

Abstract
Signaling through Notch receptors and their transcriptional effector RBP-J is essential for lymphocyte development and function, whereas its role in other immune cell types is unclear. We tested the function of the canonical Notch–RBP-J pathway in dendritic cell (DC) development and maintenance in vivo. Genetic inactivation of RBP-J in the bone marrow did not preclude DC lineage commitment but caused the reduction of splenic DC fraction. The inactivation of RBP-J in DCs using a novel DC-specific deleter strain caused selective loss of the splenic CD8− DC subset and reduced the frequency of cytokine-secreting CD8− DCs after challenge with Toll-like receptor ligands. In contrast, other splenic DC subsets and DCs in the lymph nodes and tissues were unaffected. The RBP-J–deficient splenic CD8− DCs were depleted at the postprogenitor stage, exhibited increased apoptosis, and lost the expression of the Notch target gene Deltex1. In the spleen, CD8− DCs were found adjacent to cells expressing the Notch ligand Delta-like 1 in the marginal zone (MZ). Thus, canonical Notch–RBP-J signaling controls the maintenance of CD8− DCs in the splenic MZ, revealing an unexpected role of the Notch pathway in the innate immune system.
DCs are critical components of the immune system that link its innate and adaptive arms (1). Located in tissues and lymphoid organs, DCs constantly sample the environment and recognize any invading pathogens through pattern recognition receptors such as Toll-like receptors (TLRs). Pathogen recognition and capture triggers DC maturation, which involves the up-regulation of surface MHC class II and co-stimulatory molecules, secretion of cytokines such as IL-12, and migration into T cell areas of lymphoid organs. In addition to antigen presentation to T lymphocytes, mature DCs recruit and activate multiple cell types, including B and NK lymphocytes. In the steady state, the DC system comprises resident tissue DCs, immature blood-derived DCs in the lymphoid organs, and activated DCs migrating from the tissues into lymph nodes (2). In addition to these conventional DCs, plasmacytoid DCs (PDCs) represent a distinct DC type specialized in virus recognition and secretion of type I interferons. In the mouse, the integrin αL chain CD11c serves as a relatively specific DC marker, with conventional DCs and PDCs being CD11chigh and CD11clow, respectively.
Conventional blood-derived DCs in lymphoid organs comprise two major subsets, distinguished in the mouse by the expression of surface marker CD8. In addition to the lack of CD8, CD8− DCs can be identified by the expression of the myeloid marker CD11b and of a specific marker, DC inhibitory receptor 2, recognized by mAb 33D1 (3). The development of both CD8− and CD8+ DCs in the spleen appears to be distinct from other organs, with the lineage and subset commitment occurring in the recently identified splenic DC progenitors (4). The more abundant CD8− DCs reside in the marginal zone (MZ) of the splenic lymphoid follicles and preferentially present exogenous antigens on MHC class II proteins (3). On the other hand, CD8+ DCs are believed to reside in the T cell zones, although recent studies suggest a broader distribution in the spleen (5). This DC subset is uniquely capable of the cross-presentation of cell-associated antigens via the MHC class I pathway (3, 6). Furthermore, a substantial fraction of genes differentially expressed between splenic CD8− and CD8+ DCs has been identified (3, 7). Thus, CD8− and CD8+ DCs represent genetically and functionally distinct DC subsets.
Despite the critical role of DCs in the immune response, the molecular basis of their development, subset specification, and homeostasis is poorly understood. Several important transcriptional regulators of hematopoiesis have been shown to control DC development, including NF-κB subunits, Ikaros, PU.1, Id2, interferon regulatory factor (IRF) 8, and STAT3 (for review see reference 8). Consistent with their distinct molecular and functional features, CD8− and CD8+ DCs are selectively controlled by different transcription factors. For instance, a specific block of CD8− DC development was observed in the absence of the NF-κB subunit RelB (9) and IRF family members IRF2 (10) and IRF4 (11, 12). However, the loss of these and other factors affects multiple cell types in the immune system and, therefore, may influence DCs indirectly. Thus, the DC-intrinsic roles of transcription factors and major signaling pathways remain to be established in vivo, as the task requires a system for gene targeting specifically in DCs.
The Notch signaling pathway is an evolutionarily conserved mechanism regulating the development of multiple cells and tissues (13). Notch signaling involves the interaction at the cell surface between Notch receptors and their ligands of the Delta and Jagged family, leading to γ-secretase–dependent cleavage of Notch. The intracellular domain of Notch (NotchIC) translocates into the nucleus, where it interacts with a DNA-binding transcription factor CSL (known in the mouse as RBP-J or Rbpsuh). The resulting NotchIC–RBP-J complex directly activates both general (Hes and Deltex) and tissue-specific targets of Notch signaling, thereby affecting cellular differentiation and/or homeostasis. In the adaptive immune system, Notch signaling is essential for the commitment to and early development of the T cell lineage, generation of MZ B lymphocytes, and specification of effector T cell functions (for review see reference 14). On the other hand, the role of Notch signaling in the cells of the innate immune system is poorly understood. Although a potential involvement of Notch signaling in the development and/or maturation of DCs has been suggested by in vitro studies (15–18), the exact function of Notch in the DC lineage remains unknown.
In this paper, we have developed a DC-specific Cre deleter strain and used it to inactivate RBP-J in the DC lineage in vivo. Our results indicate that Notch–RBP-J signaling is generally dispensable for DC lineage commitment, yet essential for DC homeostasis in the spleen. In particular, CD8− splenic DCs appear to receive a unique Notch signal that is required for their survival and persistence in the MZ. These data reveal a role of the Notch–RBP-J pathway in the maintenance of the innate immune system.
RESULTS
Decreased splenic DC content after RBP-J deletion in the BM
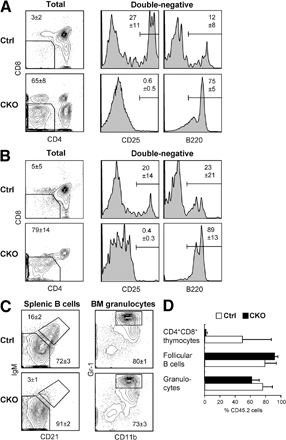
We studied DC development in the absence of RBP-J, an essential transcriptional effector of canonical Notch signaling. Initially, we used a conditional LoxP-flanked RBP-J allele (floxed; RBP-Jfl) in conjunction with an interferon-inducible Mx1-Cre transgene, which efficiently deletes RBP-J in the BM (19). 3 wk after the induction of Cre recombination by interferonogen poly(I):(C), conditional knockout (CKO; RBP-Jfl/fl Mx1-Cre+) mice manifested a block of T cell development along with increased B cell development in the thymus, confirming the functional inactivation of Notch signaling (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20062648/DC1). In the spleen, the fractions of PDCs and CD11b+ CD11c− monocytes/macrophages were elevated; on the other hand, the proportion of conventional CD11chigh MHC class II+ DCs was reduced ∼2.5-fold (Fig. 1 A). The expression of CD8 in DCs was highly variable in this experimental setting, likely because of the nonspecific effects of poly(I):(C)-induced interferon response. Nevertheless, we noticed a consistent decrease in the fraction of CD11b+ DCs corresponding to the CD8− subset.
Figure 1.

Decreased splenic DC content after RBP-J deletion in the BM. Control (RBP-Jfl/fl) or CKO (RBP-Jfl/fl Mx1-Cre+) mice were injected with poly(I):(C) to induce the deletion of RBP-J in the BM. 3 wk later, spleens were analyzed by flow cytometry, and BM was transferred to irradiated recipients. (A) Splenic DC compartment in control and CKO mice. Shown are the staining profiles of total splenocytes (or of B220+ splenocytes for PDCs), with the fraction of each population in the total splenocytes indicated (mean ± SD of three animals). (B) Splenic DC compartment in the chimeras established from control or CKO BM. Shown are the staining profiles of donor-derived (CD45.2+) splenocytes 4–5 wk after BM transfer, with the fraction of each population in the total donor-derived splenocytes indicated (mean ± SD of four to six animals). Student's test p-values are indicated for statistically significant differences.
To confirm that the observed DC defect was caused by the loss of RBP-J in hematopoietic cells, we reconstituted irradiated CD45.1+ mice with the induced CKO or control BM (CD45.2+). As expected, RBP-J–deficient BM failed to generate thymocytes and MZ B cells, whereas it extensively contributed to granulopoiesis and follicular B cell development (Fig. S1, B–D). Notably, PDC generation from the CKO BM was reproducibly enhanced, suggesting an inhibitory role of Notch signaling in PDC development. Importantly, the proportion of donor-derived splenic DCs was reduced in the CKO chimeras to the same extent as in CKO donor mice (Fig. 1 B). Because the analysis was performed relatively soon after BM transfer (4–5 wk), the distribution of CD8− and CD8+ DC subsets could not be firmly established; nevertheless, a similar decrease in the fraction of CD11b+ DCs was observed in the CKO chimeras. Collectively, these results suggest that RBP-J expression in hematopoietic cells is required to maintain DC numbers in the spleen and may be particularly important in the splenic CD8− DCs.
In contrast to the impaired generation of splenic DCs in vivo, DCs and PDCs developed normally in Flt3 ligand–supplemented cultures of CKO BM (unpublished data). Moreover, CD8− DC development from CKO BM in GM-CSF–supplemented cultures was accelerated, yielding a significantly higher proportion of DCs on day 5 (Fig. S2 A, available at http://www.jem.org/cgi/content/full/jem.20062648/DC1). The latter may be caused by the general restriction of myeloid cell fates by Notch (20), which is relieved in RBP-J–deficient BM. Although not statistically significant, a minor reduction of completely matured MHC class IIhigh DCs was observed in the CKO cultures by day 10. Thus, the Notch–RBP-J pathway appears dispensable for DC lineage commitment, suggesting that the decrease in splenic DCs in vivo may reflect their defective maintenance.
A system for gene targeting specifically in the DC lineage
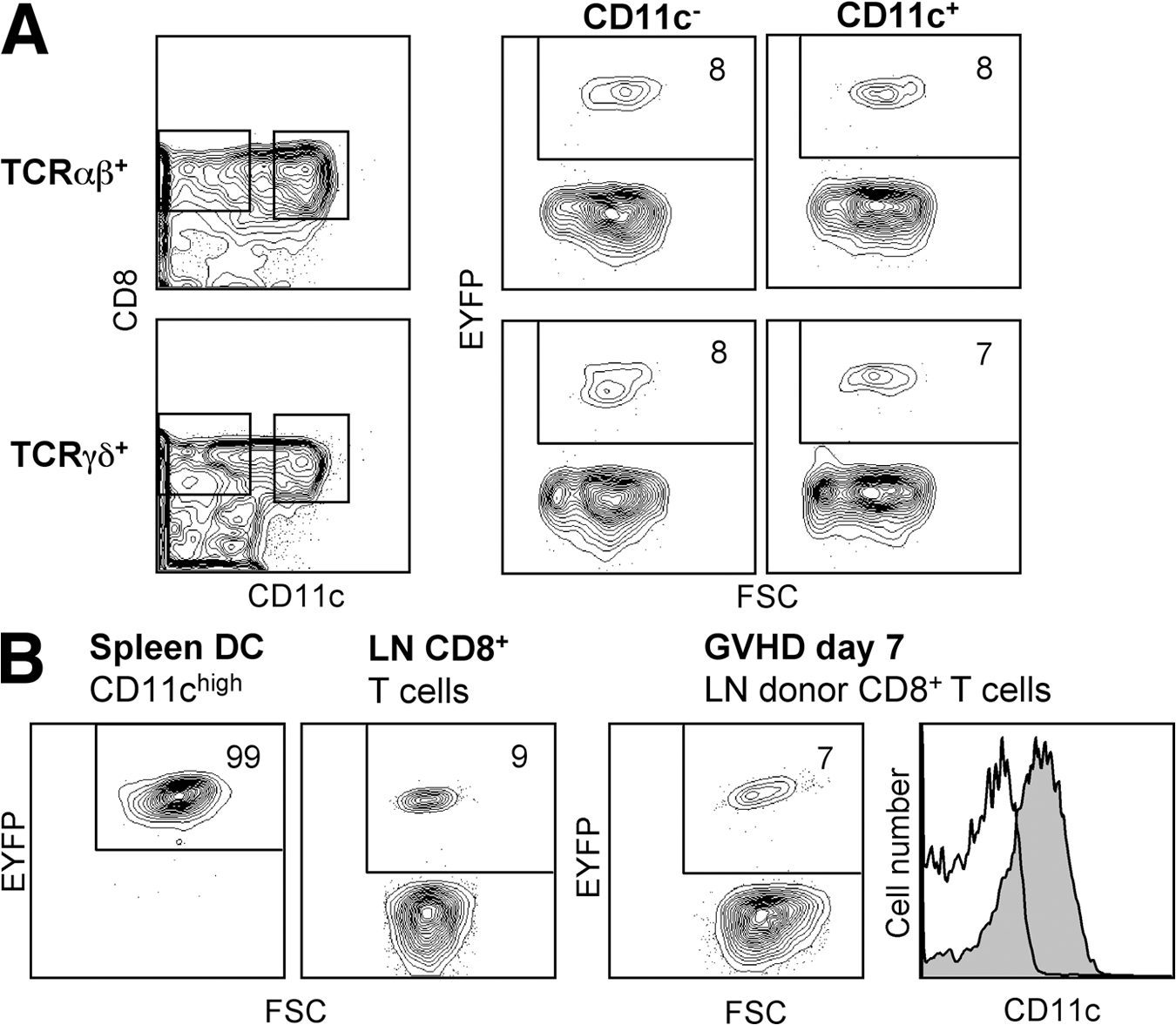
Because of the pleiotropic effects of Notch signaling in the immune system, it was essential to inactivate RBP-J specifically in the DC lineage using a DC-specific Cre deleter strain. To generate such a strain, we introduced Cre recombinase into the CD11c gene within a 160-kb bacterial artificial chromosome (BAC) genomic clone, which was used to generate transgenic mice. The resulting CD11c-Cre strain was crossed to the Rosa26-StopFlox–enhanced yellow fluorescent protein (R26-EYFP) reporter mice, in which Cre recombination activates the expression of EYFP from the ubiquitously active Rosa26 locus (21). As illustrated in Fig. 2 A, EYFP expression was observed in >95% of splenic DCs, compared with <10% of lymphocytes and <1% of myeloid cells such as granulocytes. The background recombination in lymphocytes originated in early lymphoid progenitors in the BM; notably, it was not increased in CD11clow-activated T lymphocytes (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20062648/DC1) (22). Importantly, efficient recombination was detected in all DC subsets, including CD8− and CD8+ DCs, tissue-derived DCs in the lymph nodes, and PDCs (Fig. 2 B). In addition, DCs in tissues such as the lung and epidermis were mostly EYFP+ (unpublished data). On the other hand, CD11clow immediate splenic DC progenitors (4) contained only ∼50% EYFP+ cells, suggesting that efficient recombination is initiated after DC lineage commitment. Similarly, recombination in GM-CSF–supplemented BM cultures followed the emergence of CD11c+ DCs at late stages of culture (Fig. S2 B). Furthermore, splenic myeloid cell types such as CD11c− monocytes/macrophages were EYFP negative, and only partial recombination was observed in CD11clow monocytes possibly differentiating into DCs (Fig. 2 C). The DC compartment in CD11c-Cre+ animals was indistinguishable from that of wild-type mice, suggesting that Cre expression has no adverse effects on DCs (unpublished data). Collectively, these data demonstrate an efficient and specific recombination in DCs, which is initiated after DC lineage commitment and the onset of CD11c expression.
Figure 2.

DC-specific Cre recombination in the CD11c-Cre deleter strain. Shown is the expression of the EYFP reporter gene in the CD11c-Cre+, R26-EYFP+ mice. Fluorescence profiles and percentages of EYFP+ cells are shown for the indicated cell populations (representative of three to five animals). No EYFP+ cells were detected in the absence of the Cre transgene (not depicted). (A) Major splenic cell lineages. (B) DC subsets, including splenic PDCs, CD8+ and CD8− DCs, activated DCs in the subcutaneous lymph nodes, and splenic DC progenitors. (C) Splenic myeloid cells (SSClow CD11b+), divided into four subsets based on Ly-6C and CD11c expression levels. Subset IV corresponds to differentiated CD8− DCs. FSC, forward scatter; SSC, side scatter.
DC-specific RBP-J deletion selectively impairs splenic CD8− DCs
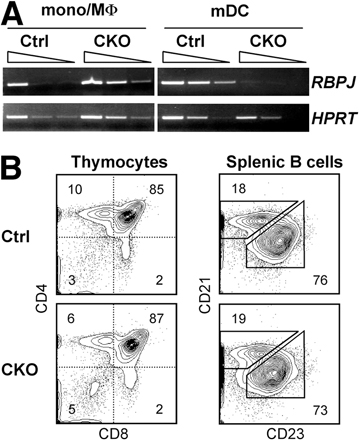
To test the cell-intrinsic role of RBP-J in DCs, we generated RBP-Jfl/fl CD11c-Cre+ CKO mice. In these mice, RBP-J was completely deleted in DCs but not in macrophages (Fig. S4 A, available at http://www.jem.org/cgi/content/full/jem.20062648/DC1); moreover, normal lymphoid development reflected the lack of widespread RBP-J deletion (Fig. S4 B). On the other hand, the splenic DC population was decreased to the same extent as in Mx1-Cre+ CKO mice and displayed a significantly reduced ratio of CD8−/CD8+ DCs (Fig. 3 A). This was caused by a threefold decrease in the absolute numbers of CD8− DCs, whereas CD8+ DC numbers were unaffected (Fig. 3 B). A similar decrease in the proportion of CD8− DCs was observed using the CD8− DC–specific mAb 33D1 (Fig. 3 C). In contrast, the fractions of activated tissue-derived DCs (CD11c+ MHC class IIhigh) and of blood-derived DCs (CD11chigh MHC class II+) were normal in the cutaneous LN, as was the ratio of CD8−/CD8+ DCs within the latter population (Fig. 3 D). Similar results were obtained in the mesenteric LN (unpublished data). Furthermore, no major abnormalities were observed in the thymic DC population, where blood-derived DCs are largely CD8+ (Fig. 3 E). We also analyzed conventional CD11c+ resident DCs and DC-like CD11chigh macrophages (23) in the lung, as both populations undergo efficient Cre recombination in CD11c-Cre mice. We found that the resident DCs in the lung (Fig. 3 F) and in the epidermis (not depicted) were largely intact in CKO mice. No enhanced maturation (as indicated by increased levels of MHC class II or co-stimulatory molecules) was observed in CKO DCs, suggesting that DC-specific RBP-J deletion does not cause overt inflammation. Collectively, these data suggest that splenic CD8− DCs are uniquely sensitive to the loss of RBP-J.
Figure 3.

The loss of splenic CD8− DCs in mice with DC-specific RBP-J deletion. Spleens from control (RBP-Jfl/fl) and CKO (RBP-Jfl/fl CD11c-Cre+) mice were analyzed for the indicated cell populations. (A) Staining profiles of PDCs and conventional DCs, with the percentages among total splenocytes indicated (mean ± SD of 3 and 17 animals per genotype, respectively). Within the DC population, the mean percentages of CD8− and CD8+ DC subsets and the CD8−/CD8+ DC ratio are shown. (B) Absolute numbers of CD8− and CD8+ DCs in individual control (squares) and CKO (triangles) mice. Horizontal lines represent the means. (C) Staining of splenic DCs with the CD8− DC–specific mAb 33D1, with the fractions of 33D1+ and 33D1− DCs among total splenocytes indicated. A threefold decrease in the absolute number of 33D1+ DCs was observed in CKO spleens (not depicted). (D and E) DC populations in the cutaneous lymph nodes (D) and the thymus (E). Shown are the staining profiles of lineage-depleted cells, with blood-derived (CD11chigh MHC class II+) and mature tissue-derived (CD11c+ MHC class IIhigh) DC populations highlighted. The percentages of these populations among the total cells of each organ are indicated (mean ± SD of four mice). Within the blood-derived DC population, the fractions of CD8− and CD8+ subsets are shown (mean ± SD of four mice). (F) Staining profiles of alveolar macrophages (CD11chigh MHC class II+) and conventional resident DCs (CD11clow MHC class IIhigh) in the lung. The percentage of each population among total lung cells is indicated (mean ± SD of four mice).
To confirm the loss of CD8− DCs at the functional level, we measured DC cytokine responses to TLR activation by flow cytometry. Initially, mice were injected i.v. with TLR agonists, resulting in a modest IL-12 expression by a small fraction of DCs 2 h later (Fig. 4 A). In this in vivo assay, the frequency of IL-12+ DCs was decreased in CKO spleens after challenge with the TLR4 agonist LPS and especially with the TLR7 agonist imiquimod, which preferentially activates CD8− DCs (24, 25). To increase the magnitude of cytokine responses by DCs, we analyzed in vitro cultures of splenocytes in which the original differences in the DC content were preserved. Splenocytes from untreated mice were incubated with TLR ligands in vitro for 6 h and stained for intracellular IL-12 and TNF-α. Although LPS elicited relatively weak cytokine responses under these conditions, imiquimod and the TLR9 agonist unmethylated CpG oligonucleotides (CpG) induced the accumulation of IL-12+ and TNF-α+ DCs (Fig. 4 B). Notably, the frequency of cytokine-secreting CD8− DCs was decreased approximately threefold in CKO cultures, whereas the frequency of cytokine-secreting CD8+ DCs was unchanged (Fig. 4 C). The decrease in cytokine-producing CD8− DCs was proportional to their reduced frequency in CKO spleens, suggesting that the loss of RBP-J–deficient splenic CD8− DCs correlates with defective DC-mediated cytokine responses.
Figure 4.

Decreased frequency of cytokine-secreting splenic DCs in the absence of RBP-J. (A) The frequency of IL-12–secreting DCs after TLR ligand challenge in vivo. Control (RBP-Jfl/fl) and CKO (RBP-Jfl/fl CD11c-Cre+) mice were injected with saline, LPS, or imiquimod, and IL-12 production in DCs was detected by intracellular staining. Shown are the representative staining profiles of MHC class II+ splenocytes and the frequency of IL-12+ DCs among total splenocytes (mean ± SD of four to five mice; P < 0.05 for both stimuli). (B and C) The frequency of cytokine-secreting DCs after TLR ligand challenge in vitro. Lymphoid lineage-depleted splenocytes were cultured in the presence of the indicated TLR ligands, and cytokine production in DCs was detected by intracellular staining for IL-12 and TNF-α. Shown are the representative staining profiles of CD11chigh MHC class II+ DCs (B) and the frequency of cytokine-secreting total and CD8− and CD8+ DCs among the splenocytes (C). The data represent the mean ± SD of three to four mice per genotype; the differences between control and CKO CD8− DCs are statistically significant (P = 0.0002 and P < 0.03 for CpG and imiquimod, respectively).
Next, we asked whether the depletion of RBP-J–deficient splenic CD8− DCs might be caused by their impaired lineage commitment, proliferation, or survival. The immediate DC progenitors, including the CD24− progenitors committed to the CD8− DC subset, were unaffected in the CKO spleens (Fig. 5 A). This is consistent with the low recombination frequency in the progenitors (Fig. 2 B) and suggests that the depletion occurs in mature CD8− DCs after their lineage commitment. Indeed, freshly isolated CKO CD8− DCs displayed an increased percentage of apoptotic cells, as measured by staining with Annexin V (Fig. 5 B). Furthermore, the rate of BrdU incorporation in CKO CD8− DCs was increased, suggesting that their faster turnover was caused by decreased survival (Fig. 5 C). Collectively, these data suggest that RBP-J regulates the maintenance of committed CD8− DCs by facilitating their survival. Similarly, the initial DC development in vitro from CKO BM was normal, whereas a moderate decrease in the fraction of total and mature MHC class IIhigh DCs was observed at late stages of culture (Fig. S2 C).
Figure 5.

Impaired survival and homeostasis of RBP-J–deficient splenic CD8− DCs. (A) Splenic DC progenitors, including CD24low DC progenitors committed to the CD8− subset, in control (RBP-Jfl/fl) and CKO (RBP-Jfl/fl CD11c-Cre+) mice. The fraction of each subset in the gated population (mean of two animals) is indicated. The fraction of CD24−/low cells corresponding to CD8− DC progenitors is indicated by the vertical dotted line. (B) The expression of the apoptotic marker phosphatidylserine in control and CKO CD8− DCs, as determined by Annexin V staining. Shown are the representative profiles of ex vivo–isolated CD8− DCs stained with Annexin V and the dead-cell marker 7-amino-actinomycin D, with the fraction of apoptotic cells indicated (mean ± SD of four mice). (C) The turnover rate of splenic CD8− DCs. Mice were fed BrdU for 4–5 d, and the fraction of BrdU+ CD8− DCs (dotted line) was determined by flow cytometry. Shown are the representative BrdU staining profiles of gated CD8− DCs and the fraction of BrdU+ CD8− DCs in control and CKO mice. Because the efficiency of BrdU incorporation and staining varied between experiments, control and CKO mice from each experiment were compared pairwise and analyzed using the paired Student's t test.
Molecular analysis of the Notch signaling pathway in DCs
To study the molecular basis of Notch signaling in DCs, we tested the expression of Notch receptors and target genes by quantitative real-time PCR (qPCR). As previously reported (26), splenic T and B lymphocytes preferentially expressed Notch1 and Notch2, respectively, but no detectable Notch4 (Fig. 6). On the other hand, wild-type CD8− DCs expressed relatively low levels of Notch1 but considerable levels of Notch2 and Notch4. The levels of Notch3 were below detection in these populations (unpublished data). The expression of at least three Notch receptors in DCs raises the possibility of genetic redundancy and may explain normal DC development in the absence of Notch1 (27). In addition, lymphocytes and CD8− DCs expressed the canonical Notch–RBP-J target genes Hes1 and Deltex1 (Dtx1); notably, the latter was present in CD8− DCs but not in monocytes/macrophages (Fig. 6).
Figure 6.

The expression of Notch pathway components in wild-type and RBP-J–deficient DCs. The expression of Notch receptors and target genes in splenic DCs was determined by qPCR. Shown are the expression levels of the indicated genes relative to the expression in wild-type CD8− DCs (indicated by the horizontal dashed line). The lack of signal above threshold is indicated by an asterisk. (left) The expression in splenic T or B lymphocytes, monocytes/macrophages (M), and CD8− DCs from wild-type mice. (right) The expression in CD8− and CD8+ DCs from control (RBP-Jfl/fl) and CKO (RBP-Jfl/fl CD11c-Cre+) mice. Results represent mean relative values ± SD of triplicate qPCR reactions. ND, not determined.
We next measured the expression of Notch receptors and targets in control and RBP-J–deficient CD8− and CD8+ DCs. Compared with CD8− DCs, control CD8+ DCs expressed higher Notch4 levels but completely lacked Dtx1 expression, in agreement with previous microarray data (7). As expected, CKO DC subsets showed a complete loss of RBP-J transcripts encoded by deleted exons (Fig. 6). Interestingly, CKO CD8− but not CD8+ DCs showed a fourfold reduction in Notch4 levels, suggesting that Notch4 expression in CD8− DCs is regulated by a positive feedback loop. Although Hes1 was moderately decreased in both RBP-J–deficient DC subsets, the specific expression of Dtx1 in CD8− DCs was completely abolished in the absence of RBP-J. Therefore, Dtx1 expression in DCs serves as a faithful indicator of Notch–RBP-J signaling, and its RBP-J–dependent expression in CD8− DCs suggests that they receive a unique Notch signal required for their maintenance in the spleen.
To explore a potential source of Notch signals for splenic CD8− DCs, we performed immunohistochemical analysis using the CD8− DC–specific antibody 33D1. As previously described (3), 33D1+ DCs in control spleens were localized primarily in the MZ (Fig. 7); moreover, their numbers were markedly reduced in CKO spleens, consistent with flow cytometry data (Fig. 3 C). Because Notch ligand Delta-like 1 (Dll1) was shown to promote DC maturation in vitro (18), we analyzed the expression of the Dll1 protein with an antibody that revealed its expression in the thymus (28). Notably, Dll1 expression was prominent outside of lymphoid follicles, including the MZ and possibly the red pulp. Unlike more compact DCs, Dll1-expressing cells morphologically resembled stromal cells such as reticular fibroblasts. Furthermore, simultaneous staining revealed a substantial overlap between 33D1 and Dll1 expression domains in the spleen. Although some 33D1+ DCs expressed Dll1 (Fig. 7, bottom, yellow cells on merged images), the majority of 33D1+ DCs (green) were clearly distinct from Dll1-positive cells (red) but were located in close contact with the latter. Thus, Dll1 expressed in the splenic MZ stroma is a likely source of Notch signals that promote the survival and maintenance of CD8− DCs.
Figure 7.

Localization of CD8− DCs and Dll1-expressing cells in the spleen. (top) Spleen sections from wild-type control (RBP-Jfl/fl) or CKO (RBP-Jfl/fl CD11c-Cre+) mice were stained for metallophilic macrophages delineating the MZ (MOMA-1+; green) and CD8− DCs (33D1; red). A serial section of control spleen was stained for MOMA1 (green) and Dll1 (red). (middle and bottom) Wild-type spleen sections were stained for CD8− DCs (33D1; green) and Dll1 (red). The location of 33D1+ cells in the MZ was confirmed by MOMA-1 staining of a serial section. Shown are the representative individual and merged images of 33D1 and Dll1 staining (middle) and a high magnification image of the area indicated by the dashed box (bottom). Bars: (top) 400 μm; (middle) 200 μm; (bottom) 100 μm.
DISCUSSION
Although Notch signaling is essential for lymphocyte development and function, its role in the DC lineage remains controversial. For instance, impaired DC generation in vitro from Notch1-deficient embryonic stem cells and Notch1-antisense hematopoietic progenitors was previously reported (17); in contrast, Notch1-deficient BM efficiently generated all DC subsets in chimeric mice (27). Gain-of-function studies demonstrated that Notch signaling is generally permissive for DC development and may facilitate DC generation and/or maturation (15, 16, 18). On the other hand, both positive (29) and negative (30, 31) effects of Notch signaling on PDC development have been reported. These discrepancies may reflect a potential redundancy of individual Notch receptors, nonphysiological effects of induced Notch activation, and/or specific features of in vitro systems used. To resolve these issues, we took an in vivo loss-of-function approach by inactivating RBP-J, an essential mediator of signaling by all Notch receptors. Indeed, deletion of RBP-J (19, 32) completely recapitulates the phenotypes of individual receptor knockouts (26, 27) and of dominant-negative inhibition of Notch signaling (33). Because no Notch-independent functions of RBP-J have been described in the mammalian system (13, 14), RBP-J deletion essentially generates a specific blockade of the canonical Notch signaling pathway.
To delete RBP-J in DCs, we used both a broad inducible deletion by Mx1-Cre transgene and a constitutive deletion specifically in DCs. For the latter purpose, we generated mice expressing Cre recombinase under the control of the DC-specific CD11c locus. Consistent with the faithful recapitulation of gene expression by large BAC-based transgenes, our CD11c-Cre transgenic strain mediated efficient recombination primarily in CD11c+ DCs, including CD11clow subsets such as PDCs. Because the recombination is initiated in committed CD11c+ DC progenitors, the system appears suitable for the studies of DC homeostasis and function, rather than of initial lineage commitment. Importantly, no increase in recombination frequency has been observed in activated CD11clow T cells, confirming the utility of the CD11c-Cre strain for functional studies of immune responses. Collectively, the CD11c-Cre strain described in this paper should facilitate genetic analysis of cell-intrinsic gene functions in DCs in vivo.
Both panhematopoietic and DC-specific deletion of RBP-J resulted in the decreased content of splenic DCs, particularly of the CD8− DC subset. This DC loss does not appear to reflect impaired DC lineage commitment, because (a) RBP-J–deficient BM gave rise to all DC subsets in vivo and in vitro, (b) essentially similar phenotypes were caused by RBP-J deletion in the BM hematopoietic progenitors (Mx1-Cre) and in committed DCs (CD11c-Cre), and (c) immediate DC progenitors in the spleen were not affected. On the other hand, the homeostasis of splenic CD8− DCs was impaired, as indicated by their reduced survival and increased turnover. The latter likely reflects an increased proliferation of the residual CD8− DCs as a compensation for their decreased survival. Indeed, splenic DCs undergo in situ proliferation controlled by lymphotoxin β receptor (34). Furthermore, a similar increase in BrdU incorporation was observed in DCs from CD47−/− mice, in which splenic CD8− DCs are depleted because of impaired migration (35). Therefore, Notch–RBP-J signaling appears to play a cell-intrinsic role in the maintenance and survival of differentiated DCs, as documented in other cell types. For example, Notch signaling is required for the metabolic homeostasis and survival of committed T cell progenitors (36), and increases the resistance of thymocytes to corticosteroid-induced cell death (37).
Although RBP-J deletion in DCs caused the loss of the splenic CD8− DC subset, the CD8+ DCs were unaffected. This selective requirement for Notch–RBP-J signaling in CD8− DCs correlated with the specific and RBP-J–dependent expression of Notch target Dtx1 by this DC population. In that respect, CD8− DCs resemble MZ B cells, a splenic B lymphocyte population that requires Notch–RBP-J signaling and expresses Dtx1 in a Notch-dependent manner (26). Although the requirement for Notch–RBP-J signaling is highly specific for CD8− DCs and MZ B cells compared with other splenic cell types, the molecular basis of this specificity remains to be elucidated. Indeed, neither CD8− DCs (Fig. 6) nor MZ B cells (26) express higher levels of Notch receptors compared with their Notch-independent counterparts such as CD8+ DCs and follicular B cells, respectively. Furthermore, both CD8− and CD8+ DCs appear to receive at least some canonical Notch signals, as indicated by reduced Hes1 expression in both DC types after RBP-J deletion (Fig. 6). One possible scenario for the observed specificity is the modulation of Notch ligand–receptor interactions by enzymes such as Fringe, which was shown to regulate Notch1 signaling in T cell development (38). Another possible scenario is a cell type–specific expression of a transcriptional cofactor for RBP-J. For instance, decreased expression of a negative RBP-J regulator MINT in MZ B cells facilitates Notch signaling in this subset compared with follicular B cells (39). Conversely, a positive Notch–RBP-J signaling cofactor might be expressed specifically in CD8− DCs and MZ B cells, a possibility that should be explored in future studies.
Another likely determinant of Notch signaling specificity in CD8− DCs and in MZ B cells is the nature and anatomic location of Notch ligands in the spleen. Notably, both cell types are located in the MZ, the compartment shown in this study to contain Dll1-expressing cells. This Notch ligand expressed on splenic stromal cells was shown to promote DC maturation in vitro, whereas another Notch ligand Jagged-1 had an inhibitory effect (18). Indeed, we found that CD8− DCs in the MZ were residing in close contact with Dll1-expressing cells, likely nonhematopoietic stromal cell types such as MZ reticular fibroblasts and/or endothelium of the marginal sinus. Moreover, Dll1 deletion in vivo caused the loss of MZ B cells (40), suggesting that Dll1 is a critical Notch ligand for B cells in the MZ. Therefore, Dll1 is a likely physiological inducer of specific Notch signals that control CD8− DC maintenance in the splenic MZ. The unique location of splenic CD8− DCs in the MZ is likely to explain their dependence on Notch–RBP-J signaling in the spleen but not in other organs (Fig. 3). This model is consistent with recent data suggesting a unique developmental pathway of splenic DCs compared with DCs in other organs (4, 41). Because CD8+ DCs were also detected in the MZ (5), further studies are required to clarify whether CD8+ DCs receive similar Notch signals. Collectively, our studies suggest that splenic MZ is an important source of Notch signals for both MZ B cells and CD8− DCs.
In conclusion, we report an unexpected role of Notch signaling in the homeostasis of splenic DCs. These results emphasize the critical role of Notch pathway not only in lymphocyte development and differentiation but also in the maintenance of the innate immune system. Notably, the loss of RBP-J–deficient splenic CD8− DCs was mirrored by the decreased frequency of cytokine-secreting DCs during challenge with TLR agonists. From the practical standpoint, these results suggest that γ-secretase inhibitors may affect DC-mediated responses when administered in vivo. This possibility further supports the utility of these agents as immunosuppressors (42) and broadens the spectrum of their potential cellular targets. For instance, the reported beneficial effects of γ-secretase inhibitors in experimental autoimmunity (42) might result from the combined modulation of T cell and DC function by these drugs. Further studies are required to elucidate Notch-mediated control of DC function in the steady state and during immune responses.
MATERIALS AND METHODS
Mice.
To generate the CD11c-Cre transgene, the 160-kb mouse genomic BAC clone RP24-361C4 (BACPAC Resources) was modified by ET recombination, as previously described (43). The clone contains the entire Itgax (CD11c) gene but lacks the 5′ end of the adjacent Itgam (CD11b) gene, preventing the overexpression of the latter. The recombination cassette containing the Cre recombinase open reading frame, followed by the bovine growth hormone (BGH) polyA signal and the FRT site-flanked prokaryotic Zeocin resistance cassette (ZeoR), replaced the coding part of the first CD11c exon, and the ZeoR cassette was subsequently removed by FLP-mediated recombination. The clone insert was released from the vector backbone using NotI digestion, gel-purified, and microinjected into fertilized oocytes. The founder line containing two copies of the transgene (as determined by quantitative Southern hybridization) was chosen for further analysis. Mice were genotyped by genomic PCR using either generic Cre primers or primers specific for the CD11c-Cre transgene (5′-ACTTGGCAGCTGTCTCCAAG-3′ and 5′-GCGAACATCTTCAGGTTCTG-3′ were specific for the CD11c promoter and Cre, respectively).
The R26-EYFP strain (21) was provided by F. Costantini (Columbia University, New York, NY). The RBP-Jfl strain (19) was provided by L. Hennighausen (National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD), with permission from T. Honjo (Kyoto University, Kyoto, Japan). The Mx1-Cre strain was previously described (44). Cre-negative RBP-Jfl/fl littermates of CKO (RBP-Jfl/fl Cre+) mice were used as controls; in preliminary experiments, wild-type CD11c-Cre+ mice were used as controls and were found indistinguishable from CD11c-Cre− animals. For inducible RBP-J deletion, adult RBP-Jfl/fl Mx1-Cre+ or control RBP-Jfl/fl mice were injected with 0.25 mg poly(I):(C) three times, with 2-d intervals, and analyzed 3 wk later. For hematopoietic reconstitution, 3 × 106 total BM cells per mouse were injected i.v. into lethally irradiated C57BL/6 mice congenic for CD45.1. The recipient mice were analyzed 4–5 wk after reconstitution. Mice were maintained in a specific pathogen-free facility and used according to the protocol approved by the Columbia University's Institutional Animal Care and Use Committee.
Flow cytometry.
Lymphoid organs and lungs were digested for 1 h at 37°C with collagenase D (Roche) in DMEM/10% FCS, filtered, and subjected to red blood cell lysis. Single-cell suspensions were stained with fluorochrome- or biotin-conjugated antibodies (obtained from eBioscience, BD Biosciences, or Miltenyi Biotec), followed by streptavidin conjugate for four to five color analysis. Where indicated in the figures, the lymphoid lineage (CD19/CD3/DX5)–negative cell fraction was isolated using magnetic beads (Miltenyi Biotec). The immediate DC progenitors were defined as previously described (4), except that the nonessential signal regulatory protein α staining was omitted. For the analysis of BrdU incorporation, mice were injected with 1 mg BrdU i.p. and kept on 0.8 mg/ml BrdU-supplemented water for 4–5 d, and splenocytes were stained for cell surface markers and BrdU using the BrdU Flow Kit (BD Biosciences). For apoptosis detection, cells were stained with antibody conjugates, FITC-conjugated Annexin V (TACS reagent; Trevigen, Inc.), and 7-amino-actinomycin D. The samples were acquired using a flow cytometer (LSR II; BD Biosciences) or sorted on a flow sorter (FACSAria; BD Biosciences) and analyzed using FlowJo software (TreeStar, Inc.). Unless indicated otherwise in the figures, cells were gated on the side scatter–low lymphoid population; light scatter and fluorescence are displayed on a linear and log10 scale, respectively.
Challenge with TLR ligands.
For in vivo assays, mice were injected i.v. with 10 μg LPS (Sigma Aldrich), 200 μg imiquimod (Invivogen), or saline and killed 2 h later. Splenocytes were collagenase-treated and incubated in DMEM/10% FCS for 3 h in the presence of 10 μg/ml brefeldin A (Sigma-Aldrich), stained for surface markers, fixed, permeabilized, and stained with Allophycocyanin (APC)-conjugated anti–mouse IL-12 or an isotype control (BD Biosciences). For in vitro assays, splenocytes from untreated mice were enriched for a lineage-negative fraction and plated at 106 cells/well in 96-well round–bottom plates. Cells were treated with saline, 1 μg/ml LPS, 100 nM CpG (Invivogen), or 3 μg/ml imiquimod for 2 h and incubated for an additional 4 h in the presence of TLR ligands and brefeldin A. Cells were stained for surface markers, fixed, permeabilized, and stained with APC-conjugated anti–mouse IL-12, anti–mouse TNF-α, or isotype controls.
Expression analysis.
Total RNA isolated from ex vivo–sorted cells was reverse transcribed and assayed by qPCR on an instrument (MX3000P; Stratagene) using SYBR green incorporation. All genes were normalized to β-actin, and relative expression was calculated using the ΔΔCT method. All primer pairs spanned introns and were validated for amplification efficiency.
Immunochemistry.
Spleens were frozen, and 5-μm cryostat sections were fixed in acetone, blocked in 3% BSA, and stained with FITC-conjugated antimetallophilic macrophages (MOMA-1; Serotec) and biotin-conjugated anti–DC inhibitory receptor 2 (33D1; eBioscience), followed by Alexa Fluor 488– or Alexa Fluor 568–conjugated streptavidin (Invitrogen), or polyclonal goat anti–mouse Dll1 (SC-9932; Santa Cruz Biotechnology, Inc.), followed by Alexa Fluor 568–conjugated anti–goat IgG (Invitrogen). Images were obtained on a microscope (model IX71; Olympus) equipped with a monochrome camera (model C8484; Hamamatsu) and processed using MicroSuite software (Olympus).
Statistical analysis.
Unless indicated otherwise, data were analyzed using the two-tailed, unpaired Student's t test.
Online supplemental material
Fig.S1 shows lymphocyte development after RBP-J deletion in Mx1-Cre CKO mice. Fig. S2 depicts in vitro DC development in the absence of RBP-J. Fig. S3 shows Cre recombination in activated T cells from CD11c-Cre mice. Fig. S4 depicts the specificity of RBP-J deletion mediated by the CD11c-Cre transgene. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20062648/DC1.
Supplemental Material
Acknowledgments
We thank P. Leder for his support; K. Calame, M. Shapiro-Shelef, E. Magnusdottir, and V. Lin for help in the generation of the CD11c-Cre strain; T. Honjo and L. Hennighausen for RBP-Jfl mice; F. Costantini for R26-EYFP mice; and E. Hou for technical help.
This work was supported in part by National Institutes of Health research grant AI067804 and a Sandler Program for Asthma Research award (to B. Reizis), as well as by National Institutes of Health training grant AI066459 (to M.L. Caton).
The authors have no competing financial interests.
Abbreviations used: BAC, bacterial artificial chromosome; CKO, conditional knockout; Dll1, Delta-like 1; EYFP, enhanced yellow fluorescent protein; IRF, interferon regulatory factor; MZ, marginal zone; PDC, plasmacytoid dendritic cell; qPCR, quantitative real-time PCR; TLR, Toll-like receptor.
References
- 1.Steinman, R.M. 2003. Some interfaces of dendritic cell biology. APMIS. 111:675–697. [DOI] [PubMed] [Google Scholar]
- 2.Shortman, K., and Y.J. Liu. 2002. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2:151–161. [DOI] [PubMed] [Google Scholar]
- 3.Dudziak, D., A.O. Kamphorst, G.F. Heidkamp, V.R. Buchholz, C. Trumpfheller, S. Yamazaki, C. Cheong, K. Liu, H.W. Lee, C.G. Park, et al. 2007. Differential antigen processing by dendritic cell subsets in vivo. Science. 315:107–111. [DOI] [PubMed] [Google Scholar]
- 4.Naik, S.H., D. Metcalf, A. van Nieuwenhuijze, I. Wicks, L. Wu, M. O'Keeffe, and K. Shortman. 2006. Intrasplenic steady-state dendritic cell precursors that are distinct from monocytes. Nat. Immunol. 7:663–671. [DOI] [PubMed] [Google Scholar]
- 5.Neuenhahn, M., K.M. Kerksiek, M. Nauerth, M.H. Suhre, M. Schiemann, F.E. Gebhardt, C. Stemberger, K. Panthel, S. Schroder, T. Chakraborty, et al. 2006. CD8alpha+ dendritic cells are required for efficient entry of Listeria monocytogenes into the spleen. Immunity. 25:619–630. [DOI] [PubMed] [Google Scholar]
- 6.den Haan, J.M., S.M. Lehar, and M.J. Bevan. 2000. CD8+ but not CD8− dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 192:1685–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edwards, A.D., D. Chaussabel, S. Tomlinson, O. Schulz, A. Sher, and C. Reis e Sousa. 2003. Relationships among murine CD11c(high) dendritic cell subsets as revealed by baseline gene expression patterns. J. Immunol. 171:47–60. [DOI] [PubMed] [Google Scholar]
- 8.Shortman, K., and S.H. Naik. 2007. Steady-state and inflammatory dendritic-cell development. Nat. Rev. Immunol. 7:19–30. [DOI] [PubMed] [Google Scholar]
- 9.Wu, L., A. D'Amico, K.D. Winkel, M. Suter, D. Lo, and K. Shortman. 1998. RelB is essential for the development of myeloid-related CD8alpha− dendritic cells but not of lymphoid-related CD8alpha+ dendritic cells. Immunity. 9:839–847. [DOI] [PubMed] [Google Scholar]
- 10.Ichikawa, E., S. Hida, Y. Omatsu, S. Shimoyama, K. Takahara, S. Miyagawa, K. Inaba, and S. Taki. 2004. Defective development of splenic and epidermal CD4+ dendritic cells in mice deficient for IFN regulatory factor-2. Proc. Natl. Acad. Sci. USA. 101:3909–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suzuki, S., K. Honma, T. Matsuyama, K. Suzuki, K. Toriyama, I. Akitoyo, K. Yamamoto, T. Suematsu, M. Nakamura, K. Yui, and A. Kumatori. 2004. Critical roles of interferon regulatory factor 4 in CD11bhighCD8alpha− dendritic cell development. Proc. Natl. Acad. Sci. USA. 101:8981–8986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tamura, T., P. Tailor, K. Yamaoka, H.J. Kong, H. Tsujimura, J.J. O'Shea, H. Singh, and K. Ozato. 2005. IFN regulatory factor-4 and -8 govern dendritic cell subset development and their functional diversity. J. Immunol. 174:2573–2581. [DOI] [PubMed] [Google Scholar]
- 13.Bray, S.J. 2006. Notch signalling: a simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 7:678–689. [DOI] [PubMed] [Google Scholar]
- 14.Maillard, I., T. Fang, and W.S. Pear. 2005. Regulation of lymphoid development, differentiation, and function by the Notch pathway. Annu. Rev. Immunol. 23:945–974. [DOI] [PubMed] [Google Scholar]
- 15.Ohishi, K., B. Varnum-Finney, R.E. Serda, C. Anasetti, and I.D. Bernstein. 2001. The Notch ligand, Delta-1, inhibits the differentiation of monocytes into macrophages but permits their differentiation into dendritic cells. Blood. 98:1402–1407. [DOI] [PubMed] [Google Scholar]
- 16.Weijzen, S., M.P. Velders, A.G. Elmishad, P.E. Bacon, J.R. Panella, B.J. Nickoloff, L. Miele, and W.M. Kast. 2002. The Notch ligand Jagged-1 is able to induce maturation of monocyte-derived human dendritic cells. J. Immunol. 169:4273–4278. [DOI] [PubMed] [Google Scholar]
- 17.Cheng, P., Y. Nefedova, L. Miele, B.A. Osborne, and D. Gabrilovich. 2003. Notch signaling is necessary but not sufficient for differentiation of dendritic cells. Blood. 102:3980–3988. [DOI] [PubMed] [Google Scholar]
- 18.Cheng, P., Y. Nefedova, C.A. Corzo, and D.I. Gabrilovich. 2006. Regulation of dendritic-cell differentiation by bone marrow stroma via different Notch ligands. Blood. 109:507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han, H., K. Tanigaki, N. Yamamoto, K. Kuroda, M. Yoshimoto, T. Nakahata, K. Ikuta, and T. Honjo. 2002. Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. Int. Immunol. 14:637–645. [DOI] [PubMed] [Google Scholar]
- 20.de Pooter, R.F., T.M. Schmitt, J.L. de la Pompa, Y. Fujiwara, S.H. Orkin, and J.C. Zuniga-Pflucker. 2006. Notch signaling requires GATA-2 to inhibit myelopoiesis from embryonic stem cells and primary hemopoietic progenitors. J. Immunol. 176:5267–5275. [DOI] [PubMed] [Google Scholar]
- 21.Srinivas, S., T. Watanabe, C.S. Lin, C.M. William, Y. Tanabe, T.M. Jessell, and F. Costantini. 2001. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huleatt, J.W., and L. Lefrancois. 1995. Antigen-driven induction of CD11c on intestinal intraepithelial lymphocytes and CD8+ T cells in vivo. J. Immunol. 154:5684–5693. [PubMed] [Google Scholar]
- 23.Padilla, J., E. Daley, A. Chow, K. Robinson, K. Parthasarathi, A.N. McKenzie, T. Tschernig, V.P. Kurup, D.D. Donaldson, and G. Grunig. 2005. IL-13 regulates the immune response to inhaled antigens. J. Immunol. 174:8097–8105. [DOI] [PubMed] [Google Scholar]
- 24.Edwards, A.D., S.S. Diebold, E.M. Slack, H. Tomizawa, H. Hemmi, T. Kaisho, S. Akira, and C. Reis e Sousa. 2003. Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 alpha+ DC correlates with unresponsiveness to imidazoquinolines. Eur. J. Immunol. 33:827–833. [DOI] [PubMed] [Google Scholar]
- 25.Doxsee, C.L., T.R. Riter, M.J. Reiter, S.J. Gibson, J.P. Vasilakos, and R.M. Kedl. 2003. The immune response modifier and Toll-like receptor 7 agonist S-27609 selectively induces IL-12 and TNF-alpha production in CD11c+CD11b+CD8− dendritic cells. J. Immunol. 171:1156–1163. [DOI] [PubMed] [Google Scholar]
- 26.Saito, T., S. Chiba, M. Ichikawa, A. Kunisato, T. Asai, K. Shimizu, T. Yamaguchi, G. Yamamoto, S. Seo, K. Kumano, et al. 2003. Notch2 is preferentially expressed in mature B cells and indispensable for marginal zone B lineage development. Immunity. 18:675–685. [DOI] [PubMed] [Google Scholar]
- 27.Radtke, F., I. Ferrero, A. Wilson, R. Lees, M. Aguet, and H.R. MacDonald. 2000. Notch1 deficiency dissociates the intrathymic development of dendritic cells and T cells. J. Exp. Med. 191:1085–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmitt, T.M., M. Ciofani, H.T. Petrie, and J.C. Zuniga-Pflucker. 2004. Maintenance of T cell specification and differentiation requires recurrent Notch receptor–ligand interactions. J. Exp. Med. 200:469–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olivier, A., E. Lauret, P. Gonin, and A. Galy. 2006. The Notch ligand delta-1 is a hematopoietic development cofactor for plasmacytoid dendritic cells. Blood. 107:2694–2701. [DOI] [PubMed] [Google Scholar]
- 30.Pelayo, R., J. Hirose, J. Huang, K.P. Garrett, A. Delogu, M. Busslinger, and P.W. Kincade. 2005. Derivation of 2 categories of plasmacytoid dendritic cells in murine bone marrow. Blood. 105:4407–4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dontje, W., R. Schotte, T. Cupedo, M. Nagasawa, F. Scheeren, R. Gimeno, H. Spits, and B. Blom. 2006. Delta-like1-induced Notch1 signaling regulates the human plasmacytoid dendritic cell versus T-cell lineage decision through control of GATA-3 and Spi-B. Blood. 107:2446–2452. [DOI] [PubMed] [Google Scholar]
- 32.Tanigaki, K., H. Han, N. Yamamoto, K. Tashiro, M. Ikegawa, K. Kuroda, A. Suzuki, T. Nakano, and T. Honjo. 2002. Notch-RBP-J signaling is involved in cell fate determination of marginal zone B cells. Nat. Immunol. 3:443–450. [DOI] [PubMed] [Google Scholar]
- 33.Maillard, I., A.P. Weng, A.C. Carpenter, C.G. Rodriguez, H. Sai, L. Xu, D. Allman, J.C. Aster, and W.S. Pear. 2004. Mastermind critically regulates Notch-mediated lymphoid cell fate decisions. Blood. 104:1696–1702. [DOI] [PubMed] [Google Scholar]
- 34.Kabashima, K., T.A. Banks, K.M. Ansel, T.T. Lu, C.F. Ware, and J.G. Cyster. 2005. Intrinsic lymphotoxin-beta receptor requirement for homeostasis of lymphoid tissue dendritic cells. Immunity. 22:439–450. [DOI] [PubMed] [Google Scholar]
- 35.Van, V.Q., S. Lesage, S. Bouguermouh, P. Gautier, M. Rubio, M. Levesque, S. Nguyen, L. Galibert, and M. Sarfati. 2006. Expression of the self-marker CD47 on dendritic cells governs their trafficking to secondary lymphoid organs. EMBO J. 25:5560–5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ciofani, M., and J.C. Zuniga-Pflucker. 2005. Notch promotes survival of pre-T cells at the beta-selection checkpoint by regulating cellular metabolism. Nat. Immunol. 6:881–888. [DOI] [PubMed] [Google Scholar]
- 37.Deftos, M.L., Y.W. He, E.W. Ojala, and M.J. Bevan. 1998. Correlating notch signaling with thymocyte maturation. Immunity. 9:777–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Visan, I., J.B. Tan, J.S. Yuan, J.A. Harper, U. Koch, and C.J. Guidos. 2006. Regulation of T lymphopoiesis by Notch1 and Lunatic fringe- mediated competition for intrathymic niches. Nat. Immunol. 7:634–643. [DOI] [PubMed] [Google Scholar]
- 39.Kuroda, K., H. Han, S. Tani, K. Tanigaki, T. Tun, T. Furukawa, Y. Taniguchi, H. Kurooka, Y. Hamada, S. Toyokuni, and T. Honjo. 2003. Regulation of marginal zone B cell development by MINT, a suppressor of Notch/RBP-J signaling pathway. Immunity. 18:301–312. [DOI] [PubMed] [Google Scholar]
- 40.Hozumi, K., N. Negishi, D. Suzuki, N. Abe, Y. Sotomaru, N. Tamaoki, C. Mailhos, D. Ish-Horowicz, S. Habu, and M.J. Owen. 2004. Delta-like 1 is necessary for the generation of marginal zone B cells but not T cells in vivo. Nat. Immunol. 5:638–644. [DOI] [PubMed] [Google Scholar]
- 41.Varol, C., L. Landsman, D.K. Fogg, L. Greenshtein, B. Gildor, R. Margalit, V. Kalchenko, F. Geissmann, and S. Jung. 2007. Monocytes give rise to mucosal, but not splenic, conventional dendritic cells. J. Exp. Med. 204:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Minter, L.M., D.M. Turley, P. Das, H.M. Shin, I. Joshi, R.G. Lawlor, O.H. Cho, T. Palaga, S. Gottipati, J.C. Telfer, et al. 2005. Inhibitors of gamma-secretase block in vivo and in vitro T helper type 1 polarization by preventing Notch upregulation of Tbx21. Nat. Immunol. 6:680–688. [PubMed] [Google Scholar]
- 43.Muyrers, J.P., Y. Zhang, G. Testa, and A.F. Stewart. 1999. Rapid modification of bacterial artificial chromosomes by ET-recombination. Nucleic Acids Res. 27:1555–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuhn, R., F. Schwenk, M. Aguet, and K. Rajewsky. 1995. Inducible gene targeting in mice. Science. 269:1427–1429. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}