

Abstract
Complement is an important component of the innate and adaptive immune response, yet complement split products generated through activation of each of the three complement pathways (classical, alternative, and lectin) can cause inflammation and tissue destruction. Previous studies have shown that complement activation through the alternative, but not classical, pathway is required to initiate antibody-induced arthritis in mice, but it is unclear if the alternative pathway (AP) plays a role in established disease. Previously, we have shown that human complement receptor of the immunoglobulin superfamily (CRIg) is a selective inhibitor of the AP of complement. Here, we present the crystal structure of murine CRIg and, using mutants, provide evidence that the structural requirements for inhibition of the AP are conserved in human and mouse. A soluble form of CRIg reversed inflammation and bone loss in two experimental models of arthritis by inhibiting the AP of complement in the joint. Our data indicate that the AP of complement is not only required for disease induction, but also disease progression. The extracellular domain of CRIg thus provides a novel tool to study the effects of inhibiting the AP of complement in established disease and constitutes a promising therapeutic with selectivity for a single complement pathway.
The complement system is required to mount an appropriate innate immune response to pathogens. It acts by facilitating phagocytosis of immune complexes and apoptotic cells and by forming a membrane attack complex resulting in cell lysis (1). Particles and pathogens in serum initiate complement activation either through the classical pathway (CP), mannose-binding lectin (MBL) pathway, or alternative pathway (AP; reference 2). Central to complement activation are the convertases, enzyme complexes that cleave the substrates C3 and C5 into their biologically active fragments, C3a, C3b, C5a, and C5b. The C3bBb dimer and C3bC3bBb multimers form the convertases of the AP and are required for amplification of complement activated through any of the three pathways, whereas C4bC2a and C3bC4bC2a are convertases of the CP and MBL pathway.
To prevent unwanted complement activation, most mammalian cells are equipped with regulators that block complement amplification on host self cells (3). In the absence of these intrinsic regulators, serum exposure results in the generation of complement split product that in turn facilitates inflammation and tissue damage (4, 5). Noncellular surfaces that lack intrinsic complement regulators are therefore especially prone to complement attack and are fully dependent on protection by soluble complement regulators in serum. Uncontrolled complement activation due to the lack of appropriate complement regulation has been associated with various chronic inflammatory diseases. Dominant in this inflammatory cascade are the complement split products C3a and C5a that function as chemoattractant and activators of neutrophils and inflammatory macrophages via the C3a and C5a receptors (6). Properdin, released from neutrophils, further amplifies the inflammatory cascade through stabilization of the AP convertase (7). Complement activation has been shown to be an important component driving inflammation in immune complex–mediated diseases, such as membranoproliferative glomerulonephritis, nephrotoxic nephritis, and arthritis (1, 8–11).
Mice that lack a functional AP through genetic deletion of factor B do not develop arthritis (12) but so far the effect of blocking the AP in established disease is unknown. Recently, we identified complement receptor of the Ig superfamily (CRIg), a macrophage complement receptor required for pathogen clearance (13). CRIg binds to C3b, the subunit of both CP and AP C5 convertases, and selectively inhibits the C3 and C5 convertases of the AP in vitro by blocking its interaction with the substrates C3 and C5 (14). In the present study, we determined whether the ability to inhibit the AP is conserved in human and mouse CRIg and tested the efficacy of CRIg in two experimental models of arthritis, one in which mice were immunized with bovine collagen type II (collagen-induced arthritis [CIA]) and another model in which antibodies to collagen type II were passively transferred (antibody-induced arthritis [AIA]). The results show that soluble CRIg acts as a potent suppressor of established inflammation in both experimental models, introducing a novel inhibitor of the AP of complement with promising therapeutic applications.
RESULTS AND DISCUSSION
Structure and function of CRIg is conserved in mouse and human
Previously, we have shown that the IgV domain of human CRIg binds to the β chain of C3b, resulting in inhibition of the AP of complement (14). Here, we present the crystal structure of murine CRIg at 1.0 Å resolution. Comparison of the human and mouse CRIg reveals that both proteins are very similar. They share 83% sequence homology within the IgV domain, and they superimpose with a root mean square deviation of 0.37 Å for 110 Cα atoms (Fig. 1 A, left and middle). To determine whether the structural requirements for C3b binding and complement inhibition are similar in human and mouse, we performed alanine substitution of residues M86 and D87 (mutant “A”) or residues H57 and Q59 (mutant “B”; Fig. 1 A, right). These residues form key contact points with the MG3 and MG6 domains of human C3b, respectively (14). Recombinant soluble versions of these mutant proteins, consisting of the extracellular domain of murine CRIg fused to the Fc portion of murine IgG1 (CRIg(A)-Fc and CRIg(B)-Fc), showed a >100-fold loss of binding affinity to C3b when compared with wild-type protein (CRIg-Fc; not depicted). The mutant proteins were folded correctly as shown by similar circular dichroism patterns (not depicted). We next determined if the loss in binding activity translates into a loss of functional activity, i.e., inhibition of the C3 and C5 convertases. Although CRIg-Fc inhibited C3 and C5 convertases of the AP in mouse serum, CRIg(A)-Fc, CRIg(B)-Fc, and the control-Fc proteins lost the ability to inhibit either the C3 or the C5 convertases (Fig. 1 B). The inhibition was selective for the convertases of the AP because CRIg-Fc did not inhibit CP activation in hemolytic assays (Fig. 1 C and Fig. S1, which is available at http://www.jem.org/cgi/content/full/jem.20070432/DC1). Collectively, these results indicate that the structural requirements for binding to and inhibition of the AP convertases, as well as the selective inhibition of the AP, are conserved in human and mouse CRIg.
Figure 1.

Structural requirements for CRIg-Fc inhibition of the AP C3 and C5 convertases in mouse serum. (A) Left: Overlay of human (white) and murine (yellow) CRIg IgV domains. Side chains of residues M86, D87 (red) and H57, and Q59 (blue) are indicated. Middle: Surface representation of murine CRIg crystal structure. Indicated in green are surface-exposed amino acid residues that are not conserved in human and murine CRIg. Right: Surface representation of muCRIg. Indicated are amino acid residues that are substituted by alanine. Red, M86 and D87 (CRIg(A)); blue, H57 and Q59 (CRIg(B)). (B) CRIg-Fc, but not CRIg(A)-Fc, CRIg(B)-Fc, or control Fc-protein, inhibits the generation of C3a des Arg (left) and C5a des Arg (right) in zymosan-activated serum. Zymosan particles were incubated for 45 min at 37°C with 4% mouse serum in the presence of Mg2+ and EGTA. C3a des Arg and C5a des Arg were detected by ELISA and expressed as percentages of values in the absence of recombinant protein. Results are representative of four independent repeats. (C) Murine CRIg-Fc inhibits complement activation through the alternative, but not classical, pathways. Rabbit erythrocytes (left) or IgM-oponized sheep erythrocytes (right) were exposed to C1q-deficient (left) or factor B–depleted (right) human serum in the presence of increasing concentrations of CRIg-Fc or control-Fc protein. Values expressed as percent hemolysis in the absence of inhibitors (mean ± SEM; n = 3).
CRIg inhibits inflammation in established arthritis
We next determined if CRIg-Fc can inhibit inflammation in vivo caused by activation of the AP of complement. We used two models of arthritis in which an intact AP is required for initiation of the disease (12, 15–17). CRIg-Fc treatment after secondary immunization with collagen (day 24) significantly (P < 0.01) reduced clinical scores, inflammation, and bone loss compared with control-Fc–treated mice (Fig. 2, A and D). A recent study has implicated a role for CRIg in Th cell–dependent B cell responses (18). In the CIA studies, systemic muCRIg-Fc did not affect B cell responses (Fig. 2 B), indicating that CRIg-Fc acts on the innate, and not adaptive, arm of the immune response in this disease model. To test whether CRIg-Fc could inhibit already established inflammation, mice were treated with wild-type or control CRIg proteins starting 42 d after primary immunization. CRIg-Fc treatment significantly (P < 0.0001) reduced clinical scores and inhibited inflammation and bone loss compared with treatment with a control fusion protein (Fig. 2, C and D), indicating that the AP of complement is required for progression of joint inflammation and bone destruction in CIA.
Figure 2.

CRIg-Fc inhibits CIA in mice. (A) Clinical scores of mice treated with CRIg-Fc or control-Fc starting 24 d after primary immunization with bovine collagen type II. (B) Anti-collagen antibody titers at day 70 are similar in mice treated with CRIg-Fc and control-Fc fusion proteins. (C) CRIg-Fc treatment inhibits established inflammation. Mice were treated with CRIg-Fc or control-Fc fusion proteins starting on day 42 after primary immunization. (D) CRIg-Fc significantly reduces inflammation (arrow in top left, histology scores in top right) and conserved joint cortical bone volume (JCBV) in the metatarsal-phalangeal and metacarpophalangeal joints (bottom) compared with mice treated with control-Fc. micro-ct renderings show representative mouse hind paws from both groups (bottom left). Histology and micro-ct were performed 70 (treatment (Rx) starting on day 24) or 84 (treatment (Rx) starting on day 42) d after primary immunization. Red arrows in A and C indicate start of treatment. Data are expressed as mean ± SEM; n = 15. *, P < 0.01; **, P < 0.0001, CRIg-Fc versus control-Fc. Bar, 100 μm.
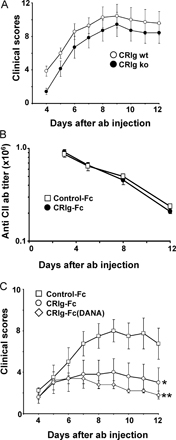
To further determine if CRIg inhibits the effector arm of immune complex–mediated inflammation, a cocktail of anti-collagen antibodies were transferred to mice, bypassing B and T cell–mediated immune responses (19). CRIg-Fc reduced clinical scores and inflammation in this model (Fig. 3, A and B), confirming that CRIg acts independently of T and B cell activity and that the AP is required for initiation of disease in AIA (12). The antiinflammatory effect of CRIg-Fc treatment was independent of the presence of endogenously expressed CRIg, as clinical scores in CRIg–wild-type and -knockout mice, injected with the anti-collagen antibodies, were similar (Fig. S2 A, available at http://www.jem.org/cgi/content/full/jem.20070432/DC1). Moreover, CRIg-Fc did not influence the levels of anti-collagen antibodies in the circulation (Fig. S2 B), indicating that CRIg-Fc did not interfere with macrophage-mediated clearance of circulating anti-collagen antibodies or antibody complexes. Finally, the inhibitory effect was not due to Fc receptor–mediated effector functions because treatment of mice with a CRIg-Fc fusion protein containing two mutations within the Fc effector domain (D265A and N297A) was equally effective compared with wild-type Fc protein in inhibiting clinical signs of arthritis (Fig. S2 C). Thus, CRIg-Fc inhibits immune complex–induced inflammation through the interaction of murine CRIg extracellular domain with C3b, a central component of the complement convertases.
Figure 3.

CRIg-Fc inhibits inflammation and bone deformation after transfer of anti-collagen antibodies. (A) CRIg-Fc significantly reduces clinical scores in a model of anti-collagen antibody–induced arthritis. A cocktail of arthritogenic antibodies (2 mg/mouse) was injected intravenously on day 0. Mice were treated daily with 4 mg/kg CRIg-Fc, TNFRII-Fc, or control-Fc proteins starting the day before antibody injection. CRIg-Fc-treatment (B, top panel and graph) reduces infiltration of inflammatory cells, predominantly neutrophils (see insets), present in the tarsal joint of the hindlimb of a control-Fc–treated mouse (B, bottom left panel and graph). (C) CRIg-Fc does not affect cytokines produced during the LPS induction phase of the arthritis, but it reduces IL-1β and IL-6 in parallel with reduced inflammation. (D) CRIg-Fc, but not CRIg(B)-Fc or TNFRII-Fc, reduces clinical scores and inflammation when treatment starts during established inflammation. CRIg-Fc, CRIg(B)-Fc, or TNFRII-Fc were given 5 d after antibody injection. Arrows in A and D indicate treatment start. Data are expressed as mean ± SEM of n = 5. Bar, 100 μm. *, P < 0.05; **, P < 0.01; ***, P < 0.001, CRIg-Fc or TNFRII-Fc versus control-Fc. Bars: B, 100 μm; D, 200 μm.
Because cytokines play an important role in inflammation associated with arthritis (20, 21), we determined whether CRIg treatment affects cytokine production in the joint. CRIg-Fc did not affect cytokine responses shortly after LPS priming (day 3, 2 h after LPS treatment), but reduced local IL-1β and IL-6 levels that reflected the degree of joint inflammation at later time points (Fig. 3 C).
To test if CRIg-Fc can inhibit established inflammation, we initiated treatment 5 d after antibody transfer. Treatment with CRIg-Fc in established disease significantly (P < 0.001) reduced clinical scores and joint inflammation (Fig. 3 D). In contrast, treatment with CRIg(B)-Fc, that has lost its ability to inhibit the AP of complement, was not efficacious. Similarly, treatment of established disease with TNFRII-Fc fusion protein did not reduce clinical scores or inflammation. Thus, while TNF-α is required for induction of disease in this arthritis model, the AP of complement is required for maintaining inflammation in the joint.
CRIg-Fc inhibits antibody-induced complement activation in the joint
We next determined if CRIg inhibited immune complex– induced inflammation through selective inhibition of the AP of complement. Anti-collagen antibodies in complex with collagen activate both the AP and CP of complement in mouse serum (22). CRIg-Fc inhibited C3 activation generated by the AP of complement (Fig. 4 A), but not C3 activation resulting from combined classical and MBL pathways (not depicted). Next to inhibiting complement activation induced by these immune complexes, CRIg-Fc significantly (P < 0.0001) inhibited complement activation in serum from arthritic mice (Fig. 4 B). As expected, control-Fc or CRIg(A)-Fc did not inhibit C3 deposition (Fig. 4 A) or anaphylatoxin generation (Fig. 4 B) after exposure of immune complexes to mouse serum. The reduced inhibitory activity of CRIg-Fc for C3 compared with C5 convertase activity in whole serum is likely due to the lower binding affinity of CRIg-Fc for the monomeric subunit of the C3 convertase (23) as compared with CRIg-Fc affinity for the dimeric C3b2 subunit of the C5 convertase (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20070432/DC1). In addition to its complement inhibitory activity in mouse serum, CRIg inhibited complement activation locally in the joint as shown by the near absence of articular C3 fragments (Fig. 4 C) and a significant reduction in the C3 activation product, C3a Des arg (Fig. 4 D). Consistent with CRIg's selective inhibition of complement activation, anti-collagen antibodies were present in the joint of both CRIg-Fc– and control-Fc–treated mice. CRIg-Fc protein colocalized with C3 fragments in the joint (Fig. 4 E), indicating that systemically delivered protein localizes to the sites of complement C3 activation. Thus, CRIg inhibits antibody-induced AP complement activation in the joint resulting in a significant reduction in inflammation. These results indicate that the inhibitory effect of CRIg-Fc in vivo was fully dependent on its binding to, and inhibition of, the AP convertases.
Figure 4.

CRIg-Fc inhibits AP complement activation induced by anti-collagen antibodies in vitro and in vivo. (A) CRIg-Fc inhibits the alternative pathway of complement induced by immune complexes in mouse serum. Collagen-coated microtitre plates were incubated with anti-collagen antibodies, and the wells were incubated with 10% mouse serum. Binding of C3 fragments to the microtitre plates was determined by ELISA. The experiment was repeated three times with similar results. (B) CRIg-Fc, but not CRIg(A)-Fc and control-Fc protein, inhibits C3a and C5a production in mouse serum. Serum obtained from CRIg-Fc–, CRIg(A)-Fc–, and control-Fc–treated mice was incubated at 37°C for 45 min, and the levels of C3a des Arg and C5a des Arg were measured by ELISA. (C) Systemic treatment with CRIg-Fc, but not control-Fc, reduces the presence of C3 fragments (green fluorescence), but not anti-collagen antibody (red fluorescence) on the cartilage surface in the metatarsal-phalangeal joints 14 d after antibody injection. (D) CRIg inhibits the production of C3a des Arg in the joints. Joints obtained at various time points after antibody injection were homogenized, and C3a des Arg was measured by ELISA. (E) CRIg-Fc and C3 fragments colocalize to the cartilage surface in the joints. Joints were obtained from mice 1 d after treatment with CRIg-Fc on day 5 after antibody injection. Data are expressed as mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.0001, CRIg-Fc versus control-Fc and CRIg(A)-Fc. Bar, 100 μm. A.U., arbritary units.
This study presents CRIg as the first complement receptor that, in its soluble form, selectively inhibits the AP in vitro and in vivo. Its mode of inhibition is different from that of other native complement inhibitors. CRIg does not have decay or cofactor activity, but instead competes with binding of C5 to the convertases of the AP, but not CP (14). Because CRIg does not influence complement activation through the CP and MBL pathway it leaves part of the complement-mediated host defense system intact. Although the extracellular domain of CRIg inhibits complement activation, we have not found evidence that the membrane-bound form of the molecule acts as an intrinsic inhibitor of complement activation. It is however likely that CRIg acts as an AP regulator of convertases bound to the macrophage cell surface during clearance of serum-opsonized particles (13). Further studies, in which the complement inhibitory function of CRIg is uncoupled from its role as a phagocytic receptor, are necessary to clarify the role of macrophage-expressed CRIg in complement regulation.
Collectively, CRIg's ability to inhibit complement activation is well conserved in human and mouse and a soluble version provides a novel tool to study the effect of therapeutic intervention of the AP of complement in preclinical models of disease. Furthermore, the recently presented structure of CRIg in complex with C3b offers the opportunity for rational design of CRIg proteins with enhanced inhibitory efficacy. Collectively, CRIg presents a promising new therapeutic to target diseases in which the AP of complement plays a prominent role, including rheumatoid arthritis, anti-phospholipid syndrome, intestinal and renal ischemia-reperfusion injury, type II membranoproliferative glomerulonephritis, and age-related macular degeneration (8, 24).
MATERIALS AND METHODS
Generation of recombinant protein.
Murine CRIg-Fc was generated as described elsewhere (13). Mutants in the extracellular domain of CRIg and mutations in the Fc effector domain of the molecule were introduced with the Quick-Change Mutagenesis kit (Stratagene) as described elsewhere (14). Circular dichroism spectra from the mutant proteins (CRIg(A)-Fc and CRIg(B)-Fc) were obtained as described previously (14). TNFRII-Fc was generated by fusing the extracellular domain of TNFRII with the Fc portion of murine IgG1. Surface plasmon resonance analysis was performed as described previously (13).
Zymosan assay for C3 and C5 convertase.
Fc-fusion proteins were diluted in GVB+/EGTA (0.1% gelatin/veronal buffer [BioWhittaker]/15 mM EGTA/24 mM MgCl2) and mixed with 4% fresh BALB/c serum and 0.1 mg/ml activated zymosan in a final volume of 100 μl for 45 min at 37°C with shaking, and the reaction stopped with 900 μl GVB containing 50 mM EDTA. Zymosan was pelleted, and the supernatants were used for mouse C3a and C5a ELISAs.
Hemolytic assays for C3 and C5 convertase.
Hemolysis assays for the AP and CP were performed as described previously (14). AP and CP hemolytic assays using assembled components were performed essentially as described previously (25). IgM-coated sheep erythrocytes were obtained from Comptech.
Arthritis models.
All animals were held under sterile pathogen-free conditions, and animal experiments were approved by the Institutional Animal Care and Use Committee of Genentech. CIA was achieved by immunization of 8–11-wk-old DBA/1J mice (The Jackson Laboratory) with bovine type II collagen as described previously (26). Mice were treated three times per week with 4 mg/kg protein when dosing was initiated on day 24, or three times per week with 12 mg/kg when dosing was initiated on day 42 after primary immunization. Antibody-induced, or passive, arthritis was performed by injection with an arthritogenic monoclonal antibody mixture (Chemicon) in 6-wk-old BALB/c mice (The Jackson Laboratory) as described previously (27). In brief, 2 mg anti-collagen type II antibodies was injected intravenously in mice, followed 3 d later by intraperitoneal injection with 25 μg LPS. Mice were treated daily with 4 mg/kg CRIg-Fc, TNFRII-Fc, or control-Fc (a monoclonal antibody to gp-120, IgG1 isotype). All dosing regiments (CIA and AIA) maintained the levels of CRIg-Fc and TNFRII-Fc between 50 and 100 μg/ml of serum. Clinical scoring was performed by trained personnel blinded to the nature of the treatment as described previously (26). For treatment of mice with already established disease, mice from the initial immunized cohort with an average clinical score of 6 were randomly assigned to the various treatment groups. Histologic evaluation of arthritis severity was performed on formalin-fixed, decalcified forelimb and hindlimb joints. Joints were scored for severity of inflammation by a pathologist blinded to the treatment using a scale of 0–5 for each paw according to Joosten et al. (28), except that synovial, bone, and cartilage changes were considered in aggregate when determining scores. X-ray micro-computed tomography (micro-ct) was performed ex vivo as described previously (26). Micro-ct estimates of joint cortical bone volume were obtained from the metatarsal-phalangeal and metacarpophalangeal joints.
Immunohistochemistry.
Frozen sections from metatarsal-phalangeal joints were prepared as described elsewhere (29). Immunohistochemistry of joint sections was performed using an FITC-conjugated polyclonal Fab fragment to mouse C3 (Cappel) and an Alexa Fluor 594–conjugated goat anti–mouse monoclonal antibody to IgG2a (Jackson ImmunoResearch Laboratories). CRIg was detected using a primary rat anti–mouse monoclonal antibody (clone 14G6; reference 13), followed by an Alexa Fluor 594–conjugated goat anti–rat polyclonal antibody (Jackson ImmunoResearch Laboratories).
Microtitre assays.
CRIg-Fc titers in the serum were determined with an ELISA using capture and detection monoclonal antibodies raised against the extracellular domain of murine CRIg (13). Anti-collagen antibody titers in the serum were determined in microtitre plates using bovine type II collagen (Chondrex) for antibody capture and biotinylated isotype-specific antibodies (Jackson ImmunoResearch Laboratories) SA-HRPO and TMB substrate (Kirkegaard and Perry Laboratories, Inc.) for detection. Complement activation induced by arthrogen antibodies was measured by adding 10 μg/ml of mouse anti-collagen antibodies to collagen-coated microtitre plates for 1 h, followed by washing three times with GVB (0.1% gelatin/veronal buffer; BioWhittaker). Fresh BALB/c serum (20% in either GVB++ [GVB/2 mM CaCl2/2 mM MgCl2] or GVB/EGTA [GVB/30 mM EGTA/48 mM MgCl2]) was added (10% final serum concentration), mixed for 1 min, and incubated at 37°C for 45 min. Complement C3 binding was detected with a goat anti–mouse horseradish peroxidase–conjugated C3 antibody (Cappel) using TMB substrate. The reaction was stopped by adding H2SO4, and optical density was read at 450 nm wavelength. Hemolysis assays specific for alternative and classical complement pathways were performed as described elsewhere (13, 14).
Cytokine and anaphylatoxin ELISAs.
Mouse cytokine analysis was performed as described elsewhere (21). Hind footpads were cut at the borderline of fur growth and frozen in liquid nitrogen. The volume of PBS used for homogenization was adjusted to 75 mg of tissue per ml of PBS. Supernatants were subjected to ELISA for murine IL-1α, TNF-α, and IL-6 (DuoSet; R&D Systems). C3a des Arg levels were quantified with capture and detection antibodies from BD Biosciences (Fig. 4 B) or with a mouse C3a des Arg ELISA kit (Bachem; Fig. 4 D). In Fig. 4 B, OD measurements were converted to arbitrary units using a standard curve generated with a stock solution of zymosan-activated mouse serum (10 mg/ml incubated for 1 h at 37°C). C5a des Arg ELISAs were established using capture and detection antibodies purchased from BD Biosciences.
Crystallization, data collection, structure solution, and refinement.
A DNA fragment encoding residues 20–137 of murine CRIg was cloned into the NdeI/BamHI sites of the pET28b expression vector (Novagen). Expression and purification were performed in the same manner as described for human CRIg (14). Crystals were grown at 19°C using the hanging drop vapor–diffusion method. 20 mg/ml of purified mCRIg in 25 mM Tris, pH 7.5, and 100 mM NaCl was mixed with equal volumes of a reservoir solution containing 0.2 M potassium tartrate and 20% PEG 3350. Crystals formed after 3 d. For data collection, crystals were dipped briefly into a solution-containing reservoir with an addition of 20% glycerol, and then flash-frozen in liquid nitrogen. Data were collected at Advanced Lightg Source beamline 5.0.2 and processed using HKL2000. Crystals of murine CRIg diffracted to 1.0 Å resolution belong to space group P212121, with cell parameters of a = 31.3, b = 50.7, and c = 62.8 Å. The structure was solved with crystals containing seleno-methionine–labeled protein using multiple anomalous dispersion and program auto-SHARP (30). Refinement using Refmac and manual adjustments with program O resulted in a model with an Rcryst of 13.1% and an Rfree of 15.1%.
Statistical testing.
All p-values were calculated with an unpaired, two-tailed Student's t test assuming equal variance.
Online supplemental material.
Fig. S1 shows that CRIg Fc inhibits AP C5 convertase initiated by assembly of a CP C5 convertase on IgM-coated sheep red blood cells. Fig. S2 shows clinical scores in CRIg wild-type and knockout mice after AIA, the influence of CRIg-Fc on antibody titers in serum, and the efficacy of CRIg-Fc with a mutation in the Fc domain. Fig. S3 illustrates the kinetics of soluble C3b dimer (C3b2) and monomer (C3b) binding to CRIg-Fc. The online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20070432/DC1.
Supplemental Material
Acknowledgments
We thank Hergen Spits and Paul Godowski for their insightful comments; and Julio Ramirez, Pamela Chan, Jean-Philippe Stephan, Fojan Zamanian, Jason Chinn, Lizette Embuscado, and Laura DeForge for their valuable contributions.
We acknowledge the support staff at beamline 5.0.2 at the Advanced Light Source. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, Materials Sciences Division of the U.S. Department of Energy under contract number DE-AC03-76SF00098 at Lawrence Berkeley National Laboratory. Coordinates and structure factors have been deposited at the Brookhaven Protein Database (2PND).
The authors have no conflicting financial interests.
References
- 1.Walport, M.J. 2001. Complement. First of two parts. N. Engl. J. Med. 344:1058–1066. [DOI] [PubMed] [Google Scholar]
- 2.Taylor, P., M. Botto, and M. Walport. 1998. The complement system. Curr. Biol. 8:R259–R261. [DOI] [PubMed] [Google Scholar]
- 3.Hourcade, D., V.M. Holers, and J.P. Atkinson. 1989. The regulators of complement activation (RCA) gene cluster. Adv. Immunol. 45:381–416. [DOI] [PubMed] [Google Scholar]
- 4.Oglesby, T.J., C.J. Allen, M.K. Liszewski, D.J. White, and J.P. Atkinson. 1992. Membrane cofactor protein (CD46) protects cells from complement-mediated attack by an intrinsic mechanism. J. Exp. Med. 175:1547–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oglesby, T.J., D. White, I. Tedja, K. Liszewski, L. Wright, J. Van den Bogarde, and J.P. Atkinson. 1991. Protection of mammalian cells from complement-mediated lysis by transfection of human membrane cofactor protein and decay-accelerating factor. Trans. Assoc. Am. Physicians. 104:164–172. [PubMed] [Google Scholar]
- 6.Mollnes, T.E., W.C. Song, and J.D. Lambris. 2002. Complement in inflammatory tissue damage and disease. Trends Immunol. 23:61–64. [DOI] [PubMed] [Google Scholar]
- 7.Lutz, H.U., and E. Jelezarova. 2006. Complement amplification revisited. Mol. Immunol. 43:2–12. [DOI] [PubMed] [Google Scholar]
- 8.Thurman, J.M., and V.M. Holers. 2006. The central role of the alternative complement pathway in human disease. J. Immunol. 176:1305–1310. [DOI] [PubMed] [Google Scholar]
- 9.Banda, N.K., D.M. Kraus, M. Muggli, A. Bendele, V.M. Holers, and W.P. Arend. 2003. Prevention of collagen-induced arthritis in mice transgenic for the complement inhibitor complement receptor 1-related gene/protein y. J. Immunol. 171:2109–2115. [DOI] [PubMed] [Google Scholar]
- 10.Weisman, H.F., T. Bartow, M.K. Leppo, H.C. Marsh Jr., G.R. Carson, M.F. Concino, M.P. Boyle, K.H. Roux, M.L. Weisfeldt, and D.T. Fearon. 1990. Soluble human complement receptor type 1: in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science. 249:146–151. [DOI] [PubMed] [Google Scholar]
- 11.Morgan, B.P., and C.L. Harris. 2003. Complement therapeutics; history and current progress. Mol. Immunol. 40:159–170. [DOI] [PubMed] [Google Scholar]
- 12.Banda, N.K., J.M. Thurman, D. Kraus, A. Wood, M.C. Carroll, W.P. Arend, and V.M. Holers. 2006. Alternative complement pathway activation is essential for inflammation and joint destruction in the passive transfer model of collagen-induced arthritis. J. Immunol. 177:1904–1912. [DOI] [PubMed] [Google Scholar]
- 13.Helmy, K.Y., K.J. Katschke Jr., N.N. Gorgani, N.M. Kljavin, J.M. Elliott, L. Diehl, S.J. Scales, N. Ghilardi, and M. van Lookeren Campagne. 2006. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell. 124:915–927. [DOI] [PubMed] [Google Scholar]
- 14.Wiesmann, C., K.J. Katschke, J. Yin, K.Y. Helmy, M. Steffek, W.J. Fairbrother, S.A. McCallum, L. Embuscado, L. DeForge, P.E. Hass, and M. van Lookeren Campagne. 2006. Structure of C3b in complex with CRIg gives insights into regulation of complement activation. Nature. 444:217–220. [DOI] [PubMed] [Google Scholar]
- 15.Hietala, M.A., I.M. Jonsson, A. Tarkowski, S. Kleinau, and M. Pekna. 2002. Complement deficiency ameliorates collagen-induced arthritis in mice. J. Immunol. 169:454–459. [DOI] [PubMed] [Google Scholar]
- 16.Solomon, S., D. Kassahn, and H. Illges. 2005. The role of the complement and the Fc gamma R system in the pathogenesis of arthritis. Arthritis Res. Ther. 7:129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trentham, D.E., A.S. Townes, and A.H. Kang. 1977. Autoimmunity to type II collagen an experimental model of arthritis. J. Exp. Med. 146:857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vogt, L., N. Schmitz, M.O. Kurrer, M. Bauer, H.I. Hinton, S. Behnke, D. Gatto, P. Sebbel, R.R. Beerli, I. Sonderegger, et al. 2006. VSIG4, a B7 family-related protein, is a negative regulator of T cell activation. J. Clin. Invest. 116:2817–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terato, K., K.A. Hasty, R.A. Reife, M.A. Cremer, A.H. Kang, and J.M. Stuart. 1992. Induction of arthritis with monoclonal antibodies to collagen. J. Immunol. 148:2103–2108. [PubMed] [Google Scholar]
- 20.Ji, H., K. Ohmura, U. Mahmood, D.M. Lee, F.M. Hofhuis, S.A. Boackle, K. Takahashi, V.M. Holers, M. Walport, C. Gerard, et al. 2002. Arthritis critically dependent on innate immune system players. Immunity. 16:157–168. [DOI] [PubMed] [Google Scholar]
- 21.Kagari, T., H. Doi, and T. Shimozato. 2002. The importance of IL-1 beta and TNF-alpha, and the noninvolvement of IL-6, in the development of monoclonal antibody-induced arthritis. J. Immunol. 169:1459–1466. [DOI] [PubMed] [Google Scholar]
- 22.Hietala, M.A., K.S. Nandakumar, L. Persson, S. Fahlen, R. Holmdahl, and M. Pekna. 2004. Complement activation by both classical and alternative pathways is critical for the effector phase of arthritis. Eur. J. Immunol. 34:1208–1216. [DOI] [PubMed] [Google Scholar]
- 23.Fishelson, Z., M.K. Pangburn, and H.J. Muller-Eberhard. 1984. Characterization of the initial C3 convertase of the alternative pathway of human complement. J. Immunol. 132:1430–1434. [PubMed] [Google Scholar]
- 24.Holers, V.M., and J.M. Thurman. 2004. The alternative pathway of complement in disease: opportunities for therapeutic targeting. Mol. Immunol. 41:147–152. [DOI] [PubMed] [Google Scholar]
- 25.Krych-Goldberg, M., R.E. Hauhart, V.B. Subramanian, B.M. Yurcisin II, D.L. Crimmins, D.E. Hourcade, and J.P. Atkinson. 1999. Decay accelerating activity of complement receptor type 1 (CD35). Two active sites are required for dissociating C5 convertases. J. Biol. Chem. 274:31160–31168. [DOI] [PubMed] [Google Scholar]
- 26.Barck, K.H., W.P. Lee, L.J. Diehl, J. Ross, P. Gribling, Y. Zhang, K. Nguyen, N. van Bruggen, S. Hurst, and R.A. Carano. 2004. Quantification of cortical bone loss and repair for therapeutic evaluation in collagen-induced arthritis, by micro-computed tomography and automated image analysis. Arthritis Rheum. 50:3377–3386. [DOI] [PubMed] [Google Scholar]
- 27.Terato, K., D.S. Harper, M.M. Griffiths, D.L. Hasty, X.J. Ye, M.A. Cremer, and J.M. Seyer. 1995. Collagen-induced arthritis in mice: synergistic effect of E. coli lipopolysaccharide bypasses epitope specificity in the induction of arthritis with monoclonal antibodies to type II collagen. Autoimmunity. 22:137–147. [DOI] [PubMed] [Google Scholar]
- 28.Joosten, L.A., E. Lubberts, P. Durez, M.M. Helsen, M.J. Jacobs, M. Goldman, and W.B. van den Berg. 1997. Role of interleukin-4 and interleukin-10 in murine collagen-induced arthritis. Protective effect of interleukin-4 and interleukin-10 treatment on cartilage destruction. Arthritis Rheum. 40:249–260. [DOI] [PubMed] [Google Scholar]
- 29.Tarpley, J.E. 2003. Preparation and sectioning of undecalcified frozen rodent long bones and joints using a tape transfer system. J. Histotechnol. 25:41–48. [Google Scholar]
- 30.Bricogne, G., C. Vonrhein, C. Flensburg, M. Schiltz, and W. Paciorek. 2003. Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr. D Biol. Crystallogr. 59:2023–2030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}