

Abstract
Healing of myocardial infarction (MI) requires monocytes/macrophages. These mononuclear phagocytes likely degrade released macromolecules and aid in scavenging of dead cardiomyocytes, while mediating aspects of granulation tissue formation and remodeling. The mechanisms that orchestrate such divergent functions remain unknown. In view of the heightened appreciation of the heterogeneity of circulating monocytes, we investigated whether distinct monocyte subsets contribute in specific ways to myocardial ischemic injury in mouse MI. We identify two distinct phases of monocyte participation after MI and propose a model that reconciles the divergent properties of these cells in healing. Infarcted hearts modulate their chemokine expression profile over time, and they sequentially and actively recruit Ly-6Chi and -6Clo monocytes via CCR2 and CX3CR1, respectively. Ly-6Chi monocytes dominate early (phase I) and exhibit phagocytic, proteolytic, and inflammatory functions. Ly-6Clo monocytes dominate later (phase II), have attenuated inflammatory properties, and express vascular–endothelial growth factor. Consequently, Ly-6Chi monocytes digest damaged tissue, whereas Ly-6Clo monocytes promote healing via myofibroblast accumulation, angiogenesis, and deposition of collagen. MI in atherosclerotic mice with chronic Ly-6Chi monocytosis results in impaired healing, underscoring the need for a balanced and coordinated response. These observations provide novel mechanistic insights into the cellular and molecular events that regulate the response to ischemic injury and identify new therapeutic targets that can influence healing and ventricular remodeling after MI.
Left ventricular remodeling after acute myocardial infarction (MI) promotes heart failure, which is an epidemic with high mortality and increasing prevalence (1). Despite advances in medical and interventional therapy for MI, many surviving patients still develop heart failure. The process of infarct healing involves a complex cascade of molecular, cellular, and physiological responses that affect prognosis by directly influencing the pathologic and structural changes in the heart (2–9). Thus, the mechanisms that regulate the myocardial response to ischemic injury continue to receive intense scrutiny.
In human and experimental MI, interruption of blood supply leads to rapid death of cardiac myocytes in the ischemic heart. Inflammatory signals recruit neutrophils to the infarct zone within 24 h, and monocytes/macrophages shortly thereafter. Together, these leukocytes degrade extracellular matrix constituents and macromolecules released by injured cells and aid clearance of dead cardiac myocytes and their debris. During this early phase, the weakened wall of the infarcted ventricle has heightened susceptibility to rupture and often causes sudden death when complicated by rupture of the ventricular septum, a papillary muscle, or of the left ventricular free wall. After ∼5 d, monocytes/macrophages and endothelial cells coordinate angiogenesis and promote blood supply in the forming granulation tissue. Type I collagen, which is synthesized by myofibroblasts, strengthens the infarct and protects it against rupture. By 2–3 wk after MI, monocytes/macrophages disappear and granulation tissue matures into a scar with cross-linked collagen as its dominant feature (for review see references [2–5]). During this process, the healing heart undergoes profound changes in ventricular geometry and function (10, 11). Optimum outcome after MI depends on a coordinated healing response that balances debris removal with repair of the myocardial extracellular matrix. Adverse remodeling and excessive inflammation can both promote heart failure (1, 12, 13).
Monocytes/macrophages persist for days in the infarct zone and contribute to inflammation, proteolysis, phagocytosis, angiogenesis, and collagen deposition (14–22). These diverse and seemingly contrasting functions position the monocyte/macrophage as a central cellular protagonist and potential therapeutic target in wound healing and tissue repair after MI (16–20, 23). The tension between host defense and repair mechanisms versus proinflammatory properties of the mononuclear phagocyte in infarcting myocardium requires caution against indiscriminate targeting of monocytes/macrophages (24, 25).
Monocytes, which are the circulating precursors of macrophages, display heterogeneity in both mouse and human (26–30). In the mouse, Ly-6Chi (Gr1hiCCR2+CX3CR1lo) monocytes efficiently infiltrate inflammatory sites and give rise to macrophages and dendritic cells in response to inflammatory stimuli (26, 27, 31–33). Their counterparts, Ly-6Clo (Gr1loCCR2−CX3CR1hi) monocytes, accumulate at inflammatory sites less efficiently and are thought to give rise to resident tissue cells. This study tested the hypotheses that Ly-6Chi and -6Clo monocyte subsets commit to specific functions while in circulation and promote disparate processes in the infarcting myocardium of mice. We report on a novel mechanism of the myocardial response to ischemic injury and identify targets for future therapies in myocardial wound healing.
RESULTS
The injured myocardium mobilizes Ly-6Chi and -6Clo monocytes in two distinct phases
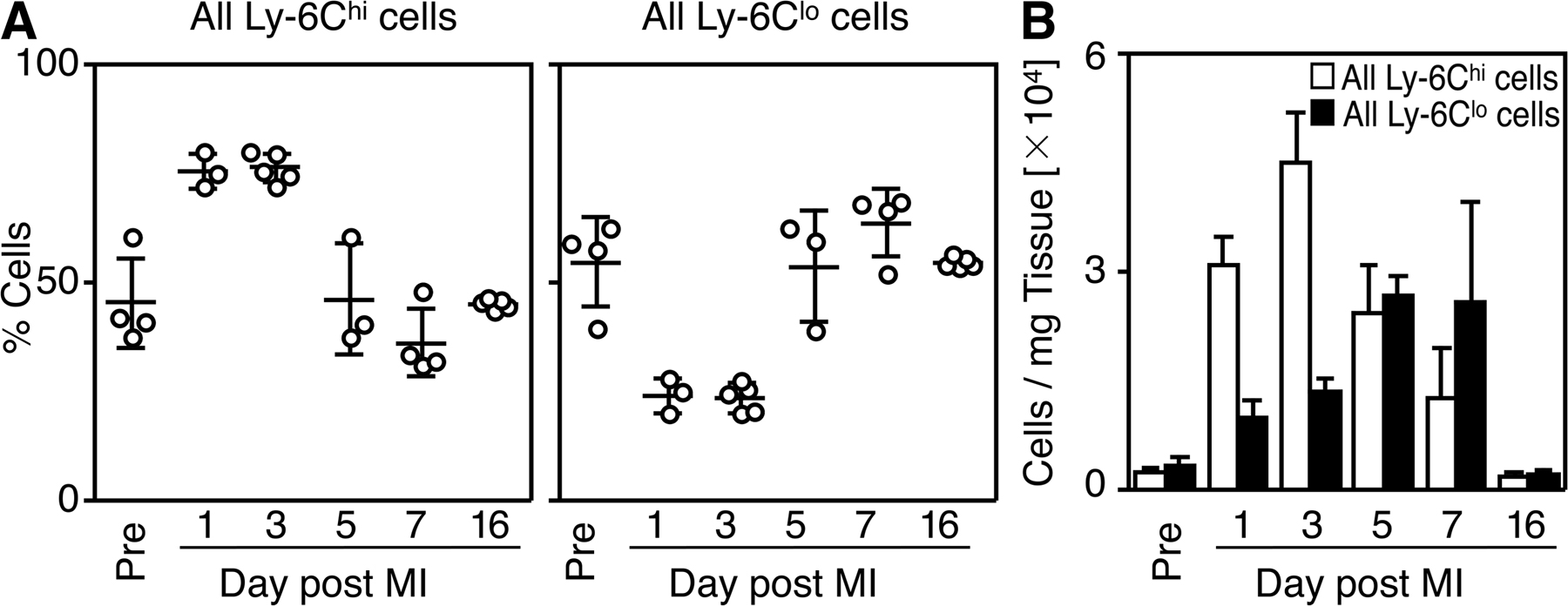
To identify the repertoire of monocytes and their lineage descendants within the injured myocardium, we induced myocardial infarction in C57BL/6 mice by permanent coronary artery ligation and analyzed single-cell suspensions of digested infarcts at different time points. Flow cytometry defined monocytes and their lineage descendants as CD11bhi (CD90/B220/CD49b/NK1.1/Ly-6G)lo mononuclear cells, as previously reported (31). These were further divided into Ly-6Chi (F4/80/I-Ab/CD11c)lo monocytes, Ly-6Clo (F4/80/I-Ab/CD11c)lo monocytes, and Ly-6Clo (F4/80/I-Ab/CD11c)hi macrophages/dendritic cells (Fig. 1 A). Delineation between monocytes and macrophages was based on the observations that (a) peripheral blood contains mostly monocytes, but not macrophages; (b) heart tissue under steady-state conditions within the CD11bhi gate contains mostly F4/80intI-Ab+CD11clo cells, but not monocytes, as assessed by separate staining; and (c) monocytes are (F4/80/I-Ab/CD11c)lo, whereas macrophages are (F4/80/I-Ab/CD11c)hi (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20070885/DC1). Longitudinal analysis identified dynamic alterations in the relative proportions of these populations. First, the ischemic myocardium included predominantly monocytes, but not macrophages/dendritic cells, at least during the first week after coronary occlusion (Fig. 1 B). Macrophages and/or dendritic cells prevailed again on day 16, after completion of the acute phase of healing, and the proportion of monocytes and macrophages/dendritic cells returned to baseline (Fig. 1 B). Second, the relative proportion of Ly-6Chi and -6Clo monocytes in injured myocardium varied strikingly over time; Ly-6Chi monocytes predominated from day 1 to 4 (∼75% of all monocytes), whereas Ly-6Clo monocytes prevailed from day 5 onward (∼75% of all monocytes, Fig. 1 C). Importantly, the biphasic Ly-6Chi:Ly-6Clo response pertained irrespective of monocyte differentiation (Fig. S2).
Figure 1.

The ischemic myocardium mobilizes Ly-6Chi and -6Clo monocytes in two distinct phases. Cell suspensions from healthy hearts or infarcts of C57BL/6 mice were stained with anti-CD11b, -CD90, -B220, -CD49b, -NK1.1, -Ly-6G, -Ly-6C, -F4/80, -I-Ab, and -CD11c mAbs. Monocytes/macrophages/dendritic cells were identified as CD11bhi (CD90/B220/CD49b/NK1.1/Ly-6G)lo. (A) Representative dot plots from individual mice within the monocyte/macrophage/dendritic cell gate depict Ly-6Chi monocytes (Ly-6Chi [F4/80/I-Ab/CD11c]lo), Ly-6Clo monocytes (Ly-6Clo [F4/80/I-Ab/CD11c]lo), and macrophages/dendritic cells (Ly-6Clo [F4/80/I-Ab/CD11c]hi) in healthy hearts and within the infarct at specified days after MI. Percentages of cells are shown as the mean ± the SEM. (B) Relative percentage of monocytes and macrophages/dendritic cells. (C) Relative percentage of Ly-6Chi and -6Clo monocytes. (D) Total number of neutrophils per milligram of tissue. (E) Total number of monocytes and macrophages/dendritic cells per milligram of tissue. (F) Total number of Ly-6Chi and -6Clo monocytes per mg tissue. (G) Time course of leukocyte infiltration to infarct. The mean and the SEM are shown. Results are pooled from 6 independent experiments with 3–10 mice per group.
As long recognized (2, 3, 5), neutrophils accumulated early in injured myocardium (∼105 cells/mg infarct tissue on day 1; Fig. 1, D and G). Monocytes started to accumulate on day 1, remained in high and relatively stable numbers until day 7 (e.g., ∼4–5 × 104 cells/mg tissue), and dropped to levels comparable to those in noninjured hearts on day 16 (∼0.3 × 104 cells/mg tissue; Fig. 1, E and G). In contrast, macrophages remained in relatively low numbers from day 1 to 7 (<1 × 104 cells/mg tissue; Fig. 1, E and G). These findings suggest an active role for undifferentiated monocytes, or macrophages not yet fully differentiated, and challenge our current thinking on the role of macrophages in the healing process. Furthermore, distinction between Ly-6Chi and -6Clo monocytes showed biphasic and contrary kinetics of mobilization during the week that followed myocardial infarction (Fig. 1, F and G). The number of Ly-6Chi monocytes in injured myocardium peaked on day 3 (∼4 × 104 cells/mg tissue) and waned thereafter (<0.5 × 104 cells/mg tissue on day 7), whereas the number of Ly-6Clo monocytes peaked only on day 7 (∼2 × 104 cells/mg tissue). Thus, myocardial infarction triggered the sequential mobilization of Ly-6Chi and -6Clo monocytes. The relative abundance of Ly-6Chi and -6Clo cells in the healing tissue thus identifies two phases of the monocytic response, hereafter referred to as phase I and II. These phases characterize relatively late events (i.e., days after MI), as opposed to events occurring within the first hours and that have been a focus of a recent study (34).
The injured myocardium sequentially recruits circulating Ly-6Chi and -6Clo monocytes through modulation of chemokine expression
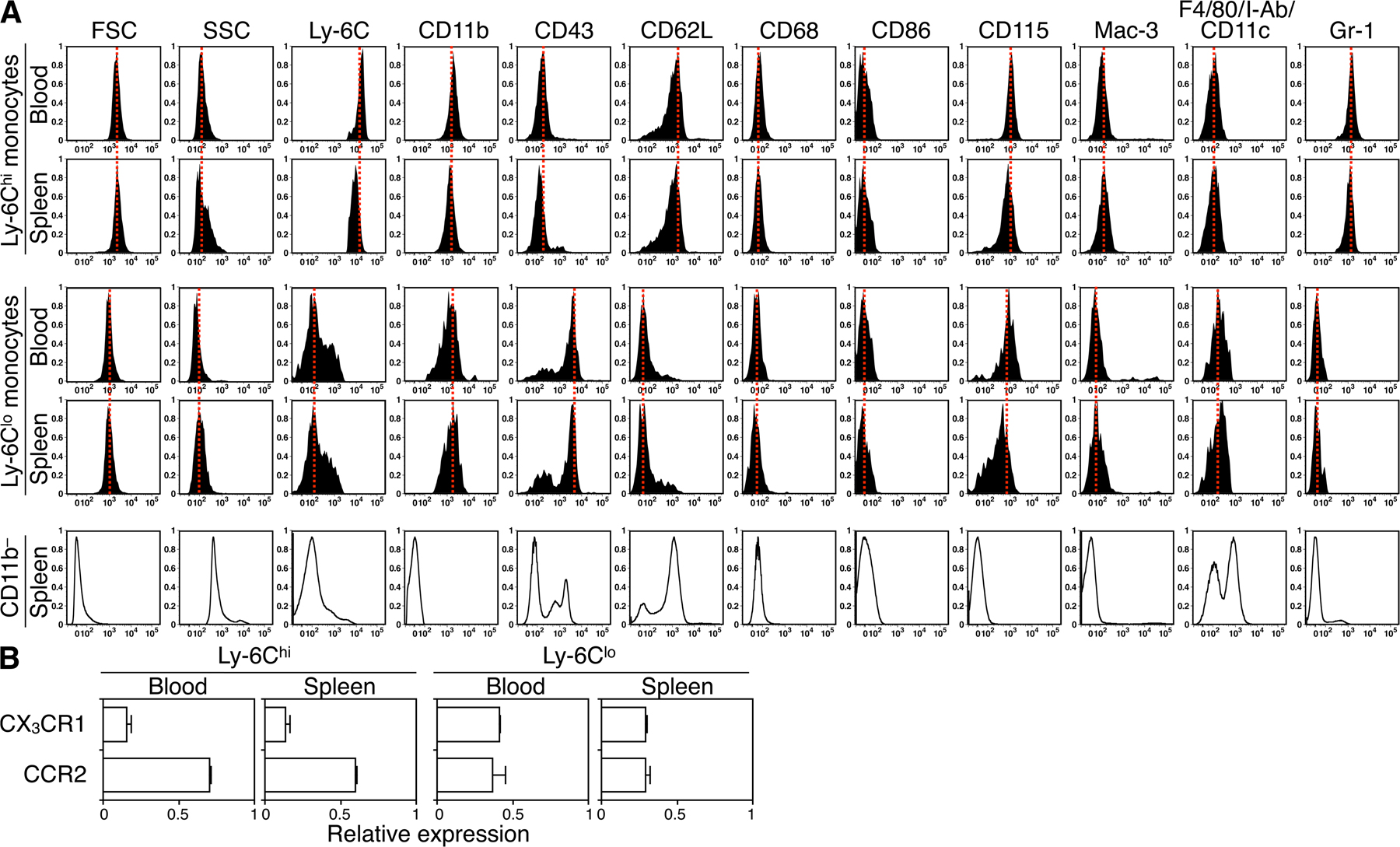
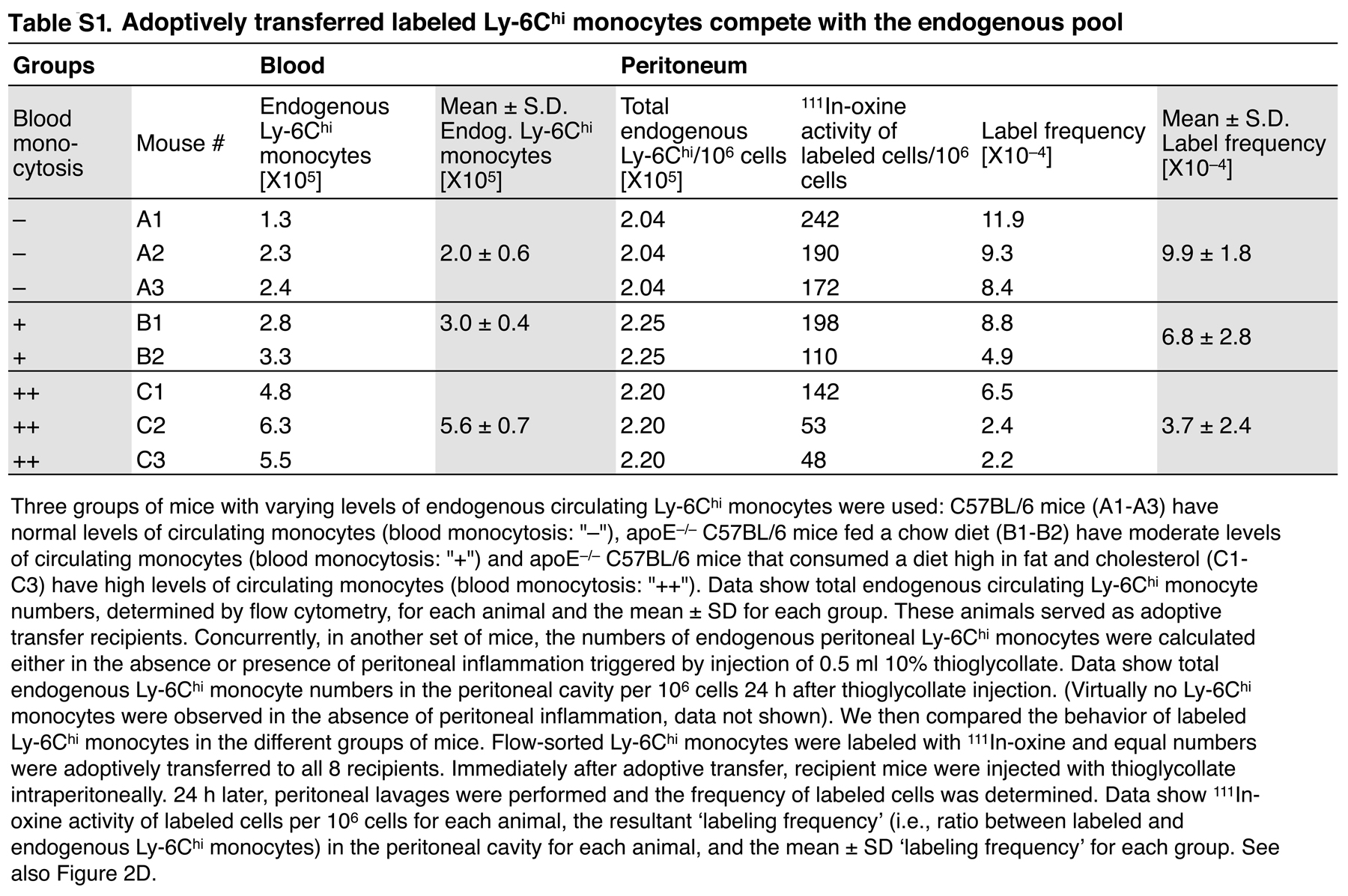
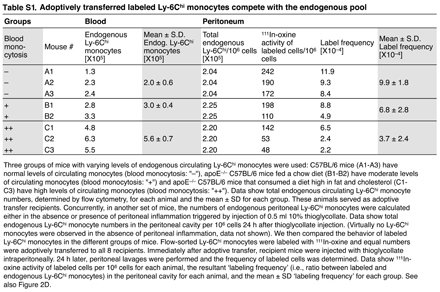
Several mechanisms might explain the selective mobilization of Ly-6Chi monocytes during phase I and Ly-6Clo monocytes during phase II. For instance, the injured myocardium may recruit Ly-6Chi cells that convert into Ly-6Clo cells at the onset of phase II. The myocardium may also recruit circulating Ly-6Chi and -6Clo cells sequentially; a biphasic recruitment of monocytes could result from alterations in the relative number of these cells in peripheral blood and/or from preferential recruitment to the target tissue. To address these issues, we sought to determine how circulating Ly-6Chi and -6Clo monocytes contribute to the pool of monocytes/macrophages in injured hearts, and we measured the following two parameters at the onset of phase I and II: (a) the relative abundance of Ly-6Chi and -6Clo monocytes in the blood, and (b) the intrinsic probability (i.e., on a cell–cell basis) of circulating Ly-6Chi and -6Clo monocytes being recruited to injured hearts. To this end, we first analyzed peripheral blood mononuclear cells ex vivo; although the total leukocyte numbers remained relatively stable over time (Fig. 2 A), myocardial infarction triggered acute Ly-6Chi monocytosis during phase I, but not during phase II, whereas the number of circulating Ly-6Clo monocytes remained unchanged (Fig. 2 B). Thus, circulating Ly-6Chi monocytes became, on average, 4.5-fold more numerous than Ly-6Clo monocytes in phase I, and then returned to relatively comparable abundance in phase II (Fig. 2 B). Second, we measured the intrinsic capacity of monocytes to accumulate in the injured myocardium. This involved labeling equal numbers of flow-sorted splenic Ly-6Chi or -6Clo monocytes with 111In-oxine, followed by intravenous injection of labeled cells into mice at the onset of either phase I or II. We used the spleen as a source of monocytes, because of the relative paucity of monocytes in mouse blood. Splenic monocytes were defined as CD11bhi (CD90/B220/CD49b/NK1.1/Ly-6G)lo (F4/80/I-Ab/CD11c)lo to exclude resident macrophages and dendritic cells, and had the same morphology as blood monocytes (31). Extensive analysis of splenic and blood monocytes also showed similar FSC, SSC, Ly-6C, CD11b, CD43, CD62L, CD68, CD86, CD115, Mac-3, F4/80/I-Ab/CD11c, Gr-1, CCR2, and CX3CR1 phenotype (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20070885/DC1), thus indicating that splenic monocytes can be used as a rich source of surrogate circulating monocytes. Excising hearts 24 h after adoptive transfer and calculating percent-injected dose per gram of tissue revealed a similar intrinsic (i.e., on a cell–cell basis without considering the endogenous, nonlabeled cell population) capacity of Ly-6Chi and -6Clo monocytes to infiltrate the injured myocardium during phase I. Conversely, Ly-6Clo monocytes showed a 4.8-fold increased capacity to enter hearts during phase II (Fig. 2 C). Because exogenously labeled, adoptively transferred monocytes compete with the endogenous pool (Fig. 2 D and Table S1), we could calculate the relative contribution of Ly-6Chi and -6Clo cells recruited to the injured myocardium by taking into account the relative proportion of circulating Ly-6Chi and -6Clo monocytes (Fig. 2 B) and their intrinsic capacity to migrate to wounded myocardium (Fig. 2 C). We found that the injured myocardium preferentially recruited Ly-6Chi cells (78%) during phase I, and Ly-6Clo cells (80%) during phase II (Fig. 2 E).
Figure 2.

The ischemic myocardium sequentially recruits circulating Ly-6Chi and -6Clo monocytes. (A) Number of total leukocytes in peripheral blood of C57BL/6 mice before MI and at the onset of phase I (day 1 after MI) and phase II (day 4). (B) Number of circulating Ly-6Chi and -6Clo monocytes at the same time points. Values in the top right quadrants indicate the ratio between the two subsets. (C) In vivo cardiac accumulation of 111In-oxine-labeled Ly-6Chi and -6Clo monocytes 24 h after adoptive transfer on day 0 (phase I) and day 4 (phase II). Values in the top right quadrants indicate the ratio between the two subsets. (D) Adoptively transferred 111In-oxine–labeled Ly-6Chi monocytes compete with endogenous Ly-6Chi monocytes. Increased numbers of endogenous circulating Ly-6Chi monocytes (x axis) decrease the ratio between transferred and endogenous Ly-6Chi monocytes (label frequency, y axis) that migrated into the peritoneal cavity of mice with peritonitis (Table S1). (E) Pie graph representing the relative proportion of Ly-6Chi and -6Clo monocyte subsets expected to accumulate in infarct tissue, based on the mean abundance of each subset in peripheral blood (B) and the intrinsic capacity of circulating cells from each subset to accumulate at infarct tissue (C). Data represent three independent experiments and are shown as the mean ± the SEM. Table S1 is available at http://www.jem.org/cgi/content/full/jem.20070885/DC1.
The mechanisms driving monocyte accumulation in hearts are different in phase I and II. The dominance of Ly-6Chi monocytes in hearts in phase I is driven by selective expansion of circulating Ly-6Chi cells. This conclusion is in accordance with the finding that Ly-6Chi monocytes accumulate abundantly in hearts after injury (the mean ± the SD from 250 ± 200 Ly-6Chi monocytes/mg of tissue before MI to 24,000 ± 6,000 on day 1 after MI), whereas Ly-6Clo monocytes do so to a much lesser extent (from 1,400 ± 1,000 Ly-6Clo monocytes/mg of tissue before MI to 5,800 ± 2,600 on day 1 after MI). In contrast, the dominance of Ly-6Clo monocytes in hearts in phase II is driven by preferential recruitment of circulating Ly-6Clo monocytes. These results also suggest that Ly-6Chi → Ly-6Clo monocyte conversion rarely occurs in the injured myocardium in phase II because the proportion of newly recruited Ly-6Clo monocytes (Fig. 2 C) matched the proportion of Ly-6Clo monocytes present in the infarct (Fig. 1 F).
Sequential recruitment of monocyte subsets to the infarct suggests coordinated orchestration by chemokines, leukocyte adhesion molecules, and their cognate receptors. We tested this hypothesis by measuring several candidate chemokines and an adhesion molecule previously implicated in directing monocyte migration (28, 33, 35). We found that MCP-1 mRNA levels peaked during phase I, MIP-1α levels increased to the same extent in phase I and II, fractalkine levels decreased only in phase I, and VCAM-1 levels increased only in phase II (Fig. 3 A). Thus, the injured myocardium expresses distinct chemokines and adhesion molecule in phases I and II. Interestingly, Ly-6Chi monocytes selectively express CCR2, a receptor for MCP-1, whereas Ly-6Clo monocytes express high levels of CX3CR1, which is a receptor for fractalkine (Fig. 3 B and reference [26]). This finding led us to propose that CCR2 or CX3CR1 support migration of Ly-6Chi cells in phase I and of Ly-6Clo cells in phase II, respectively. Testing these hypotheses involved analyzing monocyte recruitment to the wounded myocardium of CCR2−/− and CX3CR1−/− C57BL/6 mice (Fig. 3 C). In phase I, Ly-6Chi monocytes accumulated efficiently in infarcts of wild-type and CX3CR1−/− mice; however, we noted a >50-fold decrease of Ly-6Chi monocytes in infarcts of CCR2−/− mice, indicating that early Ly-6Chi monocyte accumulation depends on CCR2, but not CX3CR1. Notably, Ly-6Chi monocytes have impaired ability to leave the bone marrow of CCR2−/− mice (35, 36); however, on day 1 after MI, we observed only a 3.3-fold decrease in the number of Ly-6Chi monocytes in peripheral blood of CCR2−/− mice compared with wild-type mice; thus, decreased numbers of Ly-6Chi monocytes in infarcts result at least in part from impaired recruitment of peripheral blood monocytes. In phase II, Ly-6Chi monocytes remained in low numbers in infarcts of either wild-type, CCR2−/−, or CX3CR1−/− mice, as expected. Ly-6Clo monocytes were also found in low numbers during phase I in all mice tested. In phase II, Ly-6Clo monocytes accumulated efficiently in infarcts of wild-type and CCR2−/− mice; however, we noted a sixfold decrease of Ly-6Clo monocytes in infarcts of CX3CR1−/− mice indicating that late Ly-6Clo monocyte accumulation depends on CX3CR1, but not CCR2. Notably, we observed only a 0.9-fold decrease of circulating Ly-6Clo monocytes (and a 0.02-fold decrease of total monocytes) in CX3CR1−/− mice compared with wild-type mice on day 7 after MI, further indicating that Ly-6Clo monocyte accumulation in infarcts at this time point is dependent on CX3CR1. Our data, however, do not exclude a role of CX3CR1 in cell survival. Altogether, these data indicate sequential recruitment of monocyte subsets to the infarct through coordinated orchestration of chemokines and their cognate receptors. These data also show that Ly-6Chi→Ly-6Clo conversion in tissue is unlikely because Ly-6Chi monocytes are nearly absent in infarcts of CCR2−/− mice in phase I, although Ly-6Clo monocytes are present in phase II, and Ly-6Chi monocytes efficiently accumulate in infarcts of CX3CR1−/− mice in phase I, whereas Ly-6Clo monocytes do not in phase II.
Figure 3.

Sequential recruitment of Ly-6Chi and -6Clo monocytes depends on CCR2 and CX3CR1, respectively. (A) RT-PCR expression profile of MCP-1, MIP-1α, fractalkine, and VCAM-1 in the heart tissue before MI and during phase I (day 1) and II (day 4). (B) Monocyte subset expression profile of CCR2, CCR5, CX3CR1 (for review see reference [29]), and VLA-4 (as assessed by flow cytometry). (C) Number of Ly-6Chi and -6Clo monocytes in infarcts of wild-type, CCR2−/−, and CX3CR1−/− mice in phase I (day 1) and II (day 7). Numbers are normalized to milligrams of tissue. The mean and the SEM are shown. n = 3–5. *, P < 0.05.
Monocyte subsets exhibit differential functional properties
Having determined that Ly-6Chi and -6Clo monocytes sequentially enter the infarct, we next investigated whether these two subsets already commit for specific functions. Mice subjected to coronary ligation received intravenous injections of various molecular imaging agents 1–7 d after MI to determine phagocytosis and proteinase activity in vivo (22, 37, 38). Probes included fluorescent nanoparticles (CLIO-VT680) that are efficiently ingested by phagocytes, and activatable fluorescent sensors reporting either on cathepsin B, L, S (Prosense-680) or matrix metalloproteinases (MMPs)-2, -3, -9, and -13 (MMPsense-680) activities. 1 d later, we analyzed monocytes freshly isolated from infarcts (Fig. 4). Both Ly-6Chi and -6Clo monocytes exhibited equal phagocytic capacity in vivo. However, Ly-6Chi, but not Ly-6Clo, monocytes showed high proteinase activity, a process involved in the breakdown of extracellular matrix. Ly-6Chi, but not -6Clo, monocytes also expressed the proinflammatory cytokine TNF-α. Ly-6Chi monocytes retrieved at different time points (days 1, 4, and 7) all produced TNF-α at comparable levels (unpublished data). Conversely, Ly-6Clo monocytes selectively expressed higher levels of vascular endothelial growth factor (VEGF). Thus, Ly-6Chi monocytes exhibit proteolytic and inflammatory functions, whereas circulating Ly-6Clo monocytes have attenuated inflammatory and proangiogenic properties. The same phenotype in blood (unpublished data) and infarct for both Ly-6Chi and -6Clo monocytes indicates that these cells commit for specific functions while still in circulation and conserve these functions in the ischemic myocardium.
Figure 4.

Ly-6Chi and -6Clo monocytes commit for different functions. Relative phagocytic activity, MMP activity, cathepsin activity, TNF-α production (with or without stimulation with PMA/ionomycin), and VEGF expression in Ly-6Chi monocytes (shaded histogram), Ly-6Clo monocytes (black line), and control CD11b− cells (gray line) retrieved from the ischemic myocardium. The mean and the SEM are shown. n = 3–5. *, P < 0.05; **, P < 0.01.
In vivo relevance of the biphasic response to healing
The sequential recruitment of monocyte subsets to the infarct, combined with their differential functional properties in circulation, suggests differential roles in healing. Because both subsets are phagocytic, and because each phase occurs in a defined time frame, we elected to investigate the impact of each phase on healing by depleting circulating monocytes with clodronate-loaded liposomes (Clo-Lip). Intravenous injection of Clo-Lip depletes circulating monocytes with a nearly complete repopulation of the blood monocyte pool after 5–7 d (21, 27). Use of this technique can therefore define how kinetics and function of monocyte subsets coordinate healing. Depletion of circulating monocytes during phase I involved Clo-Lip injection immediately after MI; depletion during phase II involved Clo-Lip injection on day 3. Flow cytometry of infarct cell suspensions at the end of each phase showed a profound reduction of monocytes/macrophages, confirming that the injured myocardium actively recruits circulating monocytes (Fig. 5, A and B).
Figure 5.

In vivo relevance of the biphasic response to healing. (A) Representative dot plots from individual mice depict Ly-6Chi monocytes (bottom right), Ly-6Clo monocytes (bottom left), and macrophages/dendritic cells (top left) at the infarct after depletion of circulating monocytes with clodronate-loaded liposomes (Clo-Lip). Mice were analyzed on day 4 (Clo-Lip injection on day 0; depletion of phase I) and on day 7 (Clo-Lip injection on day 3; depletion of phase II). Control animals (Ø) did not receive Clo-Lip. Percentages of cells are shown as the mean ± the SEM. (B) Total number of monocytes per milligram of tissue at the infarct before MI, at the end of phase I (day 4) and during phase II (day 7), in the absence (–) or presence (+) of Clo-Lip. The mean and the SEM are shown. n = 3–5. (C) Immunohistochemical analysis 7 d after MI depicts representative infarct sections from undepleted (Ø), phase I–depleted (I), and phase II–depleted (II) C57BL/6 mice. Representative sections stained with anti–Mac-3, anti–NIMP-R14, Masson, α-actin, PSR, and anti-CD31 are shown. The mean and the SEM are shown. n = 7. (D) Immunohistochemistry depicts representative infarct sections from apoE−/− mice 7 d after MI. The mean and the SEM are shown. n = 5. *, P < 0.05; **, P < 0.01. Bars: (Mac-3, NIMP-R14, α-actin, and CD31) 20 μm; (PSR and Masson) 100 μm.
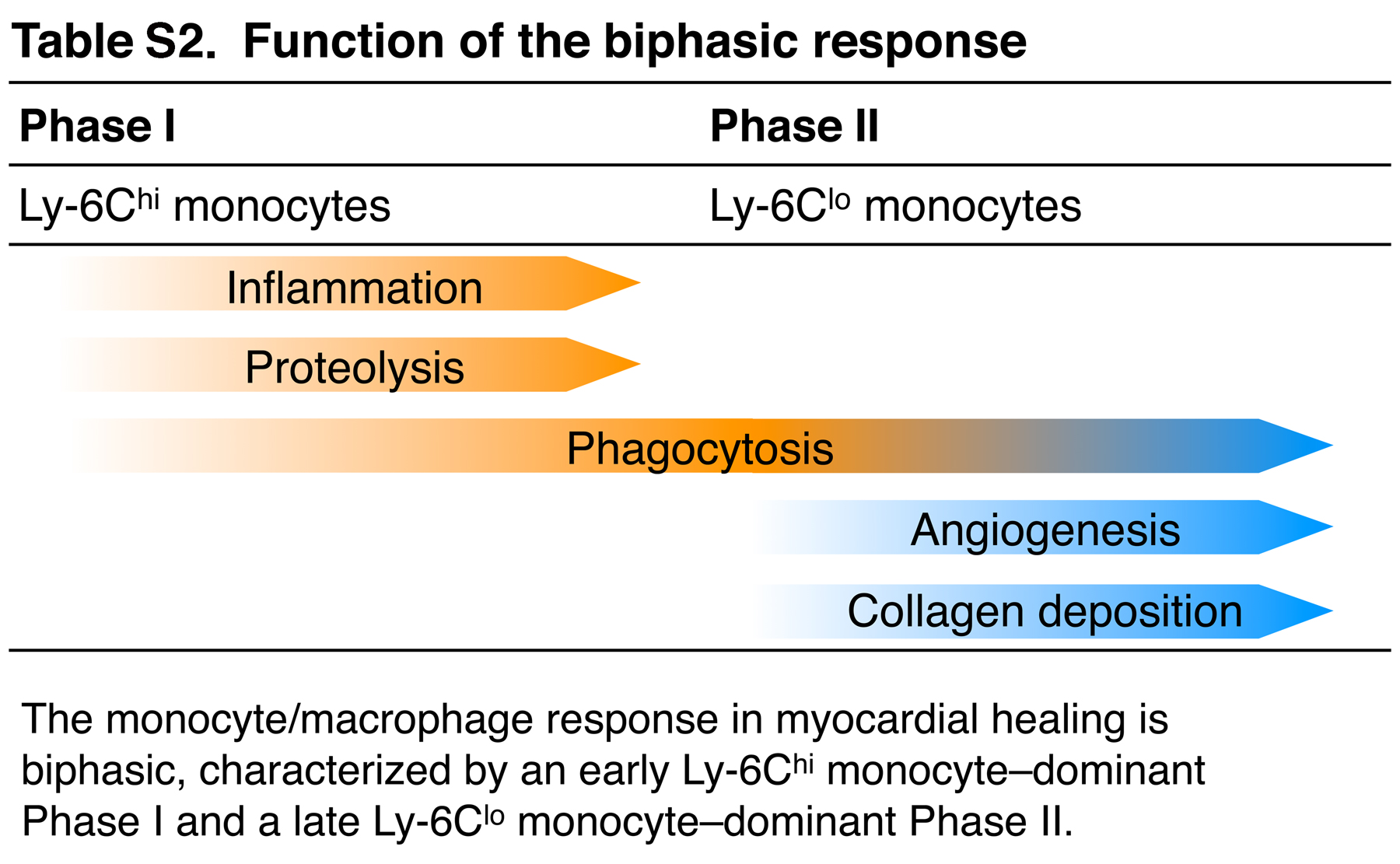
We then assessed the relevance of each phase to healing by evaluating cellular and molecular mediators in the infarct 7 d after MI (Fig. 5 C). Immunohistochemical analysis confirmed decreased numbers of Mac-3+ monocytes/macrophages in infarcts of phase I and II–depleted animals. Depletion of phase I, but not phase II, resulted in larger areas of debris and necrotic tissue, as indicated by Masson trichrome staining. Thus, removal of debris requires phase I and its accompanying influx of proteolytic and phagocytic Ly-6Chi monocytes immediately after coronary artery ligation. The increased numbers of NIMP-R14+ neutrophils in phase I–depleted animals further indicates delayed or inefficient removal of apoptotic cells and necrotic tissue (Fig. 5 C). Conversely, the hearts of mice that developed a native Ly-6Chi response in phase I, but that received Clo-Lip in phase II, showed decreased deposition of collagen as well as reduced numbers of microvascular α-actin+ smooth muscle cells and CD31+ endothelial cells. Thus, granulation tissue formation and remodeling requires noninflammatory, proangiogenic Ly-6Clo monocytes during phase II (Fig. 5 C). Of note, phase I–depleted animals also showed altered granulation tissue formation on day 7, underscoring that phase I and II must occur in sequence. Thus, the Ly-6Chi–dominant phase I involves proteolysis, inflammation, and phagocytosis for degradation and scavenging of damaged tissue, whereas the Ly-6Clo–dominant phase II promotes myofibroblast accumulation, angiogenesis, and deposition of collagen necessary for granulation tissue formation (Table S2, available at http://www.jem.org/cgi/content/full/jem.20070885/DC1).
Atherosclerosis causes most MIs in humans. Leukocyte counts rise after clinical MI, and acute monocytosis is independently associated with aneurysm formation, left ventricular dilation, and portends poor survival (39, 40). Because apolipoprotein E–deficient (apoE−/−) mice develop atherosclerosis, hyperlipidemia, and peripheral blood Ly-6Chi monocytosis when fed a diet high in fat and cholesterol (31, 33, 41, 42), we investigated infarct healing in aged apoE−/− mice. Increased immunoreactive Mac-3 staining reflected increased monocyte/macrophage recruitment to the wound, which is in accordance with studies that documented elevated peripheral blood monocyte counts in these mice (31, 33). The persistence of large areas of debris and necrotic tissue detected by Masson trichrome staining, however, indicated impaired phagocytosis. Anti-CD31 staining detected increased numbers of microvessels in apoE−/− mice, but generation of new extracellular matrix, as detected by diminished immunoreactive staining for α-actin and decreased staining with picrosirius red (PSR), was impaired (Fig. 5 D). The broad deregulation of wound healing in apoE−/− mice, as indicated by these results, underscores the requirement for a balanced and coordinated monocyte response, and raises the possibility that patients with atherosclerosis and monocytosis likewise have impaired capacity for efficient myocardial healing. Impaired healing may result from increased numbers of Ly-6Chi monocytes mobilized in infarcts that prevent/delay the initiation of phase II, or by altered functions of Ly-6Chi and/or -6Clo monocytes that directly compromise healing of the myocardium.
DISCUSSION
This study tested the hypothesis that distinct monocyte subtypes regulate left ventricular healing after MI, and documents a biphasic monocyte response to myocardial ischemic injury. Ly-6Chi monocytes accumulate via CCR2, dominate at the site of injury during the first 3 d (phase I), and scavenge necrotic debris by the combination of inflammatory mediator expression, proteolysis, and phagocytosis. Between 4 and 7 d after infarction (phase II), Ly-6Clo monocytes accumulate preferentially via CX3CR1 and promote reparative processes such as angiogenesis and extracellular matrix deposition, the classical features of granulation tissue formation. These observations enrich our mechanistic understanding of how sequential mobilization of neutrophils, monocyte/macrophages, and fibroblasts contribute to efficient healing; moreover, as a consequence of monocyte/macrophage heterogeneity, these findings reveal distinct kinetic and functional properties of monocyte subsets. The biphasic response may have broad biological relevance beyond MI, especially because monocyte subsets commit for specific function while still in circulation.
Previously, we investigated the role of monocytes in atherosclerosis (31, 43), a disease that is initiated and perpetuated by chronic inflammation (44). Continued feeding of a diet high in fat and cholesterol leads to progressive hypercholesterolemia-associated monocytosis, during which Ly-6Chi monocytes continuously expand in the blood, actively accumulate in atheromata, and differentiate into macrophages, whereas Ly-6Clo monocytes infiltrate lesions less frequently (31, 33). Inflammation that follows myocardial infarction, however, is acute and resolves within a few weeks. In this context, Ly-6Chi and -6Clo monocytes infiltrate tissue sequentially. The noninflammatory and proangiogenic properties of Ly-6Clo monocytes and their role in myocardial healing suggest that these cells actively participate in terminating inflammation. Whether these properties can be manipulated therapeutically in vivo, e.g., by obliterating Ly-6Chi monocytes, harnessing Ly-6Clo monocytes, or promoting Ly-6Chi → Ly-6Clo monocyte conversion, remains to be determined. The chemokine/chemokine receptor signature that accompanies recruitment of monocyte subsets in each phase may also constitute a therapeutic target; indeed, CCR2 antagonists are currently in clinical trials for the treatment of several chronic diseases. The possibility of selectively targeting subsets may give rise to monocyte-based therapeutics for chronic inflammatory conditions such as atherosclerosis, as well as for improvement of healing after acute myocardial infarction.
This study points to an active role for circulating Ly-6Clo monocytes during healing and argues against in situ conversion as a dominant mechanism by which Ly-6Clo cells appear in the infarct. Data in support of active recruitment of Ly-6Clo cells include the following: (a) efficient accumulation of adoptively transferred Ly-6Clo monocytes in infarcts in phase II; (b) the absence of Ly-6Chi monocytes in phase I, but presence of Ly-6Clo monocytes in phase II, in infarcts of CCR2−/− mice; and (c) presence of Ly-6Chi monocytes in phase I, but absence of Ly-6Clo monocytes in phase II, in infarcts of CX3CR1−/− mice. In contrast, according to a proposed model of the response to skeletal muscle injury by injection of a neurotoxin, Ly-6Chi, but not Ly-6Clo, monocytes accumulate in injured tissue and give rise locally to Ly-6Clo F4/80hi macrophages (45). The repair response and the ensuing recruitment of monocytes might differ in these two models. Also, we studied tissue recruitment of circulating Ly-6Clo monocytes at both early and late time points and identified mobilization of these cells only late in infarcts.
The near-absence of monocytes in infarct tissue when healing ends suggests that long-term retention is rare. Further studies need to identify whether monocytes die locally or emigrate from tissue and function beyond initial recruitment. Perhaps monocytes recently emigrated from sites of myocardial infarction can accumulate in lymphoid tissue and orchestrate adaptive immune responses. Because monocytes remain relatively undifferentiated in infarcts, they may conserve functional plasticity beyond tissue education, and commit only later into either macrophages or dendritic cells.
Elevated leukocyte counts, including neutrophilia and monocytosis, predict prognosis after MI (39, 40, 46–51). Independent of infarct size, patients with blood monocytosis display left ventricular dilatation, impaired ejection fraction, and will more likely develop aneurysms at a 6-mo followup (39, 40). These clinical findings, and the observation in hypercholesterolemic mice that peripheral monocytosis and deregulated monocyte accumulation in infarcting myocardium leads to impaired healing, underscores the need to explore the relevance of myocardial monocyte heterogeneity in the human.
Overall, this study identifies a mechanism of orchestrated mobilization of monocyte subtypes, explores the functional consequence of monocyte heterogeneity in infarcting myocardium, and proposes that monocyte-based therapeutics harness selective targeting of subsets. These observations illuminate some of the cellular and molecular mechanisms that underlie the sequence of events involved in healing of myocardial infarction; this sequence was first described at a morphological level in the 1930s (52), is fundamental to ventricular remodeling, and greatly contributes to both acute and chronic complications of MI.
MATERIALS AND METHODS
Myocardial infarction in mice.
A total of 136 female C57BL6 mice (The Jackson Laboratory), 5 apoE−/− C57BL/6 mice (The Jackson Laboratory) consuming Western diet (21.2% fat/weight; 0.2% cholesterol; Harlan Teklad, Inc.), 10 CCR2−/− C57BL/6 mice, and 9 CX3CR1−/− C57BL/6 mice were used in this study. Mice were 6–8 wk old, with the exception of apoE−/− mice that were 1 yr old. Myocardial infarction was induced by coronary ligation (53). Mice were intubated and ventilated with isoflurane supplemented with oxygen (isoflurane 1–2% vol/vol + 2 liters O2). The chest wall was shaved and a thoracotomy was performed in the fourth left intercostal space. The left ventricle was visualized, and the left coronary artery was permanently ligated with a monofilament nylon 8–0 suture at the site of its emergence from under the left atrium. The thoracotomy was closed with sutures and superglue. The institutional subcommittee on research animal care at Massachusetts General Hospital approved all animal studies.
Cells.
Mice were killed 3 h and on days 1–5, 7, and 16 after myocardial infarction (n = 3–10 mice per time point). Peripheral blood was drawn via cardiac puncture with citrate solution (100 mM Na-citrate, 130 mM glucose, pH 6.5), as anti-coagulant and mononuclear cells were purified by density centrifugation (43). Total blood leukocyte numbers were determined using acetic acid lysis solution (3% HEMA 3 Solution II, 94% ddH2O, and 3% glacial acetic acid). Spleens were removed, triturated in HBSS (Mediatech, Inc.) at 4°C with the end of a 3-ml syringe, and filtered through nylon mesh (BD Biosciences). The cell suspension was centrifuged at 300 g for 10 min at 4°C. Red blood cells were lysed with ACK lysis buffer, and the splenocytes were washed with HBSS and resuspended in HBSS supplemented with 0.2% (wt/vol) BSA and 1% (wt/vol) FCS. Infarct tissue and healthy hearts were harvested, minced with fine scissors, and placed into a cocktail of collagenase I, collagenase XI, DNase I, and hyaluronidase (Sigma-Aldrich) and shaken at 37°C for 1 h, as previously described (54). Cells were then triturated through nylon mesh and centrifuged (15 min, 500 g, 4°C). Total spleen and cardiac cell numbers were determined with Trypan blue (Mediatech, Inc.). The resulting single-cell suspensions were washed with HBSS supplemented with 0.2% (wt/vol) BSA and 1% (wt/vol) FCS. For morphologic characterizations, sorted cells were spun, resuspended in PBS, and prepared on slides by cytocentrifugation (Shandon, Inc.) at 10 g for 2 min, and stained with HEMA-3 (Thermo Fischer Scientific).
mAbs and flow cytometry.
The following antibodies were used: anti–CD90-PE, 53–2.1 (BD Biosciences), –B220-PE, RA3-6B2 (BD Biosciences),–CD49b-PE, DX5 (BD Biosciences), –NK1.1-PE, PK136 (BD Biosciences), –Ly-6G-PE, 1A8 (BD Biosciences), –Ly-6G-FITC, 1A8 (BD Biosciences), CD11b-APC, M1/70 (BD Biosciences), –F4/80-biotin, C1:A3-1 (AbD Serotec), –F4/80-FITC, C1:A3-1 (BioLegend), –CD11c-biotin, HL3 (BD Biosciences), –CD11c-FITC, HL3 (BD Biosciences), –I-Ab-biotin, AF6-120.1 (BD Biosciences), –I-Ab-FITC, AF6-120.1 (BD Biosciences), –Ly-6C-FITC, AL-21 (BD Biosciences), –Ly-6C-biotin, AL-21 (BD Biosciences), TNF-α-FITC, MP6-XT22 (AbD Serotec), anti-CD43-FITC, S7 (BD Biosciences), –CD62L-FITC, MEL-14 (BD Biosciences), –CD68-FITC, FA-11 (AbD Serotec), –CD86-biotin, GL1 (BD Biosciences), –CD115-PE, 604B5-2E11 (AbD Serotec), –Mac-3-FITC, M3/84 (BD Biosciences), –Gr-1-PeCy7, RB6-8C5 (BD Biosciences). Strep-PerCP (BD Biosciences) was used to label biotinylated antibodies. Monocytes were identified as CD11bhi (CD90/B220/CD49b/NK1.1/Ly-6G)lo (F4/80/I-Ab/CD11c)lo Ly-6Chi/lo. Macrophages/dendritic cells were identified as CD11bhi (CD90/B220/CD49b/NK1.1/Ly-6G)lo (F4/80/I-Ab/CD11c)hi Ly-6Clo. Neutrophils were identified as CD11bhi (CD90/B220/CD49b/NK1.1/Ly-6G)hi (F4/80/I-Ab/CD11c)lo Ly-6Cint. Monocyte and macrophage/dendritic cell numbers were calculated as the total cells multiplied by the percentage of cells within the monocyte/macrophage gate. Within this population, monocyte subsets were identified as (F4/80/I-Ab/CD11c)lo and either Ly-6Chi or Ly-6Clo. For calculation of total cell numbers in the heart, normalization to weight of infarct was performed. For assessment of proteolytic activity including cathepsins (22), (Prosense-680, excitation wavelength 680 ± 10 nm, emission 700 ± 10 nm, VisEn Medical) and matrix metalloproteinases 2, 3, 9, 13 (55) (MMPsense-680, excitation wavelength 680 ± 10 nm, emission 700 ± 10 nm, VisEn Medical), 5 nmol of probe in 150 μl PBS were injected into the tail vein 24 h before sacrifice. For assessment of phagocytic activity, CLIO-VT680 (excitation wavelength 680 ± 10 nm, emission 700 ± 10 nm, CMIR chemistry core), 15 mg of Fe/kg bodyweight (22), was injected into the tail vein 24 h before sacrifice. Data were acquired on an LSRII (BD Biosciences) with 685/LP and 695/40-filter configuration to detect VT680. For intracellular staining of VEGF-(AAM51B)-biotin-Strep-PerCP (AbD Serotec) and TNF-α-FITC (MP6-XT22), blood cells were permeabilized and fixed with a Cytofix/Cytoperm kit (BD Biosciences). For TNF-α staining, cells were stimulated for 5 h with 1 μg/ml PMA and 0.25 μg/ml ionomycin, and in the presence of 10 μg/ml Brefeldin A after 1 h (Sigma-Aldrich). For depletion of circulating monocytes (27), mice were injected i.v. with 0.1 ml of dichloromethylene-bisphosphonate (clodronate; Sigma-Aldrich) liposomes immediately after MI (day 0) to deplete phase I and, on day 3 after MI, to deplete phase II. Clodronate was incorporated into liposomes, as previously described (56).
Monocyte subtype tracking.
Ly-6Chi or -6Clo monocytes were sorted from the spleen of C57BL/6 mice on a FACSAria (BD Biosciences) and labeled with 111In-oxine according to the manufacturer's protocol (GE Healthcare). In brief, cells were washed with HBSS, spun, and resuspended in 111In-oxine for 15 min at 37°C, pH 6.5–7.5, and washed twice with HBSS. 111In-oxine is a widely used, FDA-approved agent that is fully biocompatible as it alters neither cell survival nor function at the doses used (31, 43, 57, 58); hence, we considered labeled monocytes to behave similarly to unlabeled monocytes. Approximately 2 × 105 Ly-6Chi or -6Clo monocytes incorporating 20 μCi were injected independently i.v. into C57BL/6 on day 0 (phase I) or 4 d (phase II) after MI. The total amount of activity injected into each animal was measured with a radioisotope calibrator (Capintec, Inc.). After 24 h, recipient animals were killed with CO2 and hearts were collected. cpm for each heart were measured using a 1480 Wizard Wallac Gamma Counter (PerkinElmer Life and Analytical Sciences, Inc.). Percent-injected dose per gram tissue (%ID/g) was calculated after correcting for decay, excretion, and tail radioactivity from occasional subtle extravasations. Accumulation was normalized to relative contribution of Ly-6Chi and -6Clo monocytes in blood at the indicated times.
Histopathological analysis.
Histopathology for assessment of healing was performed in mice killed 7 d after MI for the following groups: wild-type C57BL/6 mice, wild-type mice depleted with clodronate-loaded liposomes immediately after coronary ligation, wild-type mice depleted with clodronate-loaded liposomes 3 d after MI, and apoE−/− C57BL6 mice. Hearts were excised and rinsed in PBS and embedded in OCT (Sakura Finetek). Serial 6-μm thick sections were used for immunohistochemical staining for neutrophils (NIMP-R14, Abcam), monocytes/macrophages (Mac-3, M3/84; BD PharMingen), myofibroblasts (α-actin, RB-9010-PO; Neomarkers), and endothelial cells forming capillaries (CD31, MEC13.3; BD Biosciences). Reaction was visualized as a three-step staining procedure in combination with biotinylated secondary antibodies (BA4001; Vector Laboratories) and AEC Substrate kit (Vector Laboratories). To analyze the collagen content of the scar, sections were stained with PSR. Collagen content was visualized with polarized light (59). Necrotic debris was analyzed on Masson trichrome–stained sections. Neutrophils, macrophages, and myofibroblasts (magnification 400×) and collagen content and debris (magnification 200×) were quantified using five random fields per section and per animal with IP Lab Software (Scanalytics). The percent positive stained area was calculated in relation to the whole visual field.
Real-time RT-PCR.
Primer sequences and probes for MCP-1 (forward, GGCTCAGCCAGATGCAGTTAA; reverse, CCTACTCATTGGGATCATCTTGCT; probe, CCCCACTCACCTGCTGCTACTCATTCA [Fam-Tamra]); MIP-1α (Fwd: TCTTCTCAGCGCCATATGGA, Rev: CGTGGAATCTTCCGGCTGTA, Probe: CTGACACCCCGACTGCCTGCTG (Fam-Tamra)), Fractalkine (forward, CCAGGGTCCTTTCCTGTCTCT; reverse, TGGAGAATGCTATGGGAGTTCTG; probe, TTCACGCCAAGGACTCACCTTTGCC [Fam-Tamra]), and VCAM-1 (forward, TGTAAAAGGAAAAGAACATAACAAGGACTA; reverse, ATGGCAGGTATTACCAAGGAAGAT; probe, TTTTCGCCCGAACTCCTTGCACTC [Fam-Tamra]) were designed with PrimerExpress (ABI). Standard primers and probes for CCR2 and CX3CR1 were purchased from ABI. TriZol (Invitrogen) was used to isolate total RNA from infarct tissue samples and Oligo(dT) primers to reversely transcribe mRNA into cDNA following the manufacturer's guidelines (SuperScript; Invitrogen). Quantitative PCR was performed on an SDS 7000 system (ABI) using ABgene QPCR Rox Mix (Epsom) and standard cycling conditions, and cDNA content of samples was calculated using the standard curve method. Every sample was run in triplicate, and at least three samples per group were assessed. β-Actin was chosen as an internal calibrator.
Statistics.
Results were expressed as the mean ± the SEM. Statistical tests included unpaired, two-tailed Student's t test using Welch's correction for unequal variances and one-way analysis of variance, followed by Bonferroni's multiple comparison test. A P value of 0.05 or less was considered to denote significance.
Online supplemental material.
Fig. S1 shows that monocytes accumulate in the myocardium after MI. Fig. S2 shows the biphasic Ly-6Chi:Ly-6Clo response pertains irrespective of monocyte differentiation. Fig. S3 shows that splenic and peripheral blood monocytes show comparable phenotypes. Table S1 shows that adoptively transferred labeled Ly-6Chi compete with he endogenous pool. Table S2 shows the function of the biphasic response. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20070885/DC1.
Supplemental Material
Acknowledgments
This work was supported in part by National Institutes of Health grants U01 HL080731, P01-A154904 and R24 CA69246 (to R. Weissleder), R01HL34636 (to P. Libby) and the Donald W. Reynolds Cardiovascular Clinical Research Center grant on Atherosclerosis at Harvard Medical School (to R. Weissleder and P. Libby). F.K. Swirski was supported by a post-doctoral fellowship from the American Heart Association (0525866T). M.J. Pittet was supported by the Human Frontier Science Program Organization (LT00369/2003). The authors would like to thank Drs. Barrett Rollins (Department of Medical Oncology, Dana-Farber Cancer Institute), Andrew Luster and Joseph Elhkoury (Center for Immunology and Inflammatory Diseases, Massachusetts General Hospital and Harvard Medical School) and Israel Charo (Gladstone Institute of Cardiovascular Disease, University of California San Francisco) for providing CCR2−/− and CX3CR1−/− mice. We also thank Michael Waring (Harvard Medical School Center for AIDS Research Immunology Core at MGH and Harvard Medical School) for sorting cells, the Molecular Imaging Program (MIP-0222) for all mouse surgeries, Dr. Hanwen Zhang for assistance with radioactive cell labeling, Vincent Lok for technical assistance with immunohistochemistry, and Melissa Carlson for secretarial assistance (Center for Molecular Imaging Research, Massachusetts General Hospital and Harvard Medical School). The authors have no conflicting financial interests.
Abbreviations used: apoE, apolipoprotein E; MI, myocardial infarction; PSR, picrosirius red; VEGF, vascular endothelial growth factor.
M. Nahrendorf and F.K. Swirski contributed equally to this paper.
References
- 1.Sutton, M.G., and N. Sharpe. 2000. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 101:2981–2988. [DOI] [PubMed] [Google Scholar]
- 2.Blankesteijn, W.M., E. Creemers, E. Lutgens, J.P. Cleutjens, M.J. Daemen, and J.F. Smits. 2001. Dynamics of cardiac wound healing following myocardial infarction: observations in genetically altered mice. Acta Physiol. Scand. 173:75–82. [DOI] [PubMed] [Google Scholar]
- 3.Cleutjens, J.P., W.M. Blankesteijn, M.J. Daemen, and J.F. Smits. 1999. The infarcted myocardium: simply dead tissue, or a lively target for therapeutic interventions. Cardiovasc. Res. 44:232–241. [DOI] [PubMed] [Google Scholar]
- 4.Ertl, G., and S. Frantz. 2005. Healing after myocardial infarction. Cardiovasc. Res. 66:22–32. [DOI] [PubMed] [Google Scholar]
- 5.Frangogiannis, N.G., C.W. Smith, and M.L. Entman. 2002. The inflammatory response in myocardial infarction. Cardiovasc. Res. 53:31–47. [DOI] [PubMed] [Google Scholar]
- 6.Jugdutt, B.I. 2003. Ventricular remodeling after infarction and the extracellular collagen matrix: when is enough enough? Circulation. 108:1395–1403. [DOI] [PubMed] [Google Scholar]
- 7.Olivetti, G., J.M. Capasso, E.H. Sonnenblick, and P. Anversa. 1990. Side-to-side slippage of myocytes participates in ventricular wall remodeling acutely after myocardial infarction in rats. Circ. Res. 67:23–34. [DOI] [PubMed] [Google Scholar]
- 8.Spinale, F.G. 2002. Matrix metalloproteinases: regulation and dysregulation in the failing heart. Circ. Res. 90:520–530. [DOI] [PubMed] [Google Scholar]
- 9.Whittaker, P., D.R. Boughner, and R.A. Kloner. 1991. Role of collagen in acute myocardial infarct expansion. Circulation. 84:2123–2134. [DOI] [PubMed] [Google Scholar]
- 10.Pfeffer, J.M., M.A. Pfeffer, and E. Braunwald. 1985. Influence of chronic captopril therapy on the infarcted left ventricle of the rat. Circ. Res. 57:84–95. [DOI] [PubMed] [Google Scholar]
- 11.Pfeffer, M.A., and J.M. Pfeffer. 1987. Ventricular enlargement and reduced survival after myocardial infarction. Circulation. 75:IV93–IV97. [PubMed] [Google Scholar]
- 12.Pfeffer, M.A., and E. Braunwald. 1990. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 81:1161–1172. [DOI] [PubMed] [Google Scholar]
- 13.Nahrendorf, M., F. Wiesmann, K.H. Hiller, K. Hu, C. Waller, J. Ruff, T.E. Lanz, S. Neubauer, A. Haase, G. Ertl, and W.R. Bauer. 2001. Serial cine-magnetic resonance imaging of left ventricular remodeling after myocardial infarction in rats. J. Magn. Reson. Imaging. 14:547–555. [DOI] [PubMed] [Google Scholar]
- 14.Ducharme, A., S. Frantz, M. Aikawa, E. Rabkin, M. Lindsey, L.E. Rohde, F.J. Schoen, R.A. Kelly, Z. Werb, P. Libby, and R.T. Lee. 2000. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J. Clin. Invest. 106:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heymans, S., A. Luttun, D. Nuyens, G. Theilmeier, E. Creemers, L. Moons, G.D. Dyspersin, J.P. Cleutjens, M. Shipley, A. Angellilo, et al. 1999. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat. Med. 5:1135–1142. [DOI] [PubMed] [Google Scholar]
- 16.Dewald, O., P. Zymek, K. Winkelmann, A. Koerting, G. Ren, T. Abou-Khamis, L.H. Michael, B.J. Rollins, M.L. Entman, and N.G. Frangogiannis. 2005. CCL2/monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ. Res. 96:881–889. [DOI] [PubMed] [Google Scholar]
- 17.Hayashidani, S., H. Tsutsui, T. Shiomi, M. Ikeuchi, H. Matsusaka, N. Suematsu, J. Wen, K. Egashira, and A. Takeshita. 2003. Anti-monocyte chemoattractant protein-1 gene therapy attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation. 108:2134–2140. [DOI] [PubMed] [Google Scholar]
- 18.Kaikita, K., T. Hayasaki, T. Okuma, W.A. Kuziel, H. Ogawa, and M. Takeya. 2004. Targeted deletion of CC chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. Am. J. Pathol. 165:439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leor, J., L. Rozen, A. Zuloff-Shani, M.S. Feinberg, Y. Amsalem, I.M. Barbash, E. Kachel, R. Holbova, Y. Mardor, D. Daniels, et al. 2006. Ex vivo activated human macrophages improve healing, remodeling, and function of the infarcted heart. Circulation. 114:I94–100. [DOI] [PubMed] [Google Scholar]
- 20.Maekawa, Y., T. Anzai, T. Yoshikawa, Y. Sugano, K. Mahara, T. Kohno, T. Takahashi, and S. Ogawa. 2004. Effect of granulocyte-macrophage colony-stimulating factor inducer on left ventricular remodeling after acute myocardial infarction. J. Am. Coll. Cardiol. 44:1510–1520. [DOI] [PubMed] [Google Scholar]
- 21.van Amerongen, M.J., M.C. Harmsen, N. van Rooijen, A.H. Petersen, and M.J. van Luyn. 2007. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am. J. Pathol. 170:818–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nahrendorf, M., D.E. Sosnovik, P. Waterman, F.K. Swirski, A.N. Pande, E. Aikawa, J.L. Figueiredo, M.J. Pittet, and R. Weissleder. 2007. Dual channel optical tomographic imaging of leukocyte recruitment and protease activity in the healing myocardial infarct. Circ. Res. 100:1218–1225. [DOI] [PubMed] [Google Scholar]
- 23.Morimoto, H., M. Takahashi, A. Izawa, H. Ise, M. Hongo, P.E. Kolattukudy, and U. Ikeda. 2006. Cardiac overexpression of monocyte chemoattractant protein-1 in transgenic mice prevents cardiac dysfunction and remodeling after myocardial infarction. Circ. Res. 99:891–899. [DOI] [PubMed] [Google Scholar]
- 24.Roberts, R., V. DeMello, and B.E. Sobel. 1976. Deleterious effects of methylprednisolone in patients with myocardial infarction. Circulation. 53:I204–I206. [PubMed] [Google Scholar]
- 25.Libby, P., P.R. Maroko, C.M. Bloor, B.E. Sobel, and E. Braunwald. 1973. Reduction of experimental myocardial infarct size by corticosteroid administration. J. Clin. Invest. 52:599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geissmann, F., S. Jung, and D.R. Littman. 2003. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 19:71–82. [DOI] [PubMed] [Google Scholar]
- 27.Sunderkotter, C., T. Nikolic, M.J. Dillon, N. Van Rooijen, M. Stehling, D.A. Drevets, and P.J. Leenen. 2004. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J. Immunol. 172:4410–4417. [DOI] [PubMed] [Google Scholar]
- 28.Gordon, S., and P.R. Taylor. 2005. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 5:953–964. [DOI] [PubMed] [Google Scholar]
- 29.Gordon, S. 2007. Macrophage heterogeneity and tissue lipids. J. Clin. Invest. 117:89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ziegler-Heitbrock, L. 2007. The CD14+ CD16+ blood monocytes: their role in infection and inflammation. J. Leukoc. Biol. 81:584–592. [DOI] [PubMed] [Google Scholar]
- 31.Swirski, F.K., P. Libby, E. Aikawa, P. Alcaide, F.W. Luscinskas, R. Weissleder, and M.J. Pittet. 2007. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Invest. 117:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lumeng, C.N., J.L. Bodzin, and A.R. Saltiel. 2007. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 117:175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tacke, F., D. Alvarez, T.J. Kaplan, C. Jakubzick, R. Spanbroek, J. Llodra, A. Garin, J. Liu, M. Mack, N. van Rooijen, et al. 2007. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J. Clin. Invest. 117:185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Auffray, C., D. Fogg, M. Garfa, G. Elain, O. Join-Lambert, S. Kayal, S. Sarnacki, A. Cumano, G. Lauvau, and F. Geissmann. 2007. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 317:666–670. [DOI] [PubMed] [Google Scholar]
- 35.Tsou, C.L., W. Peters, Y. Si, S. Slaymaker, A.M. Aslanian, S.P. Weisberg, M. Mack, and I.F. Charo. 2007. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J. Clin. Invest. 117:902–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Serbina, N.V., and E.G.P. Am. 2006. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 7:311–317. [DOI] [PubMed] [Google Scholar]
- 37.Weissleder, R. 2006. Molecular imaging in cancer. Science. 312:1168–1171. [DOI] [PubMed] [Google Scholar]
- 38.Weissleder, R., and V. Ntziachristos. 2003. Shedding light onto live molecular targets. Nat. Med. 9:123–128. [DOI] [PubMed] [Google Scholar]
- 39.Maekawa, Y., T. Anzai, T. Yoshikawa, Y. Asakura, T. Takahashi, S. Ishikawa, H. Mitamura, and S. Ogawa. 2002. Prognostic significance of peripheral monocytosis after reperfused acute myocardial infarction: a possible role for left ventricular remodeling. J. Am. Coll. Cardiol. 39:241–246. [DOI] [PubMed] [Google Scholar]
- 40.Mariani, M., R. Fetiveau, E. Rossetti, A. Poli, F. Poletti, P. Vandoni, M. D'Urbano, F. Cafiero, G. Mariani, C. Klersy, and S. De Servi. 2006. Significance of total and differential leucocyte count in patients with acute myocardial infarction treated with primary coronary angioplasty. Eur. Heart J. 27:2511–2515. [DOI] [PubMed] [Google Scholar]
- 41.Plump, A.S., J.D. Smith, T. Hayek, K. Aalto-Setala, A. Walsh, J.G. Verstuyft, E.M. Rubin, and J.L. Breslow. 1992. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 71:343–353. [DOI] [PubMed] [Google Scholar]
- 42.Zhang, S.H., R.L. Reddick, J.A. Piedrahita, and N. Maeda. 1992. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 258:468–471. [DOI] [PubMed] [Google Scholar]
- 43.Swirski, F.K., M.J. Pittet, M.F. Kircher, E. Aikawa, F.A. Jaffer, P. Libby, and R. Weissleder. 2006. Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc. Natl. Acad. Sci. USA. 103:10340–10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Libby, P. 2002. Inflammation in atherosclerosis. Nature. 420:868–874. [DOI] [PubMed] [Google Scholar]
- 45.Arnold, L., A. Henry, F. Poron, Y. Baba-Amer, N. van Rooijen, A. Plonquet, R.K. Gherardi, and B. Chazaud. 2007. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 204:1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Friedman, G.D., A.L. Klatsky, and A.B. Siegelaub. 1974. The leukocyte count as a predictor of myocardial infarction. N. Engl. J. Med. 290:1275–1278. [DOI] [PubMed] [Google Scholar]
- 47.Barron, H.V., C.P. Cannon, S.A. Murphy, E. Braunwald, and C.M. Gibson. 2000. Association between white blood cell count, epicardial blood flow, myocardial perfusion, and clinical outcomes in the setting of acute myocardial infarction: a thrombolysis in myocardial infarction 10 substudy. Circulation. 102:2329–2334. [DOI] [PubMed] [Google Scholar]
- 48.Cannon, C.P., C.H. McCabe, R.G. Wilcox, J.H. Bentley, and E. Braunwald. 2001. Association of white blood cell count with increased mortality in acute myocardial infarction and unstable angina pectoris. OPUS-TIMI 16 Investigators. Am. J. Cardiol. 87:636–639. [DOI] [PubMed] [Google Scholar]
- 49.Sabatine, M.S., D.A. Morrow, C.P. Cannon, S.A. Murphy, L.A. Demopoulos, P.M. DiBattiste, C.H. McCabe, E. Braunwald, and C.M. Gibson. 2002. Relationship between baseline white blood cell count and degree of coronary artery disease and mortality in patients with acute coronary syndromes: a TACTICS-TIMI 18 (Treat Angina with Aggrastat and determine Cost of Therapy with an Invasive or Conservative Strategy-Thrombolysis in Myocardial Infarction 18 trial)substudy. J. Am. Coll. Cardiol. 40:1761–1768. [DOI] [PubMed] [Google Scholar]
- 50.Kirtane, A.J., A. Bui, S.A. Murphy, H.V. Barron, and C.M. Gibson. 2004. Association of peripheral neutrophilia with adverse angiographic outcomes in ST-elevation myocardial infarction. Am. J. Cardiol. 93:532–536. [DOI] [PubMed] [Google Scholar]
- 51.Kyne, L., J.M. Hausdorff, E. Knight, L. Dukas, G. Azhar, and J.Y. Wei. 2000. Neutrophilia and congestive heart failure after acute myocardial infarction. Am. Heart J. 139:94–100. [DOI] [PubMed] [Google Scholar]
- 52.Mallory, G.K., P.D. White, and J. Salcedo-Salgar. 1939. The speed of healing of myocardial infarction: A study of the pathologic anatomy in seventy-two cases. Am. Heart J. 18:647–671. [Google Scholar]
- 53.Kumar, D., T.A. Hacker, J. Buck, L.F. Whitesell, E.H. Kaji, P.S. Douglas, and T.J. Kamp. 2005. Distinct mouse coronary anatomy and myocardial infarction consequent to ligation. Coron. Artery Dis. 16:41–44. [DOI] [PubMed] [Google Scholar]
- 54.Galkina, E., A. Kadl, J. Sanders, D. Varughese, I.J. Sarembock, and K. Ley. 2006. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J. Exp. Med. 203:1273–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen, J., C.H. Tung, J.R. Allport, S. Chen, R. Weissleder, and P.L. Huang. 2005. Near-infrared fluorescent imaging of matrix metalloproteinase activity after myocardial infarction. Circulation. 111:1800–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van Rooijen, N., and A. Sanders. 1994. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J. Immunol. Methods. 174:83–93. [DOI] [PubMed] [Google Scholar]
- 57.Kraitchman, D.L., M. Tatsumi, W.D. Gilson, T. Ishimori, D. Kedziorek, P. Walczak, W.P. Segars, H.H. Chen, D. Fritzges, I. Izbudak, et al. 2005. Dynamic imaging of allogeneic mesenchymal stem cells trafficking to myocardial infarction. Circulation. 112:1451–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pittet, M.J., J. Grimm, C.R. Berger, T. Tamura, G. Wojtkiewicz, M. Nahrendorf, P. Romero, F.K. Swirski, and R. Weissleder. 2007. In vivo imaging of T cell delivery to tumors after adoptive transfer therapy. Proc. Natl. Acad. Sci. USA. 104:12457–12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Whittaker, P., R.A. Kloner, D.R. Boughner, and J.G. Pickering. 1994. Quantitative assessment of myocardial collagen with picrosirius red staining and circularly polarized light. Basic Res. Cardiol. 89:397–410. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}