

Abstract
Langerhans cells (LCs) are bone marrow (BM)–derived epidermal dendritic cells (DCs) that develop from precursors found in the dermis. Epidermal LCs are absent in transforming growth factor (TGF) β1-deficient mice. It is not clear whether TGFβ1 acts directly on LC precursors to promote maturation or whether it acts on accessory cells, which in turn affect LC precursors. In addition, the physiologic source of TGFβ1 is uncertain because BM chimera experiments showed that neither hematopoietic nor nonhematopoietic-derived TGFβ1 is required for LC development. To address these issues, we created mice transgenic for a bacterial artificial chromosome (BAC) containing the gene for human Langerin into which Cre recombinase had been inserted by homologous recombination (Langerin-Cre). These mice express Cre selectively in LCs, and they were bred to floxed TGFβRII and TGFβ1 mice, thereby generating mice with LCs that either cannot respond to or generate TGFβ1, respectively. Langerin-Cre TGFβRII mice had substantially reduced numbers of epidermal LCs, demonstrating that TGFβ1 acts directly on LCs in vivo. Interestingly, Langerin-Cre TGFβ1 mice also had very few LCs both in the steady state and after BM transplantation. Thus, TGFβ1 derived from LCs acts directly on LCs through an autocrine/paracrine loop, and it is required for LC development and/or survival.
Langerhans cells (LCs) are a long-lived subset of tissue DCs that reside in the epidermis (1). LCs acquire skin antigens, and then migrate to skin-draining LNs in both inflammatory and steady-state conditions (2, 3). LCs are derived from colony-stimulating factor-1 (CSF1)–dependent precursors that originate in the BM and migrate to the dermis before becoming fully differentiated and populating the epidermis (4, 5). LC development is affected by TGFβ1. BM cells cultured in granulocyte/macrophage CSF and TGFβ1 generate LC-like cells, and LCs are absent from TGFβ1−/− mice (6–8).
In vivo, TGFβ1 is secreted by leukocytes and nonhematopoietic cells, including keratinocytes, and has a pleiotropic role in the immune system (9). There are three isoforms of TGFβ, but TGFβ1 is the dominant isoform within the immune system. TGFβ1 binds to the TGFβ receptor II (TGFβRII) and ALK5 to activate Smad 2, 3, and 4.
Although it is clear that LC development requires TGFβ1, the identity of the cell types responsible for secreting TGFβ1, and whether TGFβ1 acts directly on LCs or via an intermediary cell type, is unresolved. In BM chimeric experiments, TGFβ1+/− severe combined immunodeficient BM cells transferred into irradiated TGFβ1−/− severe combined immunodeficient mice are able to produce LCs (10). Thus, TGFβ1 derived from nonhematopoietic cells in the skin, such as keratinocytes, is not required, and secretion by a cell of hematopoietic origin is sufficient for LC development. However, BM cells from TGFβ1−/− mice were also able to generate donor-derived LCs when introduced into irradiated WT recipients, suggesting that nonhematopoietic sources of TGFβ1 are sufficient to promote LC development (11). Interestingly, intradermal, but not intravenous, introduction of TGFβ1 into TGFβ1−/− mice led to LC development, which suggests that TGFβ1 acts on LC precursors within the skin (10). Thus, neither hematopoietic nor skin-derived TGFβ1 was required for LC development in these models, which leaves the source of TGFβ1 that drives LC development unresolved.
To more definitively define the mechanisms by which TGFβ promotes LC development in vivo, we developed two lines of mice that have a LC-specific deletion of either the gene for TGFβ1 or TGFβRII, thereby generating mice with LC precursors that cannot secrete or respond to the cytokine, respectively.
RESULTS AND DISCUSSION
Generation and validation of Langerin-Cre mice
To generate a mouse with selective expression of Cre recombinase in LCs, we used a genomic bacterial artificial chromosome (BAC) transgenic system similar to one we recently developed to express diphtheria toxin in LCs (12). The human genomic BAC RP11-504o1 contains the gene for Langerin, which is expressed by fully developed LCs (13, 14). Transgenic mice made with this BAC express Langerin specifically in epidermal LCs (12). The gene for a mammalian codon-optimized version of Cre was inserted into the BAC DNA just after the start ATG codon in exon I of Langerin using homologous recombination (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20071401/DC1) (15, 16). The correct insertion of Cre into exon I was confirmed by PCR (not depicted) and by restriction digest (Fig. S2). A 72-kb NotI fragment of this modified BAC was used to generate a single Langerin-Cre transgenic founder from 20 live births. Langerin-Cre mice are indistinguishable from littermate controls and have normal numbers of LCs, dermal DCs, other DC subtypes, and B and T cells in their spleen and LN (unpublished data).
To validate that Cre expression was restricted to LCs, Langerin-Cre mice were bred to Rosa26-enhanced GFP (EGFP) mice that have EGFP inserted into a ubiquitously expressed genomic locus behind floxed “stop” sequences (17). Cre activity deletes the stop sequences, thereby allowing EGFP expression only in those cells that express the Cre transgene. To quantify the number of LCs expressing the Cre transgene, we examined epidermal single-cell suspensions by flow cytometry. As expected, all epidermal MHC-II+, CD11c+ LCs from Langerin-Cre+ Rosa26-EGFPf/f mice showed expression of EGFP (Fig. 1 A, left, thick line). In contrast, EGFP was expressed only in a small subset of MHC-II+ cells in dermal single-cell suspensions (Fig. 1 A, right). These EGFP+ cells are likely either LC precursors that reside in the dermis or epidermal LCs in the process of migrating though the dermis to regional LNs. EGFP+ cells were not observed in littermate control mice.
Figure 1.

Langerin-Cre transgene is expressed by all epidermal LCs. (A) Langerin-Cre mice were bred onto the Rosa26.EGFP Cre reporter line. FACS analysis of epidermal and dermal single-cell suspensions gated on live MHC-II+ cells (Tg+, thick line; Tg−, thin line with shading). (B) Analysis of live gated cutaneous LN cells from Tg+ (left) and Tg− (right) mice showing a population of CD11cint GFP+ cells in transgenic mice. (C) GFP+-gated cells from Cre+ mice are stained for DEC-205 (left, open) and CD8 (right, open), compared with an isotype control (filled).
EGFP could also be identified on CD11cint cells within the cutaneous LNs (Fig. 1 B). These cells were CD8α− and DEC-205bright, which is consistent with their identity as LCs that have migrated to the skin-draining LN (Fig. 1 C). Importantly, we did not observe EGFP expression in any other cell present in the LN. There were no detectable EGFP+ cells in spleen, mesenteric LN, and thymus (Fig. 2 A). Thus, expression of the Cre transgene appears to be limited to epidermal LCs and to LCs that have migrated to the skin-draining LN, which is consistent with our prior experience using this BAC (12). It is important to note that endogenous mouse Langerin is expressed by several populations of CD8α+ DCs that are present throughout the secondary lymphoid tissue. We and others have shown that these cells are not derived from the epidermis (12, 18, 19). These Langerin+, CD8+ DCs were present in Langerin-Cre mice in many tissues (Fig. 2 B). Although we are not sure why expression of Cre under the control of the human Langerin BAC locus is limited to epidermal LCs and not other mouse Langerin-expressing cells, it is clear that these other Langerin-positive cells do not express the transgene and will not confound the analysis of subsequent experiments.
Figure 2.

Langerin-Cre is selectively expressed in skin-derived LCs. (A) FACS analysis of CD11c-gated live cells obtained from the indicated tissues of littermate control (top) or Langerin-Cre Rosa26.EGFP (bottom) mice stained for DEC-205. (B) Analysis of CD11c-gated live cells from the indicated tissue of Langerin-Cre mice stained for CD8α and Langerin.
LCs require the direct action of TGFβ
It is well established that TGFβ1 is required for LC development (6–8). Given that nearly all cells express receptors for TGFβ1, it is less clear whether LCs have a direct requirement for TGFβ1 or if TGFβ1 acts via another cell type, which then assists with LC development. To examine this, Langerin-Cre mice were bred to mice carrying a TGFβRII-floxed allele (20). Langerin-Cre TGFβRIIf/f mice are predicted to have LC precursors that are unable to respond to TGFβ1. Langerin-Cre TGFβRIIf/f mice appeared grossly normal and had normal numbers of lymphocytes and DCs in their spleen and LN (unpublished data). In single-cell epidermal suspensions from these mice, there was a decreased number of MHC-II+ cells compared with littermate Langerin-Cre TGFβIIf/f controls (Fig. 3 A, top). Although the number of LCs was substantially reduced, a variable number of LCs were present in the epidermis of these mice. We hypothesized that residual LCs in Langerin-Cre TGFβRIIf/f mice were cells that had not deleted both TGFβRII alleles. Indeed, all epidermal LCs present in Langerin-Cre TGFβRIIf/f mice expressed TGFβRII at levels similar to littermate controls (Fig. 3 A, bottom). Thus, those LC precursors that continued to express TGFβRII, presumably because of failure to excise both floxed loci, retained TGFβ sensitivity and developed into LCs, whereas those in which both alleles of TGFβRII were excised did not. Thus, we conclude that LCs require the direct action of TGFβ1 for their terminal development and/or survival.
Figure 3.

LC development requires the direct action of TGFβ, and escaped LCs repopulate the epidermis in clusters. (A) FACS of live gated epidermal cells from Langerin-Cre TGFβRIIf/f (right) and littermate controls (left) stained for TGFβRII (top) or isotype control (bottom). (B) Each symbol represents the ratio of the percentage of epidermal LCs in an individual Langerin-Cre TGFβRIIf/f mouse compared with a sex- and age-matched control. The percentage of LCs in Langerin-Cre TGFβRIIf/f increases with age. P < 0.05. (C) Immunofluorescence of epidermal whole mounts from Langerin-Cre TGFβRIIf/f at the indicated age or WT mice stained for MHC-II (green). The majority of fields lack LCs. Representative fields that contain LCs are shown. Bars: (Day 22) 37.5 μm; (Days 60 and 89 and WT) 75 μm.
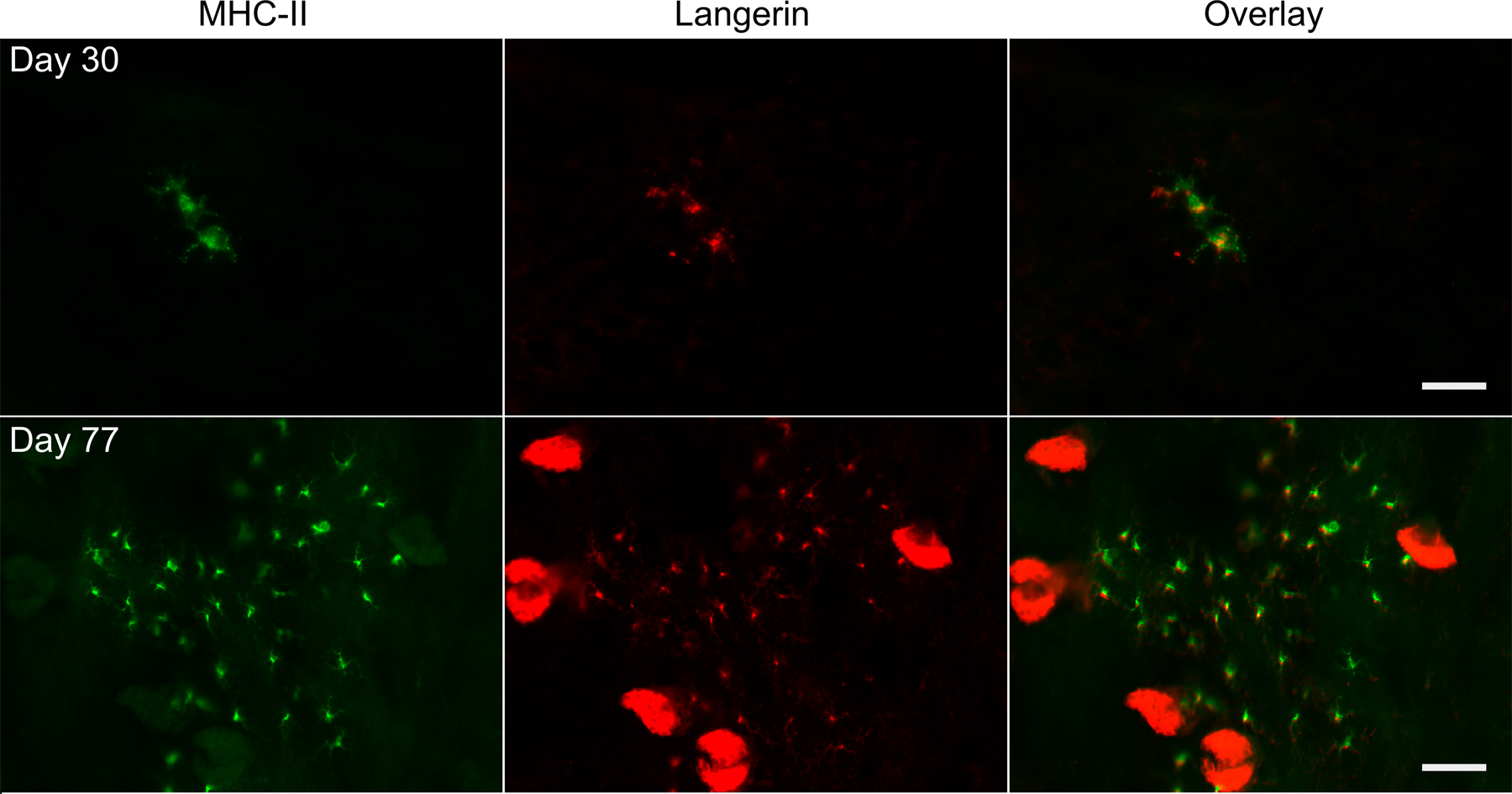
Although the percentage of “escaped” LCs present in the epidermis of Langerin-Cre TGFβRIIf/f mice was always smaller than littermate controls, it was quite variable, ranging from 0.02 to 0.59% of total epidermal cells compared with 0.46–0.80% in littermate controls. Plotting the ratio of epidermal LCs from Langerin-Cre+ versus age-matched littermate controls as a function of the age of the mouse revealed that as the mice aged, escaped LCs accumulated, and their percentage in the epidermis increased (Fig. 3 B; P < 0.05). To further investigate this finding, we examined epidermal sheets by immunofluorescence for the presence of LCs. In young, 22-d-old mice, MHC-II+ LCs were rare, and they appeared as individual cells (Fig. 3 C). In mice at 60 d of age, single cells could no longer be identified. Instead, there were clusters of LCs scattered throughout the epidermis that, by day 89, were significantly larger. To confirm that these cells were, in fact, LCs, and not another cell type that colonized the epidermal niche in the absence of LCs, epidermal sheets were stained with both anti–MHC-II and anti-Langerin (Fig. S2). All of the MHC-II+ cells in both young and old mice also expressed Langerin, thereby demonstrating that they are LCs. The fact that escaped LCs in Langerin-Cre TGFβRIIf/f mice are found as single cells in young mice, but appear in increasingly large clusters in older mice, supports the notion that fully differentiated LCs derive from locally proliferating precursors in the steady state (4, 21, 22).
LCs require autocrine/paracrine TGFβ1 for development
There are many potential sources of TGFβ1 available to the developing LC in the skin, including keratinocytes, fibroblasts, resident T cells, and LCs themselves (9, 23). To examine whether autocrine secretion of TGFβ1 participates in LC development, we bred Langerin-Cre mice to mice in which the gene for TGFβ1 was floxed, thereby generating mice with a LC-selective ablation of TGFβ1 (24). These Langerin-Cre TGFβ1f/f mice appeared grossly normal, did not have overt histologic evidence of autoimmune disease in the skin, and had normal numbers of lymphocytes and DCs in their spleen and LN (unpublished data). We consistently observed a large decrease in the number of LCs in Langerin-Cre TGFβ1f/f mice. LCs comprised 0.02–0.1% of epidermal cells in Langerin-Cre TGFβ1f/f mice compared with 0.42–1.17% in littermate controls (Fig. 4, A and B). Immunofluorescence of whole-mounted ear epidermis confirmed this result. In Langerin-Cre TGFβ1f/f mice, LCs were absent from the vast majority of the ear, but could be observed focally as MHC-II+, Langerin+ cells in a few areas (Fig. 4 C). As was seen with the Langerin-Cre TGFβRIIf/f mice, escaped LCs were found in clusters that, interestingly, were often located at the edge of the ear.
Figure 4.

Steady-state LC development requires LC-derived TGFβ1. (A) A representative FACS analysis of live gated epidermal cells from 4-wk-old Langerin-Cre TGFβ1f/f (right) compared with control mice (left). (B) Each symbol represents the percentage of LCs in an individual Langerin-Cre TGFβf/f mouse or control. (C) Immunofluorescence of epidermal whole mounts from Langerin-Cre TGFβ1f/f or WT mice stained for MHC-II (green) and Langerin (red). As with the Langerin-Cre TGFβRIIf/f, most fields lack LCs, and a representative field containing LCs is shown. Bar, 75 μm.
TGFβ1-floxed mice were constructed by insertion of loxP sites 5′ of the promoter region of Tgfb1 and 3′ of exon 1 in the first intron (24). The location of the loxP sites, however, encompasses the final exon of a hypothetical gene, LOC232987, leading to the abrogation of TGFβ1 and LOC232987 expression. To address the possibility that the absence of LCs was caused by the absence of LOC232987 and not TGFβ1, Langerin-Cre TGFβ1f/n mice were generated. TGFβn mice have GFP inserted into the first exon of Tgfb1 gene that disrupts TGFβ1 expression, but leaves LOC232987 intact (24). LCs in Langerin-Cre TGFβ1f/n mice will have both alleles of the Tgfb1 gene disrupted, but retain a functional allele of LOC232987. These mice showed a dramatic decrease in the number of epidermal LCs and were indistinguishable from Langerin-Cre TGFβ1f/f mice, thereby validating that the absence of LCs in Langerin-Cre TGFβ1f/f mice was caused by the requirement for LC-derived TGFβ1 (unpublished data).
The requirement for LC-derived TGFβ1 in LC development was unexpected, given the observation by Borkowski et al. that donor-derived LCs develop in TGFβ1−/−→WT BM chimeric mice (11). One explanation for the apparent discrepancy between our results and theirs is that LC repopulation of the epidermis after BM transplantation may not be analogous to epidermal population during ontogeny and, in contrast to our results, could occur in the absence of LC-derived TGFβ1. We examined this by creating BM chimeras using Langerin-Cre TGFβ1f/f and control littermate Cre− TGFβ1f/f mice (both CD45.2+) as BM donors and WT CD45.1 congenic B6 mice as recipients. Half of the recipients were treated with UVC 1 d after transplant, which eliminates host LCs and is known to enable the development of donor-derived LCs in the epidermis after BM transplantation (22). Hematopoietic engraftment in both sets of chimeras was confirmed by detecting CD45.2 by flow cytometry 8 wk after transplant (unpublished data). As expected, immunofluorescence of epidermal whole mounts revealed that UVC-treated recipients of control Cre− TGFβ1f/f BM developed normal numbers of CD45.2+ (donor) Langerin+ LCs throughout the epidermis (Fig. 5 A). In contrast, UVC-treated recipients of Langerin-Cre TGFβ1f/f BM had virtually no detectable LCs, and the rare LCs present were of donor origin. Recipients of Langerin-Cre TGFβ1f/f and control BM that were not irradiated with UVC retained LCs of recipient origin (Fig. 5 B), as expected (22). Thus, LC epidermal repopulation after BM transplantation recapitulated the steady state and, in our hands, is dependent on LC-derived TGFβ1.
Figure 5.

LC repopulation in chimeric mice requires LC-derived TGFβ1. BM chimeras were generated by transferring BM from either Langerin-Cre+ TGFβ1f/f mice (A and B, bottom rows) or littermate Langerin-Cre− TGFβ1f/f controls (A and B, top rows) into irradiated congenic B6.CD45.1 recipients. Recipients were either exposed to 450 mJ of UVC 1 d after transplant (A) or not irradiated (B). After 8 wk, immunofluorescence of epidermal ear whole mounts were stained for CD45.2 (green) and Langerin (red) to determine the presence of donor LCs. Representative data from three independent experiments are shown. Bar, 75 μm.
We have described the generation of Langerin-Cre, BAC-transgenic mice and demonstrated that expression of Cre in these mice is limited to epidermal and skin-derived LCs. By breeding these mice to floxed TGFβRII and TGFβ1 mice, we developed mice in which LCs are either insensitive to or unable to produce TGFβ1, respectively. The reduction of LCs in both of these strains of mice demonstrates that LC-derived TGFβ1 directly and nonredundantly promotes LC development during ontogeny and in the steady state. Our BM transplantation experiments further demonstrate a requirement for LC-derived TGFβ1, even during repopulation after UV irradiation.
The general importance of TGFβ1 in LC development was already appreciated based on the absence of LCs in TGFβ1−/− mice and the ability of TGFβ1 to enhance production of LCs in in vitro cultures (6–8). The lack of LCs in Langerin-Cre TGFβRII mice demonstrates that TGFβ1 acts directly on LCs and not indirectly via another cell type. This was a nontrivial possibility given the large number of cell types that express receptors for TGFβ. In addition, because Langerin, and hence Cre, are thought to be expressed only by mature LCs, our data indicates that LCs must require TGFβ1 after they begin to express Langerin (14). Indeed, Langerin mRNA transcripts are detectable in the skin of TGFβ1−/− mice, and local introduction of TGFβ1 can rescue LC development in TGFβ1−/− mice (10, 25). Considering these results with our data, we conclude that LC precursors become TGFβ1 dependent only once they are in the skin and begin to express Langerin. TGFβ1 signaling induces the transcription factor Id2 (26). The absence of both LCs and splenic CD8+ DCs in Id2−/− mice raised the possibility that both cell types may share a common TGFβ1-dependent precursor (26). The requirement for TGFβ1 by LCs only once they are already in the skin, however, suggests that Id2 could be required only after they have become distinct lineages.
Our observation that Langerin-Cre TGFβ1f/f mice have a greatly reduced number of LCs clearly demonstrates a nonredundant, in vivo requirement for paracrine or autocrine TGFβ1 for the development of mature LCs. Whether TGFβ1 acts transiently to foster LC development and/or is required as a survival factor is unclear. TGFβ1 is produced by epidermal keratinocytes, making it plausible that LC-derived TGFβ1 allows survival until LCs reach the epidermis, at which point keratinocytes could instead provide the necessary TGFβ1. Hematopoietic stem cells (HSCs) were thought to use an autocrine/paracrine TGFβ loop to maintain HSC quiescence (27). However, in vivo experiments, using conditionally ablated TGFβRII revealed that the absence of TGFβ signaling had no effect on HSCs, and the reliance on TGFβ in vitro was presumably caused by an indirect effect, such as the absence of soluble or environmental factors present in vivo (27). This highlights the difficulty of studying TGFβ in vitro and the importance of in vivo experimental systems. We believe this is the first documented hematopoietic cell type that is dependent on TGFβ for its development. Recent studies have also revealed an important function for T cell–derived TGFβ1 in regulating T cell function (24). Thus, regulation via TGFβ autocrine/paracrine loops may be a more common phenomenon than previously realized.
The deletion of the floxed loci in TGFβRIIf/f mice does not occur in every LC precursor, which allows some to retain sensitivity to TGFβ1 and repopulate the empty epidermal niche. One explanation for this would be that rare LC precursors in Langerin-Cre mice express insufficient levels of Cre to delete both alleles of the floxed locus. This could explain why all LCs express EGFP in Langerin-Cre Rosa-EGFP mice, which only require deletion of a single floxed allele. Other floxed genes that are not required for LC survival, such as I-Aβ, have both alleles efficiently excised by all LCs (unpublished data). Thus, it is more likely that the strong selective advantage of retaining sensitivity to TGFβ1 allows what would otherwise be a very rare cell to expand. The observation that escaped LCs in Langerin-Cre TGFβRIIf/f occur in clusters argues that fully differentiated LCs derive from locally proliferating precursors in the steady state (4, 21, 22). Moreover, the expanding size of the clusters with age suggests that LCs are proliferating within the epidermis, though proliferation in the dermis and subsequent migration into the epidermis cannot be excluded.
Langerin-Cre TGFβ1→WT UV-treated chimeras do not develop donor-derived LCs. Thus, LC-derived TGFβ1 is required for LC repopulation after UV treatment, as well as to establish a normal complement of epidermal LCs during ontogeny. The absence of LCs in Langerin-Cre TGFβF/F chimeras was particularly surprising, given the presence of donor-derived LCs in the original TGFβ1−/−→WT experiments by Borkowski et al. (11). A potentially important distinction is that in the Borkowski et al. experiments, all hematopoietic cells lacked the gene for TGFβ1, unlike Langerin-Cre TGFβ1 chimeras, in which the defect was limited to LCs. Levels of TGFβ2 are elevated in the skin of TGF1β−/− mice (11). It is possible that in the absence of hematopoietically derived TGFβ1, compensatory TGFβ2 or other TGFβ family members such as activin, which potentially could promote LC development, were induced, and thus masked the physiologic requirement for LC-derived TGFβ1 (10, 11, 28). It is also important to note that Borkowski et al. observed donor-derived LCs in control and TGFβ1−/− chimeras in the absence of UV treatment. Others have clearly shown that LCs are radio resistant, and repopulation from donor BM requires ablation of host LCs, such as that which occurs after UV treatment or cutaneous graft versus host disease (22, 29), a finding that we have confirmed. Although no obvious evidence of cutaneous autoimmunity was observed in the Borkowski study (Udey, M., personal communication), minor histocompatability differences between a partially backcrossed donor and an inbred recipient could have accounted for the presence of donor LCs in the absence of UV treatment and allowed LC development in the absence of LC-derived TGFβ1.
LC-derived TGFβ1 could also participate in other aspects of LC biology, such as maintaining an immature LC phenotype, determining whether precursors become LCs or other DC subtypes (e.g., dermal DCs), and/or regulating cutaneous immune responses. Because LCs lacking TGFβ1 fail to develop, it is much more challenging to examine the possible roles of TGFβ1 in subsequent phases of the LC lifecycle. We are in the process of generating mice that will express an inducible form of Cre in LCs, which should permit us to investigate the ongoing roles of TGFβ1 in LC biology beyond the required initial autocrine/paracrine loop we have defined in this work.
MATERIALS AND METHODS
BAC and transgenic mice.
The human BAC clone RP11-504o1 was obtained from Invitrogen. The sequence for Cre recombinase, which had optimized with mammalian codon usage (iCre; a gift from R. Sprengel, Max-Planck Institute for Medical Research, Heidelberg, Germany; reference [15]), was inserted immediately after the start ATG in exon I of Langerin using homologous recombination in Escherichia coli, as previously described (12). Recombined BAC clones were confirmed by PCR and by XhoI (New England Biolabs) restriction digest. BAC DNA for microinjection was obtained as previously described (12) and injected at 1–2 ng/μl by the Yale Genomics Services Transgenic Mouse Service into the pronuclei of (C57BL/6j x SJL/j) F2 embryos. Langerin-Cre founders were identified by PCR for RP11-50401, as previously described (12).
Mice.
Langerin-Cre mice were bred onto the following strains: Rosa26.EFGP (The Jackson Laboratory), floxed TGFβRII (a gift from H. Moses, Vanderbilt University School of Medicine, Nashville, TN; reference [20]), floxed TGFβ1 (24), and TGFβ1-GFP (unpublished data). C57BL/6 CD45.1 mice were obtained from The Jackson Laboratory. All experiments were performed with age- and sex-matched mice. Mice were housed in microisolator cages and fed autoclaved food and acidified water. The Yale Institutional Animal Care and Use Committee approved all mouse protocols.
Antibodies.
Antibodies to the following targets were used: muLangerin (929F3; provided by S. Saeland, Dendritics, Dardilly, France), CD11c (N418-FITC and APC; eBioscience), I-A/E (M5/144.15.2-FITC and biotin; BioLegend), CD11b (M1/70-PE; BD PharMingen), CD45.1 (A20-FITC; BD PharMingen), CD45.2 (104-FITC; BD PharMingen), and CD8α (Ly-2-PE; BD PharMingen). NLDC-145 (anti-DEC-205) and 24G2 (anti-FcRγ) were purified from hybridoma supernatants, as previously described (30). Conjugation to biotin or Alexa Fluor 647 was performed according to the manufacturer's directions (Invitrogen).
Flow cytometry.
Single-cell suspensions of epidermis, LN, thymus, and spleen were obtained and stained as previously described (12). Live/dead discrimination was obtained using propidium iodide (Invitrogen) or ethidium monoazide (Invitrogen). Samples were analyzed on a FACSCalibur or LSR-II flow cytometer (BD Biosciences).
Immunofluoresence.
Epidermal sheets were prepared by affixing ears to slides using double-sided adhesive (3M), followed by incubation in 21 mM EDTA in PBS for 2 h at 37° and physical removal of the dermis, as previously described (12).
Generation of chimeric mice.
BM cells were obtained from either Langerin-Cre TGFβf/f or Cre− littermate control mice. Cells were incubated in ACK buffer for erythroid cell lysis and resuspended in injection buffer (1 × PBS, 10 mM Hepes, 2.5% acid citrate dextrose anticoagulant, and 0.5% penicillin/streptomycin). Recipient B6 CD45.1 mice received 775 cGy from a cesium irradiator and were reconstituted with 107 cells/mouse. 1 d after transplantation, one cohort of mice was exposed to 450 mJ UVC light. Mice were rested for at least 8 wk before experimentation.
Statistics.
Statistical comparisons between groups were made using a standard two-tailed Student's t test. Linear regression was used to evaluate for correlation in Fig. 3 B.
Online supplemental material.
Fig. S1 shows the generation of Langerin-Cre BAC. Fig. S2 shows that escaped epidermal MHC-II+ cells in Langerin-Cre TGFβRIIf/f mice are LCs. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20071401/DC1.
Supplemental Material
Acknowledgments
We thank the Yale Animal Resources Center staff for outstanding animal care.
This work was supported by National Institutes of Health grants R01 HL66279 and R01 AR44077 (to M.J. Shlomchik) and K08 AR651092 (to D.H. Kaplan).
The authors have no conflicting financial interests.
Abbreviations used: BAC, bacterial artificial chromosome; CSF, colony-stimulating factor; EGFP, enhanced GFP; HSC, hematopoietic stem cell; LC, Langerhans cell.
References
- 1.Romani, N., S. Holzmann, C.H. Tripp, F. Koch, and P. Stoitzner. 2003. Langerhans cells - dendritic cells of the epidermis. APMIS. 111:725–740. [DOI] [PubMed] [Google Scholar]
- 2.Hemmi, H., M. Yoshino, H. Yamazaki, M. Naito, T. Iyoda, Y. Omatsu, S. Shimoyama, J.J. Letterio, T. Nakabayashi, H. Tagaya, et al. 2001. Skin antigens in the steady state are trafficked to regional lymph nodes by transforming growth factor-beta1-dependent cells. Int. Immunol. 13:695–704. [DOI] [PubMed] [Google Scholar]
- 3.Kripke, M.L., C.G. Munn, A. Jeevan, J.M. Tang, and C. Bucana. 1990. Evidence that cutaneous antigen-presenting cells migrate to regional lymph nodes during contact sensitization. J. Immunol. 145:2833–2838. [PubMed] [Google Scholar]
- 4.Larregina, A.T., A.E. Morelli, L.A. Spencer, A.J. Logar, S.C. Watkins, A.W. Thomson, and L.D. Falo Jr. 2001. Dermal-resident CD14+ cells differentiate into Langerhans cells. Nat. Immunol. 2:1151–1158. [DOI] [PubMed] [Google Scholar]
- 5.Ginhoux, F., F. Tacke, V. Angeli, M. Bogunovic, M. Loubeau, X.M. Dai, E.R. Stanley, G.J. Randolph, and M. Merad. 2006. Langerhans cells arise from monocytes in vivo. Nat. Immunol. 7:265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strobl, H., E. Riedl, C. Scheinecker, C. Bello-Fernandez, W.F. Pickl, K. Rappersberger, O. Majdic, and W. Knapp. 1996. TGF-beta 1 promotes in vitro development of dendritic cells from CD34+ hemopoietic progenitors. J. Immunol. 157:1499–1507. [PubMed] [Google Scholar]
- 7.Geissmann, F., C. Prost, J.P. Monnet, M. Dy, N. Brousse, and O. Hermine. 1998. Transforming growth factor β1, in the presence of granulocyte/macrophage colony–stimulating factor and interleukin 4, induces differentiation of human peripheral blood monocytes into dendritic Langerhans cells. J. Exp. Med. 187:961–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borkowski, T.A., J.J. Letterio, A.G. Farr, and M.C. Udey. 1996. A role for endogenous transforming growth factor β1 in Langerhans cell biology: the skin of transforming growth factor β1 null mice is devoid of epidermal Langerhans cells. J. Exp. Med. 184:2417–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li, M.O., Y.Y. Wan, S. Sanjabi, A.K. Robertson, and R.A. Flavell. 2006. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 24:99–146. [DOI] [PubMed] [Google Scholar]
- 10.Thomas, R.M., D.V. Belsito, C. Huang, L.Z. Chen Lz, I. Ormsby, W.J. Simmons, P. Cowin, J. Shaw, T. Doetschman, and G.J. Thorbecke. 2001. Appearance of Langerhans cells in the epidermis of Tgfb1(−/−) SCID mice: paracrine and autocrine effects of transforming growth factor-beta 1 and -beta 2(1). J. Invest. Dermatol. 117:1574–1580. [DOI] [PubMed] [Google Scholar]
- 11.Borkowski, T.A., J.J. Letterio, C.L. Mackall, A. Saitoh, X.J. Wang, D.R. Roop, R.E. Gress, and M.C. Udey. 1997. A role for TGFbeta1 in langerhans cell biology. Further characterization of the epidermal Langerhans cell defect in TGFbeta1 null mice. J. Clin. Invest. 100:575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaplan, D.H., M.C. Jenison, S. Saeland, W.D. Shlomchik, and M.J. Shlomchik. 2005. Epidermal langerhans cell-deficient mice develop enhanced contact hypersensitivity. Immunity. 23:611–620. [DOI] [PubMed] [Google Scholar]
- 13.Valladeau, J., O. Ravel, C. Dezutter-Dambuyant, K. Moore, M. Kleijmeer, Y. Liu, V. Duvert-Frances, C. Vincent, D. Schmitt, J. Davoust, et al. 2000. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity. 12:71–81. [DOI] [PubMed] [Google Scholar]
- 14.Tripp, C.H., S. Chang-Rodriguez, P. Stoitzner, S. Holzmann, H. Stossel, P. Douillard, S. Saeland, F. Koch, A. Elbe-Burger, and N. Romani. 2004. Ontogeny of Langerin/CD207 expression in the epidermis of mice. J. Invest. Dermatol. 122:670–672. [DOI] [PubMed] [Google Scholar]
- 15.Shimshek, D.R., J. Kim, M.R. Hubner, D.J. Spergel, F. Buchholz, E. Casanova, A.F. Stewart, P.H. Seeburg, and R. Sprengel. 2002. Codon-improved Cre recombinase (iCre) expression in the mouse. Genesis. 32:19–26. [DOI] [PubMed] [Google Scholar]
- 16.Misulovin, Z., X.W. Yang, W. Yu, N. Heintz, and E. Meffre. 2001. A rapid method for targeted modification and screening of recombinant bacterial artificial chromosome. J. Immunol. Methods. 257:99–105. [DOI] [PubMed] [Google Scholar]
- 17.Mao, X., Y. Fujiwara, A. Chapdelaine, H. Yang, and S.H. Orkin. 2001. Activation of EGFP expression by Cre-mediated excision in a new ROSA26 reporter mouse strain. Blood. 97:324–326. [DOI] [PubMed] [Google Scholar]
- 18.Douillard, P., P. Stoitzner, C.H. Tripp, V. Clair-Moninot, S. Ait-Yahia, A.D. McLellan, A. Eggert, N. Romani, and S. Saeland. 2005. Mouse lymphoid tissue contains distinct subsets of langerin/CD207 dendritic cells, only one of which represents epidermal-derived Langerhans cells. J. Invest. Dermatol. 125:983–994. [DOI] [PubMed] [Google Scholar]
- 19.Kissenpfennig, A., S. Henri, B. Dubois, C. Laplace-Builhe, P. Perrin, N. Romani, C.H. Tripp, P. Douillard, L. Leserman, D. Kaiserlian, et al. 2005. Dynamics and function of Langerhans cells in vivo dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity. 22:643–654. [DOI] [PubMed] [Google Scholar]
- 20.Chytil, A., M.A. Magnuson, C.V. Wright, and H.L. Moses. 2002. Conditional inactivation of the TGF-beta type II receptor using Cre:Lox. Genesis. 32:73–75. [DOI] [PubMed] [Google Scholar]
- 21.Holzmann, S., C.H. Tripp, M. Schmuth, K. Janke, F. Koch, S. Saeland, P. Stoitzner, and N. Romani. 2004. A model system using tape stripping for characterization of Langerhans cell-precursors in vivo. J. Invest. Dermatol. 122:1165–1174. [DOI] [PubMed] [Google Scholar]
- 22.Merad, M., M.G. Manz, H. Karsunky, A. Wagers, W. Peters, I. Charo, I.L. Weissman, J.G. Cyster, and E.G. Engleman. 2002. Langerhans cells renew in the skin throughout life under steady-state conditions. Nat. Immunol. 3:1135–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thompson, N.L., K.C. Flanders, J.M. Smith, L.R. Ellingsworth, A.B. Roberts, and M.B. Sporn. 1989. Expression of transforming growth factor- β1 in specific cells and tissues of adult and neonatal mice. J. Cell Biol. 108:661–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li, M.O., Y.Y. Wan, and R.A. Flavell. 2007. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity. 26:579–591. [DOI] [PubMed] [Google Scholar]
- 25.Takahara, K., Y. Omatsu, Y. Yashima, Y. Maeda, S. Tanaka, T. Iyoda, B.E. Clausen, K. Matsubara, J. Letterio, R.M. Steinman, et al. 2002. Identification and expression of mouse Langerin (CD207) in dendritic cells. Int. Immunol. 14:433–444. [DOI] [PubMed] [Google Scholar]
- 26.Hacker, C., R.D. Kirsch, X.S. Ju, T. Hieronymus, T.C. Gust, C. Kuhl, T. Jorgas, S.M. Kurz, S. Rose-John, Y. Yokota, and M. Zenke. 2003. Transcriptional profiling identifies Id2 function in dendritic cell development. Nat. Immunol. 4:380–386. [DOI] [PubMed] [Google Scholar]
- 27.Larsson, J., U. Blank, H. Helgadottir, J.M. Bjornsson, M. Ehinger, M.J. Goumans, X. Fan, P. Leveen, and S. Karlsson. 2003. TGF-beta signaling-deficient hematopoietic stem cells have normal self-renewal and regenerative ability in vivo despite increased proliferative capacity in vitro. Blood. 102:3129–3135. [DOI] [PubMed] [Google Scholar]
- 28.Stoitzner, P., H. Stossel, M. Wankell, S. Hofer, C. Heufler, S. Werner, and N. Romani. 2005. Langerhans cells are strongly reduced in the skin of transgenic mice overexpressing follistatin in the epidermis. Eur. J. Cell Biol. 84:733–741. [DOI] [PubMed] [Google Scholar]
- 29.Merad, M., P. Hoffmann, E. Ranheim, S. Slaymaker, M.G. Manz, S.A. Lira, I. Charo, D.N. Cook, I.L. Weissman, S. Strober, and E.G. Engleman. 2004. Depletion of host Langerhans cells before transplantation of donor alloreactive T cells prevents skin graft-versus-host disease. Nat. Med. 10:510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shlomchik, M.J., D. Zharhary, T. Saunders, S.A. Camper, and M.G. Weigert. 1993. A rheumatoid factor transgenic mouse model of autoantibody regulation. Int. Immunol. 5:1329–1341. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}