

Abstract
Mucosally ingested and inhaled antigens are taken up by membranous or microfold cells (M cells) in the follicle-associated epithelium of Peyer's patches or nasopharynx-associated lymphoid tissue. We established a novel M cell–specific monoclonal antibody (mAb NKM 16–2-4) as a carrier for M cell–targeted mucosal vaccine. mAb NKM 16–2-4 also reacted with the recently discovered villous M cells, but not with epithelial cells or goblet cells. Oral administration of tetanus toxoid (TT)– or botulinum toxoid (BT)–conjugated NKM 16–2-4, together with the mucosal adjuvant cholera toxin, induced high-level, antigen-specific serum immunoglobulin (Ig) G and mucosal IgA responses. In addition, an oral vaccine formulation of BT-conjugated NKM 16–2-4 induced protective immunity against lethal challenge with botulinum toxin. An epitope analysis of NKM 16–2-4 revealed specificity to an α(1,2)-fucose–containing carbohydrate moiety, and reactivity was enhanced under sialic acid–lacking conditions. This suggests that NKM 16–2-4 distinguishes α(1,2)-fucosylated M cells from goblet cells containing abundant sialic acids neighboring the α(1,2) fucose moiety and from non-α(1,2)-fucosylated epithelial cells. The use of NKM 16–2-4 to target vaccine antigens to the M cell–specific carbohydrate moiety is a new strategy for developing highly effective mucosal vaccines.
Membranous or microfold cells (M cells), which are located in the follicle-associated epithelium (FAE) of Peyer's patches (PPs) or nasopharynx-associated lymphoid tissue (NALT), play a pivotal role in the uptake of luminal antigens for induction of antigen-specific immune responses in both systemic and mucosal compartments (1). Unlike their neighboring columnar epithelial cells, M cells are morphologically unique because they have irregular and short microvilli for the effective uptake of ingested or inhaled antigens from luminal sites in the aerodigestive tract; they subsequently transport the sampled antigen to professional antigen-presenting cells (e.g., dendritic cells) to initiate antigen sensitization (2).
The mucosal immune system consists of two types of immunologically important sites, termed “inductive” and “effector” tissues, connected by the common mucosal immune system (3). In general, antigen sensitization occurs at inductive sites, such as PPs, after antigen uptake by M cells. Induction of antigen-specific T helper 2 (Th2) cell–mediated IgA responses and Th1 cell– and CTL-dependent immune responses then occurs at effector sites such as the lamina propria (3). However, our recent study demonstrated that the effector sites are also able to take up antigen, because antigen-sampling cells termed villous M cells are distributed in the intestinal villous epithelium (4), and antigen-specific mucosal immune responses can be induced in PP-deficient mice (5).
Although mucosal vaccination is thought to be an ideal strategy for combating mucosal infectious diseases, only a few mucosal vaccines (e.g., polio vaccine and influenza vaccine) are currently used in humans because they have lower efficacy than the currently used injectable vaccines in inducing antigen-specific immune responses (6). Because M cells possess the ability to take up luminal antigens, it is logical and attractive to develop a system of delivery of vaccine antigen to both PP-associated and villous M cells to create an effective mucosal vaccine (7). In fact, Ulex europaeus agglutinin-1 (UEA-1)–conjugated (8, 9) or σ1 protein–conjugated nasal vaccination (10, 11) induce not only strong antigen-specific plasma IgG and mucosal IgA responses but also CTL immunity, because UEA-1 specific for α(1,2) fucose specifically reacts with murine PP–associated and villous M cells (4, 12), and σ1 protein derived from reovirus specifically binds to a carbohydrate structure containing α(2,3)-linked sialic acid on the membranes of M cells (13). However, because UEA-1 also reacts strongly with goblet cells and the mucus layer covering the intestinal epithelium (14), there have been no effective oral vaccines with UEA-1 as an M cell–targeting vehicle. To overcome this obstacle, we established an M cell–specific mAb and developed a novel strategy for oral vaccination with high efficacy.
RESULTS AND DISCUSSION
Establishment of an M cell–specific monoclonal antibody (NKM 16–2-4)
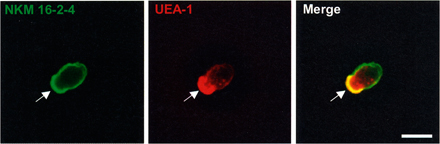
To characterize the antigen-sampling M cells for development of an effective M cell–targeted mucosal vaccine, Sprague-Dawley (SD) rats were immunized 4 times at 2-wk intervals with highly purified (>95%) UEA-1–positive cells isolated from murine PPs to establish an M cell–specific mAb. A total of 1,000 hybridomas were generated and screened by immunohistochemical analysis of intestinal tissue sections containing PPs. On the basis of the initial screening, one clone (NKM 16–2-4; rat IgG2c), which possessed specificity to M cells located in the FAE of PPs (Fig. 1 A), was selected. Half of the hybridomas showed no specificity to tissue sections; ∼40% of them showed strong reactivity to goblet cells and their secretions; and 10% showed reactivity to the microvilli in all parts of the intestinal epithelium, including M cells and neighboring columnar epithelial cells (unpublished data). These initial screening data indicated that the goblet cells contained in the immunized UEA-1–positive fraction, and their secretions, were vastly immunodominant compared with M cells. However, importantly, NKM 16–2-4 possessed no reactivity to UEA-1–positive goblet cells located in the intestinal villi (Fig. 1 A), indicating that NKM 16–2-4 is a novel mAb possessing high specificity to murine M cells. This is unlike the already known murine M cell–specific lectin UEA-1, which also reacts with goblet cells and their secretions (14). In addition, NKM 16–2-4 reacted very strongly with the apical surfaces of the M cells (Fig. 1 A), rather than the cytoplasm, suggesting that it might be able to be used as a carrier vehicle of M cell–targeted mucosal vaccine. In support of these results, flow cytometric and immuno- and lectin-cytochemical analyses demonstrated that NKM 16–2-4 specifically reacted with the surfaces of isolated UEA-1–positive M cells (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20070607/DC1), but not of UEA-1–negative epithelial cells. In addition, an electronmicroscopic analysis revealed that NKM 16–2-4 specifically reacted with typical M cells, which have short and irregular microvilli and a pocket structure containing lymphocytes and/or monocytes (Fig. 1 B). Furthermore, whole-mount staining analysis revealed that NKM 16–2-4 specifically reacted with villous M cells, in a manner similar to the reaction with PP-associated M cells (Fig. 1 C).
Figure 1.

Immunohistochemical analysis for the specificity of NKM 16–2-4. (A) Immunohistochemical analysis of PPs revealed that NKM 16–2-4 specifically reacted with UEA-1–positive M cells (red arrows), but not UEA-1–positive goblet cells (yellow arrowheads). (B) Electronmicroscopic analysis revealed that typical M cells, which had short and irregular microvilli and pocket structures containing lymphocytes and/or monocytes, specifically reacted with NKM 16–2-4. Positive reactions are shown by gold particles (18 nm). IEC, intestinal epithelial cell. (C) Whole-mount staining of PPs and villous epithelium demonstrated that, in addition to PP-associated M cells, UEA-1–positive villous M cells were specifically recognized by NKM 16–2-4. Bars, 50 μm.
M cells recognized by UEA-1 in mice are also present in the FAE of NALT, as they are in PPs, and act as antigen-sampling cells for the induction of mucosal immunity (15), although our previous finding demonstrated that the mechanism of NALT organogenesis is distinct from that of PP organogenesis (16, 17). Recently, it was reported that group A streptococcus infects its hosts through M cells (15), meaning that M cells could be defined as a portal cell subset of mucosal infection in both the gastrointestinal and respiratory tracts. A subsequent immunohistochemical analysis of NALT tissue sections revealed that NKM 16–2-4 specifically reacted with UEA-1–positive M cells, but not UEA-1–positive, morphologically typical goblet cells with secretory granules (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20070607/DC1). These results further suggest the possibility of formulating an M cell–targeted nasal vaccine with NKM 16–2-4 for protection against infectious diseases entering through the respiratory tract. Thus, in summary, the novel mAb NKM 16–2-4 specifically reacted with all subsets of M cells, but not epithelial cells or goblet cells, located in PPs, NALT, and the intestinal villi; i.e., in both the gastrointestinal and respiratory tracts (Table I).
Table I.
Immunological and biochemical characteristics of newly established mAb (NKM 16-2-4) and UEA-1 in M cells
| mAb/lectin | Specificity | Cell specificity
|
||
|---|---|---|---|---|
| M cells | Epithelial cells |
Goblet cells |
||
| NKM 16-2-4 | α(1,2) fucose- containing carbohydrate moiety |
+ | − | − |
| UEA-1 | α(1,2) fucose | + | − | + |
Use of NKM 16–2-4 to develop an M cell–targeted mucosal vaccine
Because it had been reported that the use of monoclonal antibodies to target injectable vaccine antigen to dendritic cells expressing endocytic receptor effectively initiated antigen-specific immunity (18, 19), we next addressed the characteristics of NKM 16–2-4 as a carrier vehicle of M cell–targeted mucosal vaccines. When we injected FITC-conjugated NKM 16–2-4 or FITC-conjugated control rat IgG into intestinal loops containing PPs, FITC-conjugated NKM 16–2-4 specifically attached to the apical surfaces of M cells in the dome regions of PPs within 10 min of inoculation, whereas FITC-conjugated control rat IgG did not (Fig. 2 A). Furthermore, FITC-conjugated NKM 16–2-4 was taken into the cytoplasmic regions of M cells within 30 min (Fig. 2 A) and reached the basal membrane of the M cells within 4 h, indicating that NKM 16–2-4 could likely be used as a carrier vehicle of orally administered vaccine antigen to M cells.
Figure 2.

Development of an M cell–targeted mucosal vaccine with NKM 16–2-4. (A) FITC-conjugated NKM 16–2-4, but not FITC-conjugated control rat IgG, was specifically attached to the apical surfaces of M cells in FAE of PPs within 10 min of inoculation in an intestinal loop assay. The NKM 16–2-4 was subsequently taken up into the cytoplasmic regions of M cells within 30 min and reached the basal membrane of M cells within 4 h. Bars, 10 μm. (B) TT conjugated with NKM 16–2-4 effectively induced high-level, TT-specific serum IgG and fecal IgA responses, unlike TT conjugated with control rat IgG or UEA-1. Furthermore, the levels were superior to those in mice immunized with 10 times the amount of noncoupled TT (500 μg). *, P < 0.01, Tukey's t test. (C) BT-conjugated NKM 16–2-4, but not BT-conjugated control rat IgG, induced brisk botulinum toxin–specific serum IgG and fecal IgA responses. (D) Mice orally immunized with BT-conjugated NKM 16–2-4 were protected from an i.p. challenge with 10,000× LD50 type A botulinum toxin. Data are expressed as the mean ± the SD.
To directly confirm that M cell–targeted mucosal vaccination with NKM 16–2-4 is an effective strategy for inducing high-level, antigen-specific immune responses, tetanus toxoid (TT) was selected as a prototypical vaccine antigen, as TT has been extensively used in our previous experiments to elucidate the mechanism of the antigen-specific immune responses induced in both the mucosal and systemic compartments by mucosal immunization (20). A chimeric complex of TT conjugated with NKM 16–2-4 or control rat IgG (in total, each 200 μg contained 50 μg TT per mouse) was prepared by using avidin–biotin complexes (see Materials and methods). The prepared complexes consisted of TT and NKM 16–2-4 or control rat IgG; these complexes, or noncoupled TT, were orally administered to mice, together with the mucosal adjuvant cholera toxin (CT). In addition, it has been reported that M cell–targeted mucosal vaccine with UEA-1 is effective in inducing antigen-specific humoral and cellular immunity when administered via the nasal route (8, 9); therefore, we prepared an orally administered TT-conjugated UEA-1 as a control for the efficacy of the NKM 16–2-4–based M cell–targeted mucosal vaccines. As expected, brisk TT-specific serum IgG and mucosal IgA responses were induced in mice immunized with TT-conjugated NKM 16–2-4, whereas TT-conjugated control rat IgG or 50 μg noncoupled TT induced, at best, very low TT-specific immune responses (Fig. 2 B). In addition, the level of the TT-specific immune response induced by TT-conjugated UEA-1 was lower than that induced by TT-conjugated NKM 16–2-4. These data suggest that an M cell–targeted mucosal vaccine with UEA-1 might be insufficient for antigen delivery to M cells, because the UEA-1–based vaccine is trapped by goblet cells and their secreting mucus, as well as by M cells. Furthermore, 10 times more noncoupled TT (500 μg) induced a small TT-specific immune response compared with TT-conjugated NKM 16–2-4 containing 50 μg TT (Fig. 2 B), perhaps because of the low efficacy of antigen delivery to M cells for the induction of antigen-specific immune responses. Although the levels of the antigen-specific antibody responses induced here by immunization with noncoupled TT and CT tended to be lower than those in a previous study (20), this discrepancy might have been caused by differences in the mouse haplotype or the sources of TT and CT. Despite the discrepancy, our current findings emphasize the effectiveness of the newly established NKM 16–2-4 for the targeting of vaccine antigen to M cells to induce antigen-specific immune responses.
Moreover, when mice were orally immunized with botulinum toxoid (BT) conjugated with NKM 16–2-4 or control rat IgG (in total, each 200 μg contained 50 μg BT per mouse) in the presence of CT, brisk botulinum toxin–specific serum IgG and fecal IgA responses were induced in mice immunized with BT-conjugated NKM 16–2-4, but not in those immunized with BT-conjugated control rat IgG (Fig. 2 C). In addition, the mice immunized with BT-conjugated NKM 16–2-4 survived after challenge with 200 ng (10,000× LD50) of botulinum toxin, whereas the mice immunized with BT-conjugated control rat IgG died within 3 h (Fig. 2 D). These data strongly indicate that the M cell-targeted mucosal vaccine with NKM 16–2-4 can effectively induce protective immunity with the minimum dose of vaccine antigen.
To confirm the mechanism by which the NKM 16–2-4–based M cell–targeted mucosal vaccine induces brisk antigen-specific immune responses in the systemic and mucosal compartments, and its universality, OVA was then chosen as a prototype antigen with low antigenicity. An immunocytochemical analysis revealed that Alexa Fluor 647–labeled OVA conjugated with NKM 16–2-4 and FITC-conjugated avidin specifically reacted with UEA-1–positive isolated M cells in vitro (Fig. 3 A), and intestinal loop assay clearly demonstrated that it specifically attached to the apical surfaces of M cells and was subsequently taken up into the cytoplasmic regions of M cells in vivo (Fig. 3 B). Furthermore, brisk increases in the levels of OVA-specific serum IgG were induced in mice immunized with only 200 μg OVA-conjugated NKM 16–2-4 (containing 50 μg OVA), but not with the same amount of OVA-conjugated control rat IgG (Fig. 3 C). Our previous study showed that amounts of OVA as high as 1 mg were required to induce OVA-specific immune responses (5); now, oral immunization with even small amounts of poorly immunogenic antigens (e.g., OVA) is possible by using the M cell–targeting concept with NKM 16–2-4.
Figure 3.

Effective uptake and universality of the M cell–targeted mucosal vaccine. (A) Immunocytochemical analysis showed that an M cell–targeted OVA vaccine composed of Alexa Fluor 647–conjugated OVA, FITC-conjugated avidin, and NKM 16–2-4 specifically reacted with isolated UEA-1–positive M cells. (B) In an intestinal loop assay, the M cell-targeted OVA specifically attached to the apical surfaces of M cells (red arrows) and was immediately taken up into the cytoplasmic regions of M cells. Bars, 10 μm. (C) Orally administered OVA-conjugated NKM 16–2-4 effectively induced an OVA-specific serum IgG response, whereas an OVA-conjugated control rat IgG did not. Data are expressed as the mean ± the SD.
We could not directly compare the efficacy of NKM 16–2-4–based mucosal vaccine with those of already published σ1-based mucosal vaccines (10, 11) because the latter systems have been used for nasal, but not oral, vaccines and no information is currently available on whether σ1 possesses specificity for villous M cells. However, our strategy for using NKM 16–2-4 as an M cell–targeting vehicle might be superior, because NKM 16–2-4 possesses specificity for both villous M cells and PP-associated M cells. In support of our hypothesis, our previous data showed that villous M cells are capable of taking up orally administered antigens for the induction of PP-independent, antigen-specific immune responses (4). However, it should be noted that TT- or OVA-specific immune responses were not effectively induced without the presence of the mucosal adjuvant CT, even if the antigen was targeted to M cells by using NKM 16–2-4. This finding could be explained by the observation that the gastrointestinal immune system generally operates via a sophisticated mucosal regulatory network to avoid unnecessary hyperimmune responses to the numerous orally encountered antigens in the harsh environment of the intestinal tract (3). Therefore, it is essential to use the mucosal adjuvant, which temporarily breaks the mucosal regulatory network system, to activate gastrointestinal immunity. In practical terms, further studies are needed to develop a safe mucosal adjuvant and take advantage of M cell–targeted mucosal vaccines with NKM 16–2-4.
Identification of antigens recognized by NKM 16–2-4
In attempts to elucidate the antigen-sampling mechanism of M cells for the induction of antigen-specific immune responses, a major drawback has been the lack of knowledge of the specific genes and the corresponding molecules expressed by M cells. In addition, no information regarding which murine M cell–specific glycoproteins are recognized by UEA-1 is currently available, although UEA-1 is used extensively as a specific marker of M cells in mice. Therefore, we tried to identify the membrane antigen recognized by NKM 16–2-4 by using a proteomics approach with liquid chromatography–tandem mass spectrometry (LC-MS/MS) after immunoprecipitation of an M cell lysate with NKM 16–2-4. 4 major bands (3 bands >250 kD and 1 band of ∼150 kD) were precipitated by NKM 16–2-4 (Fig. 4 A), and these were identified by LC-MS/MS as maltase glucoamylase (top three bands) and alanyl (membrane) aminopeptidase (bottom band). These two molecules, which have been reported as intestinal enzymes of 410, 275, and 260 kD (21), and 150 kD, respectively, (22) under denatured conditions, are distributed at the brush borders of epithelial cells for the final digestion of dietary nutrients (21, 22). Because they were not homologous with each other, and subsequent in situ hybridization analysis demonstrated that their mRNAs were ubiquitously and abundantly expressed in the intestinal epithelium, including in M cells (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20070607/DC1), we hypothesized that NKM 16–2-4 possesses specificity for the M cell–specific carbohydrate moiety containing α(1,2) fucose, as the precipitated antigens were commonly recognized by UEA-1 (Fig. 4 B).
Figure 4.

Identification of the antigen recognized by NKM 16–2-4. (A) Immunoprecipitation and Western blot analysis with NKM 16–2-4 were performed with an M cell lysate. 4 major bands (3 bands >250 kD and 1 band of ∼150 kD) were precipitated. A subsequent LC-MS/MS analysis identified the three top bands as maltase glucoamylase and the bottom band as alanyl (membrane) aminopeptidase. (B) Lectin blot analysis performed after immunoprecipitation with NKM 16–2-4 showed that the precipitated antigens were all recognized by UEA-1. (C) mFUT1 and mFUT2 genes were transfected into original CHO cells and CHO-derived mutant lines (Lec1, Lec2, and Lec8 cells) with a pIRES2-EGFP expression system, and the specificity of NKM 16–2-4 and UEA-1 for EGFP-expressing transfectants was analyzed. NKM 16–2-4 and UEA-1 specifically reacted with mFUT1- or mFUT2-expressing original CHO cells. The reactivity of NKM 16–2-4 but not UEA-1 to mFUT1- or mFUT2-expressing Lec2 cells was enhanced compared with that to mFUT1- or mFUT2-expressing CHO cells. On the other hand, mFUT1- or mFUT2-expressing Lec1 or Lec8 cells were not recognized at all by NKM 16–2-4.
On the basis of our hypothesis, we transfected Chinese hamster ovary (CHO) cells with the genes encoding murine fucosyl transferase 1 (mFUT1) or mFUT2, which have been identified as α(1,2) fucose transfer enzymes (23). A flow cytometric analysis revealed that both NKM 16–2-4 and UEA-1 specifically reacted with CHO cells expressing mFUT1 or mFUT2, but neither NKM 16–2-4 nor UEA-1 showed specificity for original or empty vector–transfected CHO cells (Fig. 4 C). In addition, a blocking analysis showed that pretreatment of NKM 16–2-4 with α-l-fucose did not completely abolish reactivity to mFUT1- or mFUT2-expressing transfectants, although UEA-1 reactivity to these transfectants was dramatically decreased (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20070607/DC1), indicating that the epitope recognized by NKM 16–2-4 is an mFUT1- or mFUT2-mediated carbohydrate complex containing α(1,2) fucose that is different from the UEA-1–reactive portion of α(1,2) fucose.
Because immunohistochemical analysis demonstrated that UEA-1, but not NKM 16–2-4, recognized goblet cells in the intestinal villi (Fig. 1 A), we turned to examining the differences in recognition patterns between NKM 16–2-4 and UEA-1 by using a mutant line of CHO cells to elucidate the importance of the glycosylation of M cells and goblet cells in the mucosal immune system. When the mFUT1 or mFUT2 gene was introduced into a mutant line of CHO cells (Lec2) with an inactivated CMP-sialic acid transporter (24), the reactivity of NKM 16–2-4, but not of UEA-1, was higher in these transfectants than in the mFUT1- or mFUT2-expressing original CHO cells; however, mFUT1- or mFUT2-expressing Lec1 cells with inactivated GlcNAc transferase I (i.e., a lack of N-glycans) (25) or Lec8 cells with inactivated UDP-galactose transporter (26) were not recognized at all by NKM 16–2-4. On the other hand, we observed very low reactivity of UEA-1 to mFUT1- or mFUT2-expressing Lec8 cells, although mFUT1- or mFUT2-expressing Lec1 cells were not recognized by UEA-1. This is because UEA-1 might recognize α(1,2) fucose, which is linked to very low levels of galactose on N glycans in mFUT1- or mFUT2-expressing Lec8 cells because it has been reported that Lec8 cells retain 10–20% of their galactosylation (26), and no information is currently available on whether α(1,2) fucose links to anything other than galactose. These data suggest that sialic acid might be useful in distinguishing the reactivity of NKM 16–2-4, but not UEA-1 to galactose-binding α(1,2) fucose on N-glycans, although the reactivity to α(1,2) fucose regulated by O-glycans remains unclear. Thus, our initial immunohistochemical analyses demonstrated that the specificity of NKM 16–2-4 to UEA-1–positive M cells, but not UEA-1–positive goblet cells, is attributable to the existence of abundant sialic acids neighboring the α(1,2) fucose–containing carbohydrate moiety on goblet cells, but not on M cells. With the exception of their expression patterns at the tissue level, there is currently little reliable information available on the glycobiological and molecular biological differences between mFUT1 and mFUT2 as α(1,2) fucosyltransferases (23). Therefore, further studies, especially in terms of in situ expression patterns at a cellular level at inductive sites, such as in PPs, are needed to elucidate the role of the carbohydrate moiety containing α(1,2) fucose in the mucosal immune system.
In summary, we established a novel M cell–specific mAb (NKM 16–2-4; rat IgG2c) that selectively recognizes M cells, but not goblet cells or epithelial cells, and we characterized the M cell–specific carbohydrate moiety containing α(1,2) fucose. Our strategy for M cell–targeted vaccination with NKM 16–2-4 is attractive for the development of mucosal vaccines.
MATERIALS AND METHODS
Animals.
Female BALB/c mice, Crlj: CD1-Foxn1nu mice, and SD rats between 6 and 8 wk old were obtained from CREA and Charles River Laboratories. All of them were maintained in the experimental animal facility at the Institute of Medical Science, the University of Tokyo, and experiments were performed according to the guidelines provided by the Animal Care and Use Committee of the University of Tokyo.
Establishment of an M cell–specific mAb.
The M cell–enriched fraction was prepared from murine PPs as previously described, with some modification, by using UEA-1 (4). In brief, cells isolated from murine PPs were fixed in 4% paraformaldehyde (Wako) and stained with 500 ng/ml PE-conjugated UEA-1 (Biogenesis). UEA-1–positive cells were sorted by a FACSAria cell sorter (Becton Dickinson) and injected into the footpads of SD rats (106 cells/rat) 4 times at 2-wk intervals, with TiterMax Gold (TiterMax) as an adjuvant. 4 d after the final immunization, lymphocytes isolated from the spleen and inguinal lymph nodes of the immunized rats were fused with P3X63-AG8.653 myeloma cells (CRL-1580; American Type Culture Collection) in the presence of 50% (wt/vol) polyethylene glycol 1500 (Roche). Established hybridomas were injected into Crlj: CD1-Foxn1nu mice, and mAbs were then purified from ascitic fluids by using protein G–Sepharose (GE Healthcare) and labeled with EZ-Link Sulfo-NHS-LC-biotin (Thermo Fisher Scientific), FITC (Sigma-Aldrich), or Alexa Fluor 647 (Invitrogen).
Immunohistochemical analysis.
One monoclonal antibody (NKM 16–2-4; rat IgG2c) was selected on the basis of the initial screening and its specificity to M cells determined by immunohistochemical and whole-mount staining analyses, as described previously, with some modification (4). In brief, after a blocking step with 1% BSA, 7-μm fixed frozen sections or fixed tissues containing PPs were stained with 5 μg/ml FITC-conjugated NKM 16–2-4 or FITC-conjugated isotype control (FITC-conjugated rat IgG2c; MBL International) and 1 μg/ml tetrarhodamine isothiocyanate–conjugated UEA-1 (Vector Laboratories). The sections were then counterstained with 400 ng/ml DAPI (Sigma-Aldrich) for histochemical analysis and analyzed under a confocal laser-scanning microscope (TCS SP2; Leica). For electronmicroscopic analysis, ultrathin sections (100 nm) were incubated with 1 μg/ml purified NKM 16–2-4 after blocking with 1% BSA, followed by 18-nm gold particle–conjugated goat anti–rat IgG (Jackson Immunoresearch Laboratories) diluted 1:10. Finally, the sections were stained with 4% uranyl acetate and analyzed under a transmission electron microscope (JEM100S; JEOL).
Uptake of NKM 16–2-4 by M cells.
After the mice were anesthetized with 2 mg ketamine (Sigma-Aldrich), we injected 100 μg of FITC-conjugated NKM 16–2-4 or FITC-conjugated control rat IgG (Sigma-Aldrich) into intestinal loops containing PPs, in accordance with our previous study (4). The mice were killed 10 or 30 min, or 4 h, after the inoculation, and frozen sections (7 μm) of intestinal loop were prepared and analyzed under a confocal laser-scanning microscope after counterstaining with DAPI.
M cell–targeted vaccination.
TT (provided by the Research Foundation for Microbial Diseases, Osaka University, Osaka, Japan) and type A BT (prepared according to a previous study; reference [27)]) were first treated with EZ-Link Sulfo-NHS-LC-biotin. Next, biotinylated TT or BT at 1 mg/ml was incubated with the same volume of avidin (1 mg/ml; Sigma-Aldrich). The complexes were then incubated with twice the volume of biotinylated NKM 16–2-4, biotinylated control rat IgG (Sigma-Aldrich), or biotinylated UEA-1 (Vector Laboratories; each 1 mg/ml). Mice were orally immunized with the complexes (in total, each 200 μg contained 50 μg TT or BT per mouse), noncoupled TT (50 or 500 μg per mouse), or PBS alone 3 times (once a week), together with 10 μg CT (List Biological Laboratories) as a mucosal adjuvant. 7 d after the final immunization, serum and fecal extracts were collected and analyzed for TT- or type A botulinum toxin–specific serum IgG and fecal IgA responses by ELISA, as previously described (5, 27). To examine the protective immunity, the mice were challenged via the i.p. route with 200 ng type A botulinum toxin (10,000× LD50 i.p.) diluted in 100 μl of 0.2% gelatin/PBS (27). To confirm the universality of M cell–targeted mucosal vaccine with NKM 16–2-4, OVA (Sigma-Aldrich) was conjugated with NKM 16–2-4 or control rat IgG and orally immunized together with 10 μg CT. In addition, intestinal loop assay was performed by using M cell–targeted OVA composed of Alexa Fluor 647-conjugated OVA (Invitrogen), FITC-conjugated avidin (Sigma-Aldrich), and NKM 16–2-4 or control rat IgG. Conjugation of NKM 16–2-4 or control rat IgG and the protein antigen was confirmed by sandwich ELISA (unpublished data).
Analysis of antigen recognized by NKM 16–2-4.
To identify the antigen recognized by NKM 16–2-4, we performed an immunoprecipitation assay with NKM 16–2-4 followed by an LC-MS/MS analysis. In brief, a lysate of M cells was incubated with 10 μg/ml NKM 16–2-4 or an isotype control antibody (rat IgG2c; BD Biosciences) followed by protein G–Sepharose (GE Healthcare). Immune complexes were analyzed by SDS-PAGE and Western or lectin blot with 5 μg/ml biotinylated NKM 16–2-4, 5 μg/ml biotinylated isotype control antibody (biotin-conjugated rat IgG2c; BD Biosciences), or 5 μg/ml biotinylated UEA-1 (Vector Laboratories) and ABC–AP complex (Vector Laboratories). To identify the precipitated antigen, LC-MS/MS analysis was performed after digestion with 50 nM trypsin gold (Promega).
Transfection of cells.
mFUT1 and mFUT2 genes were synthesized from mRNAs from intestinal tissue, including PPs, using specific primers (mFUT1: sense, 5′-TACTAAGCTAGCATGTGGACTCCCAGCCGGAGGCAG-3′, antisense, 5′-GCTAGCGGATCCTCAGACCAATCTAAAAAGACTGTC-3′; mFUT2: sense, 5′-ATCTAAGCTAGCATGGCGAGTGCCCAGGTACCTTTC-3′, antisense, 5′-TGCAGCGAATTCTTAGTGCTTAAGGAGTGGGGACAG-3′; NheI and BamHI [mFUT1] and NheI and EcoRI [mFUT2] restriction enzyme sites are shown by underlining) by RT-PCR and inserted into pIRES2-EGFP vector (BD Biosciences). These plasmids were then transformed into CHO-K1 cells (CCL-61; American Type Culture Collection) and three CHO-cell–derived mutant lines (Lec1, CRL-1735 [reference 25]; Lec2, CRL-1736 [reference 24]; and Lec8, CRL-1737 [reference 26]). 2 d after transfection, the cells were stained with 500 ng/ml Alexa Fluor 647–conjugated NKM 16–2-4 and 500 ng/ml PE-conjugated UEA-1, followed by the application of 10 μl/test VIA-PROVE (BD Biosciences). They were then analyzed by flow cytometry with FACSCalibur (Becton Dickinson). For blocking analysis, 500 ng/ml Alexa Fluor 647–conjugated NKM 16–2-4 or 500 ng/ml PE-conjugated UEA-1 was first pretreated with 0.5 M α-L-fucose (Wako).
Data analysis.
Data are expressed as the mean ± the SD. All analyses for statistically significant differences were performed by Tukey's t test, with P < 0.01 considered significant (denoted in the figures with an asterisk).
Online supplemental material.
Fig. S1 shows the specificity of NKM 16–2-4 to isolated UEA-1–positive M cells. Fig. S2 shows that NKM 16–2-4 specifically reacts with M cells in NALT, similar to its reaction with PP-associated M cells. Fig. S3 shows the expression of maltase glucoamylase and alanyl aminopeptidase mRNAs in PPs. Fig. S4 shows that NKM 16–2-4 reacts with different form of UEA-1–reactive portion of α(1,2) fucose. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20070607/DC1.
Supplemental Material
Acknowledgments
We thank Drs. S. Ohmi, H. Fukuda, C. Sasakawa, S. Yoshida, and M. Suzuki at the Institute of Medical Science, the University of Tokyo, for their helpful discussions and technical advice in performing the proteomics analysis and vaccine development. We also thank Dr. A. Irimura at the Graduate School of Pharmaceutical Science, the University of Tokyo, and Drs. J. Hirabayashi, A. Kuno, H. Tateno, and T. Sato at the Research Center for Medical Glycoscience, National Institute of Advanced Industrial Science and Technology, for their helpful discussions and advice in performing the glycobiological analysis.
This work was supported by Core Research for Evolutional Science and Technology of the Japan Science and Technology Corporation, by a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology and the Ministry of Health and Labor, Japan, and the Waksman Foundation, Japan.
The authors have no conflicting financial interests.
References
- 1.Kiyono, H., and S. Fukuyama. 2004. NALT- versus Peyer's-patch-mediated mucosal immunity. Nat. Rev. Immunol. 4:699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Owen, R.L. 1977. Sequential uptake of horseradish peroxidase by lymphoid follicle epithelium of Peyer's patches in the normal unobstructed mouse intestine: an ultrastructural study. Gastroenterology. 72:440–451. [PubMed] [Google Scholar]
- 3.Mestecky, J., R.S. Blumberg, H. Kiyono, and J.R. McGhee. 2003. The mucosal immune system. In 5th Fundamental Immunology. W.E. Paul, editor. Lippincott Williams & Wilkins. pp. 965–1020.
- 4.Jang, M.H., M.N. Kweon, K. Iwatani, M. Yamamoto, K. Terahara, C. Sasakawa, T. Suzuki, T. Nochi, Y. Yokota, P.D. Rennert, et al. 2004. Intestinal villous M cells: an antigen entry site in the mucosal epithelium. Proc. Natl. Acad. Sci. USA. 101:6110–6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamamoto, M., P. Rennert, J.R. McGhee, M.N. Kweon, S. Yamamoto, T. Dohi, S. Otake, H. Bluethmann, K. Fujihashi, and H. Kiyono. 2000. Alternate mucosal immune system: organized Peyer's patches are not required for IgA responses in the gastrointestinal tract. J. Immunol. 164:5184–5191. [DOI] [PubMed] [Google Scholar]
- 6.Holmgren, J., and C. Czerkinsky. 2005. Mucosal immunity and vaccines. Nat. Med. 11:S45–S53. [DOI] [PubMed] [Google Scholar]
- 7.Brayden, D.J., M.A. Jepson, and A.W. Baird. 2005. Keynote review: intestinal Peyer's patch M cells and oral vaccine targeting. Drug Discov. Today. 10:1145–1157. [DOI] [PubMed] [Google Scholar]
- 8.Manocha, M., P.C. Pal, K.T. Chitralekha, B.E. Thomas, V. Tripathi, S.D. Gupta, R. Paranjape, S. Kulkarni, and D.N. Rao. 2005. Enhanced mucosal and systemic immune response with intranasal immunization of mice with HIV peptides entrapped in PLG microparticles in combination with Ulex Europaeus-I lectin as M cell target. Vaccine. 23:5599–5617. [DOI] [PubMed] [Google Scholar]
- 9.Wang, X., I. Kochetkova, A. Haddad, T. Hoyt, D.M. Hone, and D.W. Pascual. 2005. Transgene vaccination using Ulex europaeus agglutinin I (UEA-1) for targeted mucosal immunization against HIV-1 envelope. Vaccine. 23:3836–3842. [DOI] [PubMed] [Google Scholar]
- 10.Wu, Y., X. Wang, K.L. Csencsits, A. Haddad, N. Walters, and D.W. Pascual. 2001. M cell-targeted DNA vaccination. Proc. Natl. Acad. Sci. USA. 98:9318–9323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang, X., D.M. Hone, A. Haddad, M.T. Shata, and D.W. Pascual. 2003. M cell DNA vaccination for CTL immunity to HIV. J. Immunol. 171:4717–4725. [DOI] [PubMed] [Google Scholar]
- 12.Sharma, R., U. Schumacher, and E. Adam. 1998. Lectin histochemistry reveals the appearance of M-cells in Peyer's patches of SCID mice after syngeneic normal bone marrow transplantation. J. Histochem. Cytochem. 46:143–148. [DOI] [PubMed] [Google Scholar]
- 13.Helander, A., K.J. Silvey, N.J. Mantis, A.B. Hutchings, K. Chandran, W.T. Lucas, M.L. Nibert, and M.R. Neutra. 2003. The viral σ1 protein and glycoconjugates containing α2–3-linked sialic acid are involved in type 1 reovirus adherence to M cell apical surfaces. J. Virol. 77:7964–7977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kandori, H., K. Hirayama, M. Takeda, and K. Doi. 1996. Histochemical, lectin-histochemical and morphometrical characteristics of intestinal goblet cells of germfree and conventional mice. Exp. Anim. 45:155–160. [DOI] [PubMed] [Google Scholar]
- 15.Park, H.S., K.P. Francis, J. Yu, and P.P. Cleary. 2003. Membranous cells in nasal-associated lymphoid tissue: a portal of entry for the respiratory mucosal pathogen group A streptococcus. J. Immunol. 171:2532–2537. [DOI] [PubMed] [Google Scholar]
- 16.Fukuyama, S., T. Hiroi, Y. Yokota, P.D. Rennert, M. Yanagita, N. Kinoshita, S. Terawaki, T. Shikina, M. Yamamoto, Y. Kurono, and H. Kiyono. 2002. Initiation of NALT organogenesis is independent of the IL-7R, LTßR, and NIK signaling pathways but requires the Id2 gene and CD3−CD4+CD45+ cells. Immunity. 17:31–40. [DOI] [PubMed] [Google Scholar]
- 17.Fukuyama, S., T. Nagatake, D.Y. Kim, K. Takamura, E.J. Park, T. Kaisho, N. Tanaka, Y. Kurono, and H. Kiyono. 2006. Cutting edge: uniqueness of lymphoid chemokine requirement for the initiation and maturation of nasopharynx-associated lymphoid tissue organogenesis. J. Immunol. 177:4276–4280. [DOI] [PubMed] [Google Scholar]
- 18.Bonifaz, L.C., D.P. Bonnyay, A. Charalambous, D.I. Darguste, S. Fujii, H. Soares, M.K. Brimnes, B. Moltedo, T.M. Moran, and R.M. Steinman. 2004. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J. Exp. Med. 199:815–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trumpfheller, C., J.S. Finke, C.B. Lopez, T.M. Moran, B. Moltedo, H. Soares, Y. Huang, S.J. Schlesinger, C.G. Park, M.C. Nussenzweig, et al. 2006. Intensified and protective CD4+ T cell immunity in mice with anti-dendritic cell HIV gag fusion antibody vaccine. J. Exp. Med. 203:607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu-Amano, J., H. Kiyono, R.J. Jackson, H.F. Staats, K. Fujihashi, P.D. Burrows, C.O. Elson, S. Pillai, and J.R. McGhee. 1993. Helper T cell subsets for immunoglobulin A responses: oral immunization with tetanus toxoid and cholera toxin as adjuvant selectively induces Th2 cells in mucosa associated tissues. J. Exp. Med. 178:1309–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quezada-Calvillo, R., F. Rodriguez-Zuniga, and B.J. Underdown. 2002. Partial characterization of murine intestinal maltase-glucoamylase. Biochem. Biophys. Res. Commun. 295:394–400. [DOI] [PubMed] [Google Scholar]
- 22.Look, A.T., R.A. Ashmun, L.H. Shapiro, and S.C. Peiper. 1989. Human myeloid plasma membrane glycoprotein CD13 (gp150) is identical to aminopeptidase N. J. Clin. Invest. 83:1299–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Domino, S.E., L. Zhang, and J.B. Lowe. 2001. Molecular cloning, genomic mapping, and expression of two secretor blood group α(1,2)fucosyltransferase genes differentially regulated in mouse uterine epithelium and gastrointestinal tract. J. Biol. Chem. 276:23748–23756. [DOI] [PubMed] [Google Scholar]
- 24.Eckhardt, M., B. Gotza, and R. Gerardy-Schahn. 1998. Mutants of the CMP-sialic acid transporter causing the Lec2 phenotype. J. Biol. Chem. 273:20189–20195. [DOI] [PubMed] [Google Scholar]
- 25.Chen, W., and P. Stanley. 2003. Five Lec1 CHO cell mutants have distinct Mgat1 gene mutations that encode truncated N-acetylglucosaminyltransferase I. Glycobiology. 13:43–50. [DOI] [PubMed] [Google Scholar]
- 26.Deutscher, S.L., and C.B. Hirschberg. 1986. Mechanism of galactosylation in the Golgi apparatus. A Chinese hamster ovary cell mutant deficient in translocation of UDP-galactose across Golgi vesicle membranes. J. Biol. Chem. 261:96–100. [PubMed] [Google Scholar]
- 27.Kobayashi, R., T. Kohda, K. Kataoka, H. Ihara, S. Kozaki, D.W. Pascual, H.F. Staats, H. Kiyono, J.R. McGhee, and K. Fujihashi. 2005. A novel neurotoxoid vaccine prevents mucosal botulism. J. Immunol. 174:2190–2195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}