Fig. S2 b) and lymph nodes (not depicted) were strongly altered in bim−/− as well as in bim−/−/puma−/− mice and were characterized by severely enlarged T and B cell zones. Areas of red pulp were most pronounced in the spleens of bim−/−/puma−/− animals (Fig. S2 b; and see Fig. 2 e).
Figure 2.

Splenomegaly caused by the loss of Bim is enhanced by concomitant Puma deficiency. Spleens from animals of the indicated genotypes were harvested. (a) Total cellularity determined by counting an aliquot of the single-cell suspensions using a hemocytometer and trypan blue exclusion. (b) Total cellularity of splenic T and B cells was determined by staining cell suspensions with fluorescence-conjugated antibodies specific for CD4 and CD8 or IgM and IgD, respectively. (c) The absolute numbers of activated CD44highCD62Llow T cells were determined by staining splenic T cells with antibodies specific for CD4 or CD8 in combination with antibodies to CD44 and CD62L. (d and e) Absolute numbers of granulocytes (Mac-1+Gr-1+), macrophages (Mac-1+Gr-1−; d), and nucleated erythroid cells (Ter119+; e), were determined by flow cytometric analysis using fluorescence-conjugated antibodies. Bars represent means ± SEM (error bars) of 5–10 animals of each genotype.
Impaired T lymphocyte development and thymic hyperplasia caused by the combined absence of Bim and Puma
The loss of Puma does not cause any obvious defects in lymphocyte development, whereas the absence of Bim strongly impairs T and B cell development, in part because of the failure of negative selection processes (1). Thymic cellularity is not increased in Bim-deficient or Bcl-2 tg mice, but animals of both genotypes display a substantial increase in mature CD4+8− as well as CD4−8+ single-positive T cells and an abnormal decrease in immature double-positive (DP) CD4+8+ thymocytes (Fig. 1 a). Cell counting revealed a close to twofold increase in the total thymocyte number in bim−/−/puma−/− mice compared with all other genotypes (P ≤ 0.037; Fig. 1 b). Compared with bim−/− mice, bim−/−/puma−/− mice had an even greater reduction in the percentage of CD4+8+ DP thymocytes (P = 0.0002; Fig. 1 a), but their total number was larger than that seen in bim−/− mice, approaching that found in WT animals (Fig. 1 b). In comparison to bim−/− and vav-bcl-2 tg mice, bim−/−/puma−/− mice had twice as many CD4−8− double-negative (DN) and CD4+8− as well as CD4−8+ thymocytes, amounting to a four- to sevenfold increase compared with WT mice (Fig. 1 b). A more detailed FACS analysis of CD4−8− thymocytes revealed that >90% of these DN cells were CD25−CD44− pro–T4 cells. A similar percentage of these cells was also observed in bim−/− and vav-bcl-2 tg mice (Fig. 1 c). The majority of these CD4−8−CD25−CD44− (pro-T4) cells expressed a TCR-β on their surface (Fig. 1, d and e), indicating that they had undergone β selection and may represent mature DN T cells (17). Collectively, our data demonstrate that the additional loss of Puma exacerbates the defects in T cell development in the thymus caused by the loss of Bim.
Figure 1.

Combined loss of Puma and Bim causes the accumulation of immature thymocytes. (a) Representative dot blots of stained thymocytes from animals of each genotype showing the percentages of CD4−8−, CD4+8+, CD4+8−, and CD4−8+ cells. (b) Thymic cellularity was assessed using a hemocytometer and trypan blue exclusion. The percentages of the various thymocyte subsets and total thymic cellularity were used to calculate the absolute numbers of CD4−8−, CD4+8+, CD4+8−, and CD4−8+ thymocytes. (c and d) Single-cell suspensions were stained with a combination of fluorochrome-conjugated antibodies recognizing CD4, CD8, CD25, and CD44 (c) or CD4, CD8, and TCR-β (d) to determine the percentage and overall numbers of pro–T cells (CD4−8−) and the pro–T1 (CD25−44+), pro–T2 (CD25+44+), pro–T3 (CD25+44−), and pro–T4 subsets (CD25−44−). (e) Representative histograms of TCR-β expression on CD4−8− DN thymocytes. Bars represent means ± SEM (error bars) of 5–10 animals of each genotype.
Interestingly, thymic hyperplasia was never observed in puma−/−, bim−/−, or vav-bcl-2 tg mice (Fig. S1 and Fig. 1 b). Its occurrence in bim−/−/puma−/− mice indicated that Puma, together with Bim, may antagonize a Bcl-2 prosurvival homologue that is not efficiently blocked by Bim alone and cannot be replaced by Bcl-2 overexpression. We hypothesized that the loss of puma may synergize with Bcl-2 overexpression to cause thymic hyperplasia and generated puma−/− mice expressing the vav-bcl-2 transgene. Comparing the thymi derived from 8–10-wk-old mice lacking puma with those lacking puma and overexpressing Bcl-2 did not reveal an abnormal increase in organ weight, overall cellularity, or subset composition (Fig. S3 a, available at http://www.jem.org/cgi/content/full/jem.20061552/DC1; and not depicted). Thus, Bcl-2 overexpression may be unable to fully neutralize Bim and Puma, but an increase in another prosurvival family member might be able to do so, thereby facilitating thymic hyperplasia. Bcl-xL can avidly bind to Bim and Puma (18) and delays spontaneous, anticancer drug–, or GC-induced death when overexpressed in thymocytes (19). In contrast to our expectations, double-tg offspring from intercrosses of lck-bcl-xL and vav-bcl-2 tg mice had smaller thymic organ weights and cellularity than in WT, puma−/−, and puma−/−/vav-bcl-2 tg mice (Fig. S3, a and b). This indicates that simply increasing the pool of prosurvival Bcl-2 homologues is not sufficient to cause thymic hyperplasia and may even inhibit the thymocyte proliferation of immature T cell progenitors. Remarkably, lethally irradiated Ly5.1 recipient mice reconstituted with bone marrow cells lacking both Bim and Puma had thymus sizes and cellularities indistinguishable from that of WT mice or mice reconstituted with WT bone marrow (Fig. S3 c). However, thymic subset composition reflected those of unmanipulated bim−/−/puma−/− mice (unpublished data). Therefore, we conclude that a thymocyte-independent contribution (perhaps mediated by increased thymic epithelial cell survival) in combination with the decreased apoptosis of developing thymocytes may be responsible for causing thymic hyperplasia in bim−/−/puma−/−mice.
Combined loss of Puma and Bim perturbs cellular homeostasis in the bone marrow
Analysis of bone marrow samples from the femora of WT, bim−/−, puma−/−, and bim−/−/puma−/− mice revealed that they all had equal cell numbers of hemopoietic cells. In contrast, increased cellularity was observed in vav-bcl-2 tg mice (Table S1, available at http://www.jem.org/cgi/content/full/jem.20061552/DC1). When analyzing the composition of B cell subsets, we observed that the percentage and total number of B220+CD43+sIgM− pro–B cells was elevated in animals lacking Bim or those overexpressing Bcl-2. The additional loss of Puma did not cause a further increase in pro–B cell numbers in bim−/− mice (Table S1). The percentage and number of B220+CD43−sIgM− pre–B cells did not significantly differ between genotypes (P ≥ 0.08; Table S1). Consistent with a previous study (6), we found that the numbers of sIgMlowsIgDhigh mature recirculating B cells were elevated about twofold in bim−/− mice and that the loss of Puma did not cause a further accumulation of these cells (Table S1). In contrast, Bcl-2 tg mice contained even higher cell numbers of mature recirculating B cells than bim−/− or bim−/−/puma−/− mice (Table S1), suggesting that additional BH3-only proteins besides Bim and Puma regulate B cell homeostasis. The numbers of recirculating CD4+ or CD8+ mature T cells was also elevated in the absence of Bim, but, in contrast to B cells, the additional loss of Puma drastically increased the number of these cells (Table S1). The numbers of nucleated Ter119+ erythroid cells and granulocytes were abnormally reduced in the bone marrow of bim−/−, bim−/−/puma−/−, and vav-bcl-2 tg mice (Table S1).
Combined absence of Bim and Puma causes severe splenomegaly and lymphadenopathy
Overexpression of Bcl-2 throughout the hemopoietic compartment in vav-bcl-2 tg mice causes severe splenomegaly and lymphadenopathy to an extent that exceeds the one elicited by the loss of Bim (16, 20). This indicates that the overexpression of Bcl-2 antagonizes not only Bim but also additional BH3-only proteins that are relevant for lymphocyte homeostasis. In accordance with this hypothesis, we observed that bim−/−/puma−/− mice had a clearly more extensive splenomegaly and lymphadenopathy than bim−/− mice; in fact, it was comparable with that observed in vav-bcl-2 tg mice. The combined loss of Bim and Puma caused a fourfold increase in spleen cell numbers when compared with WT spleens (P < 0.0001) and a twofold increase when compared with Bim-deficient spleens (P = 0.007). Spleens from vav-bcl-2 tg animals were comparable in size and cellularity with spleens from bim−/−/puma−/− animals (Fig. 2 a).
The percentage of immature sIgMhighsIgDlow and mature sIgMlowsIgDhigh B cells and CD4+ as well as CD8+ T cells were similar to the ones observed in spleens from bim−/− or vav-bcl-2 tg animals, but the combined loss of Puma and Bim was required to raise lymphocyte numbers to the levels observed in mice overexpressing Bcl-2 (not depicted and Fig. 2, a and b). Interestingly, bim−/−- and bim−/−/puma−/−-like vav-bcl-2 tg mice displayed an increase in the percentage of activated CD44hiCD62Llow T lymphocytes in spleens (Fig. 2 c) and lymph nodes (Table S2, available at http://www.jem.org/cgi/content/full/jem.20061552/DC1), indicating that Bim and Puma might have an overlapping role in the programmed death of activated T cells. In line with a possible role for Puma in this process, we noted that the percentage and number of T cells that had a CD44hiCD62Llow phenotype appeared mildly but repeatedly elevated in puma−/− mice (Fig. 2 c).
White blood cell numbers in bim−/− or bim−/−/puma−/− mice did not reach the levels observed in vav-bcl-2 tg mice (Table S3, available at http://www.jem.org/cgi/content/full/jem.20061552/DC1). As in the lymph nodes and spleens, mature T cells were significantly higher in double- deficient animals than in bim−/− mice (P ≤ 0.033), matching the numbers observed in vav-bcl-2 tg mice. Interestingly, the number of mature B cells was comparable between bim−/− or bim−/−/puma−/− mice but was significantly smaller than those found in vav-bcl-2 tg mice (P ≤ 0.04), explaining the difference in total white blood cell number (Table S3). This observation further supports our notion that B cell homeostasis is not regulated by Puma and that Bim may act in concert with at least one additional proapoptotic factor that can be inhibited by Bcl-2.
Mice overexpressing Bcl-2 have reduced numbers of committed myeloid progenitors in their bone marrow but an increase of these cells in the spleen, most likely as a result of extramedullary hemopoiesis (16). Similarly, mice lacking Bim accumulate myeloid cells in secondary lymphatic organs such as the spleen (20), and the loss of Puma was shown to protect bone marrow–derived myeloid precursor cells from the effects of aberrant c-myc oncogene expression and growth factor deprivation (13). Our analysis revealed that the number of Mac-1+Gr-1− macrophages was significantly increased in the spleens of bim−/−/puma−/− and vav-bcl-2 tg mice (P < 0.004) but only marginally in puma−/− or bim−/− animals (Fig. 2 d). In the peripheral blood, only vav-bcl-2 tg mice displayed a significant increase (P = 0.001) of Mac-1+Gr-1− monocytes (Table S3). These findings are consistent with our observations in bone marrow, where only Mac-1+Gr-1− monocytes/macrophages but not granulocytes were found to be elevated when Bcl-2 was overexpressed (Table S1). In contrast, granulocytes in spleens and peripheral blood were comparable in all genotypes (Fig. 2 d and Table S3). Even though they are decreased in number in the bone marrow of bim−/−, bim−/−/puma−/−, and vav-bcl-2 tg mice (Table S1), possibly as a result of the displacement by increased numbers of recirculating mature lymphocytes, these numbers are able to replenish the daily granulocyte loss in the periphery.
Consistent with the abnormal expansion of red pulp seen in the histological section of the spleens from bim−/−/puma−/− and vav-bcl-2 tg mice (Fig. S2 and not depicted), nucleated Ter119+ erythroid cells were elevated when compared with the other genotypes. The absence of bim alone caused only a minor increase in the number of Ter119+ cells, but this increase was much more pronounced when Bim and Puma were lacking or when Bcl-2 was overexpressed (Fig. 2 e). This suggests that lymphadenopathy-mediated displacement of erythroid progenitors from the bone marrow, like that observed in bim−/−, bim−/−/puma−/−, or vav-bcl-2 tg mice (Table S1), can be compensated for by extramedullary erythropoiesis in the spleen (Fig. 2 e and not depicted).
Bim and Puma cooperate in apoptosis induction in lymphocytes
The abnormal accumulation of activated T cells in the spleen and lymph nodes of bim−/−/puma−/− mice prompted us to investigate the role of these proteins in the death of activated T cells. As previously reported (6), bim−/− T cell blasts survived IL-2 deprivation in culture substantially better than T cell blasts from WT mice (Fig. 3 a). The loss of Puma provided only transient protection from IL-2 deprivation–induced death, which was most pronounced after 24 h, but T cell blasts died rapidly thereafter (Fig. 3 a). However, the combined loss of Puma and Bim protected T cell blasts from IL-2 deprivation–induced death more potently than the loss of Bim and almost as potently as Bcl-2 overexpression. This indicates that Puma, together with Bim, plays an essential role in activated T cell death (Fig. 3 a).
Figure 3.

The death of activated T cells is mediated by Bim and Puma. (a) Enriched T cells were stimulated for 72 h with 2 μg/ml ConA and 100 U/ml IL-2. Cell viability was assessed at the indicated time points after cytokine deprivation by PI exclusion and flow cytometric analysis. Means of four or more independent experiments ± SEM (error bars) are given, representing cells generated from five to six mice per genotype. (b) Ly5.1+ mice reconstituted for 8 wk with bone marrow cells derived from the indicated genotypes (all verified to contain >95% donor-derived Ly5.2+ leukocytes in peripheral blood) were challenged with SEB. The percentages of Vβ8+ and Vβ6+ T cells in peripheral blood were determined after 0, 3, 8, and 15 d. Two independent experiments using three to four animals per genotype gave similar results, so the data were pooled. (c) 3 or 6 wk after SEB challenge, animals were killed, single-cell suspensions from spleen and inguinal lymph nodes were counted, and aliquots were stained using antibodies specific for CD4 and Vβ8+ followed by flow cytometric analysis. Two independent experiments using two different donor animals per genotype gave similar results, so the data were pooled. (b and c) Mean percentages ± SD (error bars) are given.
To assess the relevance of this observation for the deletion of activated T cells in vivo, we generated a cohort of animals containing a Bim/Puma double-deficient hemopoietic system (plus all relevant controls) by reconstituting lethally irradiated C57BL/6-Ly5.1 recipients with bone marrow–derived stem cells of the desired genotype. 8 wk after successful reconstitution, these animals were challenged with the bacterial superantigen Staphylococcus enterotoxin B (SEB) that specifically activates T cells expressing a Vβ8-containing TCR. The expansion of Vβ8-bearing T cells in peripheral blood was monitored 0, 3, 8, and 15 d after SEB challenge. Consistent with previously published data (10), we observed that the expansion of Vβ8+ T cells lacking Bim (or Bim and Puma) was initially delayed compared with WT or puma−/− T cells (Fig. 3 c). This may be caused by a defect in entering the cell cycle, a phenomenon that has been well documented in lymphocytes overexpressing Bcl-2 (21). The percentages of Bim as well as Bim/Puma double-deficient Vβ8+ T cells were still elevated at day 15, whereas Puma-deficient cells regressed as rapidly as WT cells (Fig. 3 b). This was surprising given our in vitro data and suggests that a beneficial effect of Puma deficiency may only become apparent at later time points. As expected, the percentage of Vβ6+ T cells, which do not respond to SEB, did not change during the entire time course of the experiment (Fig. 3 b). When analyzing the fate of Vβ8+ T cells in lymph nodes and spleen 3 or 6 wk after SEB challenge, we again observed increased percentages in bim−/− and bim−/− puma−/− double-deficient animals (Fig. 3 c). However, we also noted that the combined loss of Puma and Bim did not protect Vβ8+ T cells more efficiently than the loss of Bim alone (Fig. 3 d). Because of the limited number of animals available to us, we were unable to determine whether a difference in the deletion efficiency may have become apparent beyond 6 wk.
Bim and Puma cooperate in stress-induced apoptosis
The extensive lymphoid hyperplasia strongly suggests that lymphocytes lacking both Bim and Puma may exhibit severe apoptosis defects. To assess quantitative differences in stress-induced apoptosis, we isolated immature DP thymocytes and mature splenic T and B cells of 8–12-wk-old WT, bim−/−, puma−/−, bim−/−/puma−/−, and vav-bcl-2 tg mice, put them into culture, and subjected them to cytokine withdrawal, treatment with the GC dexamethasone, the phorbol ester PMA, the calcium ionophore ionomycin, the broad- spectrum kinase inhibitor staurosporine, γ irradiation, the DNA-damaging drug etoposide, or the glycosylation inhibitor and ER stressor tunicamycin. Cell viability was monitored over a period of 96 h by means of annexin V/propidium iodide (PI) staining and flow cytometric analysis. Thymocytes lacking both Bim and Puma survived cytokine deprivation, γ irradiation, and treatment with staurosporine, tunicamycin, or etoposide significantly better than cells lacking only Bim or Puma and, of course, WT cells (P ≤ 0.04; Fig. 4 a and not depicted). In contrast, other death stimuli such as phorbol ester or ionomycin killed primarily in a Puma- or Bim-dependent manner, respectively (Fig. S4 a, available at http://www.jem.org/cgi/content/full/jem.20061552/DC1). Death by ligation of Fas was independent of Bim and Puma (Fig. S4 a).
Figure 4.

Bim and Puma can act synergistically in cell death activation. (a–c) FACS-sorted CD4+8+ thymocytes (a), Thy1+ splenic T cells (b), or B220+ splenic B cells (c) derived from animals of the indicated genotypes were cultured in regular medium (spontaneous death), exposed to γ irradiation (10 Gy), or cultured in the presence of 1 μg/ml etoposide, 10 μg/ml tunicamycin, 100 nM staurosporine, or 10−6 M dexamethasone. The percentages of viable cells in culture were determined by annexin V/PI staining and flow cytometric analysis. The extent of apoptosis induced specifically by different stimuli was calculated by the following equation: (induced apoptosis − spontaneous cell death)/(100 − spontaneous cell death). Means ± SEM (error bars) from four independent experiments and n ≥ 4 animals per genotype are shown.
In response to most cell death inducers tested, mature T and B cells lacking Bim and Puma behaved in a manner similar to thymocytes (Fig. 4, b and c; and not depicted). Interestingly, mature T and B cells from bim−/−/puma−/− mice were almost as resistant to cytokine deprivation or treatment with GC as cells overexpressing Bcl-2 (Fig. 4, b and c), indicating that Bim and Puma mediate most of this killing. To assess whether GC-induced cell death would entirely depend on Bim and Puma, double-deficient animals were injected i.p. with 500 μg dexamethasone. We observed that the loss of either Puma or Bim expression provided limited, albeit significant (P < 0.042), protection (Fig. 5 a). However, in the absence of Bim and Puma, the overall thymic cellularity was reduced only by ∼20% (Fig. 5 a). The numbers of DP thymocytes that was recovered in bim−/− or puma−/− single knockout animals 20 h after GC injection was substantially smaller than the number of DP thymocytes from double-deficient animals (Fig. 5 a). Similarly, more mature T and B cells were recovered from the spleens of double-deficient animals than from mice lacking Bim or Puma (Fig. 5 b). This indicates that Bim and Puma have overlapping roles in the GC-induced killing of thymocytes and mature T cells, but the combined loss of these BH3-only proteins does not confer absolute GC resistance in vivo.
Figure 5.

Bim and Puma cooperate in dexamethasone-induced T cell apoptosis in vivo. Mice of the indicated genotypes were injected i.p. with 500 μg saline or dexamethasone and killed 20 h later. Thymic cellularity was assessed using a hemocytometer and trypan blue exclusion. (a) The percentages of the CD4+8+ thymocytes and total thymic cellularity were used to calculate the absolute number of CD4+8+ thymocytes. (b) Absolute numbers of CD4+ and CD8+ splenic T cells were determined by staining single-cell suspensions with antibodies specific for CD4 and CD8 followed by flow cytometric analysis. Combined results ± SD (error bars) from two independent experiments and n ≥ 2 animals per genotype are shown.
Hyper–γ-globulinemia and autoantibody production in bim−/− and bim−/− puma−/− mice
Autoimmune glomerulonephritis observed in bim−/− mice on the mixed (129SV×C57BL/6) genetic background is caused by defects in the negative selection of autoreactive B (9) and T cells (7, 8) and excessive production of Ig from abnormally persisting plasma cells in these animals (6). However, fatal autoimmune pathology was not seen in bim−/− mice backcrossed onto the C57BL/6 genetic background (20), which is consistent with the polygenetic nature of this disease. To assess whether the combined loss of Bim and Puma may lead to autoimmune pathology on the C57BL/6 background, we monitored B cell effector function. Serum Ig levels were quantified in 8–12-wk-old animals of all genotypes. Consistent with previous observations on the mixed (C57BL/6×129SV) genetic background, the loss of Bim caused an increase in the levels of all Ig isotypes tested (IgM, IgG1, IgG2a, IgG2b, IgG3, and IgA), and this increase was similar in the extent seen in vav-bcl-2 tg mice (Fig. 6 a). The loss of Puma on its own did not cause any abnormality in serum Ig levels, with one notable exception—that of the increased levels of IgA (P = 0.05). The combined loss of both Bim and Puma did not lead to a further increase in serum IgM and IgG levels compared with that seen in bim−/− mice. Consistent with the observation in puma−/− mice, IgA levels were clearly higher in bim−/−/puma−/− animals than in bim−/− mice (P = 0.0005; Fig. 6 a), indicating that Bim and Puma coregulate the lifespan of IgA-secreting plasma cells.
Figure 6.

Enhanced Ig and autoantibody production in bim−/−, bim−/−/puma−/−, and vav-bcl-2 tg mice. (a) Ig titers in sera from 8–12-wk-old mice of the indicated genotypes were quantified by ELISA. (b) Autoantibodies to dsDNA were quantified in sera from 12-mo-old mice of the indicated genotypes by ELISA using calf thymus dsDNA for coating. (c) Representative ANA staining on rat liver sections using serum from a bim−/−/puma−/− animal. (d) Representative hematoxylin and eosin stain from kidney sections derived from a 12-mo-old bim−/−/puma−/− mouse.
Consistent with the notion that Bim-deficient B cells escape negative selection and have an abnormal tendency to escape anergy in response to challenge with autoantigens, we observed significantly increased titers of anti–double-stranded DNA (dsDNA) antibodies in serum samples of 12-mo-old bim−/− and bim−/−/puma−/− mice compared with sera from WT or puma−/− mice (P ≤ 0.05; Fig. 6 b). Sera of aged mice lacking Bim or Bim plus Puma also showed immunoreactivity with the nuclei in rat liver sections (Fig. 6 c), confirming the presence of antinuclear antibodies (ANAs). However, fatal autoimmune glomerulonephritis as observed in bim−/− mice on the mixed genetic background was not observed in any of the bim−/− (n = 15), bim−/−/puma +/− (n = 10), or bim−/−/puma−/− (n = 7) animals analyzed at 12 mo of age (Fig. 6 c and not depicted). These results indicate that Bim may be the dominant inducer of the programmed death of activated B cells, but Puma may play a minor contributory role, particularly in IgA-secreting plasma cells.
Mice lacking Bim and Puma are prone to lymphoma development
When combined with aberrant c-myc oncogene activation, the loss of Bim or the knockdown of Puma was shown to accelerate lymphomagenesis in mice (22, 23), but spontaneous tumor development has not been assessed in mice lacking Bim, Puma, or both. To investigate whether the combined loss of Bim and Puma can cause spontaneous tumor formation, we followed a cohort of bim−/−/puma−/−, bim−/−/puma +/−, bim +/−/puma−/−, bim−/−, puma−/−, and WT mice for 12 mo and performed detailed necropsy. No signs of malignancy were observed in 1-yr-old WT (0/12) or puma−/− animals (0/14), but neoplastic disease of the hemopoietic system was observed in 20% of animals lacking Bim (3/15). Consistent with an overlapping role for Bim and Puma in tumor suppression, we observed signs of malignancies in 50% (5/10) of bim−/−/puma +/− mice (but 0% of the bim +/− puma−/− mice; 0/13) and in ∼60% of bim−/−/puma−/− mice (4/7) that were still available for analysis.
Flow cytometric and histopathological assessment revealed a tumor pattern similar to the one described in aged vav-bcl-2 tg mice (24), including B220+ follicular and diffuse large B cell lymphomas (Fig. 7, a and b; and Fig. S5, available at http://www.jem.org/cgi/content/full/jem.20061552/DC1), but histiocytic sarcomas were also observed in bim−/− mice lacking one or both alleles of puma (one per genotype; Fig. 7 c). In lymphoma cases, lymph node architecture was completely effaced by either nodular/follicular or diffusely spreading neoplastic blasts, invading the lymph node capsule and, in cases of extranodal involvement, expanding onto the organ submucosae and laminae propriae with focal hypoepithelial lesions. In addition, immunohistochemical analysis of expression levels of the B cell marker B220 and the proliferating cell nuclear antigen (PCNA) revealed high levels of both proteins and high mitotic indices in all lymphoma cases tested (Figs. S5 and S6; and not depicted). Histiocytic sarcoma infiltrated spleen and lymph nodes and consisted of undifferentiated epithelioid and spindle cells with abundant mitoses and scattered tumor giant cells (Fig. 7 c). Transplantation of bone marrow from 12-mo-old bim−/− puma−/− mice with B cell lymphomas into irradiated recipients caused rapid overt disease 10 wk after reconstitution (Fig. S7). This phenomenon was never observed when bone marrow from young 8–12-wk-old double-deficient donors was used; these cells efficiently reconstituted normal hemopoiesis, but without signs of malignancy over an observation period of >20 wk (unpublished data). In addition to the malignant changes, hyperplastic infiltrations were frequently observed in the kidney, liver, and lung of aged bim−/−, bim−/−/puma +/−, and bim−/−/puma−/− mice (Fig. 7, d–f; and not depicted). Although preliminary, these observations confirm that BH3-only proteins can function as tumor suppressors and provide evidence that Bim and Puma may have overlapping roles in this process.
Figure 7.

Combined loss of Puma and Bim promotes spontaneous tumor formation. (a and b) Follicular B cell lymphoma found in the mesenteric lymph nodes of a 12-mo-old bim−/− puma−/− mouse (a) expanding into the submucosae and lamina propriae of the small gut (b). (c) One 12-mo-old bim−/−/puma−/− mouse developed a histiocytic sarcoma of the spleen. (d–f) Hyperplastic lymphocyte infiltrations were observed in the kidneys (d), liver (e), and lung (f), in part showing signs of malignancy (see lymphoepithelial lesions of the bronchus in f). Bars (a), 300 μm; (b–d), 200 μm; (e and f), 150 μm.
DISCUSSION
Previous analysis of animals lacking either Bim or Puma suggested very distinct physiological roles for the individual BH3-only proteins. Puma is considered to be the prime effector of p53 in DNA damage–induced apoptosis in cell types as diverse as lymphocytes, fibroblasts, and the central nervous system (13, 14, 25). Bim, on the other hand, seems to be most critical in the hemopoietic compartment, where it plays crucial roles in the deletion of autoreactive T and B cells, the termination of humoral immune responses, and leukocyte homeostasis in general (1). Experiments investigating cell death induction in cultured cells exposed to a wide range of apoptotic stimuli suggested functional redundancy between Bim and Puma. For example, the absence of either of these BH3-only proteins delayed apoptosis induced by cytokine withdrawal or treatment with GCs both in vitro and in vivo (6, 14, 15). Therefore, we investigated the possible functional overlap between these BH3-only proteins by generating double-deficient mice.
Bim deficiency was previously reported to cause the loss of a certain number of embryos on the mixed 129SV×C57BL/6 genetic background before embryonic day (E) 9.5 (6). This phenotype is maintained after backcrossing to C57BL/6, but the reason remains unknown (this study and unpublished data). Double-deficient animals and Bim-deficient animals were both born with a frequency between 40 and 44%, respectively, indicating that the additional loss of puma does not enhance the embryonic lethality caused by the loss of Bim. Given the low numbers of animals derived from these initial intercrosses (n = 183), more animals may be needed to allow stringent statistical analysis.
The combined loss of Bim and Puma did not markedly impair tissue homeostasis in nonhemopoietic organs such as the heart, lung, kidney, or liver, but minor abnormalities or defects in numerically small cell subsets may have escaped our attention. This may indicate that in bim−/−/puma−/− mice that successfully complete embryonic development, additional proapoptotic factors can maintain normal levels of programmed cell death. However, the combined loss of Bim and Puma did significantly enhance lymphadenopathy and splenomegaly, which are also observed in bim−/− mice (P = 0.007), and, interestingly, also created novel defects such as thymic hyperplasia (Figs. 1 and 2). The thymic hyperplasia seen in bim−/−/puma−/− mice appears to depend on a thymocyte-independent contribution, possibly increased thymic epithelial cell survival, in combination with strongly decreased thymocyte apoptosis (see bone marrow reconstitution experiments in Fig. S3). Interestingly, thymic hyperplasia was not observed in animals reconstituted with bax −/−/bak −/−-deficient bone marrow (26), and it would be interesting to know whether hyperplasia can develop in mice lacking Bax/Bak both in hemopoietic cells and the thymic stroma. Therefore, this phenotype is unique to animals lacking Bim and Puma. The combined loss of Bim and Blk (27), Bim and Bad (unpublished data), or Bim and Bmf (unpublished data) does not cause thymic hyperplasia, demonstrating that this defect is caused specifically by the combined absence of Bim and Puma and not by the combined loss of Bim and just any BH3-only protein.
All developmental defects found in bim−/−/puma−/− thymocytes were highly similar, albeit more pronounced than the ones observed in bim−/−mice (7). The percentages of CD4+8+ DP cells were further reduced in the absence of Bim and Puma than in the absence of Bim alone (Fig. 1), indicating that the enhanced accumulation of mature T cells through a presently unknown negative feedback may further impede the proliferation of immature pro–T cells. A similar reduction in CD4+8+ DP thymocytes was also seen in lethally irradiated mice reconstituted with a Bax/Bak double-deficient hemopoietic system (26).
It has previously been reported that the activation of p53 restricts the survival of DN thymocytes that fail to rearrange their TCR-β chain, which is required to secure the transition of developing thymocytes from the CD4−8− DN to the CD4+8+ DP stage (28). However, the percentage and numbers of TCR-β+ DN thymocytes did not differ between WT and puma−/− mice (Fig. 1), confirming the notion that the loss of p53 but not simply the inhibition of apoptosis downstream of p53 is required to rescue pre–T cells from the detrimental consequences of impaired or lacking pre-TCR signaling (28). Accordingly, the accumulation of DN thymocytes observed in bim−/−, bim−/−/puma−/−, or vav-bcl-2 tg mice is not caused by the survival of cells that failed β selection because most of these cells do express a TCR-β chain (Fig. 1).
In contrast to early thymocyte development, apoptosis during early B cell development appears to be regulated mainly, albeit not exclusively, by Bim. Bone marrow cellularity was found to be significantly increased only in vav-bcl-2 tg mice (P = 0.048), but the percentage and number of B220+CD43+sIgM− pro–B cells was also elevated in bim−/− mice (Table S1). This increase was not enhanced by the additional loss of Puma. Consistent with previously published data (29), pre–B cell numbers were not abnormally increased in bim−/− mice (Table S1), and the additional loss of Puma had no effect. Collectively, our findings confirm that Bcl-2 and Bim do play a crucial role in early pro–B cell survival but not in maturation downstream of the IL-7/IL-7R signaling pathway. The role of Bim in early B cell development appears to overlap with other proapoptotic factors because vav-bcl-2 tg mice show about a twofold excess of B220+CD43+sIgM− pro–B cells compared with bim−/− mice (Table S1).
The splenomegaly that is caused by the combined deficiency of Bim and Puma exceeds the one found in bim−/− mice (6) and matches the one observed in vav-bcl-2 tg mice (16). All T and B lymphocyte subsets in bim−/−/puma−/− were significantly elevated in cellularity when compared with bim−/− mice (P ≤ 0.015) but were no longer statistically different from age-matched vav-bcl-2 tg mice (Fig. 2). Therefore, the overexpression of Bcl-2 under control of the vav-gene promoter is sufficient to antagonize Bim and Puma function during lymphocyte homeostasis. In line with this hypothesis, we observed that the combined overexpression of Bcl-xL and Bcl-2 did not cause a further increase in the numbers of mature T cells (unpublished data), suggesting functional redundancy between Bcl-2 and Bcl-xL in the control of mature T cell survival.
The increase of CD44highCD62Llow T cells in bim−/−/puma−/− mice strongly suggested a nonredundant role for these BH3-only proteins in the deletion of activated T cells. Consistently, T cell blasts derived from bim−/−/puma−/− animals survived IL-2 deprivation–induced death better than bim−/− T cell blasts and, indeed, almost as strongly as those overexpressing Bcl-2 (Fig. 3 a). Although the loss of Puma on its own provided only transient protection from IL-2 deprivation, our data show that Puma can contribute to the shutdown of an immune response, particularly when Bim function is impaired. Transcriptional activation of puma in IL-2–deprived T cell blasts may be achieved independently of p53 by the forkhead transcription factor family member FOXO3a (30), which is also known to regulate the expression of Bim under these conditions (31).
However, our experiments using the injection of SEB did fail to establish clear synergy between the loss of Bim and the loss of Puma in the survival of activated Vβ8+ T cell clones (Fig. 3, b and c). Unfortunately, we were unable to follow the fate of SEB-activated T cells for >6 wk because of the limited number of animals available and, therefore, can only speculate that Bim/Puma double-deficient T cells may persist longer in vivo. Nevertheless, our findings are in line with a previous study demonstrating that the absence of Bim strongly delays but does not completely prevent the deletion of SEB-activated T cells (10).
The prolonged persistence of activated T cells lacking Bim may contribute to autoimmunity, and, on the mixed genetic background, the loss of Bim causes the production and accumulation of antibodies leading to autoimmune glomerulonephritis (6). Although bim−/− mice on an inbred C57BL/6 background do produce autoantibodies and have increased Ig titers, they no longer succumb to immune complex glomerulonephritis (reference 20; and this study). The additional loss of Puma did not restore autoimmune glomerulonephritis in bim−/−/puma−/− mice. Moreover, the levels of total IgM or IgG and anti-dsDNA autoantibodies detected in bim−/− mice were not further increased in bim−/−/puma−/− mice and were comparable with those observed in vav-bcl-2 tg mice (Fig. 6). Interestingly, however, there was a significant increase in IgA found in puma−/− mice compared with WT mice (P = 0.05) and more IgA in bim−/−/puma−/− mice compared with bim−/− mice. This may indicate that Puma, not only Bim, protects against IgA nephropathy (32).
Cell death analysis performed on isolated lymphocytes confirmed the redundant function of Bim and Puma most prominently in spontaneous but also stress-induced apoptosis. Apoptosis induced by GCs, γ irradiation, or treatment with DNA-damaging drugs, pankinase inhibitors, or ER stressors was more efficiently blocked in Bim/Puma double-deficient cells when compared with cells lacking the individual genes (Figs. 4 and 5). Studies performed on lymphoid cells from bim−/− /blk −/− (27) or bim−/− /bad −/− double-deficient mice (unpublished data) failed to demonstrate redundancy in cell death initiation between Bim and these two BH3-only proteins and, therefore, argues against the possibility that the observed effects are simply caused by increases in the pool of Bcl-2 prosurvival family members that are not bound to BH3-only proteins. Finally, the combined loss of Puma and Bim did not lead to the increased resistance of thymocytes to phorbol ester or ionomycin, indicating that the observed synergy is not a general one but is stimulus dependent (Fig. S4 a). Because the overexpression of Bcl-2 proved to be more potent in protecting lymphocytes from stress-induced cell death than the combined loss of Bim and Puma (Fig. 4), the activation of other BH3-only proteins must be assumed. However, neither Bid, Bad, Bmf, Bax, nor Bak compensatory up-regulation was observed in thymocytes lacking Bim and Puma (unpublished data). Using flow cytometric (Fig. S4, b and c) and Western blotting analysis (not depicted), we also observed that thymocytes lacking Bim and Puma efficiently activated caspase-3 and cleaved poly-ADP ribose polymerase in response to various inducers of cell death (cytokine deprivation, GC, or etoposide treatment), albeit with delayed kinetics, indicating that Bax and/or Bak can be activated effectively in the absence of Bim and Puma.
Previously published data proposed that the loss of Bim is only poorly tumorigenic on its own but does accelerate c-myc oncogene-driven lymphomagenesis in mice (23). Interestingly, the loss of Bim has been found in patients with mantle cell lymphoma and renal cell carcinoma. In contrast, although intensely investigated in carcinomas of the lung, colon, or head and neck in humans, no mutations affecting the puma locus have been detected (33). Puma must have tumor suppressor potential based on the observation that its loss or reduced expression also promotes c-myc–induced lymphomagenesis (22). Consistently, we observed spontaneously occurring malignancies in 20% of bim−/− mice within 12 mo of age. Interestingly, half of the bim−/−/puma +/− mice and ∼60% of the double-deficient animals showed signs of neoplastic growth (Fig. 7 and Figs. S5 and S6). Our observations are in line with data demonstrating that a reduction of Puma expression levels by RNA interference is able to accelerate c-myc–induced lymphomagenesis (22), as does the haploinsufficiency of Bim (23). Whether the additional loss of one allele of puma significantly increases the observed tumor incidence further will require the analysis of lager cohorts of animals. Our data indicate that the loss of Bim is more important for the development of lymphoid malignancy than the loss of Puma because bim +/−/puma−/− animals did not develop any tumors over the same observation period. Further genetic studies will be required to assess the full tumor spectrum suppressed by Bim and Puma. Disease models derived from these studies will be useful tools to assess the therapeutic potential of small molecule drugs such as BH3 mimetics that hold much promise for the treatment of cancer in the near future.
MATERIALS AND METHODS
Mice.
All animal experiments were performed in accordance with the Austrian Tierversuchsgesetz (BGBl. Nr. 501/1988 i.d.g.F) and have been granted by the Bundesministerium für Bildung, Wissenschaft, und Kultur. The generation and genotyping of the puma−/−, bim−/−, lck-bcl-xL, and vav-bcl-2 tg mice have been described previously (6, 14, 16). C57BL/6-Ly5.1 mice were purchased from Charles River Laboratories.
Cell culture and reagents.
Primary hemopoietic cells were cultured in RPMI 1640 medium (PAA Laboratories) supplemented with 13 μM folic acid, 250 μM l-glutamine (Invitrogen), 50 μM 2-mercaptoethanol, nonessential amino acids (Invitrogen), penicillin/streptomycin (Sigma-Aldrich), and 10% FCS (Invitrogen). For the induction of cell death, FLAG epitope-tagged FasL (Qbiogene) was used at 100 ng/ml together with cross-linking M2 anti-FLAG antibody (Sigma-Aldrich) at 1 μg/ml. Ionomycin (Sigma-Aldrich) was used at 1 μg/ml, PMA was used at 10 ng/ml (Sigma-Aldrich), etoposide (Sigma-Aldrich) was used at 1 or 10 μg/ml, the ER stressor tunicamycin (Sigma-Aldrich) was used at 10 μg/ml, and the GC dexamethasone was used at 10−6 or 10−7 M. Injections of saline, dexamethasone (Dexabene; Merckle Recordati), or SEB (Sigma-Aldrich) were performed i.v. in a final volume of 200 μl.
Immunofluorescence staining, flow cytometric analysis, and cell sorting.
Single-cell suspensions from the bone marrow, lymph nodes, spleen, and thymus were surface stained with monoclonal antibodies conjugated with FITC, R-PE, allophycocyanin, or biotin (Invitrogen). The monoclonal antibodies used and their specificities are as follows: RA3-6B2, anti-B220; GK1.5, anti-CD4; H129.19.6.8, anti-CD4; 53.6.72, anti-CD8; YTS 169, anti-CD8; RB6-8C5, anti–Gr-1; S7, anti-CD43; 5.1, anti-IgM; 11/26C, anti-IgD; MI/70, anti–Mac-1; Ter119, antierythroid cell surface marker; T24.31.2, anti–Thy-1; IM7, anti-CD44; PC61, anti-CD25; MEL-14, anti-CD62L; H57-59, anti–TCR-β; F23.1, anti-Vβ8; RR4-7, anti-Vβ6; A20, anti-Ly5.1; 1D4, anti-Ly5.2; and anti-CD3, 2C11. Flow cytometric analysis was performed using a FACSCalibur cell analyzer (Beckton Dickinson). Isolation of cells was performed using a FACSVantage cell sorter (Beckton Dickinson).
Immunohistochemistry.
Formalin-fixed paraffin-embedded sections of lymphomas were stained for the presence of the B cell marker B220 (RA3-6B2) or the proliferation marker PCNA (PC10) according to protocols accessible online under www.ihcworld.com.
Cell death assays.
The percentages of viable cells in culture were determined by staining cell suspensions with 2 μg/ml PI plus FITC-coupled annexin V followed by analysis in a FACScan (Becton Dickinson). Alternatively, cells were stained with trypan blue (0.1% in PBS) and analyzed in a hemocytometer (Neubauer chamber; Sigma-Aldrich). For the detection of active caspase-3, cells were fixed in 4% paraformaldehyde for 10 min, washed in PBS, and permeabilized in methanol for 1 h at −20°C. AlexaFluor488-conjugated antiactive caspase-3 antibody (Cell Signaling) was used in at a 1:100 dilution for intracellular staining in PBS/BSA.
Generation of T cell blasts.
Spleens were pushed through a steel sieve, and single-cell suspensions were subjected to RBC lysis. Cells were spun over a cushion of FCS and washed twice in medium. 107 spleen cells were incubated for 20 min on ice with a cocktail of antibodies recognizing the B cell marker B220 (RA3-6B2), the myeloid cell marker Mac-1 (MI/70), the granulocyte marker Gr-1 (S7), and the erythroid cell marker Ter119. After incubation, cells were washed in 10 ml PBS and spun over a cushion of FCS. The pellet was resuspended in 500 μl PBS/FCS solution, and cells were incubated on ice with anti–rat IgG–specific Dynabeads for 20–30 min under gentle agitation (Dynal). Negatively sorted T cells (purity of >90%) were stimulated with 2 μg/ml ConA (Sigma-Aldrich) and 100 U/ml IL-2 (PeproTech) for 3 d to generate activated T cell blasts. These cells were washed twice in PBS and subjected to cytokine withdrawal. Viability was assessed by PI exclusion and flow cytometric analysis.
Quantification of Ig titers and autoantibody production.
Ig titers in the serum from 8–12-wk-old mice were quantified using an Ig clonotyping system (Southern Biotechnology Associates, Inc.) according to the manufacturer's instructions. Serum samples were used in a dilution range from 1:1,000 to 1:160,000 depending on the isotype to secure absorbance readings in a linear range. Calf thymus DNA (Sigma-Aldrich) in 12.5 μg/ml of distilled water was allowed to bind overnight to 96-well ELISA plates (Costar) precoated for 1 h at 37°C with poly-l-lysine (0.1% in distilled water). For anti-dsDNA autoantibody detection, a horseradish peroxidase–labeled goat anti–mouse Ig(H+L)–specific antibody and ABTS as a substrate were used (Southern Biotechnology Associates, Inc.). Mouse sera were diluted 1:100 in 1% PBS/BSA. Serum derived from autoimmune-prone New Zealand white/New Zealand black F1 mice was used as a positive control.
ANA stainings were performed on 5-μm cryosections from rat liver. Serum samples were diluted 1:30 in PBS. Cryosections were fixed in acetone for 10 min and subsequently incubated with the diluted serum sample for 30 min. After washing in PBS for 30 min, sections were incubated with AlexaFluor488-conjugated goat anti–mouse IgG (Invitrogen) for an additional 30 min (1:600 in PBS). Finally, slides were washed twice in PBS for 45 min, and the sections were covered with MOWIOL mounting media and a glass coverslip. Images were acquired using a laser-scanning microscope (Axiovert; Carl Zeiss MicroImaging, Inc.).
Preparation of histological sections.
Organs were fixed in 4% paraformaldehyde in PBS, processed according to standard procedures, and stained in hematoxylin and eosin.
Statistical analysis.
Statistical analysis was performed using the unpaired Student's t test or analysis of variance where indicated and applying the StatView 4.1 software program (Abacus Concepts). P-values of <0.05 were considered to be statistically significant.
Online supplemental material.
Fig. S1 shows normal organ development in live-born Bim and Bim/Puma double knockout mice. Fig. S2 shows that bim−/−/puma−/− mice develop multiple defects postnatally. Fig. S3 shows that lymphadenopathy in bim−/−/puma−/− mice involves effects originating outside the bone marrow. Fig. S4 shows that the synergy between Bim and Puma in cell death initiation is stimulus dependent. Fig. S5 shows spontaneous tumorigenesis in mice lacking Bim and Puma, and Fig. S6 shows PCNA expression analysis in tumor samples. Fig. S7 shows the histopathological assessment of transplanted tumor material. Table S1 provides data about abnormal bone marrow composition in Bim/Puma double knockout mice. Table S2 shows that bim−/−/puma−/− mice display enlarged lymph nodes, and Table S3 provides an analysis of leukocyte composition in the peripheral blood. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20061552/DC1.
Supplemental Material
Acknowledgments
We thank J. Adams, S. Cory, P. Bouillet, S. Korsmeyer, D. Green, and M. Hahne for providing mice or reagents, P. Lukas for enabling irradiation experiments, C. Mayerl for help with immunohistochemistry, and R. Kofler and S. Geley for discussion.
This work was supported by grants and fellowships from the National Health and Medical Research Council (NHMRC; Canberra grant 257502), the National Cancer Institute (National Institutes of Health grants CA43540 and CA 80188), the Leukemia and Lymphoma Society of America (Specialized Centers of Research grant 7015), the Juvenile Diabetes Research Foundation/NHMRC (grant to A. Strasser), and the Austrian Science Fund (START and SFB021 grants to A. Villunger). V. Labi is a DOC-FFORTE Fellow of the Austrian Academy of Science.
The authors have no conflicting financial interests.
Abbreviations used: ANA, antinuclear antibody; Bad, Bcl-2 antagonist of cell death; Bak, Bcl-2 antagonist/killer; Bax, Bcl-2–associated protein X; Bcl-2, B cell lymphoma 2; BH, Bcl-2 homology; Bid, Bcl-2–interacting domain death agonist; Bim, Bcl-2–interacting mediator of cell death; Blk, Bik-like killer; Bmf, Bcl-2 modifying factor; DN, double negative; DP, double positive; dsDNA, double-stranded DNA; GC, glucocorticoid; PCNA, proliferating cell nuclear antigen; PI, propidium iodide; Puma, p53–up-regulated modulator of apoptosis; SEB, Staphylococcus enterotoxin B; tg, transgenic.
M. Erlacher's present address is Department of Pediatrics and Adolescent Medicine, Division of Pediatric Hematology and Oncology, University Hospital of Freiburg, 79104 Freiburg, Germany.
References
- 1.Strasser, A. 2005. The role of BH3-only proteins in the immune system. Nat. Rev. Immunol. 5:189–200. [DOI] [PubMed] [Google Scholar]
- 2.Zong, W.X., T. Lindsten, A.J. Ross, G.R. MacGregor, and C.B. Thompson. 2001. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 15:1481–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuwana, T., L. Bouchier-Hayes, J.E. Chipuk, C. Bonzon, B.A. Sullivan, D.R. Green, and D.D. Newmeyer. 2005. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell. 17:525–535. [DOI] [PubMed] [Google Scholar]
- 4.Certo, M., G. Moore Vdel, M. Nishino, G. Wei, S. Korsmeyer, S.A. Armstrong, and A. Letai. 2006. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 9:351–365. [DOI] [PubMed] [Google Scholar]
- 5.Willis, S.N., and J.M. Adams. 2005. Life in the balance: how BH3-only proteins induce apoptosis. Curr. Opin. Cell Biol. 17:617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bouillet, P., D. Metcalf, D.C.S. Huang, D.M. Tarlinton, T.W.H. Kay, F. Köntgen, J.M. Adams, and A. Strasser. 1999. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 286:1735–1738. [DOI] [PubMed] [Google Scholar]
- 7.Bouillet, P., J.F. Purton, D.I. Godfrey, L.-C. Zhang, L. Coultas, H. Puthalakath, M. Pellegrini, S. Cory, J.M. Adams, and A. Strasser. 2002. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature. 415:922–926. [DOI] [PubMed] [Google Scholar]
- 8.Villunger, A., V.S. Marsden, Y. Zhan, M. Erlacher, A.M. Lew, P. Bouillet, S. Berzins, D.I. Godfrey, W.R. Heath, and A. Strasser. 2004. Negative selection of semimature CD4(+)8(-)HSA+ thymocytes requires the BH3-only protein Bim but is independent of death receptor signaling. Proc. Natl. Acad. Sci. USA. 101:7052–7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Enders, A., P. Bouillet, H. Puthalakath, Y. Xu, D.M. Tarlinton, and A. Strasser. 2003. Loss of the pro-apoptotic BH3-only Bcl-2 family member Bim inhibits BCR stimulation-induced apoptosis and deletion of autoreactive B cells. J. Exp. Med. 198:1119–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hildeman, D.A., Y. Zhu, T.C. Mitchell, P. Bouillet, A. Strasser, J. Kappler, and P. Marrack. 2002. Activated T cell death in vivo mediated by pro-apoptotic Bcl-2 family member, Bim. Immunity. 16:759–767. [DOI] [PubMed] [Google Scholar]
- 11.Pellegrini, M., G. Belz, P. Bouillet, and A. Strasser. 2003. Shutdown of an acute T cell immune response to viral infection is mediated by the proapoptotic Bcl-2 homology 3-only protein Bim. Proc. Natl. Acad. Sci. USA. 100:14175–14180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shibue, T., K. Takeda, E. Oda, H. Tanaka, H. Murasawa, A. Takaoka, Y. Morishita, S. Akira, T. Taniguchi, and N. Tanaka. 2003. Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 17:2233–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeffers, J.R., E. Parganas, Y. Lee, C. Yang, J. Wang, J. Brennan, K.H. MacLean, J. Han, T. Chittenden, J.N. Ihle, et al. 2003. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 4:321–328. [DOI] [PubMed] [Google Scholar]
- 14.Villunger, A., E.M. Michalak, L. Coultas, F. Mullauer, G. Bock, M.J. Ausserlechner, J.M. Adams, and A. Strasser. 2003. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 302:1036–1038. [DOI] [PubMed] [Google Scholar]
- 15.Erlacher, M., E.M. Michalak, P.N. Kelly, V. Labi, H. Niederegger, L. Coultas, J.M. Adams, A. Strasser, and A. Villunger. 2005. BH3-only proteins Puma and Bim are rate-limiting for gamma-radiation and glucocorticoid-induced apoptosis of lymphoid cells in vivo. Blood. 106:4131–4138 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ogilvy, S., D. Metcalf, C.G. Print, M.L. Bath, A.W. Harris, and J.M. Adams. 1999. Constitutive bcl-2 expression throughout the hematopoietic compartment affects multiple lineages and enhances progenitor cell survival. Proc. Natl. Acad. Sci. USA. 96:14943–14948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Godfrey, D.I., and A. Zlotnik. 1993. Control points in early T-cell development. Immunol. Today. 14:547–553. [DOI] [PubMed] [Google Scholar]
- 18.Chen, L., S.N. Willis, A. Wei, B.J. Smith, J.I. Fletcher, M.G. Hinds, P.M. Colman, C.L. Day, J.M. Adams, and D.C. Huang. 2005. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell. 17:393–403. [DOI] [PubMed] [Google Scholar]
- 19.Grillot, D.A.M., R. Merino, and G. Nuñez. 1995. Bcl-xL displays restricted distribution during T cell development and inhibits multiple forms of apoptosis but not clonal deletion in transgenic mice. J. Exp. Med. 182:1973–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bouillet, P., S. Cory, L.-C. Zhang, A. Strasser, and J.M. Adams. 2001. Degenerative disorders caused by Bcl-2 deficiency are prevented by loss of its BH3-only antagonist Bim. Dev. Cell. 1:645–653. [DOI] [PubMed] [Google Scholar]
- 21.O'Reilly, L.A., D.C.S. Huang, and A. Strasser. 1996. The cell death inhibitor Bcl-2 and its homologues influence control of cell cycle entry. EMBO J. 15:6979–6990. [PMC free article] [PubMed] [Google Scholar]
- 22.Hemann, M.T., J.T. Zilfou, Z. Zhao, D.J. Burgess, G.J. Hannon, and S.W. Lowe. 2004. Suppression of tumorigenesis by the p53 target PUMA. Proc. Natl. Acad. Sci. USA. 101:9333–9338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Egle, A., A.W. Harris, P. Bouillet, and S. Cory. 2004. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc. Natl. Acad. Sci. USA. 101:6164–6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Egle, A., A.W. Harris, M.L. Bath, L. O'Reilly, and S. Cory. 2004. VavP-Bcl2 transgenic mice develop follicular lymphoma preceded by germinal center hyperplasia. Blood. 103:2276–2283. [DOI] [PubMed] [Google Scholar]
- 25.Wong, H.K., M. Fricker, A. Wyttenbach, A. Villunger, E.M. Michalak, A. Strasser, and A.M. Tolkovsky. 2005. Mutually exclusive subsets of BH3-only proteins are activated by the p53 and c-Jun N-terminal kinase/c-Jun signaling pathways during cortical neuron apoptosis induced by arsenite. Mol. Cell. Biol. 25:8732–8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rathmell, J.C., T. Lindsten, W.X. Zong, R.M. Cinalli, and C.B. Thompson. 2002. Deficiency in Bak and Bax perturbs thymic selection and lymphoid homeostasis. Nat. Immunol. 3:932–939. [DOI] [PubMed] [Google Scholar]
- 27.Coultas, L., P. Bouillet, K.L. Loveland, S. Meachem, H. Perlman, J.M. Adams, and A. Strasser. 2005. Concomitant loss of proapoptotic BH3-only Bcl-2 antagonists Bik and Bim arrests spermatogenesis. EMBO J. 24:3963–3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haks, M.C., P. Krimpenfort, J.H.N. van den Brakel, and A.M. Kruisbeek. 1999. Pre-TCR signaling and inactivation of p53 induces crucial cell survival pathways in pre-T cells. Immunity. 11:91–101. [DOI] [PubMed] [Google Scholar]
- 29.Oliver, P.M., M. Wang, Y. Zhu, J. White, J. Kappler, and P. Marrack. 2004. Loss of Bim allows precursor B cell survival but not precursor B cell differentiation in the absence of interleukin 7. J. Exp. Med. 200:1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.You, H., M. Pellegrini, K. Tsuchihara, K. Yamamoto, G. Hacker, M. Erlacher, A. Villunger, and T.W. Mak. 2006. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J. Exp. Med. 203:1657–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stahl, M., P.F. Dijkers, G.J. Kops, S.M. Lens, P.J. Coffer, B.M. Burgering, and R.H. Medema. 2002. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J. Immunol. 168:5024–5031. [DOI] [PubMed] [Google Scholar]
- 32.Marquina, R., M.A. Diez, M. Lopez-Hoyos, L. Buelta, A. Kuroki, S. Kikuchi, J. Villegas, M. Pihlgren, C.A. Siegrist, M. Arias, et al. 2004. Inhibition of B cell death causes the development of an IgA nephropathy in (New Zealand white x C57BL/6)F(1)-bcl-2 transgenic mice. J. Immunol. 172:7177–7185. [DOI] [PubMed] [Google Scholar]
- 33.Labi, V., M. Erlacher, S. Kiessling, and A. Villunger. 2006. BH3-only proteins in cell death initiation, malignant disease and anticancer therapy. Cell Death Differ. 13:1325–1338. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}