

Abstract
Recirculation of fluid and cells through lymphatic vessels plays a key role in normal tissue homeostasis, inflammatory diseases, and cancer. Despite recent advances in understanding lymphatic function (Alitalo, K., T. Tammela, and T.V. Petrova. 2005. Nature. 438:946–953), the cellular features responsible for entry of fluid and cells into lymphatics are incompletely understood. We report the presence of novel junctions between endothelial cells of initial lymphatics at likely sites of fluid entry. Overlapping flaps at borders of oak leaf–shaped endothelial cells of initial lymphatics lacked junctions at the tip but were anchored on the sides by discontinuous button-like junctions (buttons) that differed from conventional, continuous, zipper-like junctions (zippers) in collecting lymphatics and blood vessels. However, both buttons and zippers were composed of vascular endothelial cadherin (VE-cadherin) and tight junction–associated proteins, including occludin, claudin-5, zonula occludens–1, junctional adhesion molecule–A, and endothelial cell–selective adhesion molecule. In C57BL/6 mice, VE-cadherin was required for maintenance of junctional integrity, but platelet/endothelial cell adhesion molecule–1 was not. Growing tips of lymphatic sprouts had zippers, not buttons, suggesting that buttons are specialized junctions rather than immature ones. Our findings suggest that fluid enters throughout initial lymphatics via openings between buttons, which open and close without disrupting junctional integrity, but most leukocytes enter the proximal half of initial lymphatics.
Lymphatic vessels are key routes for the recirculation of fluid and cells that enter tissues from blood vessels. This function of lymphatics is important for maintenance of normal tissue homeostasis and in inflammatory diseases and other conditions with extensive fluid and cell efflux (1). Lymphatics are also routes for spreading cancer cells (1–3) and for antigen-presenting cells trafficking from tissues to lymph nodes in immune surveillance (4, 5). Imbalances in efflux and recirculation of fluid or cells can result in lymphedema or disturbed immune responses.
Fluid entry into lymphatics is driven largely by hydrostatic and colloidal osmotic pressure gradients (6, 7). A prevailing view is that much of the endothelium of initial lymphatics has incomplete or no intercellular junctions (8–10). The loosely apposed but overlapping borders of endothelial cells are thought to function as “primary valves” that provide unidirectional fluid flux into lymphatics (11, 12). When lymphatic endothelial cells are pulled apart by anchoring filaments tensioned by interstitial forces, lymph flows along its pressure gradient into lymphatics (8, 10, 11).
The properties of leukocyte entry into lymphatics differ from those for fluid, as leukocyte influx is a selective process. Dendritic cells, macrophages, and lymphocytes enter lymphatics, but neutrophils and erythrocytes generally do not. Leukocytes are attracted by chemokines from lymphatic endothelial cells and interact with complementary adhesion molecules that govern adhesion and migration (4, 13, 14). Yet, the specific routes cells use to cross the lymphatic endothelium are at an early stage of understanding.
Evidence that lymphatic endothelial cells make junctional proteins comes from gene profiling data, which document the expression of platelet/endothelial cell adhesion molecule–1 (PECAM-1; also known as CD31), junctional adhesion molecule–A (JAM-A), and occludin in cultured cells (14–18). Multiple adhesion molecules have also been reported at intercellular junctions in specialized “retothelial” cells of lymph node sinuses (19).
Among the unresolved questions about the entry of fluid and cells into lymphatics are the following: (a) how can the integrity of initial lymphatics be maintained if junctions are not present between endothelial cells; (b) if junctions are present, how does fluid enter without repetitive disruption of the junctions; (c) do leukocytes and fluid enter at the same sites; and (d) what is the relation of the distinctive oak leaf shape of endothelial cells of initial lymphatics (9, 20) to sites of fluid and cell entry?
Based on this background, we sought to learn whether the properties of fluid and cell entry into initial lymphatics could be explained by the specialization of junctions between endothelial cells instead of the absence of junctions. We compared the distribution and composition of junctional proteins in initial lymphatics to those of conventional intercellular junctions in collecting lymphatics and blood vessels. Of particular interest were vascular endothelial cadherin (VE-cadherin) of adherens junctions, tight junction proteins, and the endothelial adhesion molecule PECAM-1. After learning that initial lymphatics had unusual, discontinuous endothelial junctions, we tested junctional integrity and plasticity after inhibition of VE-cadherin, deletion of PECAM-1, or increased fluid and cell flux in inflammation.
The studies exploited the attributes of the mouse tracheal mucosa, where both lymphatics and blood vessels are abundant, easily visualized, and readily compared under baseline conditions or after inflammatory stimuli in wild-type or genetically altered mice (21). High resolution confocal microscopic imaging of three-dimensional whole mounts revealed discontinuous, button-like junctions (buttons) in the endothelium of initial lymphatics that contained proteins typical of both adherens junctions and tight junctions but were structurally unlike the zipper-like junctions (zippers) elsewhere. These findings suggest that regions between buttons in initial lymphatics are openings where fluid can enter without repetitive formation and dissolution of intercellular junctions.
RESULTS
Initial lymphatics of mouse trachea
In the mouse trachea, lymphatics, with rounded blind tips and simple branching, were located in the mucosa between cartilage rings (Fig. 1 A). On average, 10.8 ± 0.4 blind ends of lymphatics were present in each mucosal segment between cartilages. Lymphatic vessels were located beneath the subepithelial plexus of blood vessels (Fig. 1 B) and were two or three times the size of mucosal venules (Table I). All lymphatic vessels in the trachea expressed lymphatic vascular endothelial hyaluronan receptor–1 (LYVE-1) and Prox1, making positive identification straightforward despite the abundance of blood vessels nearby (Fig. 1, A–C). Smooth muscle and luminal valves were restricted to collecting lymphatics on the adventitial surface.
Figure 1.

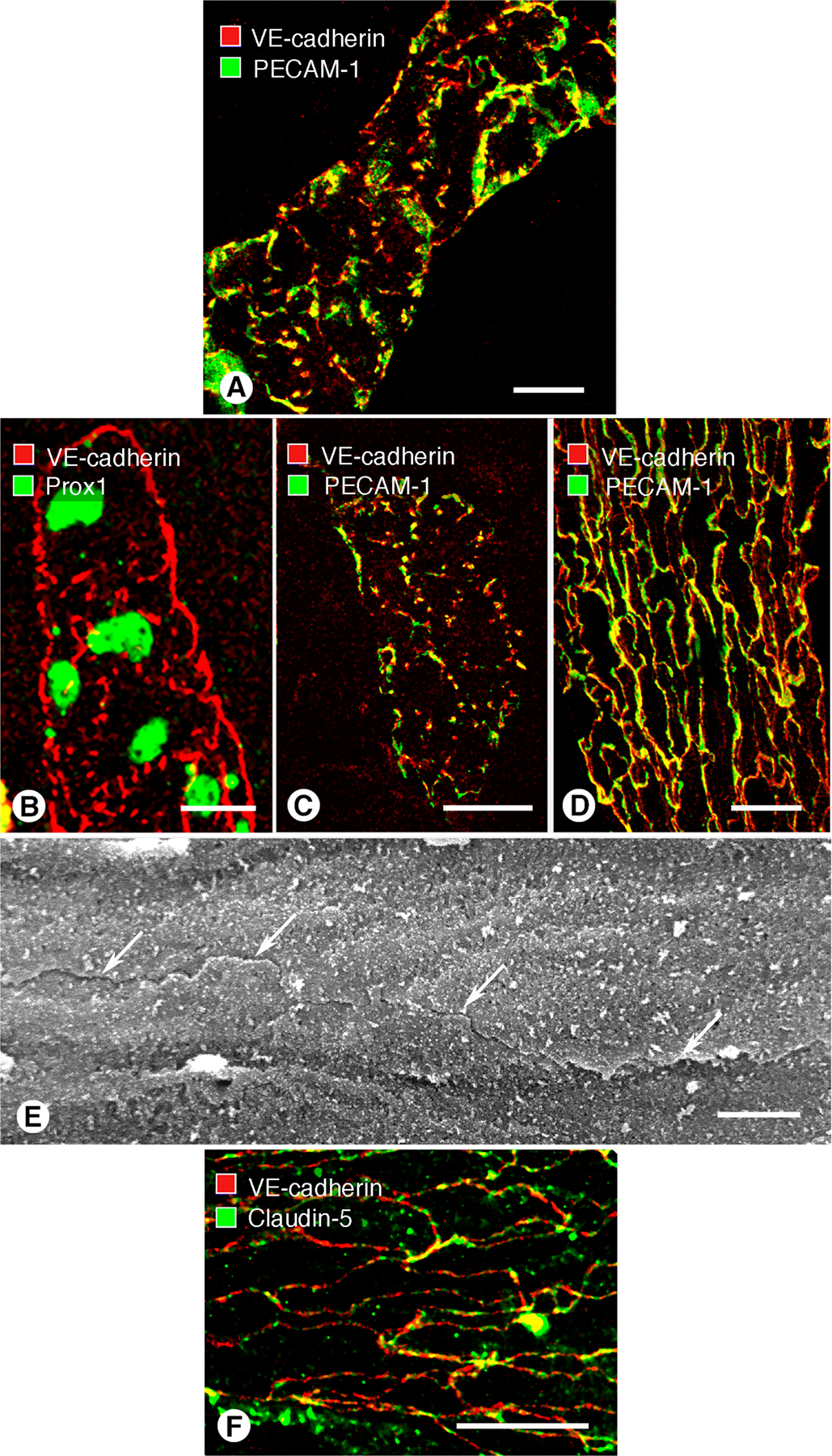
Buttons in endothelium of initial lymphatics. (A) Confocal images showing lymphatic vessels (green, LYVE-1) and blood vessels (red, PECAM-1) in whole mount of mouse trachea. Region of mucosa over horizontal cartilage (*) is mostly free of lymphatics. (B) Longitudinal section of trachea shows epithelium (green), subepithelial blood vessels (red, arrowheads), more deeply positioned initial lymphatics (diagonal arrows), collecting lymphatic (horizontal arrow), and adjacent cartilages. (C and D) Confocal images of VE-cadherin immunoreactivity (red) at discontinuous buttons in initial lymphatic (arrows; C) and continuous zippers in collecting lymphatic (D). Zippers are also present in blood capillary (arrowheads; C). Lymphatics are identified by Prox1 (green) in nuclei. (E) Distribution of 3,110 buttons along the length of 25 lymphatics in five tracheas, expressed as a function of distance from the tip. Values are presented as means ± SEM. *, P < 0.05 compared with the number at the tip (0 μm). (F and G) Confocal images showing VE-cadherin at buttons (arrows) and LYVE-1 between buttons (arrowhead) at the border of oak leaf–shaped endothelial cells of initial lymphatic. (G) Enlarged isosurface rendering of confocal image stack of boxed region in F. (H) Scanning electron microscopic image showing external surface of overlapping flaps at the junction of three endothelial cells of initial lymphatic. (I) Drawing of boxed region in H showing contributions of three endothelial cells. Bars: (A and B) 100 μm; (C, D, and F) 10 μm; (G) 5 μm; (H) 1 μm.
Table I.
Dimensions of endothelial cells of mouse lymphatics and blood vessels
| Initial lymphatics | Collecting lymphatics |
Venules | |
|---|---|---|---|
| Vessel diameter (μm) | 56 ± 1.5 (baseline) 68 ± 1.3 (LPS) | 64 ± 7 | 22 ± 1** |
| Cell length (μm) | 49 ± 1 | 66 ± 3* | 40 ± 3** |
| Cell width (μm) | 18 ± 0.8 | 14 ± 0.6* | 11 ± 0.6** |
| Cell area (μm2) | 616 ± 40 | 630 ± 40 | 312 ± 19** |
Measured in mouse tracheal whole mounts stained immunohistochemically for CD31 and LYVE-1 or VE-cadherin. All measurements were made under baseline conditions except for diameter of initial lymphatics, which were prepared under baseline conditions or 24 h after LPS. *, P < 0.05 compared with initial lymphatics; **, P < 0.05 compared with initial lymphatics and collecting lymphatics.
Buttons between endothelial cells of initial lymphatics
Immunohistochemical staining for VE-cadherin revealed conspicuous differences between initial lymphatics and collecting lymphatics in the trachea. Endothelial cells of initial lymphatics were joined by discontinuous buttons (Fig. 1 C), whereas endothelial cells of collecting lymphatics were joined by continuous zippers (Fig. 1 D), similar to those in adjacent blood vessels. Endothelial cells of lymphatics were appreciably larger than those in blood vessels (Table I).
Buttons consisted of roughly parallel linear segments of VE-cadherin, 3.2 ± 0.1 μm in length and spaced 2.9 ± 0.3 μm apart. Buttons were most abundant in the first 500 μm of tracheal lymphatics and were rare beyond 1,500 μm from the tip (Fig. 1 E). The length of button-rich regions varied from vessel to vessel, and scattered endothelial cells of some lymphatics had zippers, but the transition from buttons to zippers was typically abrupt in individual vessels. Lymphatics in other organs, including the diaphragm, urinary bladder, and skin of ear and tail, had similar buttons in initial lymphatics and zippers in collecting lymphatics (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20062596/DC1).
The unique association of buttons with initial lymphatics was confirmed by examining the distribution of VE-cadherin in the context of LYVE-1. Double staining revealed that buttons were at the perimeter of LYVE-1–positive endothelial cells with a distinctive oak leaf shape (Fig. 1 F). Cells of this shape are typical of initial lymphatics and conspicuously different from spindle-shaped endothelial cells of collecting lymphatics and blood vessels (9, 20). In initial lymphatics, LYVE-1 was concentrated at the tip of scalloped edges (flaps) of the oak leaf–shaped cells, and segments of VE-cadherin were located along the sides of flaps (Fig. 1 F). The presence of VE-cadherin at the sides of flaps explained the illusion that buttons were oriented perpendicular to the cell border (Fig. 1 C). The complementary distribution of VE-cadherin and LYVE-1 was particularly conspicuous after three-dimensional isosurface rendering of confocal image stacks (Fig. 1 G).
Scanning electron microscopic examination of the external surface of the endothelium of initial lymphatics, after exposure of the plasma membrane by alkaline hydrolysis of extracellular matrix (22, 23), revealed detailed features of the interdigitating borders of adjacent oak leaf–shaped endothelial cells (Fig. 1 H). Flaps at the scalloped borders of adjacent endothelial cells interdigitated with one another and overlapped loosely (Fig. 1, H and I). These features were not present at the continuous seams of adjacent endothelial cells of collecting lymphatics (Fig. S1) or in blood vessels.
Junctional proteins at buttons
The composition of buttons in initial lymphatics was determined by comparing the extent of colocalization of VE-cadherin and tight junction proteins. For the purposes of this study, colocalization of two junction-associated proteins was defined as pixels with fluorescence signals in two separate channels, rather than precise molecular colocalization within the junction (24). VE-cadherin at buttons colocalized with the tight junction protein occludin (Fig. 2 A). VE-cadherin at buttons also largely matched the distributions of the classical tight junction protein claudin-5 (Fig. 2 B), intracellular tight junction protein zonula occludens–1 (ZO-1; Fig. 2 C), and the recently identified tight junction–associated Ig-like transmembrane proteins endothelial cell–selective adhesion molecule (ESAM; Fig. 2 D) (25) and JAM-A (Fig. 2 E) (26). Despite striking structural differences in buttons and zippers, the two types of junctions contained the same proteins. Occludin (Fig. 2 F) and claudin-5 (Fig. S1) were continuously distributed at zippers in collecting lymphatics, where they partially colocalized with VE-cadherin.
Figure 2.

Colocalization of VE-cadherin and tight junction proteins at buttons and zippers. (A–E) Confocal images showing button-like pattern of VE-cadherin (left) paired with five different tight junction–associated proteins (middle) at endothelial junctions of initial lymphatics. Corresponding merged images (right) show that VE-cadherin colocalizes with all five tight junction proteins in buttons (A–E). (F) Continuous, zipper-like distribution of VE-cadherin (left) and occludin (middle) at endothelial junctions of collecting lymphatic; merged image (right) shows colocalization of the junctional proteins in zippers. In each case, lymphatic vessel identity was determined by vascular endothelial growth factor receptor 3 immunoreactivity. LYVE-1 or Prox1 were not used in these particular studies due to antibody species incompatibility issues. Bars: 10 μm.
Relation of PECAM-1 to VE-cadherin at buttons
Endothelial cells of initial lymphatics and collecting lymphatics, like those in blood vessels, had PECAM-1 immunoreactivity, but overall staining of lymphatics was weaker than blood vessels (Fig. 3 A). The distributions of PECAM-1 and VE-cadherin partially colocalized at buttons at the borders of oak leaf–shaped endothelial cells of initial lymphatics (Fig. 3 B) and at zippers of collecting lymphatics (Fig. 3 C). At buttons, VE-cadherin was located at the sides of flaps, and PECAM-1 tended to have a complementary distribution at the tip of flaps (Fig. 3, D–F). Arcs of PECAM-1 staining ∼3 μm in length varied from 0.5 to 2 μm in width, depending on the amount of cell overlap. By comparison, discontinuous segments of VE-cadherin staining were ∼3 μm in length and ∼0.5 μm in width.
Figure 3.

Different distributions of VE-cadherin and PECAM-1 in lymphatics. (A) Overview of PECAM-1 immunoreactivity of blood vessels (arrows) and lymphatics (arrowheads) in whole mount of mouse trachea. (B and C) Although VE-cadherin (red) and PECAM-1 (green) are both present in lymphatic endothelial cells, they do not have identical distributions in initial lymphatics (B) or collecting lymphatics (C) and colocalize only in scattered regions (yellow). (D–F) VE-cadherin (red, arrowheads) and PECAM-1 (green, arrows) have largely complementary distributions at buttons in initial lymphatics. The amount of colocalization is limited (yellow; F). Bars: (A) 100 μm; (B and C)10 μm; (D–F) 5 μm.
Contrasting properties of PECAM-1 and VE-cadherin at buttons
The complementary distributions of PECAM-1 and VE-cadherin in initial lymphatics raised the possibility that the two proteins influenced the organization of buttons. To address this issue, we asked whether loss or inactivation of either protein affected the distribution of the other protein or the organization of buttons. The effect of PECAM-1 deletion was examined in PECAM-1–null mice, which have no apparent vascular defects under baseline conditions (27). In tracheas of PECAM-1–null mice, PECAM-1 immunoreactivity was absent in blood vessels and lymphatics as expected, but VE-cadherin had a normal button-like pattern in initial lymphatics and a normal zipper-like pattern in blood vessels (Fig. 4, A–C). The pattern of VE-cadherin at buttons in PECAM-1–null mice (Fig. 4 D) was not noticeably different from wild-type mice (Fig. 4 E).
Figure 4.

Contrasting effects of loss of PECAM-1 or VE-cadherin in lymphatics. (A–E) Normal-appearing endothelial junctions in initial lymphatic (arrow) and blood vessel (arrowhead) in PECAM-1–null mice. (A–C) Normal distribution of VE-cadherin immunoreactivity at buttons in initial lymphatic and at zippers in blood vessel in PECAM-1–null mouse. Lymphatic is marked by LYVE-1 immunoreactivity (green). (D and E) Normal distribution of VE-cadherin at buttons despite absence of PECAM-1 immunoreactivity in a PECAM-1–null mouse (D) compared with complementary distributions of VE-cadherin and PECAM-1 in a wild-type mouse (E). (F–I) Disorganization of endothelial junctions in lymphatics and blood vessels 7 h after inhibition of VE-cadherin by function-blocking BV13 antibody. (F and G) Normal distribution of VE-cadherin at buttons in initial lymphatic (arrow) and at zippers in blood vessels (arrowheads) in a mouse injected with control IgG compared with disorganization of VE-cadherin and PECAM-1 immunoreactivities in initial lymphatic (arrow) and blood vessel (arrowhead) 7 h after injection of BV13 antibody (G). (H and I) Colocalization of VE-cadherin and ZO-1 at normal buttons after control IgG (H) compared with dispersion of VE-cadherin, but not ZO-1, at buttons 7 h after BV13 antibody (I). Bars: (A–C, F, G) 20 μm; (D, E, H, I) 5 μm.
Alternatively, a function-blocking antibody (BV13) was used to inactivate VE-cadherin (28). Genetic deletion of VE-cadherin leads to embryonic lethality because adherens junctions are essential for early steps of assembly of endothelial cells into blood vessels (29, 30). VE-cadherin is similarly important in the adult. The BV13 antibody causes dispersion of VE-cadherin away from junctions between endothelial cells of blood vessels accompanied by fatal leakage and hemorrhage within hours of intravascular injection (28). In our experiments, injection of BV13 antibody, unlike normal IgG of the same isotype (Fig. 4 F), led to dispersion of VE-cadherin in endothelial cells of initial lymphatics and blood vessels (Fig. 4 G). Surprisingly, BV13 also caused dispersion of PECAM-1 in lymphatics and blood vessels (Fig. 4 G) but did not alter the distribution of ZO-1 at buttons in initial lymphatics (Fig. 4, H and I).
Zippers at the tips of lymphatic sprouts
We next addressed the question of whether initial lymphatics are less differentiated regions of the lymphatic vasculature, and whether buttons represent immature versions of zippers. In this instance, we used a model of lymphatic sprouting, triggered by inoculation of Mycoplasma pulmonis into the airways of mice (21), to determine whether buttons predominate at the tip of lymphatic sprouts, as would be expected if the junctional immaturity hypothesis is valid. At 14 d after infection, new lymphatics were abundant in regions of mucosa over cartilage rings where lymphatics were normally absent (Fig. 5, A and B). The growing tips of the new lymphatics had zippers (Fig. 5, A and B, arrows) similar in appearance to those in collecting lymphatics under baseline conditions (Fig. 1 D). Most regions of lymphatics distal to the tip had oak leaf–shaped endothelial cells with buttons at the perimeter (Fig. 5 B, arrowheads). At 7 wk after infection, sprouts were less numerous, and most lymphatics had buttons (Fig. 5, C and D) resembling those in initial lymphatics under baseline conditions (Fig. 1 C).
Figure 5.

Zippers at growing tips of lymphatic sprouts. (A–D) Confocal images of tracheal mucosa after M. pulmonis infection showing lymphatic sprouts in regions that do not contain lymphatics in pathogen-free mice. (A and B) Continuous VE-cadherin–positive zippers (arrows) at growing tips of lymphatic sprouts at 14 d after M. pulmonis infection compared with discontinuous buttons in the remainder of initial lymphatics (arrowheads). Tips of lymphatic sprouts have little or no LYVE-1 immunoreactivity. (C and D) Most lymphatics identified by Prox1 immunoreactivity (arrows; C) have buttons (arrows; D) at 7 wk after infection. Some leukocytes have PECAM-1 immunoreactivity (arrowheads; D). Blood vessels have strong VE-cadherin and PECAM-1 immunoreactivities (yellow; D). Boxed regions in A and C are enlarged in B and D, respectively. Bars: (A and C) 100 μm; (B and D) 50 μm.
Sites of leukocyte entry into lymphatics
The issue of whether button-rich regions of lymphatics are preferential sites for leukocyte entry was explored by determining whether sites of cell migration coincided with buttons in a model of airway inflammation. The relationship of MHC class II (MHC II)–positive cells (mainly dendritic cells and macrophages) to lymphatics was examined 24 h after intranasal instillation of LPS. At this time, MHC II–positive cells were abundant near lymphatics (Fig. 6, A and B) and lymphatics were enlarged, with the mean diameter increased 21%, from 56 ± 1.5 μm in pathogen-free mice to 68 ± 1.3 μm after LPS (P < 0.05).
Figure 6.

Sites of leukocyte entry into initial lymphatics in airway inflammation. (A) Whole mount of mouse trachea 24 h after intratracheal LPS. MHC II–positive cell clusters (arrows, red) in or near initial lymphatics (green). (B) Enlargement of boxed region in A. Cells inside lymphatic (arrows) are more rounded than dendritic cells in trachea of pathogen-free mouse (inset). (C) Distribution of MHC II–positive cell clusters along the length of tracheal lymphatics, with the tip used as a reference. Half of the cell clusters were within 160 μm of the tip. Values are presented as means ± SEM. (D and E) Isosurface renderings of confocal images of MHC II cells (arrows) entering an initial lymphatic with buttons. (E) Enlargement of boxed region in (D). (F) MHC II–positive cells near and inside initial lymphatic of a PECAM-1–null mouse 24 h after LPS. (G) Transmission electron microscopic image of a leukocyte (pink) migrating through an intercellular junction in endothelium (green) of tracheal lymphatic with prominent junctional flap (arrow; M. pulmonis infection, 6 wk). (H and I) Confocal image (H) and isosurface rendering (I) of CD45-positive leukocytes (red, arrows) inside initial lymphatic 24 h after infection by M. pulmonis. Endothelial cell junctions are marked by VE-cadherin (red). PECAM-1, green; LYVE-1, blue. See also Video 1, available at http://www.jem.org/cgi/content/full/jem.20062596/DC1. Bars: (A) 200 μm; (B) 50 μm; (D) 10 μm; (E) 5 μm; (F) 50 μm; (G) 2 μm; (H and I) 20 μm.
Under baseline condition, most MHC II–positive cells had a dendritic phenotype (Fig. 6 B, inset) and little or no apparent association with lymphatics, but after LPS, MHC II–positive cells were more rounded and concentrated near lymphatics (Fig. 6, A and B). After LPS, 55% of initial lymphatics had clusters of four or more MHC II–positive cells inside or within 10 μm of their wall (411 lymphatics examined in five tracheas).
Further measurements made after LPS exposure showed that cell clusters were not uniformly distributed along button-rich regions of lymphatics but were preferentially found near the tips. Comparison of the position of cell clusters inside or near inflamed lymphatics with the length of each vessel, using the vessel tip as a reference point, revealed that ∼50% of cell clusters were located within the first 16% of the vessel length. The median distance for the location of cell clusters (160 μm; Fig. 6 C) was significantly less (P < 0.05) than the median length of the button-rich region of lymphatics (430 μm; Fig. 1 F), and both were significantly less than the median overall length of mucosal lymphatics in tracheas (980 μm; Fig. 6 C).
Leukocyte migration through the lymphatic endothelium was visualized by confocal microscopy (Fig. 6 B), and the transendothelial or intraluminal location of cells was confirmed by isosurface rendering of confocal image stacks and image rotation (Fig. 6, D and E; and Video 1, available at http://www.jem.org/cgi/content/full/jem.20062596/DC1). In PECAM-1–null mice exposed to LPS, leukocytes had the usual association with the proximal part of initial lymphatics (Fig. 6 F), indicating that PECAM-1 was not essential for leukocyte attraction to or migration into lymphatics. These findings suggest that migrating leukocytes preferentially entered the proximal half of the segment of lymphatics with buttons at the border of oak leaf–shaped endothelial cells.
Migration of leukocytes through endothelial junctions of lymphatics was confirmed by transmission electron microscopic examination (Fig. 6 G), but imaging of thin (two-dimensional) sections did not resolve whether cells migrated through buttons. We attempted to address this issue by confocal microscopic examination of lymphatics 24 h after M. pulmonis infection, when leukocyte migration was especially abundant. Although leukocytes clearly migrated through the endothelium of button-rich regions of lymphatics (Fig. 6, H and I), they obscured or distorted the junction at the site of transmigration, and the precise relationship between migrating leukocytes and buttons was ambiguous.
DISCUSSION
This study sought to define the structural organization and composition of endothelial junctions at sites in lymphatics where fluid and cells enter. Endothelial cells of initial lymphatics were found to be interconnected by discontinuous buttons. By comparison, collecting lymphatics downstream had continuous zippers at cell borders without openings.
Buttons in the endothelium of initial lymphatics
Identification of molecules involved in lymphatic specification, development, and pathology, through the use of molecular tools and novel animal models, has greatly advanced the understanding of the mechanism of lymphedema, immune cell trafficking, and tumor metastasis via lymphatics (Tammela, T., personal communication) (1–4, 31, 32). Multiple features of lymph and cell transport from tissues to lymphatics to lymph nodes have also been elucidated (1, 4, 7, 9), but understanding of the cellular mechanisms of fluid and cell entry into lymphatics has not advanced as far.
The distinctive oak leaf–shaped endothelial cells of initial lymphatics has been assumed to be related to fluid entry (9, 20). Flaps at loosely connected borders of these cells have been interpreted as primary valves that permit unidirectional flow of fluid into lymphatics (9, 10). Loose connection of endothelial cells would raise the question of how the structural integrity of the endothelium is maintained. The results of our studies offer a possible explanation (Fig. 7). Openings between buttons are ready candidates for the valves, and buttons at the sides of flaps would serve as anchors. Such specialized junctions that secure the sides of interdigitating flaps would permit fluid entry through openings at the tips of flaps without repetitive disassembly and reformation of endothelial junctions.
Figure 7.

Buttons in initial lymphatics border sites of fluid entry. (A) Schematic diagram showing distinctive, discontinuous buttons in endothelium of initial lymphatics and continuous zippers in collecting lymphatics. Both types of junction consist of proteins typical of adherens junctions and tight junctions. (B) More detailed view showing the oak leaf shape of endothelial cells (dashed lines) of initial lymphatics. Buttons (red) appear to be oriented perpendicular to the cell border but are in fact parallel to the sides of flaps. In contrast, most PECAM-1 expression is at the tips of flaps. (C and D) Enlarged views of buttons show that flaps of adjacent oak leaf–shaped endothelial cells have complementary shapes with overlapping edges. Adherens junctions and tight junctions at the sides of flaps direct fluid entry (arrows) to the junction-free region at the tip without repetitive disruption and reformation of junctions.
To our knowledge, intercellular junctions with the organization of buttons have not been previously described. The button-rich nature of initial lymphatics and abrupt transition to exclusively zippers in collecting lymphatics is consistent with functional differences between regions specialized for fluid uptake and regions specialized for lymph transport.
The location and mechanism of plasma extravasation from blood vessels has been extensively examined (33–37). Under baseline conditions, tight junctions between endothelial cells are crucial to normal barrier function (24, 38). In inflammation, formation of focal intercellular gaps leads to plasma leakage from venules (33, 39, 40). These gaps are structurally dissimilar to buttons in lymphatics.
Proof that buttons anchor the borders of openings for fluid entry into initial lymphatics is lacking, in part because sites of fluid entry into lymphatics have not been visualized directly. The rapid movement of tracers from the interstitium into initial lymphatics and then to collecting lymphatics blurs the spatial resolution needed for definitive localization of entry sites at the cellular level (7, 41, 42). Our preliminary experiments using fluorescent 25-nm microspheres to identify these sites confirmed this rapid movement (unpublished data). Similar factors may complicate the interpretation of results favoring the contribution of transcytotic transport to fluid entry in initial lymphatics (43).
Adherens and tight junction proteins at buttons
The finding that both VE-cadherin and tight junction–associated proteins were present in buttons seemed at first to conflict with the traditional view that lymphatic endothelium must have loose, poorly developed, or no intercellular junctions to permit entry of fluid and cells (7, 8, 44). However, the observation that both buttons and zippers have the same repertoire of junctional proteins, including occludin, claudin-5, ZO-1, ESAM, and JAM-A, indicates that the principal difference between these junctions is their organization rather than their composition. The presence of discontinuous but otherwise conventional tight junctions at buttons is consistent with their function as anchoring points along the sides of flaps and as borders for openings for fluid passage without junctional disassembly.
Importance of VE-cadherin at buttons
The consistent presence of PECAM-1 and VE-cadherin in the endothelium of initial lymphatics led us to examine the importance of each protein to the organization of buttons. PECAM-1 is known to be expressed in lymphatic endothelial cells from immunohistochemical observations in vivo (21) and in culture (14) and from gene profiling studies (14–17).
PECAM-1–null mice are viable and have a normal vasculature (27), but PECAM-1 may contribute to maintenance of vascular endothelial integrity in disease. As examples, PECAM-1–null mice have reduced transmigration of leukocytes (26, 27, 34, 45), increased bleeding times and vascular leakage (46, 47), and greater susceptibility to endotoxic shock (48). However, genetic background may contribute to the functional significance of PECAM-1, as PECAM-1 blockade or deletion predisposes FVB/n mice to chronic pulmonary inflammation and fibrosis but has little or no effect on the inflammatory response of C57BL/6 mice (45, 49). In our studies of C57BL/6 PECAM-1–null mice, we detected no change in the distribution of VE-cadherin at buttons or in the integrity of buttons in initial lymphatics. This finding is consistent with the association of PECAM-1 with openings between intercellular junctions instead of with the junctions themselves.
By comparison, inactivation of VE-cadherin at adherens junctions, by administration of function-blocking antibody BV13, resulted in dispersion of VE-cadherin at buttons and zippers in lymphatics, as shown previously for junctions in blood vessels (28). ZO-1 was not similarly affected. After BV13, PECAM-1 was dispersed at buttons in vivo but not at junctions between vascular endothelial cells in vitro (50, 51). These findings fit with the importance of VE-cadherin in the organization of buttons in initial lymphatics, but the relevance of PECAM-1 is still unclear.
Maturity of buttons in lymphatics
To address the question of whether the presence of buttons is a feature of lymphatic immaturity instead of lymphatic specialization, we examined the junctions of newly formed lymphatics where sprouting was active and junctions may be immature. In an established model of sustained inflammation accompanied by robust lymphangiogenesis (21), we found that the growing tips of lymphatic sprouts had zippers, not buttons. The presence of zippers at the tip of lymphatic sprouts indicates that continuous junctions can form rapidly and that buttons are not a feature of the most dynamic, immature region of lymphatic sprouts. However, over time all but the tips of new lymphatics had buttons largely like those present at baseline, despite elaborate expansion of the overall network of lymphatics in parallel with the increased flux of fluid and immune cells.
Sites of leukocyte entry in initial lymphatics
The presence of openings between junctions at the border of oak leaf–shaped endothelial cells raises the question of whether buttons are sites of cell entry. At first glance, openings between buttons would seem attractive routes for cell entry. The dimensions of the openings (∼3 μm) fits with the size of gaps where leukocytes migrate through the endothelium of inflamed venules (52) and with the size of pores (3 μm) that leukocytes migrate through in chemotaxis experiments in vitro (14). Although the presence of PECAM-1 at the tip of flaps is consistent with involvement in leukocyte trafficking (45), the apparently normal leukocyte migration in PECAM-1–null mice weighs against an essential role in the C57BL/6 strain. As loss of PECAM-1 results in impaired leukocyte efflux from blood vessels in mouse strains other than C57BL/6 after exposure to inflammatory stimuli (45), a role for PECAM-1 in the rate or efficiency of migration into lymphatics cannot be excluded without studies of other mouse strains.
The preferential association of leukocyte clusters with the proximal half of the region of lymphatics with buttons was an unexpected finding. The mismatch between regions of cell clusters and the overall extent of buttons indicates that precise sites of leukocyte entry are regulated by additional factors such as chemokines or adhesion molecules (4, 14, 53).
Did leukocytes enter through openings between buttons in the proximal half of the button-rich region of initial lymphatics? Our lack of success in answering this question may be caused partly by the infrequency of catching leukocytes in the act of migration and partly by the temporary deformation of junctions as migrating cells pass through them. Similarly, we cannot exclude that some cells follow a transcellular route (54). Certainly, the understanding of leukocyte migration into lymphatic vessels is in its infancy compared with what is known about leukocyte attachment and migration via intercellular and transcellular routes through the endothelium of venules (34–37, 55).
In conclusion, the borders of distinctive oak leaf–shaped endothelial cells of initial lymphatics are joined by specialized buttons (Fig. 7 A). The discontinuous feature of buttons distinguishes them from zippers in collecting lymphatics (Fig. 7 A), but both types of junctions are composed of proteins typical of adherens junctions and tight junctions found in the endothelium of blood vessels. Buttons seal the sides of flaps at the border of oak leaf–shaped endothelial cells (Fig. 7 B), leaving open the tips of flaps as routes for fluid entry without disassembly and reformation of intercellular junctions (Fig. 7, C and D). VE-cadherin is essential for maintaining the integrity of buttons, but PECAM-1, though strategically located at the tip of many flaps, is not essential for button integrity or leukocyte entry, at least not in C57BL/6 mice. Most leukocytes enter the proximal half of button-rich regions of initial lymphatics, but the exact site of entry in relation to buttons is unresolved. Collectively, our findings show that buttons are likely sites for fluid entry into initial lymphatics but are not the sole determinants of leukocyte entry.
MATERIALS AND METHODS
Mice.
Specific pathogen-free C57BL/6 mice (Charles River Laboratories) of either sex were housed under barrier conditions. PECAM-1–null mice on a C57BL/6 background, as previously described (27), were originally donated by T. Mak (Amgen Institute, Toronto, Canada). Mice were anesthetized by intramuscular injection of 100 mg/kg ketamine and 10 mg/kg xylazine. All experimental procedures were approved by the Institutional Animal Care and Use Committees of the University of California, San Francisco (UCSF) and the FIRC Institute of Molecular Oncology Foundation. All reagents were purchased from Sigma-Aldrich unless indicated otherwise.
Mouse models of inflammation.
Inflammation was induced by intranasal inoculation of mice with 250 μg LPS (type 055:B5) in 50 μl PBS (56) or 50 μl of broth containing 106 CFU of M. pulmonis organisms (strain CT8), as previously described (21). Mice were anesthetized before inoculation and allowed to recover. At 24 h after LPS or 24 h to 40 d after M. pulmonis inoculation, mice were anesthetized again for further studies. M. pulmonis organisms activate an immune response with a time course similar to other airway infections (57, 58). Robust lymphangiogenesis begins about 7 d after infection (21).
In vivo blockade of VE-cadherin.
C57BL/6 mice were injected via the tail vein with 100 μg BV13, a function-blocking rat monoclonal anti–mouse VE-cadherin antibody or with a control rat IgG (28). Tracheas were examined 3 or 7 h later. LPS-induced inflammation was not studied in mice after VE-cadherin inhibition by BV13 because of the rapid induction of progressive plasma leakage, interstitial edema, hemorrhage, and death within 24 h (28).
Immunohistochemistry.
Mice were perfused for 2 min with fixative (1% paraformaldehyde in PBS, pH 7.4) (21) from a cannula inserted through the left ventricle into the aorta. The trachea, diaphragm, urinary bladder, ear, and tail skin were removed and immersed in fixative for 1 h at 4°C. Tissues were washed and stained immunohistochemically by incubating whole mounts with one or more primary antibodies diluted in PBS containing 0.3% Triton X-100, 0.2% bovine serum albumin, 5% normal goat serum, and 0.1% sodium azide, as previously described (21). Lymphatics were LYVE-1 (rabbit polyclonal 07-538; Upstate Biotechnology), Prox1 (rabbit polyclonal AB5475; Chemicon), or vascular endothelial growth factor receptor 3 (goat polyclonal antibody AF743; R&D Systems). Adherens junction was VE-cadherin (rat clone BV13 and rabbit polyclonal antibody). Tight junctions were ESAM (rat clone 1G8.2), ZO-1 and occludin (rabbit polyclonal antibodies 40-2200 and 71-1500; Zymed Laboratories), and JAM-A and claudin-5 (rat clone BV12 and rabbit polyclonal antibody E2.8, respectively). PECAM-1 was CD31 (hamster anti–mouse PECAM-1, clone 2H8; Chemicon). Leukocytes were MHC II (rat clone M5/115.14.2; eBioscience) or CD45 (rat clone Ly-5; BD Biosciences). Secondary antibodies were labeled with FITC, Cy3, or Cy5 (Jackson ImmunoResearch Laboratories). Specimens were viewed with a fluorescence microscope (Axiophot; Carl Zeiss MicroImaging, Inc.) with a 3CCD low light red-green-blue (RGB) video camera (CoolCam; SciMeasure) or a confocal microscope (LSM-510; Carl Zeiss MicroImaging, Inc.) using AIM confocal software (version 3.2.2).
Morphometric measurements.
The vessel diameter and length, width, and area of endothelial cells were measured in real-time images of lymphatics and venules in tracheas stained for VE-cadherin and PECAM-1 by using a digitizing tablet linked to a video camera on the Axiophot microscope with 40× (NA 1.0) or 63× (NA 1.4) objectives. Cell perimeter and shape factor were measured as previously described (52). The total length of 411 mucosal lymphatics and the distance from the tip of lymphatics to the location of 228 cell clusters, consisting of four or more MHC II–positive cells inside or within 10 μm of the wall of a lymphatic, were measured in five tracheas 24 h after intranasal instillation of LPS. Tracheas were stained for MHC II and LYVE-1 immunoreactivities and imaged with 5× (NA 0.32) or 20× (NA 0.75) objectives. The distribution of 3,110 buttons was assessed along the length of five lymphatics in the trachea of each of five pathogen-free mice.
Isosurface rendering of confocal images.
Confocal RGB image stacks were imported into Imaris software (version 5.0.3; Bitplane). Voxels with fluorescence intensities above a certain threshold were assigned for each color channel. Isosurfaces were rendered from these voxels and smoothed with a Gaussian filter, creating three-dimensional reconstructions in which the spatial resolution was conserved.
Scanning and transmission electron microscopy (EM).
For scanning EM, tissues were fixed by vascular perfusion of fixative containing 2% glutaraldehyde in 100 mmol/liter of phosphate buffer, treated with 30% potassium hydroxide at 60°C for 8 min to dissolve the extracellular matrix, stained with 2% tannic acid and 1% OsO4, dehydrated with ethanol, critical point dried, coated in an osmium plasma coater (OPC60A; Filgen), and examined with a scanning electron microscope (S-5000; Hitachi) (22, 23). Transmission EM was performed as previously described (23, 52).
Statistical analysis.
Values are presented as means ± SEM with four to five mice per group, unless otherwise indicated. The significance of differences between means was assessed by analysis of variance, followed by the Dunn-Bonferroni test for multiple comparisons. P < 0.05 was considered significant. The significance of differences between distributions of buttons, clusters of dendritic cells along the length of lymphatics, and lengths of lymphatics in tracheal mucosa was analyzed by the Kolmogorov-Smirnov two-sample test.
Online supplemental material.
Fig. S1 depicts confocal and scanning electron microscopic images of junctions in initial and collecting lymphatics in other organs. Video 1 shows three-dimensional aspects of leukocytes interacting with lymphatic endothelial cell junctions. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20062596/DC1.
Supplemental Material
Acknowledgments
We thank Maria Grazia Lampugnani for assistance with experiments in Milan and helpful discussions, DongJi Zhang for production of M. pulmonis stock, and Amy Haskell for transmission EM.
This work was supported in part by grants from the National Heart, Lung, and Blood Institute (HL-24136 and HL-59157) and the National Cancer Institute (CA-82923) of the National Institutes of Health (to D. McDonald), by a grant from the Deutsche Forschungsgemeinschaft (SFB492; to D. Vestweber), and by a grant from the Associazione Italiana per la Ricerca sul Cancro and the European Community (lymphangiogenomics project grant LSHG-CT-2004-503573; to E. Dejana). J. Fuxe was supported by the Swedish Wenner-Gren Foundation and by the International Union Against Cancer American Cancer Society International Fellowship for Beginning Investigators.
The authors have no conflicting financial interests.
Abbreviations used: button, button-like junction; EM, electron microscopy; ESAM, endothelial cell–selective adhesion molecule; JAM-A, junctional adhesion molecule–A; LYVE-1, lymphatic vascular endothelial hyaluronan receptor–1; MHC II, MHC class II; PECAM-1, platelet/endothelial adhesion molecule–1; VE-cadherin, vascular endothelial cadherin; zipper, zipper-like junction; ZO-1, zonula occludens–1.
P. Baluk and J. Fuxe contributed equally to this work.
References
- 1.Witte, M.H., K. Jones, J. Wilting, M. Dictor, M. Selg, N. McHale, J.E. Gershenwald, and D.G. Jackson. 2006. Structure function relationships in the lymphatic system and implications for cancer biology. Cancer Metastasis Rev. 25:159–184. [DOI] [PubMed] [Google Scholar]
- 2.Ji, R.C. 2006. Lymphatic endothelial cells, tumor lymphangiogenesis and metastasis: New insights into intratumoral and peritumoral lymphatics. Cancer Metastasis Rev. 25:677–694. [DOI] [PubMed] [Google Scholar]
- 3.Tobler, N.E., and M. Detmar. 2006. Tumor and lymph node lymphangiogenesis–impact on cancer metastasis. J. Leukoc. Biol. 80:691–696. [DOI] [PubMed] [Google Scholar]
- 4.Randolph, G.J., V. Angeli, and M.A. Swartz. 2005. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat. Rev. Immunol. 5:617–628. [DOI] [PubMed] [Google Scholar]
- 5.Mebius, R.E., P.R. Streeter, J. Breve, A.M. Duijvestijn, and G. Kraal. 1991. The influence of afferent lymphatic vessel interruption on vascular addressin expression. J. Cell Biol. 115:85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Casley-Smith, J.R. 1985. The influence of tissue hydrostatic pressure and protein concentration on fluid and protein uptake by diaphragmatic initial lymphatics; effect of calcium dobesilate. Microcirc. Endothelium Lymphatics. 2:385–415. [PubMed] [Google Scholar]
- 7.Schmid-Schönbein, G.W. 1990. Microlymphatics and lymph flow. Physiol. Rev. 70:987–1028. [DOI] [PubMed] [Google Scholar]
- 8.Leak, L.V., and J.F. Burke. 1968. Ultrastructural studies on the lymphatic anchoring filaments. J. Cell Biol. 36:129–149. [PMC free article] [PubMed] [Google Scholar]
- 9.Zöltzer, H. 2006. Morphology and physiology of lymphatic endothelial cells. In Microvasculature Research: Biology and Pathology. D. Shepro and P.A. D'Amore, editors. Elsevier Academic Press, San Diego. 535–544.
- 10.Schmid-Schönbein, G.W. 2003. The second valve system in lymphatics. Lymphat. Res. Biol. 1:25–29. [DOI] [PubMed] [Google Scholar]
- 11.Trzewik, J., S.K. Mallipattu, G.M. Artmann, F.A. Delano, and G.W. Schmid-Schönbein. 2001. Evidence for a second valve system in lymphatics: endothelial microvalves. FASEB J. 15:1711–1717. [DOI] [PubMed] [Google Scholar]
- 12.Mendoza, E., and G.W. Schmid-Schönbein. 2003. A model for mechanics of primary lymphatic valves. J. Biomech. Eng. 125:407–414. [DOI] [PubMed] [Google Scholar]
- 13.Stoitzner, P., K. Pfaller, H. Stossel, and N. Romani. 2002. A close-up view of migrating Langerhans cells in the skin. J. Invest. Dermatol. 118:117–125. [DOI] [PubMed] [Google Scholar]
- 14.Johnson, L.A., S. Clasper, A.P. Holt, P.F. Lalor, D. Baban, and D.G. Jackson. 2006. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J. Exp. Med. 203:2763–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirakawa, S., Y.K. Hong, N. Harvey, V. Schacht, K. Matsuda, T. Libermann, and M. Detmar. 2003. Identification of vascular lineage-specific genes by transcriptional profiling of isolated blood vascular and lymphatic endothelial cells. Am. J. Pathol. 162:575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Podgrabinska, S., P. Braun, P. Velasco, B. Kloos, M.S. Pepper, and M. Skobe. 2002. Molecular characterization of lymphatic endothelial cells. Proc. Natl. Acad. Sci. USA. 99:16069–16074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sironi, M., A. Conti, S. Bernasconi, A.M. Fra, F. Pasqualini, M. Nebuloni, E. Lauri, M. De Bortoli, A. Mantovani, E. Dejana, and A. Vecchi. 2006. Generation and characterization of a mouse lymphatic endothelial cell line. Cell Tissue Res. 325:91–100. [DOI] [PubMed] [Google Scholar]
- 18.Wick, N., P. Saharinen, J. Saharinen, E. Gurnhofer, C.W. Steiner, I. Raab, D. Stokic, P. Giovanoli, S. Buchsbaum, A. Burchard, et al. 2007. Transcriptomal comparison of human dermal lymphatic endothelial cells ex vivo and in vitro. Physiol. Genomics. 28:179–192. [DOI] [PubMed] [Google Scholar]
- 19.Hammerling, B., C. Grund, J. Boda-Heggemann, R. Moll, and W.W. Franke. 2006. The complexus adhaerens of mammalian lymphatic endothelia revisited: a junction even more complex than hitherto thought. Cell Tissue Res. 324:55–67. [DOI] [PubMed] [Google Scholar]
- 20.Yoffey, J., and F. Courtice. 1970. Lymphatics, Lymph and the Lymphomyeloid Complex. Academic Press, London/New York. 942 pp.
- 21.Baluk, P., T. Tammela, E. Ator, N. Lyubynska, M.G. Achen, D.J. Hicklin, M. Jeltsch, T.V. Petrova, B. Pytowski, S.A. Stacker, et al. 2005. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J. Clin. Invest. 115:247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hashizume, H., and T. Ushiki. 2002. Three-dimensional cytoarchitecture of angiogenic blood vessels in a gelatin sheet implanted in the rat skeletal muscular layers. Arch. Histol. Cytol. 65:347–357. [DOI] [PubMed] [Google Scholar]
- 23.Inai, T., M. Mancuso, H. Hashizume, F. Baffert, A. Haskell, P. Baluk, D.D. Hu-Lowe, D.R. Shalinsky, G. Thurston, G.D. Yancopoulos, and D.M. McDonald. 2004. Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts. Am. J. Pathol. 165:35–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dejana, E. 2004. Endothelial cell-cell junctions: happy together. Nat. Rev. Mol. Cell Biol. 5:261–270. [DOI] [PubMed] [Google Scholar]
- 25.Wegmann, F., B. Petri, A.G. Khandoga, C. Moser, A. Khandoga, S. Volkery, H. Li, I. Nasdala, O. Brandau, R. Fassler, et al. 2006. ESAM supports neutrophil extravasation, activation of Rho, and VEGF-induced vascular permeability. J. Exp. Med. 203:1671–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nourshargh, S., F. Krombach, and E. Dejana. 2006. The role of JAM-A and PECAM-1 in modulating leukocyte infiltration in inflamed and ischemic tissues. J. Leukoc. Biol. 80:714–718. [DOI] [PubMed] [Google Scholar]
- 27.Duncan, G.S., D.P. Andrew, H. Takimoto, S.A. Kaufman, H. Yoshida, J. Spellberg, J. Luis de la Pompa, A. Elia, A. Wakeham, B. Karan-Tamir, et al. 1999. Genetic evidence for functional redundancy of platelet/endothelial cell adhesion molecule-1 (PECAM-1): CD31-deficient mice reveal PECAM-1-dependent and PECAM-1-independent functions. J. Immunol. 162:3022–3030. [PubMed] [Google Scholar]
- 28.Corada, M., M. Mariotti, G. Thurston, K. Smith, R. Kunkel, M. Brockhaus, M.G. Lampugnani, I. Martin-Padura, A. Stoppacciaro, L. Ruco, et al. 1999. Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc. Natl. Acad. Sci. USA. 96:9815–9820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vittet, D., T. Buchou, A. Schweitzer, E. Dejana, and P. Huber. 1997. Targeted null-mutation in the vascular endothelial-cadherin gene impairs the organization of vascular-like structures in embryoid bodies. Proc. Natl. Acad. Sci. USA. 94:6273–6278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carmeliet, P., M.G. Lampugnani, L. Moons, F. Breviario, V. Compernolle, F. Bono, G. Balconi, R. Spagnuolo, B. Oostuyse, M. Dewerchin, et al. 1999. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 98:147–157. [DOI] [PubMed] [Google Scholar]
- 31.Alitalo, K., T. Tammela, and T.V. Petrova. 2005. Lymphangiogenesis in development and human disease. Nature. 438:946–953. [DOI] [PubMed] [Google Scholar]
- 32.Lira, S.A. 2005. A passport into the lymph node. Nat. Immunol. 6:866–868. [DOI] [PubMed] [Google Scholar]
- 33.McDonald, D.M., G. Thurston, and P. Baluk. 1999. Endothelial gaps as sites for plasma leakage in inflammation. Microcirculation. 6:7–22. [PubMed] [Google Scholar]
- 34.Muller, W.A. 2003. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 24:327–334. [DOI] [PubMed] [Google Scholar]
- 35.Nourshargh, S., and F.M. Marelli-Berg. 2005. Transmigration through venular walls: a key regulator of leukocyte phenotype and function. Trends Immunol. 26:157–165. [DOI] [PubMed] [Google Scholar]
- 36.Luster, A.D., R. Alon, and U.H. von Andrian. 2005. Immune cell migration in inflammation: present and future therapeutic targets. Nat. Immunol. 6:1182–1190. [DOI] [PubMed] [Google Scholar]
- 37.Dejana, E. 2006. The transcellular railway: insights into leukocyte diapedesis. Nat. Cell Biol. 8:105–107. [DOI] [PubMed] [Google Scholar]
- 38.Tsukita, S., M. Furuse, and M. Itoh. 2001. Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2:285–293. [DOI] [PubMed] [Google Scholar]
- 39.Thurston, G., P. Baluk, and D.M. McDonald. 2000. Determinants of endothelial cell phenotype in venules. Microcirculation. 7:67–80. [PubMed] [Google Scholar]
- 40.van Nieuw Amerongen, G.P., and V.W. van Hinsbergh. 2002. Targets for pharmacological intervention of endothelial hyperpermeability and barrier function. Vascul. Pharmacol. 39:257–272. [DOI] [PubMed] [Google Scholar]
- 41.Pullinger, B., and H. Florey. 1935. Some observations on the structure and function of lymphatics: their behaviour in local oedema. Br. J. Exp. Pathol. 16:49–61. [Google Scholar]
- 42.Nagy, J.A., E. Vasile, D. Feng, C. Sundberg, L.F. Brown, M.J. Detmar, J.A. Lawitts, L. Benjamin, X. Tan, E.J. Manseau, et al. 2002. Vascular permeability factor/vascular endothelial growth factor induces lymphangiogenesis as well as angiogenesis. J. Exp. Med. 196:1497–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Azzali, G. 2003. Transendothelial transport and migration in vessels of the apparatus lymphaticus periphericus absorbens (ALPA). Int. Rev. Cytol. 230:41–87. [DOI] [PubMed] [Google Scholar]
- 44.Casley-Smith, J.R. 1965. Endothelial permeability. II. The passage of particles through the lymphatic endothelium of normal and injured ears. Br. J. Exp. Pathol. 46:35–49. [PMC free article] [PubMed] [Google Scholar]
- 45.Schenkel, A.R., T.W. Chew, and W.A. Muller. 2004. Platelet endothelial cell adhesion molecule deficiency or blockade significantly reduces leukocyte emigration in a majority of mouse strains. J. Immunol. 173:6403–6408. [DOI] [PubMed] [Google Scholar]
- 46.Mahooti, S., D. Graesser, S. Patil, P. Newman, G. Duncan, T. Mak, and J.A. Madri. 2000. PECAM-1 (CD31) expression modulates bleeding time in vivo. Am. J. Pathol. 157:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Graesser, D., A. Solowiej, M. Bruckner, E. Osterweil, A. Juedes, S. Davis, N.H. Ruddle, B. Engelhardt, and J.A. Madri. 2002. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J. Clin. Invest. 109:383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carrithers, M., S. Tandon, S. Canosa, M. Michaud, D. Graesser, and J.A. Madri. 2005. Enhanced susceptibility to endotoxic shock and impaired STAT3 signaling in CD31-deficient mice. Am. J. Pathol. 166:185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schenkel, A.R., T.W. Chew, E. Chlipala, M.W. Harbord, and W.A. Muller. 2006. Different susceptibilities of PECAM-deficient mouse strains to spontaneous idiopathic pneumonitis. Exp. Mol. Pathol. 81:23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hordijk, P.L., E. Anthony, F.P. Mul, R. Rientsma, L.C. Oomen, and D. Roos. 1999. Vascular-endothelial-cadherin modulates endothelial monolayer permeability. J. Cell Sci. 112:1915–1923. [DOI] [PubMed] [Google Scholar]
- 51.Corada, M., S. Chimenti, M.R. Cera, M. Vinci, M. Salio, F. Fiordaliso, N. De Angelis, A. Villa, M. Bossi, L.I. Staszewsky, et al. 2005. Junctional adhesion molecule-A-deficient polymorphonuclear cells show reduced diapedesis in peritonitis and heart ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA. 102:10634–10639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McDonald, D.M. 1994. Endothelial gaps and permeability of venules in rat tracheas exposed to inflammatory stimuli. Am. J. Physiol. 266:L61–L83. [DOI] [PubMed] [Google Scholar]
- 53.Randolph, G.J. 2001. Dendritic cell migration to lymph nodes: cytokines, chemokines, and lipid mediators. Semin. Immunol. 13:267–274. [DOI] [PubMed] [Google Scholar]
- 54.Azzali, G. 2006. On the transendothelial passage of tumor cell from extravasal matrix into the lumen of absorbing lymphatic vessel. Microvasc. Res. 72:74–85. [DOI] [PubMed] [Google Scholar]
- 55.Steeber, D.A., G.M. Venturi, and T.F. Tedder. 2005. A new twist to the leukocyte adhesion cascade: intimate cooperation is key. Trends Immunol. 26:9–12. [DOI] [PubMed] [Google Scholar]
- 56.Schon-Hegrad, M.A., J. Oliver, P.G. McMenamin, and P.G. Holt. 1991. Studies on the density, distribution, and surface phenotype of intraepithelial class II major histocompatibility complex antigen (Ia)–bearing dendritic cells (DCs) in the conducting airways. J. Exp. Med. 173:1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aurora, A.B., P. Baluk, D. Zhang, S.S. Sidhu, G.M. Dolganov, C. Basbaum, D.M. McDonald, and N. Killeen. 2005. Immune complex-dependent remodeling of the airway vasculature in response to a chronic bacterial infection. J. Immunol. 175:6319–6326. [DOI] [PubMed] [Google Scholar]
- 58.Legge, K.L., and T.J. Braciale. 2003. Accelerated migration of respiratory dendritic cells to the regional lymph nodes is limited to the early phase of pulmonary infection. Immunity. 18:265–277. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}