

Abstract
The molecular mechanisms underlying the initiation of innate and adaptive proallergic type 2 responses are not understood. Interleukin (IL) 25, a member of the IL-17 cytokine family, was recently reported (Owyang, A.M., C. Zaph, E.H. Wilson, K.J. Guild, T. McClanahan, H.R. Miller, D.J. Cua, M. Goldschmidt, C.A. Hunter, R.A. Kastelein, and D. Artis. 2006. J. Exp. Med. 203:843–849; Fallon, P.G., S.J. Ballantyne, N.E. Mangan, J.L. Barlow, A. Dasvarma, D.R. Hewett, A. McIlgorm, H.E. Jolin, and A.N. McKenzie. 2006. J. Exp. Med. 203:1105–1116) to be important in Th2 cell–mediated immunity to parasitic infection. However, the cellular source and targets of IL-25 are not well understood. We show that mouse IL-25 is expressed by lung epithelial cells as a result of innate immune responses to allergens. Transgenic overexpression of IL-25 by these cells leads to mucus production and airway infiltration of macrophages and eosinophils, whereas blockade of IL-25 conversely reduces the airway inflammation and Th2 cytokine production in an allergen-induced asthma model. In addition, IL-25, with a receptor more highly expressed in Th2 than other effector T cells, promotes Th2 cell differentiation in an IL-4– and signal transducer and activator of transcription 6–dependent manner. During early T cell activation, IL-25 potentiates expression of the nuclear factor of activated T cells c1 and JunB transcription factors, which possibly results in increased levels of initial IL-4 production, up-regulation of GATA-3 expression, and enhanced Th2 cell differentiation. Thus, IL-25 is a critical factor regulating the initiation of innate and adaptive proallergic responses.
Allergic response to environmental allergens is a common health problem. It is still unclear how allergens trigger the innate immune system, which in turn induces long-lasting type 2 adaptive responses. Epithelial cells, as the first cell type encountering allergens, have been shown to initiate tissue inflammatory responses by producing various cytokines and chemokines, which may create a permissive environment for the recruitment of inflammatory cells and amplification of allergic responses. CD4+ Th cells are essential regulators in chronic allergic diseases. Upon activation, Th cells undergo differentiation into functionally distinct effector subsets. Th1 cells produce IFN-γ and regulate cellular immunity, whereas Th2 cells produce IL-4, IL-5, and IL-13 and mediate humoral immunity and allergic responses (1, 2). A novel Th cell subset expressing IL-17 regulates tissue inflammatory responses (3). During Th2 cell differentiation, in which GATA-3 serves as a master transcription factor (2), IL-4 produced by activated Th cells is essential for driving Th2 cell differentiation (1). NFATc1 and JunB have been shown to be important in regulating early IL-4 production and in determining Th2 cell differentiation (4). The terminal differentiation of Th cells is determined by the innate immune system and the cytokines it produces. Despite the molecular understanding of the Th2 cell regulatory programs, whether there is a signal induced during the innate response to allergens that augments IL-4 expression by T cells and regulates Th2 cell differentiation remains unclear.
IL-25 (also known as IL-17E), a member of the IL-17 family, has been implicated in Th2 cell–mediated immunity (5, 6). IL-25 shares a receptor, named IL-17RB, with IL-17B, although it binds with much higher affinity than IL-17B (7). IL-17RB mRNA transcripts are abundantly expressed in the kidney and liver and at lower levels in the testes, brain, small intestine, and other organs (7). Overexpression of IL-25 or administration of recombinant cytokine strongly induced Th2 cytokine expression and IgE production in vivo and resulted in allergic pathologies (5, 6, 8, 9). Conversely, mice deficient in IL-25 exhibited reduced Th2 cytokine production in the draining lymph nodes, resulting in poor resistance to Trichuris infection (10, 11). The molecular mechanisms whereby IL-25 regulates type 2 immunity are still unclear. A non–B/T cell population was found to be regulated by IL-25 and produce Th2 cytokines in response to Nippostrongylus brasiliensis infection (11). However, given the broad expression of IL-17RB, additional targets of IL-25 may exist to regulate allergic responses.
In this report, we found that IL-25 mRNA was up-regulated in epithelial cells after allergen stimulation. Chronic overexpression of IL-25 in airway epithelium resulted in asthma symptoms, whereas blockade of IL-25 attenuated allergen-induced lung inflammation and Th2 cytokine expression. Moreover, we found that IL-25 directly promoted Th2 cell differentiation and GATA-3 expression in an IL-4– and STAT6-dependent manner. IL-25 enhanced NFATc1 and JunB expression to potentiate early IL-4 expression and the expansion and cytokine production of effector Th2 cells.
RESULTS AND DISCUSSION
Expression of IL-25 by lung epithelial cells initiates allergic responses
In an attempt to define the initial innate mechanism in response to allergens, we treated a mouse lung epithelial cell line, MLE12, with the allergens Aspergillus oryzae and ragweed and characterized gene expression changes by RT-PCR. In addition to several chemokine genes (not depicted), we found that IL-25 mRNA was substantially induced in response to the allergens (Fig. 1 A). The expression of IL-25 by lung epithelial cells was further confirmed using a human lung epithelial cell line, A549, and primary type II alveolar epithelial cells isolated from C57BL/6 mice (Fig. 1 A). Our data are consistent with a previous observation of up-regulation of IL-25 mRNA in lung tissue in response to A. fumigatus infection (5), suggesting that IL-25 induction in lung epithelial cells may play a role in initiating a type II immune response to allergens.
Figure 1.

IL-25 is expressed by lung epithelial cells and regulates allergic responses. (A) MLE12 cells, A549 cells, and primary type II alveolar epithelial cells stimulated with the indicated stimuli were analyzed by RT-PCR for IL-25 mRNA expression after normalization with HPRT expression. (B) Histological analysis of lung tissues from 3-mo-old IL-25 transgenic mice (b) stained with H&E was compared with that from control littermate mice (a). Bars, 500 μm. Macrophages (AM) and eosinophils (Eo) infiltrated in the airway were demonstrated by H&E and Giemsa staining (c), and mucus hyperplasia stained with PAS in 5-mo-old IL-25 transgenic-positive mice are shown (d). Bars: (c) 50 μm; (d) 200 μm. (C) RT-PCR analysis of gene expression in 3-mo-old IL-25 transgenic mice was compared with littermate controls. (D) MLE12 cells treated for 6 h with IL-25 were subject to RT-PCR analysis in comparison with the nontreated cells. (E and F) C57BL/6 mice (4–5 mice in each group) were intranasally challenged with A. oryzae allergen and OVA every other day for a total of six challenges. An anti–IL-25 mAb or control rat Ig was intraperitoneally administered at the time of each challenge. 24 h after the final challenging, mice were killed, and BAL cells were collected and analyzed for the absolute numbers of eosinophils and CD4+ T cells by total cell counting, differential cell counts, and flow cytometry analysis with mAbs to CCR3 (for eosinophils) and CD4 (BD Biosciences). BAL fluid was examined for cytokine expression by ELISA. Horizontal bars in E represent the means. Data are presented as mean values + SD and are representative of two independent experiments. HPRT, hypoxanthine-guanine phosphoribosyltransferase. *, P < 0.05.
To address the function of IL-25 produced by lung epithelial cells in vivo, we generated transgenic mice that overexpress IL-25 in lung epithelium using the CC10 promoter. Four founder lines were generated, of which two expressed high levels of IL-25 mRNA in the lung tissue and were selected for histological analysis. Transgenic mice exhibited greatly increased levels of IL-25 mRNA expression in their lung tissues and protein in the bronchoalveolar lavage (BAL) fluid (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20061675/DC1). Compared with littermate controls, IL-25 transgenic mice exhibited epithelial cell hyperplasia at 3 mo of age; after 3 mo of age, we observed goblet cell hyperplasia, epithelial cell hyperplasia, and infiltration of moderate numbers of eosin-stained enlarged macrophages and considerable numbers of eosinophils in the lung parenchyma (Fig. 1 B). Similar macrophage infiltration was found in cc10–IL-17 (12) and cc10–IL-17F (unpublished data) mice, whereas eosinophils were only seen in cc10–IL-25 mice. However, cc10–IL-13 transgenic mice were also found to have lung infiltration by macrophages and eosinophils (13). By performing periodic acid Schiff (PAS) staining, mucus-producing cells were found only in IL-25 transgenic mice and not in the control littermates (Fig. 1 B). In older IL-25 transgenic mice between 7 and 10 mo of age, substantial infiltration of CD4+ T cells and CD11C+ dendritic cells were found around blood vessels (unpublished data). IL-25 overproduction in lung epithelium thus led to allergic inflammation in the lung.
To further investigate the regulation of allergic responses by IL-25, we examined cytokine and chemokine expression profiles in lung tissues of IL-25 transgenic mice and their littermate controls. The expression of eotaxin1 (Ccl11), eotaxin2 (Ccl24), and MDC (Ccl22), which play crucial roles in the recruitment of eosinophil and Th2 cells, respectively, were detected in IL-25 transgenic mice (Fig. 1 C). Interestingly, mRNA for MUC5ac, the protein product of which constitutes a major component of respiratory lining fluid, was elevated in IL-25 transgenic mice, correlating with mucus hypersecretion in the airways of these mice. To determine whether these chemokines and mucin are direct targets of IL-25, the MLE12 cell line was treated with IL-25, and the resulting gene expression changes were determined. Expression of chemokines relevant to eosinophil (eotaxin1) and Th2 cell (regulated on activation, normal T cell expressed and secreted/CCL5 and MDC) recruitment, as well as MUC5ac, was strongly induced by IL-25 (Fig. 1 D). In addition, we also observed up-regulation of TSLP, which was recently established as a key factor in allergic inflammation (14). Collectively, these data indicate that IL-25 induced by allergens in epithelial cells can function to initiate allergic responses.
To further examine the role of IL-25 during immune responses to allergens, a panel of rat anti–mouse IL-25 mAbs was generated and screened for its abilities to inhibit the binding of IL-25 to 293 cells transfected with IL-17RB. One clone (35B) was identified as an antagonistic antibody (unpublished data), which was used in an allergen-induced experimental asthma model. C57BL/6 mice were intranasally sensitized with allergens derived from A. oryzae together with chicken OVA protein every other day for a total of six challenges, as previously described (15). The airway pathology developed in this model was previously found to be dependent on IL-4 (16). Anti–IL-25 antibody or a control rat IgG was intraperitoneally administered at the time of each challenge. 24 h after the last allergen administration, the BAL and lungs were collected. In BAL fluid, the presence of inflammatory cells was assessed by differential cell counting and flow cytometry analysis, and the levels of cytokine production were determined by ELISA. Compared with mice treated with a control rat antibody, anti–IL-25 substantially reduced the number of eosinophils, an important indicator of airway inflammation; the infiltration of CD4+ T cells was moderately reduced (Fig. 1 E). Although the production of IFN-γ in BAL was moderately increased and TNF-α was not affected, Th2 cytokines (IL-4, IL-5, and IL-13) were all significantly reduced by anti–IL-25 treatment (Fig. 1 F), indicating that the IL-25 blockade may attenuate allergic airway inflammation through the reduction of the Th2 response. However, we observed no change in the goblet cell hyperplasia by anti–IL-25 treatment, suggesting that the remaining cytokines or other factors were sufficient in this regulation. For example, IL-13 has been previously indicated as an important cytokine with a distinct role from IL-4 in regulating the development of allergic asthma symptoms, including airway hyperesponsiveness, goblet cell hyperplasia, tissue remodeling, and fibrosis (17). To further investigate the role of IL-25 in regulating T cell function during allergic responses, we restimulated splenocytes from the described experiment ex vivo with OVA protein for 3 d and measured their cytokine production. Similar to the BAL data, antigen-specific Th2 responses in cells from anti–IL-25–treated mice were reduced, especially the production of IL-5 and IL-13, whereas no alteration in the levels of IFN-γ and TNF-α was observed (unpublished data). Our data suggest that IL-25 regulates Th2 cell function during allergic responses. This result is consistent with recent studies on the important function of IL-25 in mediating antiparasitic immune responses (10, 11) and in airway inflammation (18), suggesting that allergens and large parasites may use a common pathway in activation of Th2 responses.
IL-25 promotes Th2 cell differentiation in vitro
The inhibitory effect by anti–IL-25 or IL-25 gene deletion on Th2 responses in vivo suggests a role, direct or indirect, of IL-25 in Th2 cell differentiation. Recently, high-level expression of IL-17RB by in vitro–derived Th2 cells and circulating human Th2 cell–biased memory cells in vivo has been previously reported (19–21), suggesting a possible function of IL-25 on T cells. To further investigate this idea, we first examined expression of the IL-25 receptor IL-17RB on different mouse Th cell populations. IL-17RB expression was detected on freshly isolated naive CD4+ T cells. After in vitro differentiation, Th2 but not Th1 cells maintained this expression (Fig. 2 A). Moreover, IL-17RB expression was not detected in in vitro–generated IL-17–producing Th cells (unpublished data), indicating that IL-17RB expression is restricted to Th2 cells among three effector Th cell subsets.
Figure 2.

IL-25 promotes Th2 cell differentiation in vitro. (A) mRNA expression of IL-17RB on freshly isolated naive T cells and in vitro–generated Th1 and Th2 cells was analyzed by RT-PCR. (B) Naive T cells were stimulated with anti-CD3, anti-CD28, and IL-2 in the presence of human IgG or recombinant IL-25. After 4 d, cells were restimulated with plate-bound anti-CD3 for 24 h, and cytokine production was measured by ELISA. *, P < 0.005. (C) Naive T cells were activated as described with or without anti–IFN-γ in the presence of human IgG or recombinant IL-25. After 5 d, cells were restimulated with PMA/ionomycin, and the expression of IL-4 and IFN-γ was analyzed. Numbers within the quadrants indicate the percentage of cells stained positive for each respective cytokine. (D) Naive T cells were purified and stimulated with anti-CD3, anti-CD28, anti–IFN-γ, and IL-2 in the presence of different concentrations of IL-25 for 6 d. 50 μg/ml anti–IL-25 mAb was added to the culture containing IL-25 at 2 μg/ml. The amounts of cytokines were measured by ELISA. Data are presented as mean values + SD and are representative of at least two independent experiments. HPRT, hypoxanthine-guanine phosphoribosyltransferase.
The selective expression of IL-17RB suggests that IL-25 may play a role in regulating Th2 cell differentiation. To assess this, CD44loCD62Lhi naive CD4+ T cells from C57BL/6 mice were cultured with anti-CD3, anti-CD28, and IL-2 in the presence or absence of IL-25. 4 d later, differentiated cells were restimulated with anti-CD3, and cytokine expression was examined by ELISA. Although T cells activated in the absence of IL-25 predominately differentiated into IFN-γ–producing Th1 cells, IL-25 treatment led to greatly increased levels of IL-4, IL-5, and IL-13 expression and moderately reduced amounts of IFN-γ expression by effector T cells (Fig. 2 B). By intracellular cytokine staining, we found that IL-25 treatment resulted in substantially increased numbers of IL-4–producing Th2 cells, which was more pronounced when IFN-γ was neutralized (Fig. 2 C). The effect of IL-25 on Th2 cytokine expression was dose dependent and inhibited by anti–IL-25 antibody (Fig. 2 D). Our data indicate a novel function of IL-25 in directly promoting Th2 cell differentiation.
IL-25 promotes Th2 cell differentiation in an IL-4– and STAT6-dependent manner
Th2 cell differentiation is dependent on autocrine IL-4 production by T cells. To understand the mechanism whereby IL-25 regulates Th2 cell differentiation, we assessed cytokine gene expression during T cell differentiation. Naive CD4+ T cells were activated with or without IL-25. On days 1, 2 and 3 after stimulation, cytokine gene expression was analyzed by real-time RT-PCR. IL-25 up-regulated IL-4 and IL-5 gene expression while down-regulating the expression of IFN-γ after 2 d of activation (Fig. 3 A). The induction of IL-4 and other Th2 cytokines was increasingly pronounced by day 3 of activation, suggesting that IL-25 may regulate early Th2 cytokine gene expression. To determine whether IL-25–mediated Th2 cell differentiation depends on early IL-4 production by T cells, we performed the Th cell differentiation experiment in the presence of neutralizing anti–IL-4 antibody. Anti–IL-4 completely abolished IL-4 and IL-5 production and moderately inhibited IL-13 induction, as determined by ELISA and real-time RT-PCR analysis on day 4 after activation (Fig. 3, B and C). Similarly, although IL-25 promoted Th2 cell differentiation in wild-type T cells, IL-25 was not able to induce IL-4 and IL-5 production by IL-4–deficient cells (Fig. 3 D), though residual expression of IL-13 was still detectable. Thus, IL-25–mediated Th2 cell differentiation is dependent on IL-4.
Figure 3.

IL-25 promotes Th2 cell differentiation in an IL-4-dependent manner. (A) Naive T cells were purified and stimulated with anti-CD3, anti-CD28, and IL-2 with or without IL-25. Cells were collected at the indicated time points for real-time PCR analysis. (B and C) Naive T cells were stimulated with anti-CD3, anti-CD28, and IL-2 with or without IL-25 or with the addition of anti–IL-4 mAb. After 4 d of culture, cells were washed and restimulated with anti-CD3 for 4 h to analyze the cytokine expression by real-time RT-PCR or for 24 h to measure the level of cytokine expression by ELISA. *, P < 0.05. (D) Naive T cells from IL-4–deficient mice were stimulated as described with or without IL-25 and restimulated to measure the level of cytokine expression by ELISA. Data are presented as mean values + SD and are representative of two experiments. *, P < 0.05.
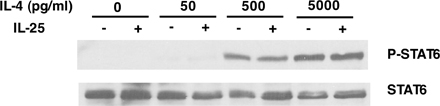
STAT proteins are selectively required in cytokine-mediated Th cell lineage differentiation. Because IL-25 promotion of Th2 cell differentiation is dependent on IL-4, we tested whether STAT6 is involved. Naive CD4+ T cells were activated with anti-CD3 and anti-CD28 with or without IL-25 or with IL-25 and anti–IL-4 neutralizing antibody for 2 d and subjected them to a Western blot analysis of STAT3, STAT5, and STAT6 phosphorylation. IL-6, IL-2, and IL-4 were included as the positive controls for STAT3, STAT5, and STAT6 phosphorylation, respectively. IL-25 treatment, although not affecting STAT3 and STAT5 phosphorylation, enhanced that of STAT6 (Fig. 4 A). Because STAT6 phosphorylation was completely abolished by an anti–IL-4 antibody (Fig. 4 A), STAT6 is likely activated by IL-4. To further address the possibility that IL-25 directly phoshorylates STAT6 or enhances IL-4 signaling, naive Th cells isolated from IL-4–deficient mice were activated with anti-CD3 and CD28 in the presence of different concentration of exogenous IL-4 with or without IL-25. In the absence of endogenous IL-4 expression, IL-25 did not activate or enhance IL-4–induced STAT6 phosphorylation (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20061675/DC1), supporting IL-25–potentiated STAT6 phosphorylation by inducing increased levels of early IL-4. Next, we further determined whether STAT6 is required for IL-25–mediated Th2 cell differentiation. Wild-type or STAT6-deficient naive Th cells were differentiated in the presence or absence of IL-25, as described in the previous paragraph. Although IL-25 enhanced IL-4 and IL-5 production in wild-type Th cells, no Th2 cytokine induction by IL-25 was observed in STAT6-deficient cells (Fig. 4 B). These data support the conclusion that the regulation of Th2 cell differentiation by IL-25 is dependent on the IL-4–STAT6 signaling pathway.
Figure 4.

IL-25 promotes Th2 cell differentiation in a STAT6-dependent manner. (A) Naive T cells were purified and stimulated with anti-CD3 and anti-CD28 with or without IL-25. IL-4, IL-2, and IL-6 were included in the treatment as positive controls for STAT6, STAT5, and STAT3 phosphorylation, respectively. Cells were collected at 48 h for Western blot analysis. (B) Purified naive T cells from STAT6-deficient and wild-type mice (BALB/c) were stimulated with anti-CD3, anti-CD28, and IL-2 with or without IL-25. After 5 d of culture, cells were restimulated with PMA/ionomycin to assess the levels of IL-4 and IL-5 and IFN-γ expression. Numbers in the quadrants/gates represent the percentage of cells stained positive for each respective cytokine. SSC, side scatter.
IL-25 induces early IL-4 expression through JunB and NFATc1
Early IL-4 production, through STAT6, plays an important role in regulating the expression of GATA-3, a master regulator of Th2 cell development. To examine the molecular mechanism whereby IL-25 regulates Th2 cell differentiation, we assessed the expression of GATA-3. Naive T cells were activated in the absence or presence of IL-25. At day 2 after activation, T cells were analyzed for the mRNA expression of GATA-3 and T-bet by real-time PCR analysis. Although no change in the T-bet expression level was detected, considerable up-regulation of GATA-3 mRNA was observed upon IL-25 stimulation (Fig. 5 A). Similarly, GATA-3 protein expression was enhanced by IL-25 at this time point (Fig. 5 A). GATA-3 mRNA and protein expression was inhibited by anti–IL-4 antibody (Fig. 5 A). These results indicate a function of IL-25 in GATA-3 regulation, which is dependent on IL-4.
Figure 5.

The function of IL-25 in early Th cell activation and in effector Th2 cells. (A) Naive T cells were stimulated with anti-CD3 and anti-CD28 with or without IL-25 or with the addition of anti–IL-4 antibody. Cells were collected at 72 h for real-time PCR or 48 h for Western blot analysis. (B) Purified naive T cells from transgenic OT-II mice were differentiated into Th1 and Th2 cells. After 5 d of culture, cells were washed and restimulated with anti-CD3 with or without IL-25 for 3 d to assess the levels of IL-4 and IFN-γ expression by intracellular cytokine staining. The percentages in the figure represent cells stained positive for each cytokine indicated. (C) Purified naive T cells from C57BL/6 mice were stimulated with anti-CD3 and anti-CD28 with the same treatment as in B. After 5 d of culture, Th1 and Th2 cells were washed and restimulated with anti-CD3 with or without IL-25 for 2 d to assess the levels of IL-4, IL-5, IFN-γ, and TNF-α expression by ELISA. Data are presented as mean values + SD. *, P < 0.05.
To understand the mechanisms whereby IL-25 regulates early IL-4 production, we also measured the expression of additional transcription regulators. NFATc1 and JunB are important in controlling early IL-4 expression during Th2 cell differentiation (4). Interestingly, we found that the protein expression of NFATc1 and JunB was substantially increased in the presence of IL-25, and this up-regulation could not be completely blocked by anti–IL-4 (Fig. 5 A). Our results suggest that IL-25 may regulate early IL-4 expression directly through potentiating the induction of NFATc1 and JunB, which leads to IL-4–dependent up- regulation of GATA-3 expression.
Regulation of effector Th2 cells by IL-25
Because IL17RB expression is restricted to Th2 cells, we next examined whether IL-25 plays an additional role in polarized Th2 cells. Naive T cells isolated from OT-II TCR transgenic mice were differentiated into Th1 or Th2 cell lineages. After 5 d of polarization, Th1 and Th2 cells were restimulated with anti-CD3 in the absence or presence of IL-25. As shown by intracellular cytokine analysis of IL-4 and IFN-γ expression, IL-25 treatment resulted in an increased number of IL-4–expressing Th2 cells while having no effect on IFN-γ–secreting Th1 cells (Fig. 5 B). Consistently, IL-25 also enhanced the production of IL-4 and IL-5 in Th2 cells generated from naive T cells isolated from C57BL/6 mice but not IFN-γ and TNF-α production in Th1 cells (Fig. 5 C). Thus, IL-25 functions not only by potentiating early IL-4 production to promote Th2 cell differentiation but also by enhancing the expansion and cytokine production of effector Th2 cells.
In summary, we found that IL-25, up-regulated in lung epithelial cells exposed to allergens, functions to regulate proallergic responses in vivo. More importantly, we discovered a direct function of IL-25 on Th cells in promoting Th2 cell differentiation and enhancing Th2 cytokine production in polarized Th2 cells. We identified NFATc1 and JunB as the transcription factors directly regulated by IL-25 to increase early IL-4 production, which results in enhanced GATA-3 expression and Th2 cell differentiation. This action of IL-25 resembles those of inducible T cell co-stimulator (22) and OX40 (23). As a result of polarized differentiation, Th2 cells selectively express IL-17RB and respond to IL-25 for their expansion and cytokine production. All of these data support an important function of IL-25 in type II immunity; IL-25 may serve as a therapeutic target for diseases such as allergic asthma. In addition, recent findings by Owyang et al. using a gut infection model indicated that IL-25 is required for limiting type 1 inflammation (10). The mechanisms we identified in this report whereby IL-25 regulates type II responses may apply to nonallergic systems and may be beneficial in protecting hosts from uncontrolled immune-mediated pathology.
MATERIALS AND METHODS
Generation of IL-25 recombinant protein and anti–IL-25 mAbs.
Recombinant mouse IL-25–Ig protein was prepared as we previously reported (24). Anti–IL-25 mAbs were generated as previously described (25).
Generation and analysis of IL-25 transgenic mice.
Full-length cDNA encoding mouse IL-25 was cloned into the cc10 promoter construct (a gift from J. Elias, Yale University, New Haven, CT), as we previously did for the IL-17 transgenic mouse (12). The IL-25 transgene construct was microinjected into C57BL/6 mice at the Transgenic Mouse Facility at the University of Washington. Four IL-25 transgenic founders were obtained and bred with B6 mice. Two transgenic lines expressing high IL-25 mRNA levels were further characterized. The level of IL-25 expression was determined by RT-PCR and ELISA using an ELISA kit (R&D Systems). For histological analysis of IL-25 transgenic mice, 6–40-wk-old IL-25 transgenic mice or littermate controls were killed, and individual lungs were fixed in 10% buffer formalin or frozen. Hematoxylin and eosin (H&E), Giemsa, and PAS staining were performed by Histology Consultation Services, Inc.
Lung epithelial cell culture.
Mouse lung epithelial (MLE12) and human lung epithelial (A549) cell lines were obtained from American Type Culture Collection. Primary lung epithelial cells were isolated from C57BL/6 mice, as previously described (26), after depletion with anti-CD32/CD16 and anti-CD45 antibodies (Miltenyi Biotec). Cells were treated with 200 ng/ml A. oryzae protease or ragweed. After 6 h of stimulation, cells were collected and lysed, and mRNA was extracted by using TRIzol reagent (Invitrogen).
Induction and analysis of an asthma model.
C57BL/6 mice were induced with airway inflammation by A. oryzae for six total challenges, as previously described (15). Anti–IL-25 or 100 μg of a control rat Ig was given intraperitoneally at the time of challenge. 24 h after the last challenge, mice were killed for analysis of BAL fluid. BAL cells were collected and further subjected to differential cell counts and flow cytometry analysis by staining with mAb to CCR3 for eosinophils and CD4 (BD Biosciences). BAL fluid was measured for cytokines by ELISA. All animal studies were approved by the MD Anderson Cancer Center Institutional Animal Care and Use Committee.
Th cell differentiation.
Lymph nodes and spleens of various mice were FACS-sorted based on the CD4+CD62LhiCD44lo surface phenotypes (96–99% purity) and stimulated with 2 μg/ml of plate-bound anti-CD3 plus 5 μg/ml of soluble anti-CD28 and 50 U/ml of human IL-2 in the presence of 2 μg/ml of human Ig or IL-25Ig or 500 ng/ml of human IL-25 (R&D Systems). After 4 d of restimulation, cells were washed and restimulated with 2 μg/ml of plate-bound anti-CD3 for 24 h, and culture supernatants were collected and analyzed for cytokines by ELISA. Naive T cells from OT-II mice purified by FACS sorting were differentiated into Th1 or Th2 cells, as we previously reported (27). After 5 d of stimulation, Th1 and Th2 cells were restimulated with 0.5 μg/ml anti-CD3 with or without IL-25 for an additional 3 d. For intracellular cytokine analysis, cells were restimulated with 500 ng/ml ionomycin and 50 ng/ml PMA in the presence of GolgiStop (BD Biosciences) for 5 h. Cells were permeabilized with a Cytofix/Cytoperm kit (BD Biosciences) and analyzed for the expression of IFN-γ, IL-4, and IL-5.
RT-PCR analysis.
Total RNA extracted using TRIzol reagent was used to generate cDNA using oligo-dT, random hexamers, and SuperScript RT II (Invitrogen). For quantitation of cytokine and transcription factor gene expression, cDNA samples were amplified in iQ SYBR Green Supermix (Bio-Rad Laboratories). The primer pairs used for RT-PCR are shown in Table S1 (available at http://www.jem.org/cgi/content/full/jem.20061675/DC1).
Immunoblot analysis.
Cell lysates prepared as previously reported (22) were subjected to Western blot analysis with anti–phospho-STAT6 (Tyr641), anti–phospho-STAT5 (Tyr694), anti–phospho-STAT3 (Tyr705), STAT3, STAT5a, and STAT6 antibodies (provided by K. Arima and E. Esashi, University of Texas MD Anderson Cancer Center, Houston, TX) and antiactin (Sigma-Aldrich), anti–GATA-3, and anti-JunB (Santa Cruz Biotechnology, Inc.) antibodies. The signal was detected with ECL reagent (Pierce Chemical Co.).
Statistical analysis.
Data are presented as mean values + SD. Data were analyzed using the Student's t test (n = 2 groups). P < 0.05 was considered significant.
Online supplemental material.
Fig. S1 shows an analysis of IL-25 expression by real-time PCR and ELISA in transgenic mice and control littermates. Fig. S2 depicts increased STAT6 phosphorylation in IL-4– deficient T cells in response to exogenous IL-4 but not IL-25 treatment. Table S1 provides the primer pairs used for RT-PCR analysis. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20061675/DC1.
Supplemental Material
Acknowledgments
We thank Julie Duong, Smita Aggarwal, and Ying Wang for assistance; Dr. Jack Elias for the cc10 promoter construct; Dr. Kazuhiko Arima for Western blot reagents; and the entire Dong lab for their help and discussion.
Supported in part by National Institutes of Health grants (to C. Dong). P. Angkasekwinai received a fellowship from the Royal Thai government, Y.-J. Liu received a Senior Sandler Asthma Research Award, and C. Dong is a Cancer Research Institute Investigator, an American Lung Association Career Investigator, and a Trust Fellow of the MD Anderson Cancer Center.
The authors have no conflicting financial interests.
References
- 1.Murphy, K.M. 1998. T lymphocyte differentiation in the periphery. Curr. Opin. Immunol. 10:226–232. [DOI] [PubMed] [Google Scholar]
- 2.Dong, C., and R.A. Flavell. 2000. Cell fate decision: T-helper 1 and 2 subsets in immune responses. Arthritis Res. 2:179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dong, C. 2006. Diversification of T-helper-cell lineages: finding the family root of IL-17-producing cells. Nat. Rev. Immunol. 6:329–334. [DOI] [PubMed] [Google Scholar]
- 4.Dong, C., and R.A. Flavell. 2000. Control of T helper cell differentiation—in search of master genes. Sci. STKE. 2000:PE1. [DOI] [PubMed]
- 5.Hurst, S.D., T. Muchamuel, D.M. Gorman, J.M. Gilbert, T. Clifford, S. Kwan, S. Menon, B. Seymour, C. Jackson, T.T. Kung, et al. 2002. New IL-17 family members promote Th1 or Th2 responses in the lung: in vivo function of the novel cytokine IL-25. J. Immunol. 169:443–453. [DOI] [PubMed] [Google Scholar]
- 6.Fort, M.M., J. Cheung, D. Yen, J. Li, S.M. Zurawski, S. Lo, S. Menon, T. Clifford, B. Hunte, R. Lesley, et al. 2001. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 15:985–995. [DOI] [PubMed] [Google Scholar]
- 7.Lee, J., W.H. Ho, M. Maruoka, R.T. Corpuz, D.T. Baldwin, J.S. Foster, A.D. Goddard, D.G. Yansura, R.L. Vandlen, W.I. Wood, and A.L. Gurney. 2001. IL-17E, a novel proinflammatory ligand for the IL-17 receptor homolog IL-17Rh1. J. Biol. Chem. 276:1660–1664. [DOI] [PubMed] [Google Scholar]
- 8.Pan, G., D. French, W. Mao, M. Maruoka, P. Risser, J. Lee, J. Foster, S. Aggarwal, K. Nicholes, S. Guillet, et al. 2001. Forced expression of murine IL-17E induces growth retardation, jaundice, a Th2-biased response, and multiorgan inflammation in mice. J. Immunol. 167:6559–6567. [DOI] [PubMed] [Google Scholar]
- 9.Kim, M.R., R. Manoukian, R. Yeh, S.M. Silbiger, D.M. Danilenko, S. Scully, J. Sun, M.L. DeRose, M. Stolina, D. Chang, et al. 2002. Transgenic overexpression of human IL-17E results in eosinophilia, B-lymphocyte hyperplasia, and altered antibody production. Blood. 100:2330–2340. [DOI] [PubMed] [Google Scholar]
- 10.Owyang, A.M., C. Zaph, E.H. Wilson, K.J. Guild, T. McClanahan, H.R. Miller, D.J. Cua, M. Goldschmidt, C.A. Hunter, R.A. Kastelein, and D. Artis. 2006. Interleukin 25 regulates type 2 cytokine-dependent immunity and limits chronic inflammation in the gastrointestinal tract. J. Exp. Med. 203:843–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fallon, P.G., S.J. Ballantyne, N.E. Mangan, J.L. Barlow, A. Dasvarma, D.R. Hewett, A. McIlgorm, H.E. Jolin, and A.N. McKenzie. 2006. Identification of an interleukin (IL)-25–dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J. Exp. Med. 203:1105–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park, H., Z. Li, X.O. Yang, S.H. Chang, R. Nurieva, Y.H. Wang, Y. Wang, L. Hood, Z. Zhu, Q. Tian, and C. Dong. 2005. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 6:1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu, Z., R.J. Homer, Z. Wang, Q. Chen, G.P. Geba, J. Wang, Y. Zhang, and J.A. Elias. 1999. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J. Clin. Invest. 103:779–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu, Y.J. 2006. Thymic stromal lymphopoietin: master switch for allergic inflammation. J. Exp. Med. 203:269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kheradmand, F., A. Kiss, J. Xu, S.H. Lee, P.E. Kolattukudy, and D.B. Corry. 2002. A protease-activated pathway underlying Th cell type 2 activation and allergic lung disease. J. Immunol. 169:5904–5911. [DOI] [PubMed] [Google Scholar]
- 16.Corry, D.B., G. Grunig, H. Hadeiba, V.P. Kurup, M.L. Warnock, D. Sheppard, D.M. Rennick, and R.M. Locksley. 1998. Requirements for allergen-induced airway hyperreactivity in T and B cell-deficient mice. Mol. Med. 4:344–355. [PMC free article] [PubMed] [Google Scholar]
- 17.Wynn, T.A. 2003. IL-13 effector functions. Annu. Rev. Immunol. 21:425–456. [DOI] [PubMed] [Google Scholar]
- 18.Tamachi, T., Y. Maezawa, K. Ikeda, S. Kagami, M. Hatano, Y. Seto, A. Suto, K. Suzuki, N. Watanabe, Y. Saito, et al. 2006. IL-25 enhances allergic airway inflammation by amplifying a TH2 cell-dependent pathway in mice. J. Allergy Clin. Immunol. 118:606–614. [DOI] [PubMed] [Google Scholar]
- 19.Wang, Y.H., T. Ito, B. Homey, N. Watanabe, R. Martin, C.J. Barnes, B.W. McIntyre, M. Gilliet, R. Kumar, Z. Yao, and Y.J. Liu. 2006. Maintenance and polarization of human TH2 central memory T cells by thymic stromal lymphopoietin-activated dendritic cells. Immunity. 24:827–838. [DOI] [PubMed] [Google Scholar]
- 20.Chtanova, T., S.G. Tangye, R. Newton, N. Frank, M.R. Hodge, M.S. Rolph, and C.R. Mackay. 2004. T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. J. Immunol. 173:68–78. [DOI] [PubMed] [Google Scholar]
- 21.Bosco, A., K.L. McKenna, C.J. Devitt, M.J. Firth, P.D. Sly, and P.G. Holt. 2006. Identification of novel Th2-associated genes in T memory responses to allergens. J. Immunol. 176:4766–4777. [DOI] [PubMed] [Google Scholar]
- 22.Nurieva, R.I., J. Duong, H. Kishikawa, U. Dianzani, J.M. Rojo, I. Ho, R.A. Flavell, and C. Dong. 2003. Transcriptional regulation of th2 differentiation by inducible costimulator. Immunity. 18:801–811. [DOI] [PubMed] [Google Scholar]
- 23.So, T., J. Song, K. Sugie, A. Altman, and M. Croft. 2006. Signals from OX40 regulate nuclear factor of activated T cells c1 and T cell helper 2 lineage commitment. Proc. Natl. Acad. Sci. USA. 103:3740–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun, M., S. Richards, D.V. Prasad, X.M. Mai, A. Rudensky, and C. Dong. 2002. Characterization of mouse and human B7-H3 genes. J. Immunol. 168:6294–6297. [DOI] [PubMed] [Google Scholar]
- 25.Prasad, D.V., T. Nguyen, Z. Li, Y. Yang, J. Duong, Y. Wang, and C. Dong. 2004. Murine B7-H3 is a negative regulator of T cells. J. Immunol. 173:2500–2506. [DOI] [PubMed] [Google Scholar]
- 26.Wang, J., F. Gigliotti, S. Maggirwar, C. Johnston, J.N. Finkelstein, and T.W. Wright. 2005. Pneumocystis carinii activates the NF-kappaB signaling pathway in alveolar epithelial cells. Infect. Immun. 73:2766–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chung, Y., X. Yang, S.H. Chang, L. Ma, Q. Tian, and C. Dong. 2006. Expression and regulation of IL-22 in the IL-17-producing CD4+ T lymphocytes. Cell Res. 16:902–907. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}