

Abstract
B cell homeostasis is regulated by multiple signaling processes, including nuclear factor-κB (NF-κB), BAFF-, and B cell receptor signaling. Conditional disruption of genes involved in these pathways has shed light on the mechanisms governing signaling from the cell surface to the nucleus. We describe a novel mouse strain that expresses solely and excessively a naturally occurring splice variant of CYLD (CYLDex7/8 mice), which is a deubiquitinating enzyme that is integral to NF-κB signaling. This shorter CYLD protein lacks the TRAF2 and NEMO binding sites present in full-length CYLD. A dramatic expansion of mature B lymphocyte populations in all peripheral lymphoid organs occurs in this strain. The B lymphocytes themselves exhibit prolonged survival and manifest a variety of signaling disarrangements that do not occur in mice with a complete deletion of CYLD. Although both the full-length and the mutant CYLD are able to interact with Bcl-3, a predominant nuclear accumulation of Bcl-3 occurs in the CYLD mutant B cells. More dramatic, however, is the accumulation of the NF-κB proteins p100 and RelB in CYLDex7/8 B cells, which, presumably in combination with nuclear Bcl-3, results in increased levels of Bcl-2 expression. These findings suggest that CYLD can both positively and negatively regulate signal transduction and homeostasis of B cells in vivo, depending on the expression of CYLD splice variants.
The tumor suppressor gene CYLD was identified in patients with familial cylindromatosis, which is an autosomal dominant predisposition to multiple neoplasms of the skin appendages. The tumors are believed to arise from the eccrine or apocrine cells of the skin (1). Approximately 70% of the cylindromas exhibit loss of heterozygosity on chromosome 16q containing the CYLD gene (2–6).
Sequence analysis of the CYLD gene predicts a protein with several functional domains, including three CAP-Gly domains and a deubiquitination domain at the C terminus (7–9). In vitro studies showed that CYLD contains binding sites for TRAF2 and NEMO (7), and that the deubiquitinating activity of CYLD is directed to lysine 63 (K63)–linked ubiquitin (Ub) (7). Linkage of Ub through K63 assembles a new molecular platform, allowing the recruitment of proteins involved in signal transduction (10–12), whereas Ub chains linked through K48 destine proteins for proteasomal degradation (13, 14). Suppression of CYLD expression by RNA interference causes an increase in the activity of the transcription factor NF-κB, which is activated by TNFα signaling, thus inhibiting apoptosis (7, 8, 15). The increase of NF-κB activation in CYLD knockdown cells was attributed to defective removal of K63 Ub chains from TRAF2 and NF-κB essential modulator (NEMO) by CYLD (7–9).
In a mechanistically analogous manner to its effect on TRAF2, CYLD was found to inhibit signaling from Toll-like receptor 2 (TLR2) by removing K63-linked Ub from TRAF6 and TRAF7 (16). Interestingly, it was shown that after TLR2 activation, CYLD inhibits the phosphorylation of TRAF6 and TRAF7, which, in turn, suppresses MKK3 and MKK6, leading to reduced MAP kinase p38 phosphorylation (17). In addition to p38 signaling, CYLD negatively regulates the stress-activating MAP kinase family, the JNK kinases. CYLD inactivation resulted in hyperactivation of JNK upon TNFα, IL-1β, LPS, and anti-CD40 treatment (17). This hyperactivation is believed to be a result of negatively regulating the activation of MKK7, which is the kinase responsible for JNK activation (17).
To investigate the physiological role of CYLD in vivo, we generated mouse strains with targeted modifications in the CYLD gene. In addition to a complete knockout of CYLD, exon 7 of the CYLD gene was flanked by loxP sites in a second mouse strain to result in conditional, excessive, and solitary expression of a shorter splice variant of the CYLD protein, hereafter termed sCYLD. The sCYLD protein is devoid of both TRAF2 and NEMO binding sites. In this article, we present data describing the profound alterations in the immune system of these mice (CYLDex7/8), which is characterized by lymphomegaly and splenomegaly and a striking increase in B cell numbers. Overexpression of sCYLD further bestows B lymphocytes with increased IgG1 production and enhances survival capabilities of the B cell.
RESULTS
Generation of CYLDex7/8 mutant mice
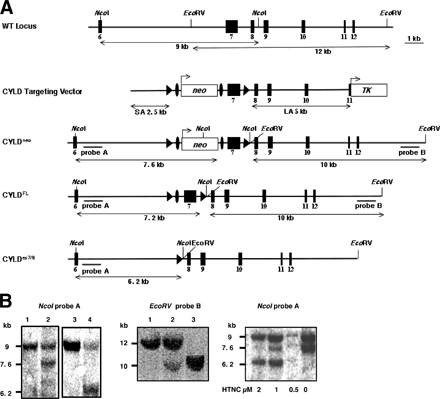
CYLD mutant mice were generated applying standard gene-targeting techniques in mouse embryonic stem cells (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20070318/DC1). Using Cre/loxP technology, three different mouse strains were generated. The first strain, CYLDneo, contains the neo resistance gene upstream of exon 7, decreasing transcription of CYLD (Fig. S1). The second strain, CYLDFL, harbors two loxP sites flanking exon 7 of CYLD (Fig. S1). Finally, CYLDex7/8 mice lacking exon 7 after Cre-mediated recombination through transient transfection of the targeted embryonic stem cells with a Cre-expressing plasmid (Fig. S1). CYLDex7/8 mice were born at the expected Mendelian frequencies and survived normally when housed under pathogen-free conditions.
Germline deletion of exon 7 should lead to splicing from exon 6 to 8 resulting in an out-of-frame translation of CYLD (Fig. 1 A). RT-PCR was applied to cDNA from mouse embryonic fibroblasts (MEFs) of the indicated genotypes using primers located in exon 6 and 9 of the CYLD transcript to verify the absence of the WT allele in CYLDex7/8 MEFs. This analysis revealed an unexpected shorter amplified product besides the expected band from the full-length transcript (FL-CYLD) in WT MEFs. This shorter product represents an alternative splice variant of CYLD lacking exons 7 and 8, termed sCYLD. CYLDex7/8 MEFs are devoid of the full-length transcript, but express the sCYLD splice variant excessively (Fig. 1 B). Using CYLD-specific antibodies, sCYLD protein could be detected in different tissues and cells of WT animals (Fig. 1 C). To investigate whether deletion of WT CYLD (FL-CYLD) in CYLDex7/8 mice results in higher expression levels of sCYLD, protein extracts were prepared from purified B cells of WT and CYLDex7/8 mice, revealing high expression of sCYLD in CYLDex7/8 cells compared with WT cells (Fig. 1 D).
Figure 1.

The identification of a new splice variant of CYLD. (A) Schematic representation of exons 6–9 of the CYLD gene, with respective transcripts. (B) RT-PCR of cDNA generated from MEFs of the indicated genotypes using primer pair P1 and P2. WT full-length CYLD transcript corresponds to the 770-bp band, whereas the alternatively spliced CYLD transcript is 244 bp. Bands were isolated, subcloned, and verified by sequencing. The intermediate band could be determined as an unspecific band. (C) Western blot analysis of lysates from WT tissue and cells. (lane 1) Spleen; (lane 2) T cells; (lane 3) LNs; (lane 4) thymus; (lane 5) B cells. Actin was used as loading control. (D) Western blot analysis of lysates from WT and CYLDex7/8 B cells using anti-CYLD antibody. Actin was used as loading control. (E) Schematic representation of the different CYLD constructs used for transfection experiments in HeLa cells. Highlighted are the important domains of CYLD. FL-CYLD (full-length CYLD), C/S-mut CYLD (full-length CYLD carrying a mutation from C to S to generate a catalytically inactive form), and sCYLD alternatively spliced CYLD lacking exons 7 and 8. (F) HeLa cells were transfected with plasmids encoding either His-FL-CYLD or for His-sCYLD together with either plasmid pcDNA or plasmid encoding for TRAF2. Transfected cells were lysed and coimmunoprecipitated with anti-His antibody for CYLD, separated on an 8% SDS-PAGE, and transferred to PVDF membrane. The membranes were incubated with TRAF2 and CYLD antibodies, respectively. (G) MEFs were transfected with TRAF2-FLAG cDNA and TRAF2 was immunoprecipitated with anti-FLAG antibody. Thereafter, TRAF2 was separated on a 4–12% SDS-PAGE and transferred to PVDF membrane. The membrane was incubated with Ub-specific antibody. (lane 1) Mock-transfected WT MEFs; (lane 2) WT MEFs transfected with TRAF2-FLAG; (lane 3) CYLDex7/8 MEFs transfected with TRAF2-FLAG. Data presented are representative of three different experiments. (H) HeLa cells were transfected with the indicated CYLD construct encoding plasmids together with plasmids coding for Flag-Bcl-3 and His-Ubiquitin. Protein lysates of transfected cells were used for immunoprecipitations with anti-His antibody, and Western blots were incubated with antibodies against the His and the Flag tags. Flag-Bcl-3 was used as a loading control for the Bcl-3 levels in the different cell types.
Previously, CYLD was identified as a deubiquitinating enzyme, removing K63-conjugated Ub molecules from TRAF2, TRAF6, and NEMO (7–9). However, the putative TRAF2 and NEMO binding sites are absent in the sCYLD protein (Fig. 1 E). To verify whether sCYLD is, indeed, unable to bind TRAF2, HeLa cells were transfected with expression plasmids encoding either FL-CYLD or sCYLD together with TRAF2-encoding or control plasmids. As shown in Fig. 1 F, FL-CYLD protein, but not sCYLD, coimmunoprecipitated with TRAF2, indicating that in contrast to FL-CYLD, sCYLD does not bind TRAF2. However, in an 8% SDS-PAGE, the difference in size between FL-CYLD and sCYLD could not be unambiguously demonstrated (Fig. 1 F). In agreement, immunoprecipitated TRAF2 from extracts of CYLDex7/8 MEFs was highly ubiquitinated compared with TRAF2 immunoprecipitated from WT cells (Fig. 1 G). To investigate whether sCYLD is still able to function as a deubiquitinating enzyme, similar transfection experiments were applied to determine whether both CYLD proteins are capable of removing K63-conjugated Ub from Bcl-3, which is a novel substrate for CYLD (18). The Bcl-3 binding site in CYLD is predicted to reside outside the area absent in sCYLD (Fig. 1 E). We cotransfected HeLa cells with expression vectors encoding His-tagged Ub and Flag-tagged Bcl-3, together with plasmids coding either for FL-CYLD, or for a catalytically inactive form of CYLD (C/S-CYLD) or for sCYLD. Fig. 1 H shows that both sCYLD and FL-CYLD are able to remove Ub from Bcl-3. Collectively, our results demonstrate that sCYLD is not able to deubiquitinate TRAF2, but still removes K63-linked Ub from Bcl-3.
Lymphoid system defects in CYLDex7/8 mice
CYLDex7/8 mice exhibited considerably larger spleens, LNs, and Peyer's patches (Fig. 2 A and not depicted). Careful microscopic examination revealed a disorganized structure of the spleen, where the distinct B cell and T cell zones of the follicle were not uniform as in WT spleens (Fig. 2 B). Importantly, such abnormalities of the secondary immune organs were not reported in mice with complete inactivation of the CYLD gene (18) (unpublished data).
Figure 2.

Enlarged spleen and Peyer's patches (PP) in CYLDex7/8 mice. (A) Spleens and PP dissected from WT and CYLDex7/8 mice were compared. Ruler indicates the size of the organs (centimeters). (B) Cryostat sections from WT and CYLDex7/8 spleens were immunostained for B and T cell follicles with anti-B220 (blue) and anti-CD3ε (brown). Bars, 500 μm.
The CYLDex7/8 mice displayed massively enlarged secondary lymphoid organs as early as 4 wk of age, whereas other tissues appeared normal (unpublished data). To investigate the mechanisms underlying these enlarged lymphoid organs, we examined lymphocyte populations of BM, spleen, LNs, and peritoneal cavity (PC). Analysis of B220+ cells in the BM of CYLDex7/8 mutant mice shows a small decrease in the percentage of the immature B lymphocyte compartments (Fig. 3 A, top). To analyze early B cell development, B220+IgM− cells were analyzed for the expression of the pro-B cell marker, c-Kit. We observed an increase in the pro-B cell fraction, indicating that deletion of the FL-CYLD leads to a partial block in early B cell differentiation (Fig. 3 A, bottom).
Figure 3.

B cell populations in CYLDex7/8 mice. Flow cytometry of 8-wk-old WT, CYLDex7/8, and CYLDex7/8;wt littermate controls stained with the indicated antibodies. (A) Analysis of BM populations. (bottom) Cells gated on IgM− lymphocytes. (B) Analysis of B/T cell ratio in spleen (Sp) and inguinal LNs (LN). (C) Decreased percentage of immature (CD21lowHSAhigh) B cells in spleens of CYLDex7/8 mice. Shown are cells gated on CD19+ lymphocytes. (D) Increased percentage of MZ (CD21highCD23low) B cells in CYLDex7/8 spleens. Cells were gated on CD19+ lymphocytes. (bottom) The mean fluorescence intensity of CD23 and CD21 expression of WT and CYLDex7/8 B cells. (E) Histological analysis of spleen sections from CYLDex7/8 and WT mice using MOMA1 (blue) and IgM antibody (brown). (top) 10× magnification; (bottom) 20× magnification. Bars: (top) 400 μm; (bottom) 100 μm. (F) B cells isolated from the PC were analyzed for B-1 and B-2 B cell populations. Cells were gated on CD19+ B cells. In all graphs, data are representative of five independent experiments with five mice of each genotype. Data are shown for cells gated on the lymphocyte gate, excluding dead cells using Topro3. Numbers in the graphs indicate the percentage of cells from the gated population.
Strikingly, CYLDex7/8 mice, but not heterozygous and control mice, demonstrate a dramatic accumulation of B cells in all secondary lymphoid organs tested (Fig. 3 B and Table I; not depicted for Peyer's patches). The proportion and absolute number of immature B cells was reduced in CYLDex7/8 mice (Fig. 3 C and Table I), although they displayed significantly higher total B cell numbers (Table I).
Table I.
Lymphocyte population sizes
| Spleen | ||||
|---|---|---|---|---|
| Genotype | Total | B cells | Immature | MZ |
| WT (14) | 79.4 (8.2) | 46.7 (4.8) | 7.7 (0.8) | 2.4 (0.3) |
| CYLDex7/8;wt (9) | 96.9 (13.0) | 49.3 (6.6) | 6.2 (0.9) | 2.9 (0.4) |
| CYLDex7/8 (14) | 138.7 (21.6)a | 111.1 (17.3)b | 4.8 (0.7)a | 10.3 (1.6)b |
| CYLDko (5) | 84.7 (8.4) | 42.4 (4.1) | 14.4 (1.4) | 4.6 (0.4) |
| CD19-Cre/CYLDFL/FL (5) | 134.5 (12.6)a | 100.8 (9.5)a | 11.1 (1.0) | 10.1 (0.9)a |
| Peritoneal cavity | ||||
| Genotype | B-1 | B-2 | ||
| WT (4) | 1.4 (0.3) | 0.9 (0.2) | ||
| CYLDex7/8;wt (4) | 1.1 (0.1) | 1.6 (0.1) | ||
| CYLDex7/8 (4) | 0.3 (0.2)a | 2.1 (0.2)a | ||
| CYLDko (5) | 1.5 (0.3) | 0.3 (0.1) | ||
| CD19-Cre/CYLDFL/FL (5) | 0.4 (0.1)a | 0.8 (0.2) |
The mean size of lymphocyte populations was calculated based on their frequency as determined by flow cytometry (Fig. 2). Total numbers (× 106) are indicated (the SEM is shown in parentheses). Data are the mean of the number of mice indicated in parentheses after the mice genotypes. Values are the mean ± the SEM.
P ≤ 0.05.
P ≤ 0.001 versus control.
As seen in Fig. 3 D and Table I, the proportion, as well as the absolute number, of marginal zone (MZ; CD21highCD23low) and follicular (CD21inCD23lo) B cells were increased by two- to threefold in CYLDex7/8 mice compared with WT mice. Interestingly, expression of CD21 and CD23 by B cells isolated from CYLDex7/8 mice was elevated compared with control B cells (Fig. 3 D). The latter was accompanied by an increase in cell size of the CYLD mutant B cells (unpublished data). To analyze the structure of MZ B cells, spleen sections from CYLDex7/8 and littermate control mice were stained with anti-MOMA1 antibody, which binds to metallophilic macrophages, marking the border of the MZ. As shown in Fig. 3 E, MOMA1+ cells can be detected in the spleens of CYLDex7/8; however, the MZ appeared to be disordered and discontinuous compared with WT spleens. Furthermore, the amount of MOMA1+ cells was increased without forming a closed ring around the follicle. Notably, the B cell region located outside of the MOMA1+ cells corresponding to the MZ B cells was larger in the mutant spleens compared with controls (Fig. 3 E). Interestingly, the expansion of MZ and follicular B cells was accompanied by a decrease in the proportion and absolute number of B-1a (CD19highCD5+CD23−) and B-1b (CD19highCD5−CD23−) B cells in the PC (Fig. 3 F and Table I). These results suggest that either overexpression of sCYLD or disruption of FL-CYLD is critical for regulating the maturation of BM B cells and the distribution of peripheral B cells.
To address whether these differences in the B cell compartment resulted from the overexpression of sCYLD, we analyzed another CYLD mutant strain, CYLDko, in which the neomycin resistance gene was introduced into the first coding exon of the CYLD gene, thus preventing its expression (18). Fig. S2 A (available at http://www.jem.org/cgi/content/full/jem.20070318/DC1) shows that the B cell development in the BM was unchanged in CYLDko mice. Furthermore, in contrast to CYLDex7/8 mice, CYLDko mice showed neither differences in B/T cell ratio nor an elevation of MZ and follicular B cells in the spleen nor a decrease of B-1 B cells in the PC (Fig. S2, B–D). Moreover, B cell numbers in these mice were comparable to WT B cell numbers (Table I). Thus, we conclude that the absence of the FL-CYLD, in tandem with the overexpression of sCYLD, is responsible for the increased proportion of B cells in the lymphoid organs of CYLDex7/8 mice.
Skewed humoral immune response of CYLDex7/8 mice
To determine whether the expansion of B cells in CYLDex7/8 mice affects the immune response, we evaluated serum antibody levels in CYLDex7/8 and WT mice. The sera of CYLDex7/8 mice contained significantly lower IgM and IgG3 antibody titers compared with WT littermates, whereas all other tested antibody classes, including IgG1, IgG2a, IgG2b, and IgA, were elevated in CYLDex7/8 mice (Fig. 4 A). The latter antibody isotypes are more frequently secreted by class-switched B cells after encountering T cell–dependent (TD) antigens. Therefore, we next investigated TD antigen responses of CYLDex7/8 B cells and control B cells. To this end, mice were challenged with nitrophenol-conjugated chicken γ-globulin (NP-CG) and immune responses were measured by ELISA. Upon in vivo challenge with NP-CG, CYLDex7/8 mice developed significantly lower antibody titers of both IgM and IgG3 isotypes, whereas secretion of NP-specific IgG1 and IgG2a antibodies was enhanced compared with WT mice (Fig. 4 B). In addition, CYLDex7/8 mice developed higher titers of NP-specific Igλ antibodies, whereas the NP-specific Igκ antibody levels were similar to the levels in WT mice (Fig. 4 B, top). Previously, it was shown that WT mice harbor a relatively high number of NP-specific precursor cells (∼1%) (19). To test whether these findings hold true for another TD antigen with lower precursor frequency, we immunized mice with KLH. CYLDex7/8 mice developed significantly higher levels of KLH-specific IgG1 antibodies after immunization compared with WT mice (Fig. S3 A, available at http://www.jem.org/cgi/content/full/jem.20070318/DC1). CYLDex7/8 mice and littermate controls were also immunized with the T cell–independent (TI) antigen NP-Ficoll, resulting in similar levels of NP-specific IgM and IgG3 antibodies in CYLDex7/8 and WT mice (Fig. 4 C).
Figure 4.

Serum immunoglobulin levels of CYLDex7/8 mice. Sera from 8-wk-old CYLDex7/8 mice (n = 5) and WT littermates (n = 5) were analyzed by ELISA for various immunoglobulin levels. (A) Total immunoglobulin levels of CYLDex7/8 and WT mice. (B) TD immune responses. 8-wk-old CYLDex7/8 and WT littermate mice were immunized with NP28-CG. Blood was taken at the indicated days after immunization. The NP-specific antibody titers were analyzed by ELISA. (C) TI immune responses. 8-wk-old mice were immunized with NP-Ficoll. Blood was taken at the indicated days and analyzed for TI antibody isotypes with capture antigen NP30-BSA by ELISA. (D) FACS analysis of IgG1/IgM-positive B cells isolated from Peyer's patches with the indicated genotypes. (E) Germinal center B cells from Peyer's patches of KLH-immunized mice with the indicated genotypes using cell surface markers for FAS/PNA (bottom). In D and E, cells were gated as CD19+ lymphocytes negative for Topro3.
To examine whether the high IgG1 antibody levels observed in nonimmunized mice, as well as in the immunized mutant mice, were a consequence of increased antibody production by plasma cells or whether CYLDex7/8 B cells switch more efficiently to other antibody classes, B cells from WT and CYLDex7/8 mice were isolated and, subsequently, incubated for 5 d ex vivo with optimal concentrations of LPS and IL-4. A higher proportion of CYLD mutant B cells switched to the IgG1+ isotype compared with WT B cells (Fig. S3 B). B cells in the Peyer's patches are constantly encountered by the gut flora, and therefore yield higher proportions of class-switched B cells. Peyer's patches from CYLDex7/8 mice contain a threefold increased B cell population rich in IgG1-expressing B cells compared with WT mice (Fig. 4 D). Additionally, these results were strengthened by an increased proportion of PNA+Fas+ B cells in CYLDex7/8 Peyer's patches, which is indicative of germinal center B cells (Fig. 4 E). The hyper TD antigen-specific response observed in CYLDex7/8 mice suggests that sCYLD is important for negatively regulating TD, but not TI, responses.
The B cell phenotype in CYLDex7/8 mice is B cell intrinsic
As all cells in CYLDex7/8 mice lacked exon 7, it was unclear whether the defect in B cells was intrinsic to B cells or secondary because of defects in other cell types. Therefore, we crossed CYLDFL/FL mice to CD19-Cre mice (20, 21), leading to Cre-mediated excision of exon 7 of CYLD solely in B cells. The resulting mice, CD19-Cre/CYLDFL/FL exhibited enlarged secondary immune organs, including spleen, LNs, and Peyer's patches similar to the CYLDex7/8 mice that harbor the mutation in the germline (unpublished data). To analyze B cell development in these mice, BM cells were stained for B220 and IgM. As seen in Fig. 5 A (top), BM cells of CD19-Cre/CYLDFL/FL mice show a small decrease in the percentage of immature cells, similar to BM of CYLDex7/8 mice. B220+IgM− cells were gated and analyzed for the expression of the pro-B cell marker, c-Kit. As for the CYLDex7/8 mice, an accumulation of c-Kit–positive cells in the BM of CD19-Cre/CYLDFL/FL was detected (Fig. 5 A, bottom). Importantly, also in CD19-Cre/CYLDFL/FL mice, the total number of B cells was increased by two- to threefold (Fig. 5 B and Table I) compared with WT mice. All other characteristics of the B lymphocytes detected in CYLDex7/8 mice were also manifested in CD19-Cre/CYLDFL/FL mice, including a reduction in the percentage of transitional B cells, a reduction of immature B cells (Fig. 5 C), and an increase in the total number of MZ B cells (Fig. 5 D and Table I). Finally, in both CD19-Cre/CYLDFL/FL and CYLDex7/8 mice, the ratio of B-1 to B-2 B cells in the PC was inverted (Fig. 5 E). Together, these data indicate that the B cell defects seen in the CYLDex7/8 mice are cell autonomous.
Figure 5.

B cell–intrinsic phenotype of the CYLDex7/8 mice. Flow cytometry analysis of cells from the BM, spleen, LN, and PC. Cells of 8-wk-old CD19-Cre/CYLDFL/FL, CYLDex7/8, and WT controls, stained with the indicated antibodies. The genotypes of the mice shown are indicated on top of the figure. (A) Analysis of BM populations. (bottom) Cells gated on Ig− lymphocytes. (B) B/T cell ratio of spleen cells. (C) Immature B cell populations in the spleen. IgM+CD23− cells, which are defined as transitional 1 (T1) cells, and IgM+CD23+ B cells (T2/T3 and mature cells) are shown. (D) FACS analysis for spleen MZ B cells (CD21highCD23low). (E) B-1 and B-2 B cells in the PC of the indicated mice. Shown are all cells gated on the lymphocyte gate, excluding dead cells using Topro3. (B–D) All cells are further gated on CD19+. Data are representative of five independent experiments. Data were acquired and analyzed as described in Fig. 3.
Overexpression of sCYLD results in prolonged B cell survival
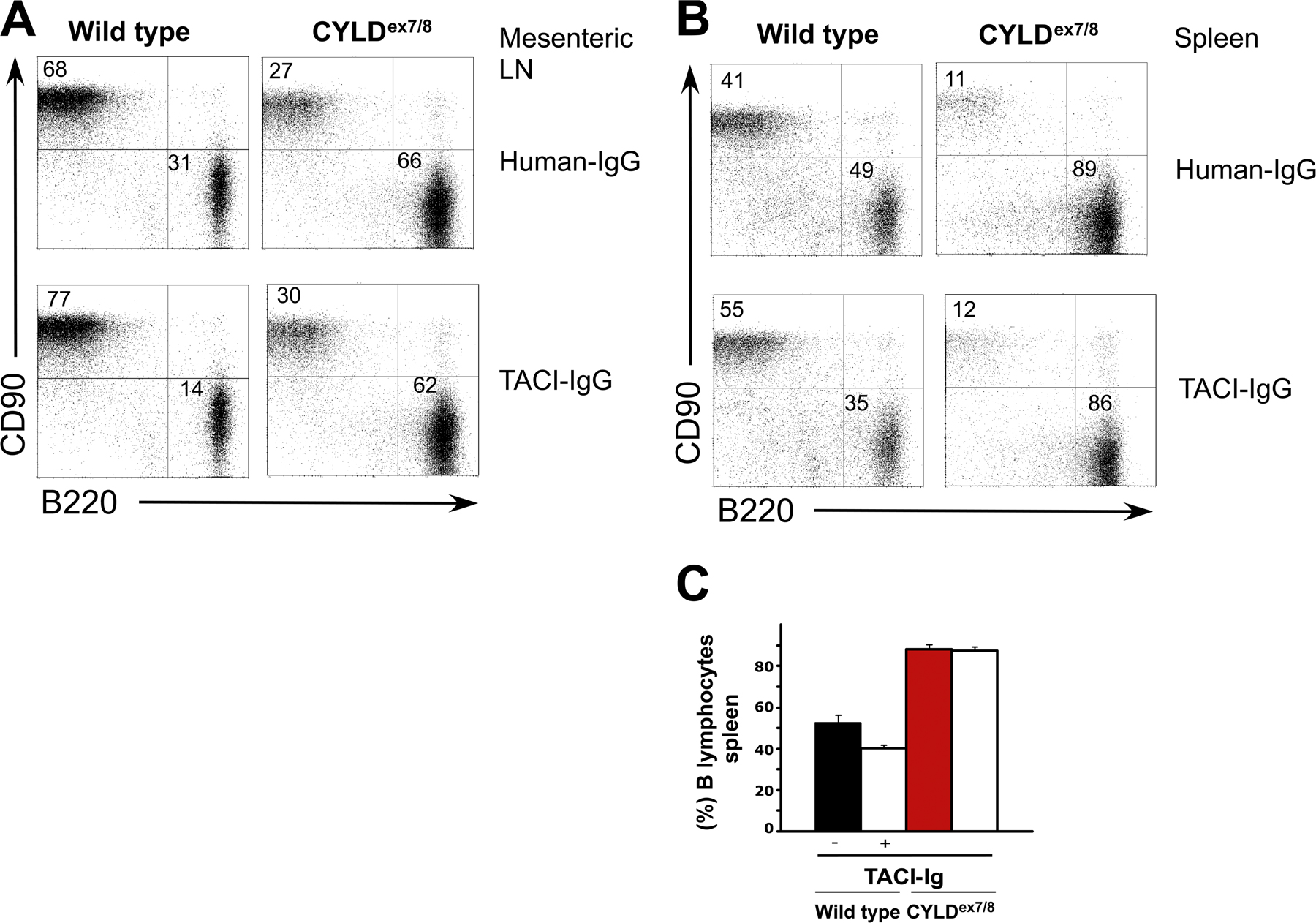
Bcl-2 transgenic mice exhibit enhanced B cell survival in vitro; this is associated with an increased size of secondary immune organs and B cell compartments in vivo (22), which is similar to CYLDex7/8 mice. To examine the survival capacities, B cells from CYLDex7/8 and WT mice were cultured in enriched medium and monitored daily for survival by cell counting and FACS analysis. The CYLD mutant B cells exhibited prolonged survival compared with WT B cells (Fig. 6 A, top). After 4 d of culture, the number of living CYLDex7/8 B cells was increased by twofold compared with WT B cells. Furthermore, the survival properties of CYLDex7/8 B cells were compared with those of WT B cells cultured in medium supplemented with the B cell survival factor BAFF. BAFF treatment led to improved survival of both WT and mutant B cells (Fig. 6 A, bottom). However, at all time points, CYLDex7/8 B cells responded better to BAFF treatment compared with control B cells. Additionally, BAFF triggering of CYLDex7/8 B cells resulted in the up-regulation of the activation markers CD40 and CD86, respectively, in comparison to WT B cells, whereas CpG treatment had similar effects on both B cell cultures (Fig. 6 B). To investigate whether the increased numbers of mature B cells in CYLDex7/8 mice was related to response to BAFF in vivo, CYLD mutant and control mice were treated with either TACI- or human-Ig as control. It was shown that TACI-Ig blocks BAFF signaling, and that this treatment resulted in a dramatic decrease in B cell numbers (23). In treated WT mice, we detected a significant decrease in the percentage of mature B cells in the mesenteric LN (Fig. 6 C and Fig. S4 A, available at http://www.jem.org/cgi/content/full/jem.20070318/DC1) and spleen (Fig. S4, B and C). In contrast, the percentage of B cells in CYLDex7/8 mice was hardly affected by the TACI-Ig treatment (Fig. 6 C and Fig. S4). No differences in the total numbers of T cells in CYLDex7/8 and WT mice were observed upon TACI treatment, showing that, indeed, TACI-Ig acts specifically on B lymphocytes (unpublished data).
Figure 6.

In vitro stimulation and survival of CYLDex7/8 B cells. (A) Purified (4 × 106) CYLDex7/8 and WT B cells were cultured in medium alone (without stimulation) or in medium supplemented with 100 μg BAFF. Numbers of surviving cells were determined by counting viable cells and by FACS analysis. Each time point represents the average of six samples ± the SEM. (B) Expression of activation markers CD40 (left) and CD86 (right) before and after stimulation of splenic B cells from the indicated genotypes with either BAFF or CpG. (C) CYLDex7/8 and WT mice (n = 3) were treated twice a week for 1 wk with 20 μg TACI-Ig or Hu-Ig control protein. Comparison of percentages of mesenteric LN B cells from mice treated with control Hu-Ig (−) and TACI-Ig (+) is shown. Values are the mean ± the SEM. *, P ≤ 0.05. (D) Relative expression of Bcl-2 from purified splenic B cells from mice of the indicated genotypes (n = 3) was determined by quantitative real-time PCR and intracellular staining. Values are the mean ± the SEM. *, P ≤ 0.05. (E) TNF-induced apoptosis. Embryonic fibroblasts isolated from mice of the indicated genotypes were pretreated with PMA or left untreated before induction of apoptosis by the simultaneous addition of TNFα and CHX. Shown is the percentage of dead (AnnexinV+Topro3+) and apoptotic cells (AnnexinV+Topro3−) from the total cells in the culture. (F) In vitro proliferation assay of purified splenic B cells from CYLDex7/8 and WT control mice. B cells were isolated and loaded with CFSE. Thereafter, cells were triggered for 4 d with the indicated stimuli with and without BAFF. Shown are cells gated on lymphocyte and blast cell gate using Topro3 to exclude dead cells. Cells were further gated using CD45R (B220) surface marker.
To investigate the increased viability of CYLDex7/8 B cells in vitro, the expression of the antiapoptotic gene Bcl-2 was assessed by quantitative real-time PCR. CYLDex7/8 B cells express fivefold more Bcl-2 mRNA compared with WT B cells (Fig. 6 D). These results were further verified at protein level by intracellular staining with Bcl-2–specific antibody (Fig. 6 D). In addition, we found that primary fibroblasts isolated from CYLDex7/8 mutant mice are more resistant to TNFα-induced apoptosis in the absence of protein synthesis (Fig. 6 E). Therefore, it is tempting to speculate that the deletion of the FL-CYLD, concomitant with the overexpression of sCYLD, conveys increased resistance to apoptosis, leading to the enlarged B cell compartment in CYLDex7/8 mice.
Next, we determined the responses of CYLDex7/8 and WT B cells to activating stimuli by incubating CFSE-labeled B cell cultures with optimal concentrations of anti-IgM F(ab)′2 and anti-CD40 antibodies, or with anti-RP105, LPS, or BAFF. As shown in Fig. 6 F, B cells from CYLDex7/8 mice responded similarly to the different stimuli compared with WT B cells. Although the addition of BAFF to culture medium resulted in a more activated status of the mutant B cells compared with the WT B cells (Fig. 6 B), supplementing the culture with BAFF together with LPS failed to increase proliferation (Fig. 6 F). Therefore, we conclude that sCYLD contributes to the survival capacity of B cells, but does not affect B cell proliferation.
Elevated expression of NF-κB and TRAF proteins in resting CYLDex7/8 B cells
Previously, CYLD was shown to act on different proteins involved in the NF-κB (7–9) and mitogen-activated protein kinase (MAPK) signaling pathways (17, 24). To study whether the enlargement of the B cell compartment in CYLDex7/8 mice results from alterations in the expression levels of proteins involved in the NF-κB signaling pathway, whole-cell lysates of purified B cells from three CYLDex7/8 mutant mice and three control mice were subjected to Western blot analysis. This analysis revealed increased protein levels of TRAF2 in the CYLD mutant B cells compared with WT B cells (Fig. 7 A). Additionally, the protein levels of TRAF3 and NEMO were determined, showing that TRAF3 levels were increased, whereas NEMO levels were unchanged (Fig. 7 A). The levels of IκBα protein were markedly elevated in CYLDex7/8 B cells, although its phosphorylation on conserved serine residues was unaltered. Most interestingly, NFκB2/p100 protein levels primarily involved in the noncanonical NF-κB pathway were dramatically increased, whereas the levels of its processed form, p52, were only slightly increased (Fig. 7 A). Furthermore, we could show that the RelB protein levels were also highly increased in the cytoplasm, as well as the nucleus, of CYLDex7/8 B cells (Fig. 7 B). The elevated protein levels of TRAF2, TRAF3, p100, RelB, and IκBα in CYLDex7/8 B cells likely resulted from increased protein stability rather than from increased transcriptional activity, as quantitative real-time PCR revealed no significant differences in relative expression of these genes (Fig. S5, available at http://www.jem.org/cgi/content/full/jem.20070318/DC1). To investigate whether the noncanonical NF-κB pathway is affected in CYLDex7/8 B cells, purified B lymphocytes of control and CYLDex7/8 mice were cultivated with optimal concentrations of BAFF for the indicated time points (Fig. 7 C). At all time points analyzed, we found increased p100 levels, but no significant changes in the levels of its processed form, p52, in CYLDex7/8 B cells when compared with controls (Fig. 7 C).
Figure 7.

Analysis of signal transduction of CYLDex7/8 mutant splenic B cells. (A) Whole-cell extracts were examined by Western blot using antibodies against the indicated proteins. Shown are three Western blots from B cell lysates of three different WT (lanes 1–3) and three different CYLDex7/8 (lanes 4–6) mice isolated in the same experiment. AKT-specific antibody was used as loading control. (B; left) cytoplasmic and (right) nuclear extracts of nonactivated B cells isolated from the indicated genotypes were probed with RelB-specific antibody. H2B and actin-specific antibodies were used as loading control. (C) MACS-purified B cells were rested for 4 h and subsequently stimulated with optimal concentrations of BAFF for the indicated time points and subjected to Western blot analysis using the indicated antibody. (D) Extracts from CYLDex7/8 and WT B cells treated with an anti-CD40 antibody, LPS, or anti-BCR for different time points were analyzed with antibodies against IκBα and pIκBα. Actin-specific antibody was used as loading control. (E) EMSA was performed with extracts of B cells rested for 4 h after isolation. Cells were then activated by anti-BCR for the indicated time points before extracts were prepared, incubated with NF-κB–specific labeled probe, and separated on native PAGE. (F) B cell lysates from CYLDex7/8 and WT controls were stimulated with LPS for the indicated time points, and CBA was performed using beads loaded with antibodies specific for p-p38 and pERK1/2. (G) Western blot was performed for p-p38 and pERK from B cells of the indicated genotypes stimulated with anti-BCR for the indicated time points. Actin-specific antibody was used as loading control.
To measure canonical NF-κB activity in CYLDex7/8 B cells, splenic B cells were stimulated with anti-CD40, LPS, and anti-BCR (Fig. 7 D). We did not observe significant differences in IκBα degradation upon triggering of CYLDex7/8 and WT B cells with the aforementioned stimuli (Fig. 7 D). Similarly, we did not detect changes in IκB degradation and phosphorylation after anti-BCR treatment (Fig. 7 D). As a direct measure of NF-κB activity, we performed electrophoretic mobility shift assay (EMSA) using NF-κB consensus-binding oligonucleotides with nuclear extracts from resting B cells stimulated with optimal concentrations of anti-BCR (Fig. 7 E). Nonstimulated CYLDex7/8 B cells exhibited detectable NF-κB activation, which was not observed in WT B cells (Fig. 7 E). However, the NF-κB–activating response to anti-IgM was unaltered at the different time points of stimulation in CYLDex7/8 B cells compared with WT B cells. This result suggests that the basal activity of NF-κB may reflect an increased response to a yet unknown endogenous activator.
To estimate the effect of the sCYLD in B cells on MAPK signaling, we performed cytometric bead assay (CBA) to determine quantitatively phosphorylated MAPKs, which can be visualized using FACS analysis. In this assay, LPS stimulation resulted in increased phosphorylation of MAPK p38 in CYLDex7/8 B cells compared with WT B cells (Fig. 7 F). Similar results were observed by Western blotting of anti-BCR–stimulated B cells (Fig. 7 G). In contrast, we observed reduced phosphorylation of ERK1 and ERK2 upon LPS stimulation (Fig. 7 F). This could be further confirmed with Western blot analysis (Fig. 7 G).
In keratinocytes isolated from CYLDko mice, Bcl-3 associates with the NF-κB proteins p52 and p50 and promotes increased transactivation of cyclin D1 compared with WT cells (18). Although we have shown that sCYLD is able to interact with Bcl-3 and to remove K63-conjugated Ub, we were interested to see whether Bcl-3 is present in the nuclei of B cells from CYLDex7/8 mice without activation. To this end, we prepared cytospins of CYLDex7/8, CYLDex7/8;wt, CYLDko, and WT B cells. Fig. 8 (A and B) demonstrates that Bcl-3 was predominantly located in the nucleus, or in close proximity to the nuclei, of untreated CYLDex7/8 B cells, whereas in WT B cells, Bcl-3 was primarily found in the cytoplasm. Calculating the percentage of B cells of the various genotypes containing nuclear Bcl-3 revealed that 60% of the CYLDex7/8 B cells contain nuclear Bcl-3, whereas B cells from WT, CYLDex7/8;wt, and CYLDko exhibit 8, 3, and 2.5% of Bcl-3 in the nucleus, respectively (Fig. 8 C). Furthermore, we could corroborate these results using Western blot analysis of extracts from CYLDex7/8 mice (Fig. 8 D). By quantitative real-time PCR, we could show that the increased protein levels of Bcl-3 in the B cells of the CYLDex7/8 mice are not a result of higher Bcl-3 gene expression (Fig. S5). Interestingly, mice that are deficient for CYLD do not show spontaneous translocation of Bcl-3 to the nucleus. These results are reminiscent of the previously published Bcl-3 transgenic mice (25), in which overexpression of Bcl-3 in B cells resulted in lymphadenopathy, splenomegaly, and altered immunoglobulin production, similar to what was observed in CYLDex7/8 mice.
Figure 8.

Bcl-3 accumulation in CYLDex7/8 B cells. (A) Cytospins were prepared from B cells isolated from CYLDex7/8, CYLDex7/8;wt, CYLDko, and WT mice and immunostained with anti–Bcl-3 and counterstained with Hoechst 33258. Shown are unstimulated B cells (top) and B cells treated with anti-BCR (bottom). Bar, 5 μm. (B) Same as in A, but showing more cells. Bar, 5 μm. (C) Quantification of nuclear Bcl-3 from unstimulated B cell cytospins from the indicated genotypes. For each individual genotype, 400 cells were counted and quantified for nuclear Bcl-3 localization. The frequency of nuclear Bcl-3 localization in CYLDex7/8 B cells were statistically significant (P < 0.001) compared with B cells isolated from WT, CYLDex7/8;wt, and CYLDko mice. (D) Lysates of MACS-purified B cells from the indicated genotypes subjected to Western blot analysis using Bcl-3 antibody. Duplicates for each mouse strain are shown. Actin was used as a loading control.
DISCUSSION
In this article, we report the function of a splice variant of CYLD devoid of the TRAF2 and NEMO binding sites. Expression of this shorter form of CYLD (sCYLD) with a simultaneous deficiency of the full-length (FL) protein gave rise to a dramatic accumulation of mature B lymphocytes in all secondary lymphoid organs in vivo. This B cell accumulation is a consequence of increased survival, rather than increased proliferation, putatively resulting from aggravated Bcl-2 expression caused by various alterations in signal transduction pathways, such as NF-κB and MAPK signaling. In contrast, mice that are entirely devoid of CYLD (CYLDko mice) do not show any abnormalities in the B cell compartment. Therefore, we reason that sCYLD has other functions in B cell homeostasis than FL-CYLD. Under physiological conditions FL-CYLD expression might be diminished, whereas the expression of sCYLD is elevated. Indeed, we detected various expression levels of both CYLD forms in different tissues and cell types. Therefore, we suggest that also under normal conditions, the unique function of sCYLD can be manifested.
The CYLD-deficient mice generated by Reiley et al. (26) demonstrate an elevated number of B lymphocyte, which is explained by a compensatory effect caused by the reduction in the T-lymphocyte number (26). In CYLDex7/8 mutant mice, the generalized accumulation of mature B cells seems to be B cell intrinsic because CD19-Cre/CYLDFL/FL mice, in which only the B cells express sCYLD and concomitantly lack the FL-CYLD protein, show a dramatically enlarged B cell compartment. We further observed that CYLDex7/8 mice have an increased number of Igλ-positive B cells compared with WT mice. This might be caused by the partial block in B cell development, which is demonstrated by an increase in the number of c-Kit–positive B cells in the BM. A block during B cell development may allow the developing B cells to edit their light chain locus, as seen in the process of B cell editing (27), which, indeed, results in higher numbers of λ-positive B cells.
We observed increased protein levels of several molecules involved in the NF-κB signaling pathway in CYLDex7/8 B cells, including IκBα. However, the degradation and phosphorylation of IκBα is as efficient in the CYLDex7/8 B cells as in the WT B cells upon stimulation, showing that the canonical NF-κB activation is unaltered. Consistently, the relative expression of IκBα, p100, and RelB, which are all under the regulation of the NF-κB pathway, was unaltered as determined by quantitative real-time PCR, thus pointing to increased protein stability resulting from differences in posttranslational modifications. Nevertheless, EMSA revealed elevated binding of nuclear transcription factors to the NF-κB consensus probe in unstimulated B cells.
We could demonstrate elevated protein levels of TRAF2 and TRAF3 in CYLDex7/8 B cells, which is a consequence of impaired posttranslational modification because relative expression revealed by real-time PCR was unaltered. Consistently, TRAF2 was highly ubiquitinated in CYLDex7/8 MEFs, presumably by K63-linked Ub chains, as shown by Western blot analysis. Therefore, it is tempting to speculate that K63-ubiquitinated TRAF2 exhibit increased protein stability and possibly have impaired functionality. This is supported by the aggravated TRAF3 protein levels in CYLDex7/8 B cells because TRAF2 was shown to be a negative regulator of TRAF3 upon stimulation with CD40 (28). B lymphocytes deficient for TRAF2 demonstrate a phenotype similar to that of CYLDex7/8 B cells with increased TRAF3 protein levels, but also show constitutive processing of p100 to p52, which is contrary to CYLDex7/8 B cells (29). In contrast, however, TRAF3-deficient cells also display constitutive processing of p100 to p52, resulting in embryonic lethality. This phenotype can be rescued in vivo by simultaneous deficiency of p100 (30). Therefore, elevated TRAF3 levels should lead to increased p100 levels. Consistently, CYLDex7/8 B cells exhibit increased p100 protein levels, but not elevated processing to p52. In addition, elevated TRAF2 and TRAF3 levels might promote MAPK p38-mediated isotype switching to IgG1, as essential functions for these adaptor molecules in CD40-induced class switch recombination and Ig production were reported (31, 32). Indeed, CYLDex7/8 B cells switch more efficiently to IgG1, but whether this is a result of the p38 hyperactivation observed in CYLDex7/8 B cells or a result of impaired ERK activation still needs further investigation.
Furthermore, CYLDex7/8 B cells exhibit a significant accumulation of Bcl-3 in the nucleus that cannot be explained by an inability of sCYLD to associate with Bcl-3, as we could show that sCYLD binds and removes K63-linked ubiquitin chains from Bcl-3. Western blot analysis of B lymphocytes using Bcl-3 antibody revealed aggravated Bcl-3 protein levels, whereas on the RNA level, expression of Bcl-3 was unaltered in CYLDex7/8 B cells. Ultimately, CYLDex7/8 mice show a very similar phenotype to mice overexpressing Bcl-3 in B lymphocytes (25). Both transgenic mice show lymphadenopathy and splenomegaly and exhibit distorted follicles in the spleen, resulting from an enlarged B cell compartment. Further, both mouse strains showed an increase in IgG1 and decreased levels of IgM and IgG3 in the sera compared with WT mice. In addition, enhanced B cell survival, accumulation of MZ B cells, as well as increased B cell size, and the increased expression of CD21/CD23 observed in CYLDex7/8 mice are also found in Bcl-3 transgenic mice.
The elevated expression of Bcl-2 in CYLDex7/8 B cells may be a consequence of the dramatic overexpression of the NF-κB proteins p100 and RelB, which might activate transcription of Bcl-2 in a complex with Bcl-3 upon signaling through the noncanonical NF-κB pathway (33). Thus, nuclear Bcl-3 in combination with p100 can induce the expression of Bcl-2 (33) and might account for the elevated B cell survival observed in the CYLDex7/8 mice, as shown previously for Bcl-2 transgenic mice (22, 34). Concomitantly, CYLDex7/8 B cells are less dependent on BAFF signaling by a yet unsolved mechanism. This was demonstrated by ineffective BAFF blocking by TACI-Ig. Collectively, our data suggest that the newly identified splice variant sCYLD regulates B cell survival mediated through a variety of signaling disarrangements.
MATERIALS AND METHODS
Generation of CYLD mutant mice.
The generation of CYLD mutant mice is described in the Supplemental materials and methods. All animal experiments were in accordance with the guidelines of the Central Animal Facility Institution of the University of Mainz.
RNA analysis.
For RNA analysis, total RNA was isolated using Trizol reagent (Invitrogen) according to the manufacturer's instruction. The following primers were used for RT-PCR amplification of mouse CYLD: P1, 5′-CTCAGACCCTGGAAGTAGAA-3′; P2, 5′-TTGTAATGGCCCTGGATGCC-3′. Quantitative real-time PCR of Bcl-2, p100, RelB, IkBα, TRAF2, TRAF3, and Bcl-3 was performed using primers from QIAGEN, as described on their homepage (https://www1.qiagen.com/GeneGlobe/Default.aspx).
Immunostaining of purified B cells.
Cytospins of purified B cells were fixed in acetone for 10 min at −20°C and stained for Bcl-3 according to standard methods, using polyclonal rabbit IgG Bcl-3 antibody. All incubation steps of the staining procedure were performed at room temperature. The cytospins were counterstained with Hoechst 33258. Cytospins were analyzed by confocal microscopy.
Histological analysis.
For histological staining of B and T cells and germinal centers, frozen 6-μm spleen sections were thawed, air dried, fixed in acetone, and stained for 1 h at room temperature in a humidified chamber with biotinylated rat anti-CD19 (BD Biosciences), rat anti-B220, anti-CD3e, and anti-MOMA1, respectively (eBioscience), followed by horseradish peroxidase–conjugated secondary antibody and alkaline phosphatase–conjugated streptavidin.
Flow cytometry.
Single-cell suspensions were prepared from the different organs. Red blood cells of LN and spleen were lysed in cell suspension with tris-ammonium chloride, pH 7.2. Cells were incubated with combinations of antibodies to cell surface determinants, conjugated to PE, FITC, Cy-Cychrome, or biotin. Antibodies specific to the following surface markers were purchased from BD Biosciences: CD5, CD19, CD21 (7G6), CD23, HSA (CD24), CD25, CD90.2, CD43, and IgD. Anti-CD45R (B220; clone RA3-6B2) and anti-IgM (R33-24-12) were prepared in our laboratory. Biotinylated cells were visualized with streptavidin conjugated to PE or Cy-Cychrome (BD Biosciences). Bcl-2 was visualized using anti–Bcl-2 antibody (Santa Cruz Biotechnology) and secondary donkey anti–rabbit sera conjugated to FITC (Jackson ImmunoResearch Laboratories). All samples were acquired on a FACSCalibur (BD Biosciences), and results were analyzed with CellQuest software (BD Biosciences). Absolute numbers of splenocyte subpopulations were calculated based on their percentage and the total number of splenocytes.
Immunostaining and transfection.
The coding regions of murine Bcl-3, TRAF2, FL-CYLD, or sCYLD were cloned after RT-PCR using specific primers from the total RNA of activated B cells. PCR products were cloned in pcDNA3.1 vector (Invitrogen). HeLa cells and MEFs were transfected with the different constructs or histidine-Ub (His-Ub), as described in Fig. 1, using LipofectAmine2000 (Invitrogen). For anti–Bcl-3 or anti-TRAF2 immunoprecipitation, lysates were precleaned for 30 min at 4°C. The protein content was determined and compensated for equal content in all supernatants. For testing protein input, a small part of the resulting lysates was gel separated and immunoblotted (protein input), and the remaining part was used for immunoprecipitation. Immunoblots were developed with the ECL Plus reagent (GE Healthcare) according to the manufacturer's guidelines.
Immunization.
8-wk-old WT and CYLDex7/8 mice were immunized intraperitoneally with 20 μg of NP28-CCG or 100 μg of KLH for TD response, or 20 μg of NP-Ficoll for TI responses. On days 0, 7, 14, 21, and 28, serum was collected from peripheral blood. Circulating antibodies were measured by isotype- and antigen-specific ELISA. Captured antibodies were detected with enzyme-conjugated secondary antibodies.
TACI-Ig treatment.
CYLDex7/8 and control mice were treated via i.p. injection with 20 μg of TACI-Ig (Alexis Biochemicals) or human IgG (Hu-Ig) twice a week for a total of 1 wk.
B cell preparation and B cell proliferation assay.
Total mouse splenic B cells were purified through negative depletion with CD43 beads (Miltenyi Biotech). The purity (>95%) was determined by flow cytometry. For in vitro proliferation studies, B cells were labeled with CFSE, and 2 × 105 cells/well were incubated in RPMI medium plus 10% FCS or untreated or treated with 5 μg/ml anti-CD40, 5 μg/ml anti-IgM, 2 μg/ml LPS, and 5 ng/ml LPS plus IL-4 with and without 100 ng/ml BAFF for 4 d.
Measurement of TNFα-induced cell death.
MEFs (106 per well) were plated onto 6-well plates and cultured for 24 h in DME containing 10% FCS. Subsequently, the cells were stimulated with either 10 ng/ml TNFα with and without 300 ng/ml cycloheximide. Triplicate samples of TNFα-induced cell death were measured by FACS analysis using Topro3 and Annexin V.
Western blot.
To prepare whole-cell lysates, B cells were lysed in 1% Triton X-100 with protease inhibitors. MACS-purified B cells were prepared. Protein lysates (30 μg) were separated by 10% SDS-PAGE and subsequently transferred to PVDF membranes. Protein blots were probed with antibodies to phospho-IκBα, IκBα, phospho-ERK, ERK, phospho-p38, p38, p100, and Bcl-2 (all from Cell Signaling Technology), Bcl-3, NEMO, TRAF2, TRAF3, and RelB (from Santa Cruz Biotechnology), and with antibodies to actin (Sigma-Aldrich), AKT, or H2B (Cell Signaling Technology) as internal loading controls. CYLD-specific antibodies were prepared by immunization of rabbits with recombinant CYLD fragments. Nuclear extracts were prepared using high-salt buffer, and proteins were subjected to Western blotting. CBA was performed according to the manufacturer's guidelines.
Statistical analysis.
Values are presented as the means ± the SEM or SD, with the number of independent experiments. Statistical differences were determined using the Student's t test.
Online supplemental material.
Fig. S1 shows the targeting strategy to generate the CYLDex7/8 mice. Fig. S2 describes the B cell population of CYLDko mice. Fig. S3 describes the KLH-specific immune responses of CYLDex7/8 mice and class switch recombination in culture. Fig. S4 shows the B cell populations in CYLDex7/8 mice treated with TACI-Ig and Fig. S5 summarizes the relative RNA expression of different molecules using real time PCR. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20070318/DC1.
Supplemental Material
Acknowledgments
We thank Claudia Uthoff-Hachenberg, Annette Shrestha, Sonja Becker, Nicole Voltz, and Marina Snetkova for excellent technical assistance. Further, we thank Dr. Marc Schmidt-Supprian and Sonja Reissig for discussion and Dr. Hansjörg Schild and Andrew Croxford for critically reading the manuscript.
This work was funded by the FP6 Marie Curie Research Training Network MRTN-CT-2004-005632, the Deutsche Forschungsgemeinschaft grants SFB548 and SFB432 and by funds from the Boehringer Ingelheim Stiftung to A. Waisman. J. Song was a fellow of the MD/PhD program of the Center for Molecular Medicine Cologne, Cologne, Germany.
The authors have no conflicting financial interests.
Abbreviations used: CBA, cytometric bead assay; EMSA, electrophoretic mobility shift assay; MAPK, mitogen-activated protein kinase; MEF, mouse embryonic fibroblast; MZ, marginal zone; NEMO, NF-κB essential modulator; NP-CG, nitrophenol-conjugated chicken γ-globulin; PC, peritoneal cavity; TD, T cell–dependent; TI, T cell–independent; TLR, Toll-like receptor.
N. Hövelmeyer and F.T. Wunderlich contributed equally to this paper.
R. Massoumi's present address is Division of Experimental Pathology, Malmö University Hospital, Malmö, Sweden.
References
- 1.Abenoza, P., and A.B. Ackerman. 1990. Neoplasms with Eccrine Differentiation. Lea and Febiger, Philadelphia.
- 2.Biggs, P.J., P. Chapman, S.R. Lakhani, J. Burn, and M.R. Stratton. 1996. The cylindromatosis gene (cyld1) on chromosome 16q may be the only tumour suppressor gene involved in the development of cylindromas. Oncogene. 12:1375–1377. [PubMed] [Google Scholar]
- 3.Biggs, P.J., R. Wooster, D. Ford, P. Chapman, J. Mangion, Y. Quirk, D.F. Easton, J. Burn, and M.R. Stratton. 1995. Familial cylindromatosis (turban tumour syndrome) gene localised to chromosome 16q12-q13: evidence for its role as a tumour suppressor gene. Nat. Genet. 11:441–443. [DOI] [PubMed] [Google Scholar]
- 4.Takahashi, M., E. Rapley, P.J. Biggs, S.R. Lakhani, D. Cooke, J. Hansen, E. Blair, B. Hofmann, R. Siebert, G. Turner, et al. 2000. Linkage and LOH studies in 19 cylindromatosis families show no evidence of genetic heterogeneity and refine the CYLD locus on chromosome 16q12-q13. Hum. Genet. 106:58–65. [DOI] [PubMed] [Google Scholar]
- 5.Thomson, S.A., S.A. Rasmussen, J. Zhang, and M.R. Wallace. 1999. A new hereditary cylindromatosis family associated with CYLD1 on chromosome 16. Hum. Genet. 105:171–173. [DOI] [PubMed] [Google Scholar]
- 6.Verhoef, S., C.T. Schrander-Stumpel, V.D. Vuzevski, A. Tempelaars, L.A. Jansen, G.A. Malfeyt, T.L. Ceelen, D. Lindhout, D.J. Halley, and A.M. van den Ouweland. 1998. Familial cylindromatosis mimicking tuberous sclerosis complex and confirmation of the cylindromatosis locus, CYLD1, in a large family. J. Med. Genet. 35:841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kovalenko, A., C. Chable-Bessia, G. Cantarella, A. Israel, D. Wallach, and G. Courtois. 2003. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature. 424:801–805. [DOI] [PubMed] [Google Scholar]
- 8.Brummelkamp, T.R., S.M. Nijman, A.M. Dirac, and R. Bernards. 2003. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature. 424:797–801. [DOI] [PubMed] [Google Scholar]
- 9.Trompouki, E., E. Hatzivassiliou, T. Tsichritzis, H. Farmer, A. Ashworth, and G. Mosialos. 2003. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 424:793–796. [DOI] [PubMed] [Google Scholar]
- 10.Amit, S., and Y. Ben-Neriah. 2003. NF-kappaB activation in cancer: a challenge for ubiquitination- and proteasome-based therapeutic approach. Semin. Cancer Biol. 13:15–28. [DOI] [PubMed] [Google Scholar]
- 11.Deng, L., C. Wang, E. Spencer, L. Yang, A. Braun, J. You, C. Slaughter, C. Pickart, and Z.J. Chen. 2000. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 103:351–361. [DOI] [PubMed] [Google Scholar]
- 12.Wang, C., L. Deng, M. Hong, G.R. Akkaraju, J. Inoue, and Z.J. Chen. 2001. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 412:346–351. [DOI] [PubMed] [Google Scholar]
- 13.Pickart, C.M. 2001. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70:503–533. [DOI] [PubMed] [Google Scholar]
- 14.Weissman, A.M. 2001. Themes and variations on ubiquitylation. Nat. Rev. Mol. Cell Biol. 2:169–178. [DOI] [PubMed] [Google Scholar]
- 15.Jono, H., J.H. Lim, L.F. Chen, H. Xu, E. Trompouki, Z.K. Pan, G. Mosialos, and J.D. Li. 2004. NF-kappaB is essential for induction of CYLD, the negative regulator of NF-kappaB: evidence for a novel inducible autoregulatory feedback pathway. J. Biol. Chem. 279:36171–36174. [DOI] [PubMed] [Google Scholar]
- 16.Yoshida, H., H. Jono, H. Kai, and J.D. Li. 2005. The tumor suppressor cylindromatosis (CYLD) acts as a negative regulator for toll-like receptor 2 signaling via negative cross-talk with TRAF6 AND TRAF7. J. Biol. Chem. 280:41111–41121. [DOI] [PubMed] [Google Scholar]
- 17.Reiley, W., M. Zhang, and S.C. Sun. 2004. Negative regulation of JNK signaling pathway by the tumor suppressor CYLD. J. Biol. Chem. 279:55161–55167. [DOI] [PubMed] [Google Scholar]
- 18.Massoumi, R., K. Chmielarska, K. Hennecke, A. Pfeifer, and R. Fassler. 2006. Cyld inhibits tumor cell proliferation by blocking bcl-3-dependent NF-kappaB signaling. Cell. 125:665–677. [DOI] [PubMed] [Google Scholar]
- 19.Lalor, P.A., G.J. Nossal, R.D. Sanderson, and M.G. McHeyzer-Williams. 1992. Functional and molecular characterization of single, (4-hydroxy-3-nitrophenyl)acetyl (NP)-specific, IgG1+ B cells from antibody-secreting and memory B cell pathways in the C57BL/6 immune response to NP. Eur. J. Immunol. 22:3001–3011. [DOI] [PubMed] [Google Scholar]
- 20.Rickert, R.C., K. Rajewsky, and J. Roes. 1995. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. 376:352–355. [DOI] [PubMed] [Google Scholar]
- 21.Rickert, R.C., J. Roes, and K. Rajewsky. 1997. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 25:1317–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strasser, A., S. Whittingham, D.L. Vaux, M.L. Bath, J.M. Adams, S. Cory, and A.W. Harris. 1991. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc. Natl. Acad. Sci. USA. 88:8661–8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gross, J.A., J. Johnston, S. Mudri, R. Enselman, S.R. Dillon, K. Madden, W. Xu, J. Parrish-Novak, D. Foster, C. Lofton-Day, et al. 2000. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 404:995–999. [DOI] [PubMed] [Google Scholar]
- 24.Reiley, W., M. Zhang, X. Wu, E. Granger, and S.C. Sun. 2005. Regulation of the deubiquitinating enzyme CYLD by I{kappa }B kinase gamma-dependent phosphorylation. Mol. Cell. Biol. 25:3886–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ong, S.T., M.L. Hackbarth, L.C. Degenstein, D.A. Baunoch, J. Anastasi, and T.W. McKeithan. 1998. Lymphadenopathy, splenomegaly, and altered immunoglobulin production in BCL3 transgenic mice. Oncogene. 16:2333–2343. [DOI] [PubMed] [Google Scholar]
- 26.Reiley, W.W., M. Zhang, W. Jin, M. Losiewicz, K.B. Donohue, C.C. Norbury, and S.C. Sun. 2006. Regulation of T cell development by the deubiquitinating enzyme CYLD. Nat. Immunol. 7:411–417. [DOI] [PubMed] [Google Scholar]
- 27.Pelanda, R., S. Schwers, E. Sonoda, R.M. Torres, D. Nemazee, and K. Rajewsky. 1997. Receptor editing in a transgenic mouse model: site, efficiency, and role in B cell tolerance and antibody diversification. Immunity. 7:765–775. [DOI] [PubMed] [Google Scholar]
- 28.Hostager, B.S., S.A. Haxhinasto, S.L. Rowland, and G.A. Bishop. 2003. Tumor necrosis factor receptor-associated factor 2 (TRAF2)-deficient B lymphocytes reveal novel roles for TRAF2 in CD40 signaling. J. Biol. Chem. 278:45382–45390. [DOI] [PubMed] [Google Scholar]
- 29.Grech, A.P., M. Amesbury, T. Chan, S. Gardam, A. Basten, and R. Brink. 2004. TRAF2 differentially regulates the canonical and noncanonical pathways of NF-kappaB activation in mature B Cells. Immunity. 21:629–642. [DOI] [PubMed] [Google Scholar]
- 30.He, J.Q., S.K. Saha, J.R. Kang, B. Zarnegar, and G. Cheng. 2006. Specificity of TRAF3 in its negative regulation of the noncanonical NF-kappa B pathway. J. Biol. Chem. 282:3688–2418. [DOI] [PubMed] [Google Scholar]
- 31.Grammer, A.C., J.L. Swantek, R.D. McFarland, Y. Miura, T. Geppert, and P.E. Lipsky. 1998. TNF receptor-associated factor-3 signaling mediates activation of p38 and Jun N-terminal kinase, cytokine secretion, and Ig production following ligation of CD40 on human B cells. J. Immunol. 161:1183–1193. [PubMed] [Google Scholar]
- 32.Jabara, H., D. Laouini, E. Tsitsikov, E. Mizoguchi, A. Bhan, E. Castigli, F. Dedeoglu, V. Pivniouk, S. Brodeur, and R. Geha. 2002. The binding site for TRAF2 and TRAF3 but not for TRAF6 is essential for CD40-mediated immunoglobulin class switching. Immunity. 17:265–276. [DOI] [PubMed] [Google Scholar]
- 33.Viatour, P., M. Bentires-Alj, A. Chariot, V. Deregowski, L. de Leval, M.P. Merville, and V. Bours. 2003. NF-kappa B2/p100 induces Bcl-2 expression. Leukemia. 17:1349–1356. [DOI] [PubMed] [Google Scholar]
- 34.Strasser, A., A.W. Harris, D.L. Vaux, E. Webb, M.L. Bath, J.M. Adams, and S. Cory. 1990. Abnormalities of the immune system induced by dysregulated bcl-2 expression in transgenic mice. Curr. Top. Microbiol. Immunol. 166:175–181. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}