

Abstract
Most treatments that prevent autoimmune diabetes in nonobese diabetic (NOD) mice require intervention at early pathogenic stages, when insulitis is first developing. We tested whether dendritic cell (DC)–expanded, islet antigen–specific CD4+ CD25+ suppressor T cells could treat diabetes at later stages of disease, when most of the insulin-producing islet β cells had been destroyed by infiltrating lymphocytes. CD4+ CD25+ CD62L+ regulatory T cells (T reg cells) from BDC2.5 T cell receptor transgenic mice were expanded with antigen-pulsed DCs and IL-2, and were then injected into NOD mice. A single dose of as few as 5 × 104 of these islet-specific T reg cells blocked diabetes development in prediabetic 13-wk-old NOD mice. The T reg cells also induced long-lasting reversal of hyperglycemia in 50% of mice in which overt diabetes had developed. Successfully treated diabetic mice had similar responses to glucose challenge compared with nondiabetic NOD mice. The successfully treated mice retained diabetogenic T cells, but also had substantially increased Foxp3+ cells in draining pancreatic lymph nodes. However, these Foxp3+ cells were derived from the recipient mice and not the injected T reg cells, suggesting a role for endogenous T reg cells in maintaining tolerance after treatment. Therefore, inoculation of DC-expanded, antigen-specific suppressor T cells has considerable efficacy in ameliorating ongoing diabetes in NOD mice.
Nonobese diabetic (NOD) mice develop spontaneous autoimmune diabetes, particularly in females. Lymphocytes begin to infiltrate the pancreatic islets between 4 and 9 wk of age, whereas overt diabetes caused by destruction of insulin-producing β cells (defined by the inability to maintain normal blood glucose levels) does not occur until 11–25 wk of age. Many treatments are able to prevent diabetes in NOD mice when given weeks before diabetes develops (1). However, few treatments have been found that can inhibit diabetes development at the time of onset or reverse diabetes after hyperglycemia is observed (2–6). These two later pathogenic stages parallel potential intervention points in human type 1 diabetes. Identifying tolerance mechanisms that work at these points is important both for understanding the disease and for translating findings into clinical studies and results. In humans, antigen nonspecific anti-CD3 has been used with some efficacy (2, 7). Adding antigen specificity may improve treatment, and this issue is beginning to be addressed in mouse models, where treatment of diabetes is seen with lower doses of anti-CD3 if given along with islet peptides (6).
Recently, we have shown that antigen-pulsed DCs and IL-2 can expand functional antigen-specific T reg cells (8–10). To study T reg cells in the context of NOD mice, we have used CD4+ CD25+ TCR transgenic T cells from the BDC2.5 line (11). BDC2.5 T cells recognize a peptide that mimics an unidentified natural antigen in islet β cells (12). DC-expanded, BDC2.5 islet-specific T reg cells are >100-fold more potent in preventing diabetes than T reg cells derived from a polyclonal NOD T cell repertoire, indicating the importance of antigen specificity for T reg cell function (9). Islet-specific BDC T reg cells can also be expanded using beads coated with either mitogenic antibodies or tetramers, but it has not been determined if these bead-expanded T reg cells express high levels of Foxp3 protein (a transcription factor important for T reg cell function [13]) or if these cells are as potent as DC-expanded T reg cells for inhibition of diabetes (14, 15).
In some instances, islet-specific T reg cells have been shown to treat later stages of diabetes. In one experiment, in addition to treatment with T reg cells, islet transplant was used to rescue insulin production (14). In another experiment, the T reg cells were artificially induced by transfecting the T cells with Foxp3 (16). Both of these systems have drawbacks for potential translation into human therapy, the former because of the limited availability of islets for transplantation and the later because of the demands of human gene therapy. Therefore, it is important to establish the efficacy of naturally occurring islet-specific T reg cells in a cell therapy model and to use these cells without the addition of other demanding strategies.
In this study, we show that DC-expanded, islet-specific T reg cells can block diabetes months after initiation of insulitis and restore long-term normoglycemia when given to recent-onset diabetic mice. When compared with untreated diabetic controls, the diabetic mice successfully treated with T reg cells respond better in a glucose tolerance test and have increased numbers of Foxp3+ cells when compared with untreated diabetic mice. Interestingly, the Foxp3+ cells in treated mice are not from the injected T reg cells but derive from the recipient T cell repertoire.
RESULTS
After expansion with DCs, CD4+ CD25+ CD62L+ cells from BDC2.5 mice retain high Foxp3 and CD62L expression and divide in the pancreatic lymph node
Previously, we showed that bone marrow–derived DCs loaded with cognate peptide were effective at inducing expansion of CD4+ CD25+ T cells (8, 9). In this study, we further separated this population before expansion, according to expression of the lymph node homing and memory/activation marker CD62L, into CD4+ CD25+ CD62L+ and CD4+ CD25+ CD62L− (Fig. 1 A). CD62L+ cells can home to lymph nodes, which may be important for T reg cell function (17, 18). After expansion with DCs, the majority of CD62L+ cells maintained high levels of CD62L and Foxp3. DC-expanded CD62L− T cells expressed similar levels of Foxp3 but lower CD62L when compared with the CD62L+ cells (Fig. 1, B and C).
Figure 1.

Characterization of DC-expanded T reg cells from BDC2.5 NOD mice. (A) CD25-enriched cells from NOD BDC2.5 mice before (top) and after sorting for CD4+ CD25+ CD62L− (middle) or CD4+ CD25+ CD62L+ (bottom) cells. (B) Foxp3 expression on CD4+ CD25+ CD62L+ (top) or CD4+ CD25+ CD62L− (bottom) cells before and after expansion with DCs. (C) CD62L expression on CD4+ CD25+ CD62L− (left) and CD4+ CD25+ CD62L+ (right) BDC T cells after expansion with DCs. (D) DC-expanded BDC T reg cells were labeled with CFSE and injected into either 5-wk-old or 12-wk-old NOD females. CFSE and anti-BDC clonotype stainings are shown on cells gated on size and CD4 from lymph nodes or pancreatic lymph nodes. The percentage of cells in the indicated gates is shown. Results are representative of at least three experiments.
BDC2.5 T cells proliferate and expand in the pancreatic lymph node, but not other lymph nodes (14, 19), most likely because antigens from islet β cells are presented by DCs in the draining pancreatic lymph node (20). To check the in vivo activity of DC-expanded CD4+ CD25+ cells, these cells were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) and transferred into NOD mice. BDC2.5 T cells were followed using a clonotype antibody specific for the BDC2.5 TCR. 10 d later, few BDC clonotype+ cells remained in the peripheral lymph nodes, and most had not diluted CFSE. In contrast, in the pancreatic lymph node, clonotype+ T cells accumulated, and a substantial fraction had undergone one or more divisions, as indicated by lower CFSE fluorescence (Fig. 1 D). Therefore, like conventional T cells, the DC-expanded T reg cells home to and proliferate in antigen-draining lymph nodes, presumably in response to DCs presenting cognate antigen (21).
Some previous studies have shown that the number of T reg cells in NOD mice decreases with age (22, 23). An impaired ability of T reg cells to proliferate in vivo is one explanation to account for this observation. Other studies have observed constant numbers of T reg cells in different ages of NOD female mice (22, 24). In our observation, the ability of the DC-expanded BDC T reg cells to proliferate in response to antigen in the pancreatic lymph node did not substantially alter with the age of the host. At 5 wk, which correlates with the onset of insulitis, and at 12 wk, which correlates with onset of destructive diabetes, similar levels of CFSE dilution were observed (Fig. 1 D). Therefore, no alteration in environment occurs in the pancreatic lymph node during diabetes pathogenesis that inhibits the ability of T reg cells to proliferate.
Islet-specific T reg cells block spontaneous diabetes when given just before the time of diabetes onset
We tested whether these islet-specific T reg cells from BDC2.5 mice were effective at blocking the development of spontaneous diabetes in 13-wk-old female NOD mice. This time point is late in the pathogenic process, when diabetes begins and islet inflammation has usually progressed for at least 6 wk (25). We found that one dose of 3 × 105 DC-expanded BDC2.5 CD4+ CD25+ CD62L+ T cells could significantly delay diabetes when given at 13 wk; mice receiving these cells only began to slowly develop diabetes after 30 wk. In contrast, the CD4+ CD25+ CD62L− population had no effect (Fig. 2 A). In a second prevention experiment, smaller numbers of DC-expanded CD4+ CD25+ CD62L+ cells from BDC2.5 mice were given to prediabetic mice. Groups receiving 2 × 105, 5 × 104, or 104 cells had a significant delay in diabetes. Again, DC-expanded CD4+ CD25+ CD62L− cells had no effect on diabetes development (Fig. 2 B). In addition, CD62L+ T reg cells from NOD mice expanded with DCs and anti-CD3 were not able to inhibit diabetes development (Fig. 2 C), showing the need for islet-antigen specificity for tolerance induction, as observed in the NOD.scid transfer model (9). Therefore, small numbers of islet-specific CD62L+ T reg cells can block diabetes when given just before the mice normally develop diabetes.
Figure 2.

CD25+ CD62L+ T cells from BDC2.5 mice protect from diabetes development when given to prediabetic mice. 13-wk-old nondiabetic NOD females were given the indicated numbers of DC-expanded CD4+ CD25+ CD62L+ or CD4+ CD25+ CD62L− T cells from BDC2.5 (A–C) or NOD (C) mice, and diabetes was monitored weekly. The number of mice in each group is indicated in parentheses. In A, P = 0.0373 for PBS versus CD4+ CD25+ CD62L+ cells.
Treatment of diabetic mice with islet-specific T reg cells
We next turned to newly diabetic mice with elevated blood glucose levels. We determined the effectiveness of using a combination of islet-specific T reg cells and short-term insulin replacement using insulin pellets, which release insulin for ∼3 wk. In addition, we used the gut-derived glucagon-like peptide 1 (GLP-1), which stimulates insulin secretion (26) and lowers circulating glucose levels in type 2 diabetes patients and healthy human subjects (27, 28). Recently, it has been reported that GLP-1 promotes the growth and survival of the pancreatic β cells and decreases the metabolic requirement for insulin secretion (29).
In the first treatment experiment, recent-onset diabetic NOD mice were given insulin and GLP-1 for 3 wk to maintain normal glucose levels, while the induction of immune tolerance by the injected suppressor T cells takes place. Groups were matched for the range of starting blood glucose. In all four control mice that received GLP-1 but not T reg cells, the blood glucose rose again soon after exogenous insulin was not available. In contrast, in three out of five mice that received T reg cells and GLP-1, the blood glucose readings stayed below 200 mg/dL for an additional 90 d (Fig. 3 A).
Figure 3.

DC-expanded BDC T reg cells reverse hyperglycemia in recent-onset diabetic mice. (A) Diabetic female NOD mice were given insulin pellets and 1.5 × 106 BDC T reg cells, where indicated at days 0 and 14, and GLP-1. Blood glucose levels were measured on the days indicated. Each symbol represents an individual mouse in the treatment protocol indicated on the panel. (B) Diabetic female NOD mice were given BDC T reg cells (left) or BDC T reg cells and exendin-4 (right). Insulin pellets were given to maintain blood glucose levels below 250 mg/dl for 42 d, at which time all pellets were removed (as indicated by the arrows). Blood glucose levels were measured on the days indicated (for clarity, blood glucose levels during insulin treatment are not shown). The ratio of successfully treated/total in each group is shown in parentheses. Different symbols represent individual mice treated as indicated in the panel. (C) Initial blood glucose levels in diabetic mice that responded (left) or did not respond (right) to treatment with T reg cells and GLP-1 or exendin-4. Horizontal lines represent the means. (D) Summary of recent-onset diabetic NOD mice treated with BDC T reg cells. Successfully treated mice maintained long-term normoglycemia without exogenous insulin treatment. P = 0.004 for BDC T reg cells + exendin-4 or GLP-1 versus exendin-4 or GLP-1; P = 0.0429 for BDC T reg cells versus exendin-4 or GLP-1.
Because of a concern of potential residual exogenous insulin secretion from the pellets after the presumed cessation of its release at the 3-wk interval, insulin pellets were removed after 40 d in subsequent experiments. In addition, exendin-4, a more stable analogue of GLP-1, was given i.p. where indicated (30). Similar results to the ones shown in Fig. 3 A were obtained (Fig. 3 D and Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20061631/DC1). To determine the importance of adding exendin-4, recent-onset diabetic mice were given T reg cells alone or T reg cells and exendin-4. T reg cells alone could restore normoglycemia (Fig. 3, B and D). In our colony, diabetic NOD mice given no treatment do not become normoglycemic for long periods of time. An example of blood glucose levels from one cohort of NOD female mice followed from 12–25 wk is shown in Fig. S2.
Interestingly, mice successfully treated with T reg cells and exendin-4 started the experiment with a statistically significant lower mean level of initial blood glucose than those mice in which the same treatment failed; responders had a mean level of 365 mg/dl as compared with 467 mg/dl for nonresponders (Fig. 3 C). This suggests that the amount of residual β cell mass may be limiting the ability to treat diabetic mice via immune tolerance. Because groups were matched for initial blood glucose, as expected, mice given exendin-4 alone had the same mean initial blood glucose level (421 mg/dl) as mice given T reg cells and exendin-4 (including responders and nonresponders).
In all, a total of 41 mice were followed long term after treatment, and 14 out of 30 mice receiving T reg cells were successfully treated, whereas all 11 mice that did not receive T reg cells failed to maintain long-term normoglycemia (Fig. 3 D and not depicted). We conclude that diabetes can be reversed after onset by treatment with a combination of CD4+ CD25+ CD62L+ T reg cells from BDC2.5 mice and insulin, with or without GLP-1 receptor agonists.
Characterization of insulin response to glucose challenge and insulitis in T reg cell–treated mice
Maintenance of normoglycemia among diabetic mice treated with insulin, GLP-1, and islet-specific T reg cells suggests some restoration of β cell function. One measure of β cell function that has been shown to correlate with β cell mass is a glucose tolerance test: determining the time for mice to return to baseline blood glucose levels after i.p. glucose injection; better β cell function (and greater β cell mass) correlates with a faster response (31). We compared T reg cell–treated mice with three control groups: 12–15-wk-old nondiabetic, 30-wk-old nondiabetic, and recently diabetic NOD mice. The younger nondiabetic control mice exhibit a faster insulin response, as indicated by a more rapid return of blood glucose levels to baseline, than either the older nondiabetic or diabetic control mice. The response in the T reg cell–treated mice was similar to the prediabetic mice; one example of each group is shown in Fig. 4 A. This response can be represented by the area under the curve: a more rapid response corresponding to increased insulin secretion will provide a lower area. When summarized over numerous experiments, the response of T reg cell–treated mice to glucose challenge was significantly lower than the diabetic controls and not significantly different from 12–15-wk-old prediabetic NOD females (Fig. 4 B), suggesting that the treated mice regained the ability to secrete enough insulin to control the elevated glucose levels that occur after feeding, potentially via increased β cell mass.
Figure 4.

T reg cell–treated mice have a normal response to glucose challenge. Glucose tolerance test in T reg cell–treated diabetic mice. Blood glucose levels were followed for 90 min after i.p. glucose injection into female NOD mice either successfully treated after diabetes onset with BDC T reg cells (closed squares), 12–15-wk-old (open circles) or 30 wk-old nondiabetic mice (closed circles), or diabetic mice (closed diamonds). One example of each group is shown in (A). For all mice tested, the area under the curve as shown in A was calculated, and the means of each group are indicated by the horizontal lines (B). P ≤ 0.0001 for both T reg cell–treated versus diabetic mice and 12–15-wk-old nondiabetic versus diabetic mice; P = 0.027 for 30-wk-old nondiabetic versus diabetic mice.
The level of insulitis in DC-expanded T reg cell–treated mice was also compared with diabetic or 12–15- or 30-wk- old nondiabetic NOD females. In all groups, a mix of healthy and infiltrated islets was observed (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20061631/DC1). Comparable lymphocytic infiltrates were found in the salivary glands of all mice tested (unpublished data).
Islet-specific T reg cell–treated mice contain both pathogenic cells and increased T reg cells
Successfully treated mice were further characterized to look for phenotypes that correlate with treatment. Foxp3 is a transcription factor that can be used to identify T reg cells, especially in inflammatory conditions where some CD4+ CD25+ cells are recently activated T cells (13). Both the percentage of CD4+ cells that were Foxp3+ and the total number of Foxp3+ CD4+ T cells was significantly higher in lymph nodes of T reg cell–treated mice as compared with 12–15-wk-old NOD females, either diabetic or nondiabetic. This difference was especially pronounced in the draining pancreatic lymph node, where the T reg cell–treated mice had as high as 30% Foxp3+ cells within the CD4+ T cell population. The mean percentage was 25% in treated mice as compared with 15% in diabetic or 14% in nondiabetic controls (Fig. 5, A and B; and not depicted).
Figure 5.

Pathogenic and T reg cells in lymphoid organs of T reg cell–treated diabetic mice. (A) Cells from pancreatic lymph nodes, peripheral lymph nodes, or spleens of female NOD mice either nondiabetic (top) or successfully treated with BDC T reg cells (bottom) were harvested and stained for CD4-FITC, CD25-APC, and Foxp3-PE. The plots shown have been gated first on size and then on CD4+ cells. The percentage of cells in the two gates shown (CD25+ Foxp3− and Foxp3+) are indicated on the plots. (B) Summary of the percentage of Foxp3+ cells of CD4+ cells (as gated in A) for BDC T reg cell–treated (closed squares), diabetic (closed diamonds), or nondiabetic (open circles) NOD female mice. Horizontal lines represent the means. In both pancreatic and peripheral lymph nodes, P < 0.004 for T reg cell–treated versus either diabetic or nondiabetic mice. (C) Spleen cells from either newly diabetic mice (closed circles), diabetic mice successfully treated with BDC T reg cells (triangles), or nondiabetic mice (open circles) were transferred to NOD.scid females. Development of diabetes was then monitored by urine glucose. The number of mice in each group is indicated in parentheses.
To determine if pathogenic T cells were still present in the treated mice, spleen cells from treated mice were transferred to NOD.scid mice, and diabetes development was followed. Mice injected with spleen cells from diabetic control mice developed diabetes between 30 and 80 d after transfer, and NOD.scid mice injected with spleen cells from T reg cell–treated mice developed diabetes in a similar timeframe (Fig. 5 C). Therefore, mice successfully treated with DC-expanded T reg cells retain pathogenic cells capable of causing diabetes but also have increased numbers of T reg cells.
Foxp3+ cells in mice successfully treated with T reg cells derive from the diabetic recipient
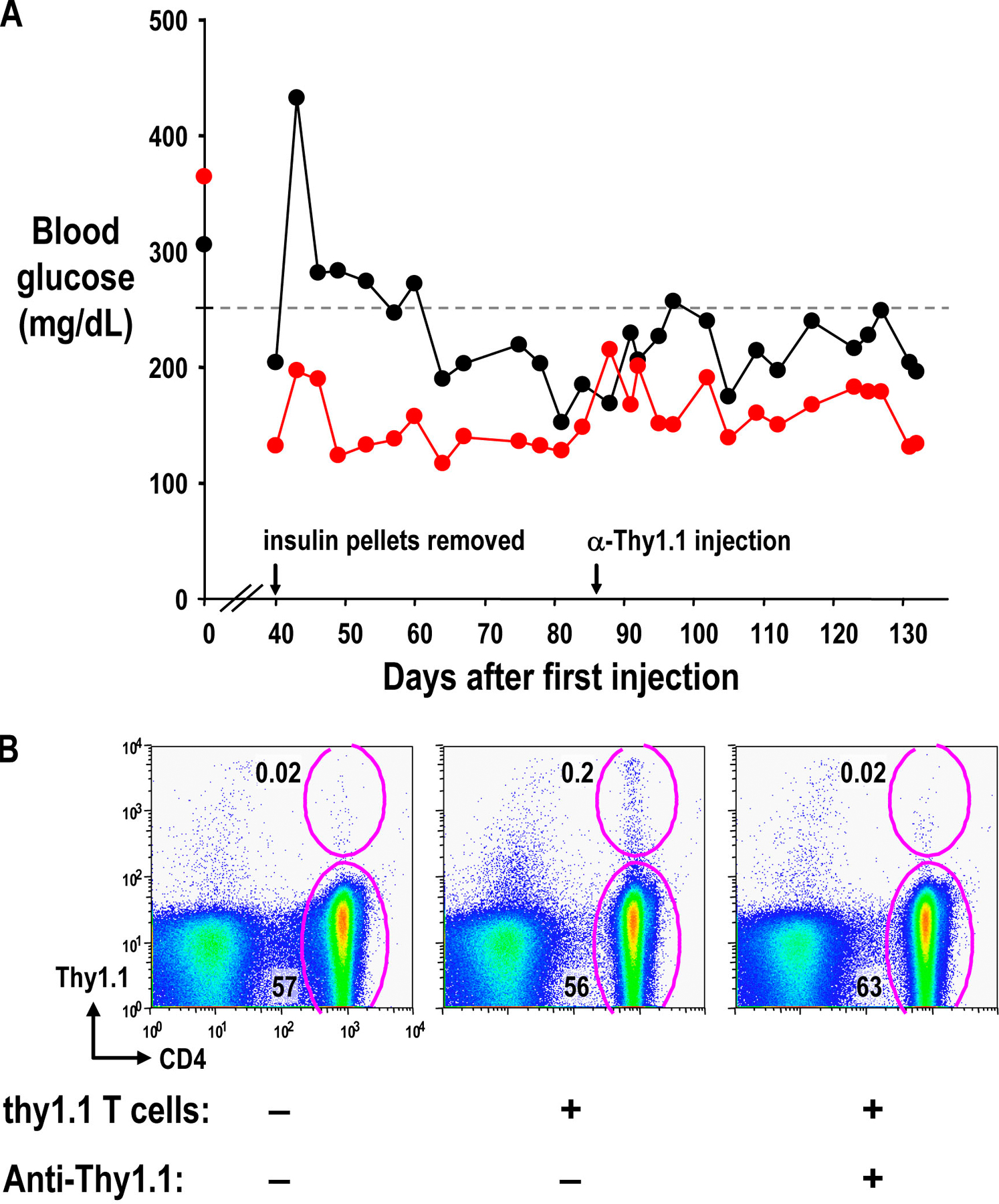
To determine whether the increase in Foxp3+ cells was caused by the expansion of injected cells, we injected DC-expanded T reg cells from BDC NOD Thy1.1 mice and exendin-4 into diabetic NOD Thy1.2 mice according to the treatment protocol described in Fig. 3 B. 10 d after the insulin pellets were removed, mice that responded to T reg cell treatment (with blood glucose levels >250 mg/dl) were characterized for expression of Thy1.1 and Foxp3. However, in those treated mice almost none of the CD4+ Foxp3+ cells in lymphoid organs were Thy1.1+, and CD4+ T cells in the pancreas of treated mice were Thy1.1−, indicating that they derived from the diabetic host and not the injected T reg cells (Fig. 6, A and B; and not depicted). In a similar experiment, no notable population of Thy1.1 T cells was found at a later time point after successful treatment with Thy1.1+ T reg cells (unpublished data). As a positive control, DC-expanded CD62L+ or CD62L− T reg cells from BDC2.5 Thy1.1 mice were injected into prediabetic NOD Thy1.2 mice. After 1 wk, CD4+ Thy1.1+ T cells could be found in both the pancreatic lymph node and the pancreas (Fig. 6, C and D). In addition, pancreas sections from Thy1.2+ diabetic mice treated with Th1.1+ T reg cells were stained with antibodies specific for CD4, Foxp3, and Thy1.1, and numerous CD4+ Foxp3+ Thy1.1− cells were found in the lymphocytic infiltrate (Fig. 6 E). As a positive control for Thy1.1 staining, the lymph node from a Thy1.1-expressing mouse was stained with the same antibodies (Fig. 6 F); isotype control staining gave minimal signals in all channels (not depicted). Therefore, during long-term successful treatment, the donor T reg cells can no longer be found in the recipients, even at the disease sites.
Figure 6.

Thy1.1+ BDC T reg cells do not persist after successful treatment of diabetes. (A and B) Diabetic NOD Thy1.2 mice successfully treated with 106 Thy1.1 BDC T reg cells were harvested 50 d after T reg cell injection (10 d after insulin pellets were removed). The percentage of lymphocytes in the pancreatic lymph node (A) or the pancreas (B) that are either CD4+ Thy1.1+ or CD4+ Thy1.1− are shown. (C and D) 12-wk-old nondiabetic Thy1.2 NOD mice were injected with nothing (left) or 106 CD62L− (middle) or CD62L+ (right) T reg cells from BDC Thy1.1 mice. The percentages of lymphocytes 7 d later in the pancreatic lymph node (C) or the pancreas (D) that are either CD4+ Thy1.1+ or CD4+ Thy1.1− are shown. (E and F) Pancreas from diabetic NOD Thy1.2 mice successfully treated with Thy1.1 BDC T reg cells (E) or pancreatic lymph node of prediabetic NOD Thy1.1 mice (F) were stained for CD4 (green), Foxp3 (red), and Thy1.1 (blue).
We also depleted Thy1.1+ cells from two mice that had been successfully treated with Thy1.1+ T reg cells. This treatment did not lead to diabetes, because blood glucose levels did not substantially change for 7 wk after depletion (Fig. S4 A, available at http://www.jem.org/cgi/content/full/jem.20061631/DC1). As the Thy1.1+ cells were already almost undetectable, it was difficult to prove that depletion was successful. However, we could use the same Thy1.1-specific antibody to deplete Thy1.1+ cells from control Thy1.2 mice injected with 5 × 106 CD4+ Thy1.1+ cells (Fig. S 4 B).
In addition, CD4+ CD25high cells were sorted from treated mice, and these T reg cells were able to suppress proliferation of CD4+ CD25− T cells from BDC2.5 mice when stimulated with anti-CD3 but not with BDC peptide, suggesting that the majority of these cells were not BDC specific (unpublished data). Collectively, this suggests that the injected T reg cells were not sustained in large numbers but allowed endogenous T reg cells to maintain a favorable ratio of Foxp3+ cells to Foxp3− cells.
DISCUSSION
Treatment of autoimmunity at late pathogenic stages
Individuals with a high risk for developing diabetes can be identified by genetic markers and the presence of high affinity islet autoantibodies (32, 33), and at the time of diagnosis, type 1 diabetes patients retain ∼10% of their normal β cell mass (34). Therefore, inducing T cell tolerance and halting immune-mediated destruction at these two stages of disease may contribute to successful therapy for type 1 diabetes patients. In this study, we addressed T reg cells as inducers of tolerance, but in contrast to most previous studies, we assessed the capacity of T reg cells to treat established autoimmunity rather than prevent it. In the one previous example of natural T reg cells treating spontaneous diabetes, 107 islet-specific T reg cells were used, an amount almost 10-fold higher than that used in this study (14). We show that islet-specific T reg cells, but not polyclonal T reg cells, can be used to treat autoimmune diabetes in NOD mice months after insulitis has begun and even after overt diabetes has developed. These T reg cells have been expanded in number by DCs presenting a specific peptide. This type of expansion of T reg cells by DCs also happens in vivo (9) and represents an important control of the number and, potentially, function of T reg cells.
In the prediabetic mice, CD62L+ but not CD62L− T reg cells were able to inhibit diabetes development (Fig. 2). The CD62L+ T reg cells may be more efficacious, because the CD62L− population may contain more recently activated T cells; alternatively, it is possible that, because CD62L is a homing marker for lymph node trafficking, the T reg cells must home to lymph nodes to gain efficacy. Supporting this second possibility, 7 d after transfer of 106 Thy1.1+ T reg cells sorted initially on either CD62L+ or CD62L− cells, fewer Thy1.1+ cells were found in the pancreatic lymph node of mice injected with CD62L− T reg cells (Fig. 6 C). Interestingly, CD6L− cells were also found in lower numbers in the pancreas (Fig. 6 D), suggesting that CD62L is necessary for T reg cells to home first to the lymph node before trafficking to the inflamed tissue.
Limited evidence exists for the therapeutic role of T reg cells in other autoimmune models. In two different mouse models of colitis induced by injection of pathogenic T cells into lymphopoenic mice, T reg cells given after development of severe intestinal inflammation could reverse colitis (35, 36). In experimental autoimmune encephalitis, although transfer of T reg cells has not been shown to reverse disease, spontaneous remission correlated with increased numbers of endogenous T reg cells in the central nervous system, suggesting a role for T reg cells in the natural recovery of established autoimmunity (37).
Restoration of insulin production in new-onset diabetes
Rescue of insulin production is another important aspect of treatment of diabetes. Islet cell transplant can increase insulin production but requires demanding immunosuppressive drugs to maintain transplantation tolerance. In addition, donors are limited, making this an option for only a small fraction of type 1 diabetic patients (38). In our experiments, 50% of newly diabetic mice were successfully treated with T reg cells without islet transplant. Interestingly, the T reg cell–treated diabetic mice resemble prediabetic mice in both blood glucose levels and response to glucose challenge, suggesting considerable restoration of β cell function. Those mice in which blood glucose remained high may have had too few remaining insulin-producing cells such that even with tolerance induction, these mice would not recover. Significantly lower starting blood glucose levels in mice in which the treatment worked supports the idea that β cell mass, and not tolerance induction, was limiting the success rate of treatment with islet-specific T reg cells (Fig. 3 C).
We also tested the effects of GLP-1, because it can increase β cell proliferation and decrease β cell apoptosis, as well as have metabolic effects that lower insulin requirements, probably allowing β cells to rest (39). However, addition of GLP-1 or exendin-4 did not improve the fraction of diabetic mice successfully treated with T reg cells, in contrast to some recently reported findings (5, 40). One possible explanation for the observed differences is that GLP-1 cannot promote differentiation from precursors; in this case, if mice did not have enough β cells remaining, GLP-1 would not increase the efficacy of T reg cell treatment. In addition, the absence of GLP-1 effects may be caused by the extremely short in vivo half-life of GLP-1 (41). Although exendin-4 is more stable in vivo, it is a peptide of nonmammalian origin, and its continuous administration may lead to the production of neutralizing antibodies. New GLP-1 analogues with increased half-lives may help clarify this issue. β cell growth factors may be important for the restoration of endogenous insulin production without islet transplant as long as autoimmunity can be contained with T reg cells. For example, the addition of gastrin, which has also been shown to increase β cell proliferation, may improve the efficacy of T reg cell treatment of diabetes (42, 43).
Expansion of T reg cells in diabetic recipients
Diabetic mice treated with T reg cells have higher numbers of Foxp3+ cells in lymph nodes compared with either prediabetic or diabetic NOD females. Although immediately after injection BDC T reg cells divide and expand in the pancreatic lymph node, when examined 50 d after treatment, the increased Foxp3+ cells in the treated mice derive almost entirely from the recipient diabetic mice and not from the injected cells. This was seen in both the pancreatic lymph node and the pancreas. We suspect that the initial expansion of islet-specific T reg cells sets up a regulatory environment in the pancreas and draining lymph node that encourages either expansion or differentiation of endogenous T reg cells to balance out the increased numbers of pathogenic cells likely found in diabetic mice. The ability of splenocytes from successfully treated mice to transfer diabetes suggests that a dominant, nondeletional tolerance mechanism is important. Interestingly, it was previously shown that transient expression of TGF-β in the pancreas of NOD mice leads to protection for diabetes development and induction of T reg cells (44).
In some other experimental systems, injected T reg cells are necessary for maintenance of tolerance. For example, antigen-induced Thy1.1+ T reg cells prevent autoimmune ovarian disease. In this system, considerable numbers of the Thy1.1+ T reg cells are maintained long term, but disease returns if the Thy1.1+ T cells are depleted (45). Similar results were found in a mouse model of multiple sclerosis (46). However, induction of new T reg cells by other T reg cells has been proposed and may involve TGF-β or other cytokines, as has been shown in vitro (47). In transplantation, anti-CD4 blockade in mice devoid of natural T reg cells leads to acceptance of allogeneic skin grafts. This tolerance is TGF-β dependent and involves the induction of Foxp3+ cells, but doesn't directly show one T reg cell population inducing a new population of T reg cells (48, 49).
Our findings represent the first demonstration of a defined set of natural T reg cells inducing new T reg cells in vivo. The injected T reg cells could have a direct effect on expansion or differentiation of new T reg cells through cytokine or co-stimulatory molecules. It is also possible that the injected T reg cells “license” the DCs in the pancreas or draining lymph node in a way that improves the ability of these DCs to expand or differentiate new T reg cells. These results also show that T reg cells can be expanded from diabetic mice, which may allow the design of antigen-specific therapies in new-onset diabetic patients.
In summary, the experiments described in this paper show that T cell tolerance induced by DC-expanded, islet-specific T reg cells can both increase endogenous T reg cells and reverse autoimmune diabetes in NOD mice. This antigen-specific therapy can now be added to the short list of mostly nonspecific methods for reversing diabetes, including anti-CD3 and TGF-β, which may work through induction of T reg cells (50). These results, in conjunction with our previous report, reiterate the role for DCs in expanding disease-treating T reg cells. Given the fact that DCs expand disease-suppressive, antigen-specific T reg cells, a study of these interacting cells could be informative.
MATERIALS AND METHODS
Mice.
NOD mice and BDC2.5 TCR transgenic mice on the NOD genetic background were purchased from the Jackson Laboratory and bred under specific pathogen-free conditions at the Rockefeller University. Mice of both sexes were used at the indicated ages according to our institutional guidelines. Protocols were approved by the Institutional Animal Care and Use Committee.
Antibodies and staining.
Antibodies specific for: Gr1 (RB6-8C5), CD4 (H129.19), CD86 (GL1), CD25 (7D4), CD62L (MEL-14), CD25 (PC61), CD4 (RM4-5), and Thy1.1 (OX-7); and isotype controls, 7-amino-actinomycin D, and PE-streptavidin were purchased from BD Biosciences. Streptavidin–Pacific blue was purchased from Invitrogen. Anti-Foxp3 (FJK-16s) conjugated to PE or APC was purchased from eBioscience. A hybridoma expressing the anticlonotype antibody specific for the BDC2.5 TCR (BDC) was provided by O. Kanagawa (Washington University, St. Louis, MO), and the antibody was purified and biotinylated. All staining was done according to standard protocols.
Purification and DC-based expansion of regulatory cells.
Bone marrow–derived DCs from NOD mice were matured with LPS and enriched for CD86+ cells using magnetic beads, as described previously (9). T cells were purified from lymph nodes and spleens from BDC2.5 NOD mice by first labeling with anti-CD4–FITC, CD62L-APC, and CD25-biotin, followed by streptavidin-PE and enriching for CD25+ cells with magnetic beads coated with PE-specific antibodies (Miltenyi Biotec). CD4+ CD25+ CD62L+ or CD4+ CD25+ CD62L− cells were then sorted to >98% purity. The T cells and DCs were cultured at a ratio of 1:1, as previously described (9), for 6–8 d with 100 ng/ml BDC mimetope peptide (termed 1040–55; sequence RVRPLWVRME) (12) and 200 U/ml rHuIL2 (Chiron Corp). Unless otherwise indicated, the T reg cells used in this study are CD4+ CD25+ CD62L+ cells from BDC2.5 NOD mice expanded with DCs.
Diabetes experiments.
Treatment of prediabetic mice was as follows. 13-wk-old nondiabetic female NOD mice were given the numbers of DC-expanded T reg cells via i.v. injection indicated in the figures, and diabetes was monitored by urine glucose once per week. Diabetes was defined as three consecutive positive readings.
Treatment of diabetic NOD mice was as follows. To have an average of 5–10 newly diabetic mice at any one time, we have followed a cohort of ∼200–300 NOD females between 12–30 wk old. Diabetic NOD females were identified by urine glucose and confirmed by a blood glucose reading >250 mg/dl. Within each experiment, these starting blood glucose levels were matched between groups. Insulin pellets were then implanted s.c. as needed to maintain blood glucose <250 mg/dl for 40 d and removed. Each pellet contains 13 mg of insulin and continuously releases ∼0.1 unit/ implant (LinShin Canada, Inc.). In the groups indicated, mice were injected on days 0 and 14 with 0.8–1.5 × 106 BDC T reg cells by i.v. injection and with 50 ng exendin-4 (Bachem) on days 0, 1, 2, 7, 8, and 9 by i.p. injection. The subsequent development of diabetes was monitored by blood glucose readings two to three times per week. All mice denoted in the figures as responders, successfully treated, or T reg treated were followed for a minimum of 2 mo and for as long as 6 mo after removal of exogenous insulin. For the experiment shown in Fig. 3 A, NOD females with diabetes were identified by urine glucose within 5 d of onset and given insulin via pellets that secrete continuously for ∼3 wk. All mice received i.p. injection of 5 μg GLP-1 (Bachem) on days 2, 3, 5, 11, 16, 23, 25, and 29. Within 3 d of receiving the insulin pellet, one group was injected i.v. with 1.5 × 106 DC-expanded CD4+ CD25+ CD62L+ cells isolated from BDC2.5 mice.
Depletion with anti-Thy1.1.
100 μg anti-Thy1.1 (clone HIS51) was given i.v., and expression of Thy1.1 after depletion was monitored using anti-Thy1.1 (clone OX-7) at the times indicated in the figures.
Glucose tolerance test.
Mice were kept without food for 15 h and were given 2 mg of glucose per gram of bodyweight i.p. Blood glucose was monitored at the intervals indicated in Fig. 4 A for 90 min.
Histology.
Insulitis and sialitis were checked by observing lymphocytic infiltrates in the pancreatic islets or salivary glands using hematoxylin and eosin sections. Insulitis was scored as follows: no insulitis, periinsulitis, intrainsulitis with <70% of the islet infiltrated, or intrainsulitis with >70% of the islet infiltrated.
Immunofluorescence.
Tissue was frozen in optimal cutting temperature compound. 10-μm sections were air dried and fixed in acetone. Sections were stained with anti-Thy1.1–FITC (Ox-7), anti-FITC–Alexa Fluor 488 (Invitrogen), Foxp3-PE, anti–rat–Alexa Fluor 546 (Invitrogen), and CD4–Alexa Fluor 647 (L3T4; BD Biosciences) or isotype controls and blocked with 5% mouse serum and 2–5% rat serum. Images were acquired on an inverted laser scanning confocal microscope (LSM 510; Carl Zeiss MicroImaging, Inc.) using a 40× objective.
Statistics.
Comparisons of groups in Fig. 3 D were analyzed by Fisher's exact test. Groups in Fig. 2 were compared by the Mann-Whitney U test. Groups in Fig. 3 C, Fig. 4 B, and Fig. 5 B were compared using a t test.
Online supplemental material.
Fig. S1 shows an additional example of treatment of diabetic mice with T reg cells and exendin-4. Fig. S2 shows blood glucose levels of female NOD mice from 13 to 25 wk of age. Fig. S3 shows insulitis levels in T reg cell–treated diabetic mice. Fig. S4 shows the results of depletion of Thy1.1+ cells with anti-Thy1.1. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20061631/DC1.
Supplemental Material
Acknowledgments
We would like to thank Klara Velinzon for expert cell sorting and the Rockefeller University Bio-Imaging Resource Center (Alison North, Shivaprasad Bhuvanendran, and Mathieu Marchand), Maggi Pack, and Juliana Idoyaga for help with immunostaining.
This work was supported by program project grants from the Juvenile Diabetes Research Foundation International and the National Institutes of Health (AI 51573) to R. Steinman, and by a career award from the National Institutes of Health (5K01DK071586) to K. Tarbell.
The authors have no conflicting financial interests.
Abbreviations used: CFSE, carboxyfluorescein diacetate succinimidyl ester; GLP-1, glucagon-like peptide 1; NOD, nonobese diabetic.
X. Luo's present address is Depts. of Medicine and Surgery, Northwestern University Feinberg School of Medicine, Chicago, IL 60611.
References
- 1.Shoda, L.K., D.L. Young, S. Ramanujan, C.C. Whiting, M.A. Atkinson, J.A. Bluestone, G.S. Eisenbarth, D. Mathis, A.A. Rossini, S.E. Campbell, et al. 2005. A comprehensive review of interventions in the NOD mouse and implications for translation. Immunity. 23:115–126. [DOI] [PubMed] [Google Scholar]
- 2.Herold, K.C., W. Hagopian, J.A. Auger, E. Poumian-Ruiz, L. Taylor, D. Donaldson, S.E. Gitelman, D.M. Harlan, D. Xu, R.A. Zivin, and J.A. Bluestone. 2002. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N. Engl. J. Med. 346:1692–1698. [DOI] [PubMed] [Google Scholar]
- 3.Chatenoud, L., E. Thervet, J. Primo, and J.F. Bach. 1994. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc. Natl. Acad. Sci. USA. 91:123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luo, X., H. Yang, I.S. Kim, F. Saint-Hilaire, D.A. Thomas, B.P. De, E. Ozkaynak, T. Muthukumar, W.W. Hancock, R.G. Crystal, and M. Suthanthiran. 2005. Systemic transforming growth factor-beta1 gene therapy induces Foxp3+ regulatory cells, restores self-tolerance, and facilitates regeneration of beta cell function in overtly diabetic nonobese diabetic mice. Transplantation. 79:1091–1096. [DOI] [PubMed] [Google Scholar]
- 5.Ogawa, N., J.F. List, J.F. Habener, and T. Maki. 2004. Cure of overt diabetes in NOD mice by transient treatment with anti-lymphocyte serum and exendin-4. Diabetes. 53:1700–1705. [DOI] [PubMed] [Google Scholar]
- 6.Bresson, D., L. Togher, E. Rodrigo, Y. Chen, J.A. Bluestone, K.C. Herold, and M. von Herrath. 2006. Anti-CD3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing Tregs. J. Clin. Invest. 116:1371–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keymeulen, B., E. Vandemeulebroucke, A.G. Ziegler, C. Mathieu, L. Kaufman, G. Hale, F. Gorus, M. Goldman, M. Walter, S. Candon, et al. 2005. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N. Engl. J. Med. 352:2598–2608. [DOI] [PubMed] [Google Scholar]
- 8.Yamazaki, S., T. Iyoda, K. Tarbell, K. Olson, K. Velinzon, K. Inaba, and R.M. Steinman. 2003. Direct expansion of functional CD25+ CD4+ regulatory T cells by antigen-processing dendritic cells. J. Exp. Med. 198:235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tarbell, K.V., S. Yamazaki, K. Olson, P. Toy, and R.M. Steinman. 2004. CD25+ CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J. Exp. Med. 199:1467–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamazaki, S., M. Patel, A. Harper, A. Bonito, H. Fukuyama, M. Pack, K.V. Tarbell, M. Talmor, J.V. Ravetch, K. Inaba, and R.M. Steinman. 2006. Effective expansion of alloantigen-specific Foxp3+ CD25+ CD4+ regulatory T cells by dendritic cells during the mixed leukocyte reaction. Proc. Natl. Acad. Sci. USA. 103:2758–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katz, J.D., B. Wang, K. Haskins, C. Benoist, and D. Mathis. 1993. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 74:1089–1100. [DOI] [PubMed] [Google Scholar]
- 12.Judkowski, V., C. Pinilla, K. Schroder, L. Tucker, N. Sarvetnick, and D.B. Wilson. 2001. Identification of MHC class II-restricted peptide ligands, including a glutamic acid decarboxylase 65 sequence, that stimulate diabetogenic T cells from transgenic BDC2.5 nonobese diabetic mice. J. Immunol. 166:908–917. [DOI] [PubMed] [Google Scholar]
- 13.Hori, S., T. Nomura, and S. Sakaguchi. 2003. Control of regulatory T cell development by the transcription factor foxp3. Science. 299:1057– 1061. [DOI] [PubMed] [Google Scholar]
- 14.Tang, Q., K.J. Henriksen, M. Bi, E.B. Finger, G. Szot, J. Ye, E.L. Masteller, H. McDevitt, M. Bonyhadi, and J.A. Bluestone. 2004. In vitro–expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J. Exp. Med. 199:1455–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Masteller, E.L., M.R. Warner, Q. Tang, K.V. Tarbell, H. McDevitt, and J.A. Bluestone. 2005. Expansion of functional endogenous antigen-specific CD4+CD25+ regulatory T cells from nonobese diabetic mice. J. Immunol. 175:3053–3059. [DOI] [PubMed] [Google Scholar]
- 16.Jaeckel, E., H. von Boehmer, and M.P. Manns. 2005. Antigen-specific FoxP3-transduced T-cells can control established type 1 diabetes. Diabetes. 54:306–310. [DOI] [PubMed] [Google Scholar]
- 17.Szanya, V., J. Ermann, C. Taylor, C. Holness, and C.G. Fathman. 2002. The subpopulation of CD4+CD25+ splenocytes that delays adoptive transfer of diabetes expresses l-selectin and high levels of CCR7. J. Immunol. 169:2461–2465. [DOI] [PubMed] [Google Scholar]
- 18.You, S., G. Slehoffer, S. Barriot, J.F. Bach, and L. Chatenoud. 2004. Unique role of CD4+CD62L+ regulatory T cells in the control of autoimmune diabetes in T cell receptor transgenic mice. Proc. Natl. Acad. Sci. USA. 101:14580–14585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoglund, P., J. Mintern, C. Waltzinger, W. Heath, C. Benoist, and D. Mathis. 1999. Initiation of autoimmune diabetes by developmentally regulated presentation of islet cell antigens in the pancreatic lymph nodes. J. Exp. Med. 189:331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turley, S., L. Poirot, M. Hattori, C. Benoist, and D. Mathis. 2003. Physiological β cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J. Exp. Med. 198:1527–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang, Q., J.Y. Adams, A.J. Tooley, M. Bi, B.T. Fife, P. Serra, P. Santamaria, R.M. Locksley, M.F. Krummel, and J.A. Bluestone. 2006. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat. Immunol. 7:83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gregori, S., N. Giarratana, S. Smiroldo, and L. Adorini. 2003. Dynamics of pathogenic and suppressor T cells in autoimmune diabetes development. J. Immunol. 171:4040–4047. [DOI] [PubMed] [Google Scholar]
- 23.Pop, S.M., C.P. Wong, D.A. Culton, S.H. Clarke, and R. Tisch. 2005. Single cell analysis shows decreasing FoxP3 and TGFβ1 coexpressing CD4+CD25+ regulatory T cells during autoimmune diabetes. J. Exp. Med. 201:1333–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berzins, S.P., E.S. Venanzi, C. Benoist, and D. Mathis. 2003. T-cell compartments of prediabetic NOD mice. Diabetes. 52:327–334. [DOI] [PubMed] [Google Scholar]
- 25.Andre, I., A. Gonzalez, B. Wang, J. Katz, C. Benoist, and D. Mathis. 1996. Checkpoints in the progression of autoimmune disease: lessons from diabetes models. Proc. Natl. Acad. Sci. USA. 93:2260–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mojsov, S., G.C. Weir, and J.F. Habener. 1987. Insulinotropin: glucagon-like peptide 1 [7-37] co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J. Clin. Invest. 79:616–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nathan, D.M., E. Screiber, H. Fogel, S. Mojsov, and J.F. Habener. 1992. Insulinotropic actions of glucagon-like peptide-I [7-37] administered to diabetic and non-diabetic human subjects. Diabetes Care. 15:270–276. [DOI] [PubMed] [Google Scholar]
- 28.Gutniak, M., C. Orskov, J.J. Holst, B. Ahren, and S. Efendic. 1992. Antidiabetogenic effect of glucagon-like peptide-I [7-36] amide in normal subjects and patients with diabetes mellitus. N. Engl. J. Med. 326:1316–1322. [DOI] [PubMed] [Google Scholar]
- 29.Li, Y., T. Hansotia, B. Yusta, F. Ris, P.A. Halban, and D.J. Drucker. 2003. Glucagon-like peptide-1 receptor signaling modulates beta cell apoptosis. J. Biol. Chem. 278:471–478. [DOI] [PubMed] [Google Scholar]
- 30.Xu, G., D.A. Stoffers, J.F. Habener, and S. Bonner-Weir. 1999. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes. 48:2270–2276. [DOI] [PubMed] [Google Scholar]
- 31.Amrani, A., S. Durant, M. Throsby, J. Coulaud, M. Dardenne, and F. Homo-Delarche. 1998. Glucose homeostasis in the nonobese diabetic mouse at the prediabetic stage. Endocrinology. 139:1115–1124. [DOI] [PubMed] [Google Scholar]
- 32.Achenbach, P., K. Koczwara, A. Knopff, H. Naserke, A.G. Ziegler, and E. Bonifacio. 2004. Mature high-affinity immune responses to (pro)insulin anticipate the autoimmune cascade that leads to type 1 diabetes. J. Clin. Invest. 114:589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gianani, R., and G.S. Eisenbarth. 2005. The stages of type 1A diabetes: 2005. Immunol. Rev. 204:232–249. [DOI] [PubMed] [Google Scholar]
- 34.Couper, J.J., I. Hudson, G.A. Werther, G.L. Warne, J.M. Court, and L.C. Harrison. 1991. Factors predicting residual beta-cell function in the first year after diagnosis of childhood type 1 diabetes. Diabetes Res. Clin. Pract. 11:9–16. [DOI] [PubMed] [Google Scholar]
- 35.Mottet, C., H.H. Uhlig, and F. Powrie. 2003. Cure of colitis by CD4+CD25+ regulatory T cells. J. Immunol. 170:3939–3943. [DOI] [PubMed] [Google Scholar]
- 36.Liu, H., B. Hu, D. Xu, and F.Y. Liew. 2003. CD4+CD25+ regulatory T cells cure murine colitis: the role of IL-10, TGF-beta, and CTLA4. J. Immunol. 171:5012–5017. [DOI] [PubMed] [Google Scholar]
- 37.McGeachy, M.J., L.A. Stephens, and S.M. Anderton. 2005. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J. Immunol. 175:3025–3032. [DOI] [PubMed] [Google Scholar]
- 38.Naftanel, M.A., and D.M. Harlan. 2004. Pancreatic islet transplantation. PLoS Med. 1:e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Egan, J.M., A. Bulotta, H. Hui, and R. Perfetti. 2003. GLP-1 receptor agonists are growth and differentiation factors for pancreatic islet beta cells. Diabetes Metab. Res. Rev. 19:115–123. [DOI] [PubMed] [Google Scholar]
- 40.Yang, Z., M. Chen, J.D. Carter, C.S. Nunemaker, J.C. Garmey, S.D. Kimble, and J.L. Nadler. 2006. Combined treatment with lisofylline and exendin-4 reverses autoimmune diabetes. Biochem. Biophys. Res. Commun. 344:1017–1022. [DOI] [PubMed] [Google Scholar]
- 41.Deacon, C.F., A.H. Johnsen, and J.J. Holst. 1995. Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo. J. Clin. Endocrinol. Metab. 80:952–957. [DOI] [PubMed] [Google Scholar]
- 42.Rooman, I., and L. Bouwens. 2004. Combined gastrin and epidermal growth factor treatment induces islet regeneration and restores normoglycaemia in C57Bl6/J mice treated with alloxan. Diabetologia. 47:259–265. [DOI] [PubMed] [Google Scholar]
- 43.Brand, S.J., S. Tagerud, P. Lambert, S.G. Magil, K. Tatarkiewicz, K. Doiron, and Y. Yan. 2002. Pharmacological treatment of chronic diabetes by stimulating pancreatic beta-cell regeneration with systemic co-administration of EGF and gastrin. Pharmacol. Toxicol. 91:414–420. [DOI] [PubMed] [Google Scholar]
- 44.Peng, Y., Y. Laouar, M.O. Li, E.A. Green, and R.A. Flavell. 2004. TGF-β regulates in vivo expansion of Foxp3-expressing CD4+CD25+ regulatory T cells responsible for protection against diabetes. Proc. Natl. Acad. Sci. USA. 101:4572–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samy, E.T., L.A. Parker, C.P. Sharp, and K.S. Tung. 2005. Continuous control of autoimmune disease by antigen-dependent polyclonal CD4+CD25+ regulatory T cells in the regional lymph node. J. Exp. Med. 202:771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hori, S., M. Haury, J.J. Lafaille, J. Demengeot, and A. Coutinho. 2002. Peripheral expansion of thymus-derived regulatory cells in anti-myelin basic protein T cell receptor transgenic mice. Eur. J. Immunol. 32:3729–3735. [DOI] [PubMed] [Google Scholar]
- 47.Zheng, S.G., J.H. Wang, J.D. Gray, H. Soucier, and D.A. Horwitz. 2004. Natural and induced CD4+CD25+ cells educate CD4+CD25− cells to develop suppressive activity: the role of IL-2, TGF-β, and IL-10. J. Immunol. 172:5213–5221. [DOI] [PubMed] [Google Scholar]
- 48.Waldmann, H., E. Adams, P. Fairchild, and S. Cobbold. 2006. Infectious tolerance and the long-term acceptance of transplanted tissue. Immunol. Rev. 212:301–313. [DOI] [PubMed] [Google Scholar]
- 49.Cobbold, S.P., R. Castejon, E. Adams, D. Zelenika, L. Graca, S. Humm, and H. Waldmann. 2004. Induction of foxP3+ regulatory T cells in the periphery of T cell receptor transgenic mice tolerized to transplants. J. Immunol. 172:6003–6010. [DOI] [PubMed] [Google Scholar]
- 50.Bisikirska, B., J. Colgan, J. Luban, J.A. Bluestone, and K.C. Herold. 2005. TCR stimulation with modified anti-CD3 mAb expands CD8+ T cell population and induces CD8+CD25+ Tregs. J. Clin. Invest. 115:2904–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}