

Abstract
Experimental autoimmune myocarditis (EAM) appears after infectious heart disease, the most common cause of dilated cardiomyopathy in humans. Here we report that mice lacking T-bet, a T-box transcription factor required for T helper (Th)1 cell differentiation and interferon (IFN)-γ production, develop severe autoimmune heart disease compared to T-bet −/− control mice. Experiments in T-bet −/− IL-4−/− and T-bet −/− IL-4Rα−/− mice, as well as transfer of heart-specific Th1 and Th2 cell lines, showed that autoimmune heart disease develops independently of Th1 or Th2 polarization. Analysis of T-bet −/− IL-12Rβ1−/− and T-bet −/− IL-12p35−/− mice then identified interleukin (IL)-23 as critical for EAM pathogenesis. In addition, T-bet −/− mice showed a marked increase in production of the IL-23–dependent cytokine IL-17 by heart-infiltrating lymphocytes, and in vivo IL-17 depletion markedly reduced EAM severity in T-bet −/− mice. Heart-infiltrating T-bet −/− CD8+ but not CD8− T cells secrete IFN-γ, which inhibits IL-17 production and protects against severe EAM. In contrast, T-bet −/− CD8+ lymphocytes completely lost their capacity to release IFN-γ within the heart. Collectively, these data show that severe IL-17–mediated EAM can develop in the absence of T-bet, and that T-bet can regulate autoimmunity via the control of nonspecific CD8+ T cell bystander functions in the inflamed target organ.
Cardiovascular disease is a leading cause of morbidity and mortality in the Western world. Dilated cardiomyopathy (DCM), often resulting from Coxsackievirus B3 and Chlamydia-triggered myocarditis, may be present in up to one quarter of all cases of heart failure (1, 2). Notably, many affected individuals develop heart antigen–specific autoantibody responses (3), and immunosuppressive therapy can improve heart function in DCM patients who display no evidence of viral or bacterial genomes in heart biopsies (4). Interestingly, peripheral blood lymphocytes from patients with DCM could adoptively transfer disease to SCID mice lacking T and B cells (5). These observations suggest that postinfectious autoimmune mechanisms promote disease development.
Experimental autoimmune myocarditis (EAM) is a mouse model of postinfectious myocarditis and cardiomyopathy (6). EAM can be induced in susceptible mouse strains by immunization with self-peptides derived from the myosin α heavy chain (MyHC-α). EAM is a CD4+ T cell–mediated disease (6–8), but the relative contributions of the CD4+ Th1 and Th2 subsets are unclear (9–13). The majority of infiltrating cells in EAM are macrophages, suggesting that Th1 signals predominate. Signaling through the key Th2 cytokine IL-4 is dispensable for the development of EAM (10). Furthermore, the p40 subunit of the Th1-driving cytokine IL-12 and the β1 receptor subunit of the IL-12R are essential for disease progression (10, 11). However, in vivo IL-4 blockade has been shown to reduce disease severity (12). At the same time, loss of either IFN-γ or its receptor results in increased disease severity, implying that this key Th1 cytokine is a negative regulator of EAM (10, 11, 13).
As IFN-γ signaling may not be absolutely required for the development of Th1 responses (14), we tested the disease susceptibility of mice lacking T-bet, a T-box transcription factor essential for Th1 lineage differentiation (15, 16). T-bet −/− CD4+ T cells display a profound defect in IFN-γ production in vitro, and T-bet expression is critical for the development of inflammatory autoimmune diseases, such as experimental autoimmune encephalomyelitis, Crohn's disease, type 1 diabetes, and atherosclerosis (17–21).
Here we show that in marked contrast to other autoimmune models, mice lacking T-bet develop severe EAM. MyHC-α–specific CD4+ T cells trigger autoimmune myocarditis regardless of Th1 or Th2 commitment. Analysis of T-bet −/− IL-12Rβ1 −/− and T-bet −/− IL-12p35 −/− mice and antibody depletion experiments showed that IL-23 and IL-17 are critical for EAM pathogenesis. Loss of T-bet results in increased IL-17 production in the inflamed heart, and this tissue-specific repression depends on CD8+ T cell–mediated bystander suppression. Our data indicate that T-bet expressed in the CD8+ T cell compartment is a negative regulator of autoimmune heart disease.
RESULTS
T-bet −/− mice develop severe myocarditis
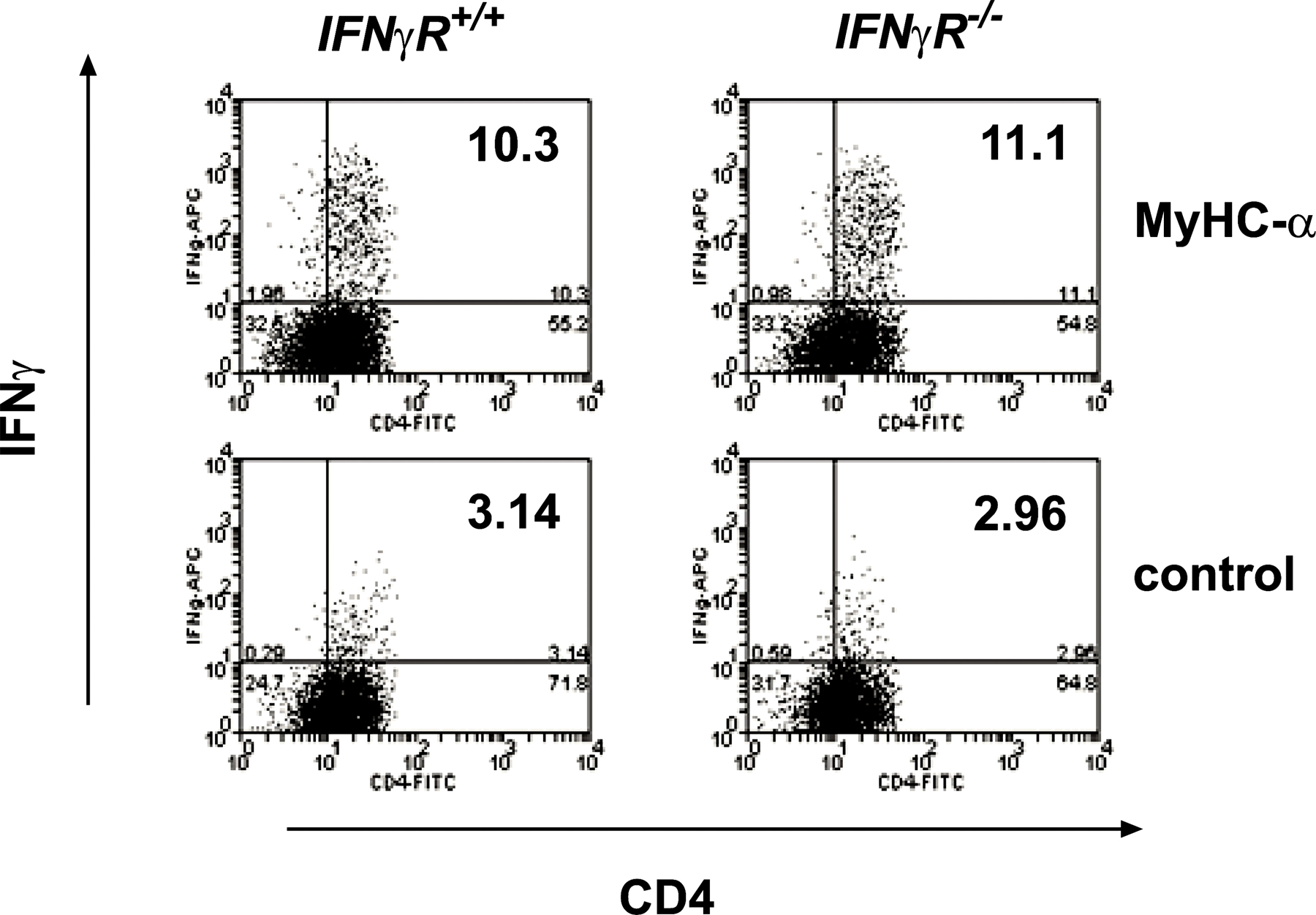
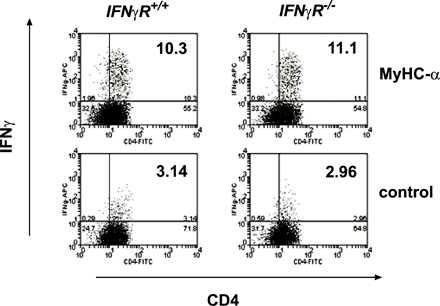
IFN-γ production characterizes Th1 cells, and IFN-γ secreted from Th1 cells strongly activates macrophages and actively represses transcription of IL-4 (22). Absence of IFN-γ receptor signaling, however, does not exclude Th1 commitment in EAM because CD4+ T cells from MyHC-α–immunized IFN-γR −/− mice were highly competent to produce IFN-γ upon in vitro restimulation (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20052222/DC1). We therefore clarified the role of Th1 responses in EAM using mice genetically deficient for T-bet, a T-box transcription factor essential for Th1 cell lineage commitment (15). Surprisingly, T-bet −/− mice developed severe EAM after immunization with MyHC-α emulsified in CFA (Fig. 1, A and B, and Table I) as well as upon injection of activated MyHC-α–loaded, T-bet +/+ bone marrow–derived DCs (BMDCs) (Table I).
Figure 1.

T-bet−/− mice develop EAM of increased severity. T-bet +/+ and T-bet −/− mice were immunized with MyHC-α peptide in CFA and killed 21 d later. (A) Severity scores of individual diseased T-bet +/+ versus T-bet −/− mice: ***, P < 0.000001. (B) Hematoxylin and eosin–stained sections from hearts of immunized mice. MyHC-α–immunized T-bet +/+ (top left), MyHC-α–immunized T-bet −/− (top right), T-bet −/− immunized with CFA alone (bottom left), and MyHC-α–immunized T-bet −/− IL-4 −/−.
Table I.
Myocarditis prevalence and severity in MyHC-α–immunized mice
| Mice (genotype) |
Immunization | Disease prevalence (no. diseased/no. treated) |
Mean score of diseased mice |
|---|---|---|---|
| T-bet −/− | MyHC-α/CFA | 18/18 | 3.56a e h l r |
| T-bet +/+ | MyHC-α/CFA | 16/19j m | 2a b c d g k p |
| IL-4 −/− | MyHC-α/CFA | 3/4 | 1.67b f |
| IL-4Rα −/− | MyHC-α/CFA | 3/4 | 2c i |
| T-bet −/− IL-4 −/− | MyHC-α/CFA | 4/4 | 3.75d e f |
| T-bet −/− IL-4Rα −/− | MyHC-α/CFA | 5/5 | 3.8g h i |
| IL-12Rβ1 −/− | MyHC-α/CFA | 0/4j n | n.a. |
| IL-12p35 −/− | MyHC-α/CFA | 5/5 | 1.6k l q |
| T-bet −/− IL-12Rβ1 −/− | MyHC-α/CFA | 0/6m n o | n.a. |
| T-bet −/− IL-12p35 −/− | MyHC-α/CFA | 6/6 | 3.67p q r |
| T-bet +/+ | MyHC-α–pulsed T-bet +/+ DCs | 4/5 | 1.5s t |
| T-bet −/− | MyHC-α–pulsed T-bet +/+ DCs | 5/5 | 2.6s |
| T-bet +/+ | MyHC-α–pulsed T-bet −/− DCs | 3/3 | 1.67t |
| IL-12p35 +/+ | MyHC-α–pulsed IL-12p35 +/+ DCs | 4/4 | 1.87u |
| IL-12p35 +/+ | MyHC-α–pulsed IL-12p35 −/− DCs | 4/4 | 2.25u |
Hematoxylin and eosin sections were scored as described in Materials and methods. CFA/MyHC-α–immunized mice were killed on day 21, and DC-immunized mice were killed on day 10. Statistical significance was assessed using the Mann-Whitney U test unless otherwise indicated. n.a., not applicable.
P < 0.000001, T-bet −/− versus T-bet +/+.
P < n.s, T-bet +/+ versus IL-4 −/−.
P < n.s, T-bet +/+ versus IL-4Rα −/−.
P < 0.0008, T-bet +/+ versus T-bet −/− IL-4 −/−.
P < n.s., T-bet −/− versus T-bet −/− IL-4 −/−.
P < 0.029, IL-4 −/− versus T-bet −/− IL-4 −/−.
P < 0.0002, T-bet +/+ versus T-bet −/− IL-4Rα −/−.
P < n.s., T-bet −/− versus T-bet −/− IL-4Rα −/−.
P < 0.036, IL-4Ra −/− versus T-bet −/− IL-4Ra −/−.
P < 0.004, T-bet +/+ versus IL-12Rβ1 −/− (Fisher's exact test).
P < n.s, T-bet +/+ versus IL-12p35 −/−.
P < 0.0001, T-bet −/− versus IL-12p35 −/−.
P < 0.0005, T-bet +/+ versus T-bet −/− IL-12Rβ1 −/− (Fisher's exact test).
P < n.s, IL-12Rβ1 −/− versus T-bet −/− IL-12Rβ1 −/− (Fisher's exact test).
P < 0.000007, T-bet −/− versus T-bet −/− IL-12Rβ1 −/− (Fisher's exact test).
P < 0.002, T-bet +/+ versus T-bet −/− IL-12p35 −/−.
P < 0.009, IL-12p35 −/− versus T-bet −/− IL-12p35 −/−.
P < n.s, T-bet −/− versus T-bet −/− IL-12p35 −/−.
P < 0.0317, T-bet +/+ (T-bet +/+ DC-immunized) versus T-bet −/− (T-bet +/+ DC-immunized).
P < n.s, T-bet +/+ (T-bet +/+ DC-immunized) versus T-bet +/+ (T-bet −/− DC-immunized).
P < n.s., IL-12p35+/+ (IL-12p35 +/+ DC-immunized) versus IL-12p35+/+ (IL-12p35 −/− DC-immunized).
Inflammatory infiltrates in the hearts of immunized T-bet +/+ and T-bet −/− mice consisted of granulocytes and mononuclear cells, including macrophages and lymphocytes. T-bet −/− hearts showed slightly greater eosinophilia and less fibrosis as compared with T-bet +/+ hearts, suggesting a Th2-biased response (not depicted). Immunohistochemical analysis, however, showed no apparent differences in relative numbers of CD68+ macrophages, CD4+ T cells, and CD8+ T cells between diseased T-bet −/− and T-bet +/+ hearts (Fig. 2). FACS analysis indicated that proportions of CD4+ versus CD8+ heart-infiltrating cells were also comparable in T-bet +/+ and T-bet −/− mice (not depicted).
Figure 2.

Characterization of heart-infiltrating cells in T-bet−/− mice. Frozen heart sections from MyHC-α–immunized T-bet +/+ and T-bet −/− mice were stained to determine proportions of infiltrating macrophages (CD68, right) and lymphocytes (CD4, second from left; CD8, second from right).
The increase in EAM severity observed in IFN-γ −/− mice has been explained by impaired apoptosis of activated CD4+ T cells as well as by unrestricted expansion of CD4+CD44high T cells (23). However, we saw no evidence of impaired cell death in CD4+ T cells isolated from MyHC-α–immunized T-bet −/− mice, nor did we observe any increase in the numbers of peripheral CD4+CD44high cells in these mice (not depicted). Furthermore, most T-bet −/− mice, but none of the IFN-γR −/− animals, slowly recovered from disease within 2 mo after immunization (not depicted), suggesting that loss of T-bet does not mimic IFN-γ deficiency in EAM.
T-bet −/− mice develop an autoreactive Th2 response
Consistent with the idea of CD4+ T cell–mediated disease and enhanced disease severity in T-bet −/− mice, in vitro recall responses of T-bet −/− CD4+ T cells to MyHC-α were increased in comparison with T-bet +/+ controls (Fig. 3 A). To exclude the possibility that T-bet does not act as a Th1 transcription factor in BALB/c mice, we measured IFN-γ and IL-4 production in T-bet −/− and T-bet +/+ CD4+ T cells. In fact, CD4+ T cells from immunized T-bet −/− mice produced no IFN-γ but significant amounts of IL-4 relative to T-bet +/+ CD4+ T cells upon in vitro restimulation (Fig. 3 B). Furthermore, diseased T-bet −/− mice displayed a complete lack of myosin-specific IgG2a, a Th1-dependent antibody isotype (Fig. 3 C). Collectively, these data indicate that Th1 differentiation is not required for the development of autoimmune myocarditis and confirm a Th2 phenotype of MyHC-α–specific T-bet −/− CD4+ T cells.
Figure 3.

CD4− T cell responses and cytokine production patterns of immunized T-bet−/− versus T-bet+/+ mice. CD4+ T cells from T-bet +/+ (blue bars) and T-bet −/− mice (red bars) were restimulated with either MyHC-α or control OVA peptide. (A) Proliferative responses after 48 h, and (B) IFN-γ and IL-4 production in supernatants after 40 h of restimulation, representative of three independent experiments. (C) Serum MyHC-α autoantibody responses from immunized T-bet +/+ (blue) and T-bet −/− (red) mice at day 21. *, P < 0.05; **, P < 0.01.
EAM develops regardless of Th1 or Th2 differentiation
IL-4 is a key determinant of Th2 differentiation (24). To test the possibility that the exaggerated production of IL-4 seen from T-bet −/− CD4+ T cells could increase disease severity, we crossed T-bet mutant mice onto IL-4– and IL-4Rα–deficient strains. Intriguingly, both T-bet −/− IL-4 −/− and T-bet −/− IL4-Rα −/− mice developed severe EAM relative to T-bet +/+ and IL-4 −/− or IL4-Rα −/− controls. However, disease in T-bet −/− IL-4 −/− and T-bet −/− IL-4Rα −/− mice was comparable in severity to that seen in T-bet −/− mice (Fig. 1 B and Table I). This demonstrates that the exacerbated disease seen in T-bet −/− mice is not due to increased IL-4 signaling.
To directly examine the pathogenic role of MyHC-α–specific CD4+ T cells, we derived MyHC-α–specific CD4+ lines from immunized T-bet −/− and T-bet +/+ mice. T-bet +/+ CD4+ T cell lines were skewed toward a Th1 phenotype, as they produced large amounts of IFN-γ, but no IL-4, upon restimulation. In contrast, T-bet −/− lines were Th2 skewed, as they produced no IFN-γ yet generated high levels of IL-4 (Fig. 4 A). Adoptive transfer of both T-bet +/+ and T-bet −/− lines induced myocarditis of comparable disease severity in T-bet +/+ recipient mice (Fig. 4 B). These data suggest that EAM can develop regardless of whether Th1 or Th2 signals predominate in MyHC-α–specific CD4+ T cells.
Figure 4.

Th1 as well as Th2 CD4+ T cell lines trigger autoimmune myocarditis. (A) MyHC-α–specific lines were generated by subjecting CD4+ splenocytes from immunized T-bet +/+ (blue) or T-bet −/− (red) mice to multiple rounds of antigen restimulation followed by rest in minimal IL-2. Supernatant was collected for ELISA 48 h after the third antigen restimulation. ***, P < 0.0001. One representative experiment is shown. (B) Both MyHC-α–specific Th1 cell lines (left) and Th2 cell lines (right) are pathogenic in wild-type recipient mice. Hematoxylin and eosin–stained sections: bar: 50 μm.
Myocarditis induction in T-bet −/− mice is IL-23 dependent
IL-12 is produced in large amounts by activated DCs and macrophages, and it strongly induces Th1 differentiation (25). By inducing the up-regulation of IL-12Rβ2, T-bet can increase Th1 cell sensitivity to IL-12 and thereby stabilize Th1 lineage commitment (26, 27). In addition, IL-12p40 −/− and IL-12Rβ1 −/− mice are resistant to EAM (10, 11). However, IL-12 shares the p40 subunit with the related cytokine IL-23, and the IL-23 receptor heterodimer contains IL-12Rβ1. Indeed, many of the immunological and pathogenic functions initially attributed to IL-12 have now been described as IL-23 dependent (28). We therefore sought to examine whether the severe EAM that develops in the absence of T-bet could be regulated by either IL-12 or IL-23. We crossed T-bet −/− mice onto an IL-12p35–deficient strain, which specifically lacks IL-12. IL-12p35 −/− mice developed disease of a severity comparable to wild-type mice, whereas T-bet −/− IL-12p35 −/− mice developed severe EAM at a level similar to T-bet −/− mice (Table I). DCs are a major source of IL-12 production (28); however, activated and MyHC-α–pulsed IL-12p35 −/− BMDCs were as capable as IL-12p35 +/+ counterparts of inducing myocarditis in wild-type recipients (Table I). We then examined the effect of loss of T-bet on an IL-12Rβ1 −/− strain, which lacks both the IL-12 and the IL-23 receptors. Strikingly, IL-12Rβ1 −/− and T-bet −/− IL-12Rβ1 −/− mice were resistant to EAM (Table I). These data show that IL-23, rather than IL-12, is a key pathogenic cytokine in EAM. Moreover, they indicate that loss of IL-23 signaling can abrogate the development of exacerbated EAM seen in T-bet −/− mice.
Increased IL-17 in the heart, but not peripheral lymphoid tissue, of immunized T-bet −/− mice
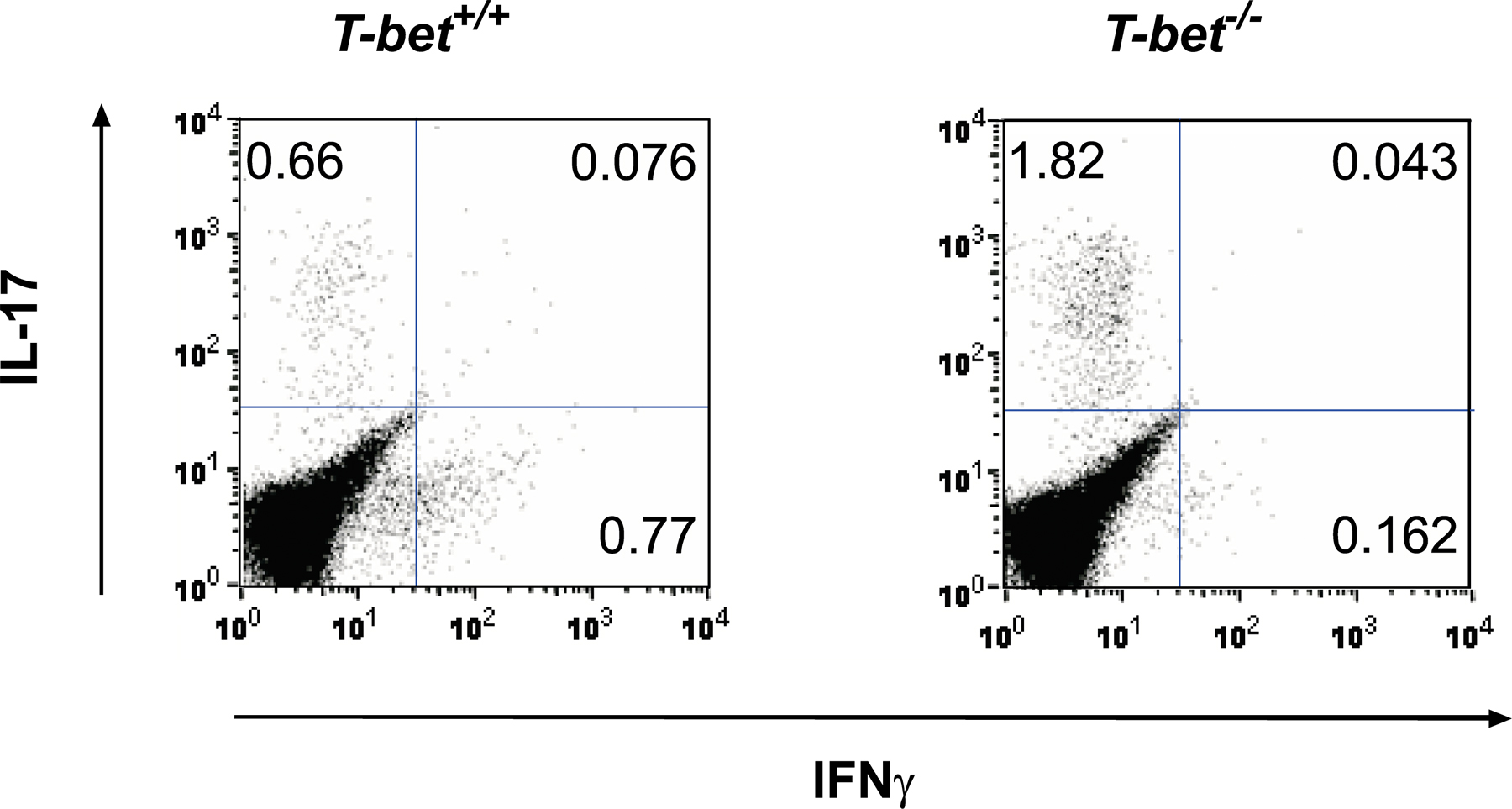
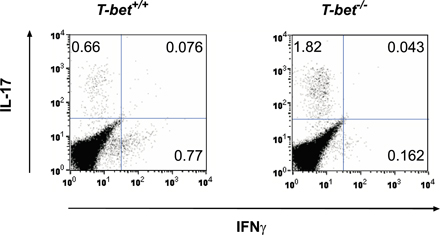
Recent findings suggest that IL-23 plays an important role in the expansion of an IL-17–producing subset of CD4+ T cells (ThIL-17) in vitro (29, 30). These cells play an essential role in the infectious response (31, 32) as well as in autoimmunity (33–35). Further data have shown that the ThIL-17 lineage is distinct from Th1, and that IFN-γ and T-bet can repress CD4+ T cell production of IL-17 (30, 35). In light of our finding that deficiency in IL-23 signaling could rescue T-bet −/− mice from severe EAM, we examined whether increased generation of ThIL-17 cells could explain the exacerbated disease seen in T-bet −/− mice. Restimulation of CD4+ T cells from both T-bet +/+ and T-bet −/− mice resulted in small numbers of IL-17+CD4+ T cells that were IFN-γ− (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20052222/DC1). Despite the identification of ThIL-17 as a distinct CD4+ effector T cell lineage, a proportion of IL-17+ T cells can still produce IFN-γ (30). Indeed, our MyHC-α–specific, pathogenic T-bet +/+ and T-bet −/− CD4+ T cell lines, which produce elevated levels of IFN-γ and IL-4, respectively, both secreted significant but comparable amounts of IL-17 (Fig. 4 A). These findings suggest that although IL-17 production from MyHC-α–specific CD4+ T cells may play a role in EAM in the absence of classical Th1 and Th2 signals, T-bet does not repress the generation of such cells.
Given that IL-17 promotes the infiltration of neutrophils to local sites of inflammation (36), we asked whether heart-infiltrating T cells are competent to release IL-17 in immunized mice. Surprisingly, heart-infiltrating CD3+ T cells from immunized T-bet −/− mice made significantly more IL-17 than those taken from T-bet +/+ counterparts (Fig. 5 A). In vivo depletion of IL-17 in immunized T-bet −/− mice markedly reduced EAM severity relative to nondepleted T-bet −/− controls (Fig. 5 B). These data suggest that the heightened disease severity seen in T-bet −/− mice may be explained by an increase in T cell–mediated IL-17 production in the heart.
Figure 5.

Heart-infiltrating T-bet−/− CD3+ T cells display an altered cytokine profile, and IL-17 depletion improves EAM severity in T-bet−/− mice. (A) Cytokine production profile of T-bet −/− heart-infiltrating T cells. Infiltrating CD3+ cells were isolated from the hearts of immunized T-bet +/+ (blue) or T-bet −/− (red) mice and restimulated with anti-CD3/anti-CD28 for 24 h. Production of indicated cytokines was assessed by supernatant ELISA. *, P < 0.005. A representative of two individual experiments is shown. (B) T-bet −/− mice were immunized with 50 μg MyHC-α and injected with either 100 μg anti–mouse IL-17 or with 100 μl PBS on days 9, 12, and 15. Mice were killed on day 17. Hematoxylin and eosin–stained sections: bar, 200 μm.
Normal function of T-bet −/− BMDCs and regulatory T (T reg) cells
Peripheral CD4+ cells from T-bet +/+– and T-bet −/−–immunized mice produced comparable levels of IL-17 (Fig. S2 and Fig. 4). We therefore examined whether loss of T-bet in a different compartment might be responsible for the increase in EAM severity observed in T-bet −/− mice. Because T-bet expression is up-regulated in stimulated DCs (37, 38), we first evaluated the pathogenicity of activated, MyHC-α–loaded T-bet −/− DCs using a model of DC-induced autoimmune myocarditis (7, 39). Stimulated and MyHC-α–pulsed T-bet −/− BMDCs were as pathogenic as T-bet +/+ BMDCs (Fig. 6 A and Table I). T-bet −/− BMDCs produced similar amounts of IL-6, TNF-α, and IL-12p40, and up-regulated MHC class II and CD80 to comparable levels, as wild-type controls upon in vitro stimulation (not depicted). Thus, in our model system of DC-induced EAM, loss of T-bet in DCs does not enhance autoimmune heart disease.
Figure 6.

T-bet−/− DCs and CD4+CD25+ T reg cells function normally. (A) T-bet +/+ mice were immunized with activated, MyHC-α–pulsed T-bet +/+ or T-bet −/− BMDCs. (B) T-bet +/+ CD4+CD25− T cells (Teff) were stimulated with anti-CD3 with or without T-bet +/+ or T-bet −/− CD4+CD25+ cells (Treg). Proliferation was measured at 72 h by [3H]thymidine incorporation. Data are representative of three independent experiments.
We next examined the potential role of T-bet in the maintenance of peripheral tolerance. CD4+CD25+ T reg cells suppress proliferation of CD4+CD25− effector T cells in vitro and in vivo (40). Furthermore, Afanasyeva et al. (23) recently described impaired apoptosis of CD4+CD25+ T cells in IFN-γ−/− mice using the EAM model. T-bet −/− CD4+CD25+ T reg cells were as capable as their T-bet +/+ counterparts of suppressing the proliferation of CD4+CD25− effector cells (Fig. 6 B). This data, as well as that of others (17), indicates that loss of T-bet does not affect the suppressive function of CD4+CD25+ T reg cells in vitro.
Loss of T-bet in the CD8+ compartment exacerbates EAM
Besides CD4+ T cells, CD8+ cells are also found in the heart infiltrates of diseased mice (Fig. 2), and T-bet is expressed in CD8+ T cells (16, 20, 41). It is unclear whether CD8+ cells promote or modulate EAM pathogenesis (42, 43). A direct pathogenic role for CD8+ T cells has been shown in transgenic mice expressing a class I–restricted OVA-derived peptide specifically in cardiomyocytes (43). In nontransgenic mice, however, EAM is CD4+ T cell dependent and CD8+ T cells do not mediate disease (7). In contrast, it appears that the loss of CD8+ T cells exacerbates rather than protects against EAM as CD8-deficient mice develop exacerbated disease (42).
We first examined the functional capabilities of T-bet −/− CD8+ T cells by several methods. CD8+ T cells from immunized T-bet +/+ and T-bet −/− mice did not respond in vitro to irradiated splenocytes pulsed with whole myosin (not depicted), excluding antigenic spreading to class I–restricted myosin epitopes as a pathogenic mechanism. Furthermore, T-bet −/− CD8+ T cells mediated cytotoxic lysis of allogeneic targets to a similar degree as T-bet +/+ counterparts (Fig. S3 A, available at http://www.jem.org/cgi/content/full/jem.20052222/DC1).
IFN-γ has been shown to inhibit the generation of IL-17+ CD4+ T cells in vitro (30, 35). We therefore assessed the ability of T-bet −/− CD8+ T cells to produce IFN-γ. In agreement with previous findings (16), we found that peripheral T-bet −/− CD8+ T cells display apparently normal IFN-γ production upon polyclonal stimulation, regardless of whether they were taken from naive mice (Fig. S3 B) or from immunized ones (not depicted).
As T-bet −/− heart-infiltrating lymphocytes produced exacerbated IL-17, we examined IFN-γ production from T-bet +/+ and T-bet −/− T cells isolated from the hearts of immunized mice. Intriguingly, we found that heart-infiltrating CD3+ T cells from immunized T-bet +/+ mice generated substantial amounts of IFN-γ when stimulated with anti-CD3/anti-CD28, whereas heart infiltrating CD3+ cells from their T-bet −/− counterparts did not (Fig. 5 A). This finding differed from the situation in the periphery: anti-CD3/anti-CD28–stimulated splenocytes from immunized T-bet −/− mice could be induced to make IFN-γ (Fig. S3 C). Furthermore, heart-infiltrating T-bet +/+, but not T-bet −/−, CD8+ T cells also released IFN-γ in a TCR-independent manner when cultured in conditioned supernatants from LPS-activated BMDCs (not depicted). Importantly, CD3+CD8+, and not CD3+CD8−, T cells were the only heart-infiltrating cells capable of releasing IFN-γ in an antigen-independent manner in T-bet +/+ mice (Fig. 7 A). This finding suggests that T cell–specific production of IFN-γ in the inflamed heart is restricted to the CD8+ compartment; furthermore, this function is T-bet dependent. Of note, we found no differences in the production of the immunosuppressive cytokines TGF-β (Fig. 5 A) and IL-10 (not depicted) between heart-infiltrating T-bet +/+ and T-bet −/− CD3+ T cells.
Figure 7.

Heart-infiltrating T-bet−/− CD8+ T cells display impaired IFN-γ production, and loss of T-bet in CD8+ T cells exacerbates myocarditis. (A) Heart-infiltrating mononuclear cells from immunized mice were restimulated with anti-CD3/anti-CD28, stained for CD3, CD8, and intracellular IFN-γ, and analyzed by flow cytometry. Blue boxes and histograms, T-bet +/+; red boxes and histograms, T-bet −/−. A representative of two individual experiments is shown. (B) SCID mice were reconstituted with either T-bet +/+ CD4+ plus T-bet +/+ CD8+ or T-bet +/+ CD4+ plus T-bet −/− CD8+ and immunized. Mice were killed 21 d later. Hematoxylin and eosin–stained sections of hearts from mice receiving T-bet −/− CD8+ cells (right) versus mice receiving T-bet +/+ CD8+ cells (left) are shown. Bars, (top) 1 mm; (bottom) 100 μm.
A role for tissue-resident CD8+ T cells in mediating noncognate bystander suppression has recently been described in a model of allergic airway inflammation (44). In this model, bystander suppression depended on IFN-γ production by lung resident memory CD8+ T cells. Together with the fact that IFN-γ negatively regulates autoimmune myocarditis (10, 11, 13), our findings suggest that loss of the IFN-γ–producing capacity of T-bet −/− CD8+ T cells might play an important role in the enhanced disease severity in T-bet −/− mice. To resolve this question in vivo, we simultaneously immunized SCID mice with the autoimmune MyHC-α peptide and reconstituted them with naive T-bet +/+ CD4+ cells plus either T-bet +/+ or T-bet −/− naive CD8+ cells. Intriguingly, mice that received T-bet −/− CD8+ cells developed significantly more severe disease than those receiving T-bet +/+ CD8+ cells (Fig. 7 B and Table II). One explanation for these findings could be that T-bet −/− CD8+ T cells have a proliferative defect in vivo, thereby leading to excessive expansion of cotransferred T-bet +/+ CD4+ T cells and subsequently to exacerbated disease. To exclude this possibility, we CFSE labeled T-bet +/+ and T-bet −/− CD8+ and transferred them to SCID mice. T-bet +/+ and T-bet −/− CD8+ cells diluted CFSE to comparable levels upon transfer (not depicted), demonstrating that T-bet −/− CD8+ cells proliferate normally in lymphopenic hosts. Taken with the knowledge that genetic ablation of the CD8+ T cell compartment exacerbates autoimmune myocarditis (42), our results demonstrate that CD8+ T cells display down-regulatory properties rather than disease-promoting roles in EAM, and that these suppressive functions are impaired in the absence of T-bet.
Table II.
Myocarditis prevalence and disease severity in SCID mice reconstituted with T-bet +/+ CD4+ T cells and T-bet +/+ versus T-bet −/− CD8 T cells
| Recipient | Reconstitution | Immunization | Disease prevalence (no. diseased/no. treated) |
Mean score of diseased mice |
|---|---|---|---|---|
| SCID | T-bet +/+ CD4+ T cells | MyHC-α/CFA | 4/15 | 1a |
| T-bet +/+ CD8+ T cells | ||||
| SCID | T-bet +/+ CD4+ T cells | MyHC-α/CFA | 6/13 | 2.3a |
| T-bet −/− CD8+ T cells |
SCID mice were reconstituted with T-bet +/+ CD4+ cells and either T-bet +/+ or T-bet −/− CD8+ cells (2:1 ratio of CD4+/CD8+). Recipients were immunized with MyHC-α at the time of reconstitution and killed 21 d later.
P < 0.02 (Mann Whitney U).
In conclusion, expression of T-bet in heart-infiltrating CD8+ T cells appears critical for their ability to mediate antigen-nonspecific bystander suppression of local inflammation in the EAM model. This observation parallels the impaired release of IFN-γ and the increased production of IL-17 from heart-infiltrating T-bet −/− T cells.
DISCUSSION
To date, it has been unclear whether EAM is a Th1- or Th2-mediated disease. Although mice lacking IL-4 signaling could develop EAM, the same was also true for mice deficient in IFN-γ or IFN-γR (10, 11, 13). However, our data and that of others (45) indicate that Th1 responses can progress in the absence of IFN-γ signaling. We therefore examined the development of EAM in mice lacking the T-box transcription factor T-bet, which is critical for Th1 differentiation (15) and essential for CD4+ cell tissue infiltration in Th1-mediated models of inflammation (46). Our findings showed that in sharp contrast to other autoimmune disease models (17–21, 47), T-bet −/− mice developed EAM of exacerbated severity relative to T-bet +/+ controls. Although T-bet −/− mice were previously described to develop transiently increased disease severity relative to wild-type controls in a model of Staphylococcus aureus–induced arthritis (48), these mice also suffered from greatly increased bacterial load, making it likely that increased arthritis severity was due to impaired infectious clearance rather than to autoimmune mechanisms. We therefore describe the first model in which T-bet negatively regulates autoimmune disease.
As expected, immunized T-bet −/− mice developed defective Th1 responses, as measured by the presence of MyHC-α–specific, IgG2a isotype autoantibodies and by IFN-γ production by restimulated CD4+ T cells. We then found that T-bet −/− IL-12p35 −/− mice were also highly susceptible to EAM, excluding the possibility that IL-12 can promote severe Th1-driven autoimmunity in the absence of T-bet. At the same time, Th1- and Th2-polarized, MyHC-α–specific CD4+ T cell lines were equally effective in transferring disease to healthy recipients, and both T-bet −/− IL-4 −/− and T-bet −/− IL-4Rα −/− mice still developed severe myocarditis. Therefore, our results describe EAM as an autoimmune disease that can develop independently of Th1/Th2 dysregulation. However, we cannot exclude that Th1 or Th2 polarization might contribute to the broad pattern of different morphologic phenotypes and clinical courses in patients with inflammatory heart disease of different causes (1, 49).
Interestingly, IL-23 signaling was absolutely required for EAM development, as IL-12Rβ1 −/− and T-bet −/− IL-12Rβ1 −/− mice, but not IL-12p35 −/− and T-bet −/− IL-12p35 −/− mice, were protected from disease. IL-23 is essential for the generation of IL-17–producing CD4+ T cells in vivo, and IL-17 is a pathogenic cytokine critical for the progression of experimental autoimmune encephalomyelitis (34). IL-17–producing CD4+ T cells (ThIL-17) have been described as representing an effector cell type independent from Th1 or Th2 cells (30, 35, 50). Although recent evidence argues against a role for IL-23 in the initiation of ThIL-17 differentiation (51), it is clear that IL-23 is at least essential for the expansion or survival of ThIL-17 cells in vivo. We therefore examined whether loss of T-bet could increase IL-17 production, and by extension disease severity, in our EAM model. However, both CD4+ T cells from immunized T-bet +/+ and T-bet −/− mice, as well as in vitro–expanded and pathogenic MyHC-α–specific Th1 T-bet +/+ and Th2 T-bet −/− CD4+ T cell lines, produced comparable levels of IL-17. This suggested that although IL-17 production might characterize pathogenic CD4+ T cells in EAM, T-bet did not repress IL-17 production in the peripheral CD4+ compartment.
However, in contrast to peripheral T cells, we observed that IL-17 production from heart-infiltrating T cells was markedly greater in immunized T-bet −/− mice as compared with T-bet +/+ controls. IL-17 can act on stromal endothelial cells in inflamed tissue to induce secretion of neutrophil-attracting factors, such as IL-8, CXCL1, and GM-CSF (36). This suggests that the heart-localized up-regulation of IL-17 production in T-bet −/− mice could be pathologically relevant. Indeed, depletion of IL-17 in vivo markedly reduced EAM severity in T-bet −/− mice. Recent reports have suggested that loss of T-bet results in increased IL-17 production from CD4+ T cells in vitro (30) and from restimulated draining LN cells taken from MOG peptide–immunized mice (35). Our data strongly suggest a direct pathogenic role for IL-17+CD4+ T cells in EAM. Furthermore, they establish for the first time that T-bet–mediated repression of IL-17 can modulate the severity of an autoimmune disease.
IFN-γ can inhibit the production of IL-17 from CD4+ T cells. We therefore analyzed the capacity of heart-infiltrating T cells to produce IFN-γ. In T-bet +/+ mice, heart-infiltrating CD3+CD8+, but not CD3+CD8−, cells made IFN-γ upon polyclonal stimulation. Intriguingly, CD8+ T cells appear to be responsible for almost all of the IFN-γ released within the diseased heart of wild-type mice in autoimmune myocarditis. This secretion might act to suppress local production of IL-17. Furthermore, CD8+ T cells from both immunized T-bet +/+ and T-bet −/− mice could not proliferate in response to whole myosin, suggesting that heart-infiltrating CD8+ T cells in EAM secrete IFN-γ in an antigen-nonspecific manner. Importantly, T-bet −/− heart-infiltrating T cells were wholly deficient in their capacity to produce IFN-γ yet were still able to produce other cytokines, such as TGF-β and TNF-α. This absence of infiltrating lymphocyte-specific production of IFN-γ parallels the observed increase in IL-17 production in T-bet −/− hearts.
Although IFN-γ has been well described as the prototypical Th1 effector cytokine, it is becoming clear that it also possesses immunomodulatory properties. IFN-γ can induce APC expression of immunoregulatory molecules, such as inducible nitric oxide synthase and indoleamine-2,3-dioxygenase, and IFN-γ is essential for the generation of T reg cells with in vivo suppressive function (52). IFN-γ production is suppressed in arthritic joints despite significant T cell infiltration, suggesting that local down-regulation of IFN-γ production contributes to autoimmune inflammation in rheumatoid arthritis (53), and IFN-γ −/− and IFN-γR −/− mice are highly susceptible to severe EAM (10, 11, 13). Bystander, noncognate CD8+-mediated production of IFN-γ has also been implicated in the suppression of inflammation; CD8+ T cells directed against an influenza-derived epitope were able to protect against OVA-mediated allergic airway inflammation (44). We found that although cotransfer of T-bet +/+ CD8+ T cells with T-bet +/+ CD4+ T cells resulted in minimal disease in immunized SCID recipients, transfer of T-bet −/− CD8+ T cells and T-bet +/+ CD4+ T cells caused significantly more severe EAM. These findings point toward a critical T-bet–dependent role of CD8+ T cells in modulating autoimmune myocarditis severity.
Our findings that organ-infiltrating but not peripheral T-bet −/− CD8+ T cells lack the capacity to release IFN-γ complement recent findings that antigen-specific, pancreatic LN–localized T-bet −/− CD8+ T cells lose the capacity to release IFN-γ and to mediate disease in the rat insulin promoter–lymphocytic choriomeningitis virus transgenic model of autoimmune diabetes (20). In contrast to autoimmune myocarditis, however, the transgenic autoimmune diabetes model depends on a T-bet– and IFN-γ–dependent T cell response against an MHC class I–restricted antigen. Nevertheless, it might be that the common defect in IFN-γ production by organ-infiltrating T-bet −/− CD8+ T cells results in impaired cytotoxicity in an MHC class I–restricted immune response, yet leads to impaired bystander suppression in an MHC class II–mediated disease model. We propose that the defect of heart-infiltrating T-bet −/− CD8+ T cells to release IFN-γ impairs their capacity to suppress local inflammation in EAM.
Although we and others have found that peripheral T-bet −/− CD8+ T cells can produce IFN-γ, the question arises as to why heart-infiltrating CD8+ cells require T-bet expression to produce IFN-γ. Peripheral TCR-transgenic T-bet −/− CD8+ cells display defective IFN-γ production upon stimulation with specific peptide, suggesting that upon strong TCR stimulation, other factors can compensate for the loss of T-bet in driving IFN-γ (54). Recently, the transcription factor Eomes was identified as an inducer of IFN-γ in CD8+ T cells (54, 55). Although Eomes and other factors may have functions that overlap with T-bet in peripheral CD8+ cells, our data show that T-bet is clearly essential for IFN-γ production from heart-infiltrating CD8+ cells. It is possible that upon migration to the heart, CD8+ T cells undergo a process of epigenetic reprogramming that renders T-bet essential for IFN-γ generation. Alternately, heart-infiltrating CD8+ cells may arise from a subset of peripheral CD8+ T cells that absolutely require T-bet for the production of IFN-γ. Further research is required to distinguish between these and other scenarios.
Clinical studies suggest that inflammation is a major factor contributing to the pathogenesis of cardiovascular diseases. Here we have shown that mice lacking T-bet are highly susceptible to autoimmune myocarditis. Polymorphisms in the gene encoding T-bet as well as its promoter have been associated with differential susceptibility to asthma in human subjects (56–58). Epidemiological studies have found a significant association between idiopathic DCM and a history of asthma (59, 60), making it tempting to speculate that loss of T-bet may provide a common molecular mechanism underlying these pathologies.
Intriguingly, induction of EAM appears to be independent of classical Th1 or Th2 effector responses and is rather mediated by a subset of CD4+ T cells characterized by IL-17 production. Indeed, the ablation of Th1 signaling in T-bet −/− mice results in severe EAM, in part by the abolition of a T-bet–dependent, CD8+ T cell–mediated mechanism of inflammatory suppression. T-bet plays an essential role in the pathogenesis of multiple autoimmune diseases (17–21, 47) and has thus been proposed as a potential target of immunomodulatory therapy (61). However, the findings presented here imply that care must be taken in manipulating Th1 and Th2 responses in the treatment of autoimmune diseases so as to avoid potentially devastating side effects.
MATERIALS AND METHODS
Mice.
T-bet −/− mice have been described previously (62) and were a gift from L. Glimcher (Harvard University, Boston, MA). IL-4−/−, IL-4Rα−/−, IFN-γR−/−, IL-12Rβ1−/−, IL-12p35−/−, and CB17.SCID mice were purchased from The Jackson Laboratory. All mice were backcrossed to the BALB/c strain for >10 generations and were used at 6–8 wk of age. All experiments were conducted in accordance with Canadian, Austrian, or Swiss federal laws and institutional guidelines.
Immunization protocols.
Mice were immunized with 100 μg of the MyHC-α peptide (MyHC-α614–634) Ac-SLKLMATLFSTYASAD-OH emulsified 1:1 in PBS/CFA (1 mg/ml; H37Ra; Difco Laboratories) as described previously (63). For DC immunization, BMDCs were pulsed for 2 h with 10 μg/ml MyHC-α peptide and stimulated for another 4 h with 0.1 μg/ml LPS (O111:B4; Sigma-Aldrich) and 5 μg/ml anti-CD40 (BD Biosciences) (7). Recipient mice received 2.5 × 105 pulsed and activated BMDCs i.p. for three consecutive days and were killed 10 d after the first injection. In IL-17 depletion experiments, mice were immunized with 50 μg MyHC-α/CFA, injected with either anti–mouse IL-17 (R&D Systems) or PBS on days 9, 12, and 15, and killed on day 17.
Histopathology, immunohistochemistry, and detection of autoantibodies.
Myocarditis was scored on hematoxylin and eosin–stained sections using grades from 0 to 4: 0, no inflammatory infiltrates; 1, small foci of inflammatory cells between myocytes; 2, larger foci of >100 inflammatory cells; 3, >10% of a cross section involved; 4, >30% of a cross section involved (7). For immunohistochemistry, OCT (Sakura Finetek) - embedded frozen heart sections were fixed in acetone and processed for antibody staining according to standard protocols. The following antibodies were used: anti-CD4 (YTS 191), anti-CD8 (YTS 169; BD Biosciences), and anti-CD68 (FA-11; Serotec). Autoantibody responses were determined by ELISA as described previously (7) using alkaline phosphatase–labeled goat anti–mouse IgM and IgG subclass antibodies (SouthernBiotech).
T cell functions.
CD4+ and CD8+ cells were purified using magnetic beads (CD4+ and CD8+ T cell isolation kits; Miltenyi Biotec). 5–10 × 104 CD4+ T cells were restimulated for 72 h on 2 μg/ml MyHC-α peptide–pulsed, irradiated (20 Gy) syngeneic splenocytes (2 × 105). In some experiments, CD4+ or CD8+ T cells were stimulated with either 1 μg/ml each of plate-bound anti-CD3 and anti-CD28 (BD Biosciences) or with 1 μg/ml soluble anti-CD3 on 2 × 105 irradiated syngeneic splenocytes. For T reg cell assays, CD4+ T cells were sorted into CD4+CD25− (effector T cell) and CD4+CD25+(T reg cell) populations using a high-speed cell sorter (FACSAria; BD Biosciences). 5 × 104 effector T cells were cocultured with T reg cells on 2 × 105 irradiated syngeneic splenocytes in the presence or absence of 1 μg/ml soluble anti-CD3 (BD Biosciences) for 72 h. [3H]thymidine incorporation was measured as a readout for proliferation responses. For some analyses of heart-infiltrating T cell cytokine production, cells were either stimulated in the presence of soluble anti-CD3/anti-CD28 or cultured for 72 h in supernatants from mature, LPS-stimulated BMDC cultures (conditioned medium). IFN-γ levels in conditioned medium were always below the detection limits of our ELISA kits (<50 pg/ml). Cytokine levels were measured using commercially available Quantikine ELISA kits for detection of IFN-γ, TGF-β1, TNF-α, IL-4, IL-6, IL-10, IL-12p40, and IL-17 (R&D Systems). For analysis of in vivo expansion, CD8+ T cells were incubated for 10 min with 10 μM CFSE (Invitrogen) at 37°C before injection.
MyHC-α–specific Th1 or Th2 CD4+ lines and adoptive transfer.
CD4+ T cells were purified from diseased T-bet +/+ or T-bet −/− mice and cultured with irradiated (20 Gy) splenocytes at a 1:2 ratio in the presence of 2 μg/ml MyHC-α for 7 d. Cells were then washed and rested in the presence of 10 U/ml of recombinant IL-2 for an additional 7 d. This pulse/rest cycle was repeated at least three times. Finally, CD4+ T cells were restimulated for 4 d before i.p. injection of 107 CD4+ T cells per syngeneic recipient. For reconstitution of SCID mice, naive CD4+ and CD8+ T cells were isolated and 5 × 106 CD4+ and 2.5 × 106 CD8+ T cells were injected i.p. into same-day immunized recipients.
FACS analysis.
Cell suspensions were stained using fluorochrome-conjugated mouse-specific antibodies against CD3, CD4, CD8α, CD8β, CD11c, CD80, MHC class II (I-Ad), CD25, IFN-γ, and IL-17. All antibodies were purchased from BD Biosciences. Before intracellular staining, cells were restimulated for 4 h with 50 ng/ml PMA and 500 ng/ml ionomycin in the presence of 1 μg/ml GolgiPlug (BD Biosciences). Intracellular staining was conducted using the Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer's protocols. Samples were analyzed on a FACSCalibur cell sorter (BD Biosciences) using Weasel (Walter and Eliza Hall Institute, Melbourne, Australia) or FlowJo (TreeStar) software.
Cytotoxicity assays.
T-bet +/+ and T-bet −/− (H-2d) BALB/c mice were primed by i.p. injection of allogeneic (H-2b) spleen-derived DCs. CD8+ T cells were isolated from primed mice using a biotinylated anti-CD8β antibody (BD Biosciences) and anti-biotin microbeads (Miltenyi Biotec). After in vitro restimulation on irradiated H-2b splenocytes, cytotoxic CD8+ T cells were used as effector cells in a calcein release assay using calcein-loaded EL-4 H-2b target cells. Spontaneous calcein release (SR) was determined by the addition of medium, and maximal release (MR) was determined by the addition of lysis buffer to target cells. Calcein release was measured after 2 h of incubation in a fluorescence multiwell plate reader (SpectraMax Gemini XS; Molecular Devices). Percent-specific lysis was calculated as (sample release-SR)/(MR-SR)×100%.
Isolation of heart-infiltrating cells.
Heart-infiltrating lymphocytes were isolated as described previously (64) with the following modifications: Collagenase D (Worthington Biochemical Corporation) was used at a concentration of 0.895 mg/ml, and tissue suspensions were passed sequentially through 70- and 40-μm cell strainers (BD Falcon), and finally through 15-μm self-assembled strainers (Sefar AG).
Statistics.
The Mann-Whitney U test was used for the evaluation of severity scores. Dichotomous data were analyzed using Fisher's exact test. Proliferation responses and cytokine levels were compared using ANOVA and Student's t test. Statistical analysis was conducted using Prism 4 software (GraphPad Software).
Online supplemental material.
Fig. S1 shows that CD4+ T cells taken from MyHC-α–immunized IFN-γR −/− mice at day 21 are capable of producing IFN-γ upon antigenic restimulation. Fig. S2 shows that a small number of CD4+ T cells from both T-bet +/+ and T-bet −/− MyHC-α–immunized mice develop into IL-17 producers upon antigenic restimulation. Fig. S3 A shows that T-bet +/+ and T-bet −/− CD8+ T cells can mediate cytotoxic lysis of allogeneic target cells to a comparable degree, and Fig. S3 B shows that naive peripheral T-bet +/+ and T-bet −/− CD8+ T cells produce similar levels of IFN-γ upon polyclonal stimulation. Fig. S3 C shows that whole splenocytes from MyHC-α–immunized T-bet −/− mice can produce significant, albeit reduced, levels of IFN-γ upon both antigenic and anti-CD3/anti-CD28 restimulation.
Supplemental Material
Acknowledgments
We thank Dr. Laurie Glimcher for providing T-bet −/− mice, Heidi Bodmer and Gabriele Stengl for technical assistance, and Ludger Klein, Gregory Neely, Toshikatsu Hanada, and Elisabeth Vollmann for critical discussions.
This work is supported by the Institute for Molecular Biotechnology of the Austrian Academy of Sciences (to J.M. Penninger), the Austrian National Bank (to J.M. Penninger), the Austrian FWF (SFB grant F23 to J.M. Penninger), the Swiss National Foundation (to U. Eriksson), and the Novartis Foundation (to U. Eriksson). M. Rangachari was supported by the European Union (grant EURO-RA) as well as by scholarships from the University of Toronto and the National Sciences and Engineering Research Council of Canada. U. Eriksson holds a Swiss National Foundation professorship.
The authors have no conflicting financial interests.
Abbreviations used: BMDC, bone marrow–derived DC; DCM, dilated cardiomyopathy; EAM, experimental autoimmune myocarditis; MyHC-α, myosin α heavy chain; T reg, regulatory T.
J.M. Penninger and U. Eriksson contributed equally to this work.
References
- 1.Eriksson, U., and J.M. Penninger. 2005. Autoimmune heart failure: new understandings of pathogenesis. Int. J. Biochem. Cell Biol. 37:27–32. [DOI] [PubMed] [Google Scholar]
- 2.Koelsch, S.P.S., G. Hufnagel, and B. Maisch. 2004. The European study of epidemiology and treatment of cardiac inflammatory diseases (ESETCID)—epidemiological results after 6 years. In Annual Meeting of the AHA, New Orleans.
- 3.Caforio, A.L., E. Bonifacio, J.T. Stewart, D. Neglia, O. Parodi, G.F. Bottazzo, and W.J. McKenna. 1990. Novel organ-specific circulating cardiac autoantibodies in dilated cardiomyopathy. J. Am. Coll. Cardiol. 15:1527–1534. [DOI] [PubMed] [Google Scholar]
- 4.Frustaci, A., C. Chimenti, F. Calabrese, M. Pieroni, G. Thiene, and A. Maseri. 2003. Immunosuppressive therapy for active lymphocytic myocarditis: virological and immunologic profile of responders versus nonresponders. Circulation. 107:857–863. [DOI] [PubMed] [Google Scholar]
- 5.Omerovic, E., E. Bollano, B. Andersson, V. Kujacic, W. Schulze, A. Hjalmarson, F. Waagstein, and M. Fu. 2000. Induction of cardiomyopathy in severe combined immunodeficiency mice by transfer of lymphocytes from patients with idiopathic dilated cardiomyopathy. Autoimmunity. 32:271–280. [DOI] [PubMed] [Google Scholar]
- 6.Fairweather, D., Z. Kaya, G.R. Shellam, C.M. Lawson, and N.R. Rose. 2001. From infection to autoimmunity. J. Autoimmun. 16:175–186. [DOI] [PubMed] [Google Scholar]
- 7.Eriksson, U., R. Ricci, L. Hunziker, M.O. Kurrer, G.Y. Oudit, T.H. Watts, I. Sonderegger, K. Bachmaier, M. Kopf, and J.M. Penninger. 2003. Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat. Med. 9:1484–1490. [DOI] [PubMed] [Google Scholar]
- 8.Bachmaier, K., N. Neu, L.M. de la Maza, S. Pal, A. Hessel, and J.M. Penninger. 1999. Chlamydia infections and heart disease linked through antigenic mimicry. Science. 283:1335–1339. [DOI] [PubMed] [Google Scholar]
- 9.Nicholson, L.B., and V.K. Kuchroo. 1996. Manipulation of the Th1/Th2 balance in autoimmune disease. Curr. Opin. Immunol. 8:837–842. [DOI] [PubMed] [Google Scholar]
- 10.Eriksson, U., M.O. Kurrer, W. Sebald, F. Brombacher, and M. Kopf. 2001. Dual role of the IL-12/IFN-gamma axis in the development of autoimmune myocarditis: induction by IL-12 and protection by IFN-gamma. J. Immunol. 167:5464–5469. [DOI] [PubMed] [Google Scholar]
- 11.Afanasyeva, M., Y. Wang, Z. Kaya, E.A. Stafford, K.M. Dohmen, A.A. Sadighi Akha, and N.R. Rose. 2001. Interleukin-12 receptor/STAT4 signaling is required for the development of autoimmune myocarditis in mice by an interferon-gamma-independent pathway. Circulation. 104:3145–3151. [DOI] [PubMed] [Google Scholar]
- 12.Afanasyeva, M., Y. Wang, Z. Kaya, S. Park, M.J. Zilliox, B.H. Schofield, S.L. Hill, and N.R. Rose. 2001. Experimental autoimmune myocarditis in A/J mice is an interleukin-4-dependent disease with a Th2 phenotype. Am. J. Pathol. 159:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eriksson, U., M.O. Kurrer, R. Bingisser, H.P. Eugster, P. Saremaslani, F. Follath, S. Marsch, and U. Widmer. 2001. Lethal autoimmune myocarditis in interferon-gamma receptor-deficient mice: enhanced disease severity by impaired inducible nitric oxide synthase induction. Circulation. 103:18–21. [DOI] [PubMed] [Google Scholar]
- 14.Rosloniec, E.F., K. Latham, and Y.B. Guedez. 2002. Paradoxical roles of IFN-gamma in models of Th1-mediated autoimmunity. Arthritis Res. 4:333–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szabo, S.J., S.T. Kim, G.L. Costa, X. Zhang, C.G. Fathman, and L.H. Glimcher. 2000. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 100:655–669. [DOI] [PubMed] [Google Scholar]
- 16.Szabo, S.J., B.M. Sullivan, C. Stemmann, A.R. Satoskar, B.P. Sleckman, and L.H. Glimcher. 2002. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 295:338–342. [DOI] [PubMed] [Google Scholar]
- 17.Bettelli, E., B. Sullivan, S.J. Szabo, R.A. Sobel, L.H. Glimcher, and V.K. Kuchroo. 2004. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J. Exp. Med. 200:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lovett-Racke, A.E., A.E. Rocchini, J. Choy, S.C. Northrop, R.Z. Hussain, R.B. Ratts, D. Sikder, and M.K. Racke. 2004. Silencing T-bet defines a critical role in the differentiation of autoreactive T lymphocytes. Immunity. 21:719–731. [DOI] [PubMed] [Google Scholar]
- 19.Matsuoka, K., N. Inoue, T. Sato, S. Okamoto, T. Hisamatsu, Y. Kishi, A. Sakuraba, O. Hitotsumatsu, H. Ogata, K. Koganei, et al. 2004. T-bet upregulation and subsequent interleukin 12 stimulation are essential for induction of Th1 mediated immunopathology in Crohn's disease. Gut. 53:1303–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Juedes, A.E., E. Rodrigo, L. Togher, L.H. Glimcher, and M.G. von Herrath. 2004. T-bet controls autoaggressive CD8 lymphocyte responses in type 1 diabetes. J. Exp. Med. 199:1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buono, C., C.J. Binder, G. Stavrakis, J.L. Witztum, L.H. Glimcher, and A.H. Lichtman. 2005. T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Proc. Natl. Acad. Sci. USA. 102:1596–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Venkataraman, C., S. Leung, A. Salvekar, H. Mano, and U. Schindler. 1999. Repression of IL-4-induced gene expression by IFN-gamma requires Stat1 activation. J. Immunol. 162:4053–4061. [PubMed] [Google Scholar]
- 23.Afanasyeva, M., D. Georgakopoulos, D.F. Belardi, D. Bedja, D. Fairweather, Y. Wang, Z. Kaya, K.L. Gabrielson, E.R. Rodriguez, P. Caturegli, et al. 2005. Impaired up-regulation of CD25 on CD4+ T cells in IFN-gamma knockout mice is associated with progression of myocarditis to heart failure. Proc. Natl. Acad. Sci. USA. 102:180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murphy, K.M., and S.L. Reiner. 2002. The lineage decisions of helper T cells. Nat. Rev. Immunol. 2:933–944. [DOI] [PubMed] [Google Scholar]
- 25.Watford, W.T., M. Moriguchi, A. Morinobu, and J.J. O'Shea. 2003. The biology of IL-12: coordinating innate and adaptive immune responses. Cytokine Growth Factor Rev. 14:361–368. [DOI] [PubMed] [Google Scholar]
- 26.Mullen, A.C., F.A. High, A.S. Hutchins, H.W. Lee, A.V. Villarino, D.M. Livingston, A.L. Kung, N. Cereb, T.P. Yao, S.Y. Yang, and S.L. Reiner. 2001. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science. 292:1907–1910. [DOI] [PubMed] [Google Scholar]
- 27.Afkarian, M., J.R. Sedy, J. Yang, N.G. Jacobson, N. Cereb, S.Y. Yang, T.L. Murphy, and K.M. Murphy. 2002. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat. Immunol. 3:549–557. [DOI] [PubMed] [Google Scholar]
- 28.Trinchieri, G. 2003. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 3:133–146. [DOI] [PubMed] [Google Scholar]
- 29.Aggarwal, S., N. Ghilardi, M.H. Xie, F.J. de Sauvage, and A.L. Gurney. 2003. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J. Biol. Chem. 278:1910–1914. [DOI] [PubMed] [Google Scholar]
- 30.Harrington, L.E., R.D. Hatton, P.R. Mangan, H. Turner, T.L. Murphy, K.M. Murphy, and C.T. Weaver. 2005. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 6:1123–1132. [DOI] [PubMed] [Google Scholar]
- 31.Happel, K.I., M. Zheng, E. Young, L.J. Quinton, E. Lockhart, A.J. Ramsay, J.E. Shellito, J.R. Schurr, G.J. Bagby, S. Nelson, and J.K. Kolls. 2003. Cutting edge: roles of Toll-like receptor 4 and IL-23 in IL-17 expression in response to Klebsiella pneumoniae infection. J. Immunol. 170:4432–4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rutitzky, L.I., J.R. Lopes da Rosa, and M.J. Stadecker. 2005. Severe CD4 T cell-mediated immunopathology in murine schistosomiasis is dependent on IL-12p40 and correlates with high levels of IL-17. J. Immunol. 175:3920–3926. [DOI] [PubMed] [Google Scholar]
- 33.Murphy, C.A., C.L. Langrish, Y. Chen, W. Blumenschein, T. McClanahan, R.A. Kastelein, J.D. Sedgwick, and D.J. Cua. 2003. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J. Exp. Med. 198:1951–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langrish, C.L., Y. Chen, W.M. Blumenschein, J. Mattson, B. Basham, J.D. Sedgwick, T. McClanahan, R.A. Kastelein, and D.J. Cua. 2005. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 201:233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park, H., Z. Li, X.O. Yang, S.H. Chang, R. Nurieva, Y.H. Wang, Y. Wang, L. Hood, Z. Zhu, Q. Tian, and C. Dong. 2005. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 6:1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKenzie, B.S., R.A. Kastelein, and D.J. Cua. 2006. Understanding the IL-23-IL-17 immune pathway. Trends Immunol. 27:17–23. [DOI] [PubMed] [Google Scholar]
- 37.Lugo-Villarino, G., R. Maldonado-Lopez, R. Possemato, C. Penaranda, and L.H. Glimcher. 2003. T-bet is required for optimal production of IFN-gamma and antigen-specific T cell activation by dendritic cells. Proc. Natl. Acad. Sci. USA. 100:7749–7754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lighvani, A.A., D.M. Frucht, D. Jankovic, H. Yamane, J. Aliberti, B.D. Hissong, B.V. Nguyen, M. Gadina, A. Sher, W.E. Paul, and J.J. O'Shea. 2001. T-bet is rapidly induced by interferon-gamma in lymphoid and myeloid cells. Proc. Natl. Acad. Sci. USA. 98:15137–15142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rangachari, M., U. Eriksson, E.H. Vollmann, and J.M. Penninger. 2005. A novel model for pathogenesis of autoimmune heart failure: the role of dendritic cells. In The Innate Immune System: Strategies for Disease Control, 1285. M. Taniguchi, S. Akira, and T. Nakayama, editors. International Congress Series, Netherlands. 192–201.
- 40.Gavin, M., and A. Rudensky. 2003. Control of immune homeostasis by naturally arising regulatory CD4+ T cells. Curr. Opin. Immunol. 15:690–696. [DOI] [PubMed] [Google Scholar]
- 41.Sullivan, B.M., A. Juedes, S.J. Szabo, M. von Herrath, and L.H. Glimcher. 2003. Antigen-driven effector CD8 T cell function regulated by T-bet. Proc. Natl. Acad. Sci. USA. 100:15818–15823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Penninger, J.M., N. Neu, E. Timms, V.A. Wallace, D.R. Koh, K. Kishihara, C. Pummerer, and T.W. Mak. 1993. The induction of experimental autoimmune myocarditis in mice lacking CD4 or CD8 molecules. J. Exp. Med. 178:1837–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grabie, N., M.W. Delfs, J.R. Westrich, V.A. Love, G. Stavrakis, F. Ahmad, C.E. Seidman, J.G. Seidman, and A.H. Lichtman. 2003. IL-12 is required for differentiation of pathogenic CD8+ T cell effectors that cause myocarditis. J. Clin. Invest. 111:671–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marsland, B.J., N.L. Harris, M. Camberis, M. Kopf, S.M. Hook, and G. Le Gros. 2004. Bystander suppression of allergic airway inflammation by lung resident memory CD8+ T cells. Proc. Natl. Acad. Sci. USA. 101:6116–6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haring, J.S., V.P. Badovinac, M.R. Olson, S.M. Varga, and J.T. Harty. 2005. In vivo generation of pathogen-specific Th1 cells in the absence of the IFN-gamma receptor. J. Immunol. 175:3117–3122. [DOI] [PubMed] [Google Scholar]
- 46.Lord, G.M., R.M. Rao, H. Choe, B.M. Sullivan, A.H. Lichtman, F.W. Luscinskas, and L.H. Glimcher. 2005. T-bet is required for optimal proinflammatory CD4+ T-cell trafficking. Blood. 106:3432–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neurath, M.F., B. Weigmann, S. Finotto, J. Glickman, E. Nieuwenhuis, H. Iijima, A. Mizoguchi, E. Mizoguchi, J. Mudter, P.R. Galle, et al. 2002. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn's disease. J. Exp. Med. 195:1129–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hultgren, O.H., M. Verdrengh, and A. Tarkowski. 2004. T-box transcription-factor-deficient mice display increased joint pathology and failure of infection control during staphylococcal arthritis. Microbes Infect. 6:529–535. [DOI] [PubMed] [Google Scholar]
- 49.Burian, J., P. Buser, and U. Eriksson. 2005. Myocarditis: the immunologist's view on pathogenesis and treatment. Swiss Med. Wkly. 135:359–364. [DOI] [PubMed] [Google Scholar]
- 50.Wynn, T.A. 2005. T(H)-17: a giant step from T(H)1 and T(H)2. Nat. Immunol. 6:1069–1070. [DOI] [PubMed] [Google Scholar]
- 51.Veldhoen, M., R.J. Hocking, C.J. Atkins, R.M. Locksley, and B. Stockinger. 2006. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 24:179–189. [DOI] [PubMed] [Google Scholar]
- 52.Wood, K.J., and B. Sawitzki. 2006. Interferon gamma: a crucial role in the function of induced regulatory T cells in vivo. Trends Immunol. 27:183–187. [DOI] [PubMed] [Google Scholar]
- 53.Kinne, R.W., E. Palombo-Kinne, and F. Emmrich. 1997. T-cells in the pathogenesis of rheumatoid arthritis villains or accomplices? Biochim. Biophys. Acta. 1360:109–141. [DOI] [PubMed] [Google Scholar]
- 54.Pearce, E.L., A.C. Mullen, G.A. Martins, C.M. Krawczyk, A.S. Hutchins, V.P. Zediak, M. Banica, C.B. DiCioccio, D.A. Gross, C.A. Mao, et al. 2003. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science. 302:1041–1043. [DOI] [PubMed] [Google Scholar]
- 55.Glimcher, L.H., M.J. Townsend, B.M. Sullivan, and G.M. Lord. 2004. Recent developments in the transcriptional regulation of cytolytic effector cells. Nat. Rev. Immunol. 4:900–911. [DOI] [PubMed] [Google Scholar]
- 56.Tantisira, K.G., E.S. Hwang, B.A. Raby, E.S. Silverman, S.L. Lake, B.G. Richter, S.L. Peng, J.M. Drazen, L.H. Glimcher, and S.T. Weiss. 2004. TBX21: a functional variant predicts improvement in asthma with the use of inhaled corticosteroids. Proc. Natl. Acad. Sci. USA. 101:18099–18104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Akahoshi, M., K. Obara, T. Hirota, A. Matsuda, K. Hasegawa, N. Takahashi, M. Shimizu, K. Nakashima, L. Cheng, S. Doi, et al. 2005. Functional promoter polymorphism in the TBX21 gene associated with aspirin-induced asthma. Hum. Genet. 117:16–26. [DOI] [PubMed] [Google Scholar]
- 58.Raby, B.A., E.S. Hwang, K. Van Steen, K. Tantisira, S. Peng, A. Litonjua, R. Lazarus, C. Giallourakis, J.D. Rioux, D. Sparrow, et al. 2006. T-bet polymorphisms are associated with asthma and airway hyperresponsiveness. Am. J. Respir. Crit. Care Med. 173:64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sengstock, D.M., O. Obeidat, V. Pasnoori, P. Mehra, K.R. Sandberg, and P.A. McCullough. 2002. Asthma, beta-agonists, and development of congestive heart failure: results of the ABCHF study. J. Card. Fail. 8:232–238. [DOI] [PubMed] [Google Scholar]
- 60.Coughlin, S.S., M. Szklo, K. Baughman, and T.A. Pearson. 1989. Idiopathic dilated cardiomyopathy and atopic disease: epidemiologic evidence for an association with asthma. Am. Heart J. 118:768–774. [DOI] [PubMed] [Google Scholar]
- 61.Chinen, J., and W.T. Shearer. 2003. Basic and clinical immunology. J. Allergy Clin. Immunol. 111:S813–S818. [DOI] [PubMed] [Google Scholar]
- 62.Finotto, S., M.F. Neurath, J.N. Glickman, S. Qin, H.A. Lehr, F.H. Green, K. Ackerman, K. Haley, P.R. Galle, S.J. Szabo, et al. 2002. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science. 295:336–338. [DOI] [PubMed] [Google Scholar]
- 63.Eriksson, U., M.O. Kurrer, N. Schmitz, S.C. Marsch, A. Fontana, H.P. Eugster, and M. Kopf. 2003. Interleukin-6-deficient mice resist development of autoimmune myocarditis associated with impaired upregulation of complement C3. Circulation. 107:320–325. [DOI] [PubMed] [Google Scholar]
- 64.Afanasyeva, M., D. Georgakopoulos, D.F. Belardi, A.C. Ramsundar, J.G. Barin, D.A. Kass, and N.R. Rose. 2004. Quantitative analysis of myocardial inflammation by flow cytometry in murine autoimmune myocarditis: correlation with cardiac function. Am. J. Pathol. 164:807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}