

Abstract
Inflammatory conditions can lead to debilitating and persistent pain. This hyperalgesia reflects sensitization of peripheral terminals and facilitation of pain signaling at the spinal level. Studies of peripheral systems show that tissue injury triggers not only inflammation but also a well-orchestrated series of events that leads to reversal of the inflammatory state. In this regard, lipoxins represent a unique class of lipid mediators that promote resolution of inflammation. The antiinflammatory role of peripheral lipoxins raises the hypothesis that similar neuraxial systems may also down-regulate injury-induced spinal facilitation of pain processing. We report that the lipoxin A4 receptor is expressed on spinal astrocytes both in vivo and in vitro and that spinal delivery of lipoxin A4, as well as stable analogues, attenuates inflammation-induced pain. Furthermore, activation of extracellular signal-regulated kinase and c-Jun N-terminal kinase in astrocytes, which has been indicated to play an important role in spinal pain processing, was attenuated in the presence of lipoxins. This linkage opens the possibility that lipoxins regulate spinal nociceptive processing though their actions upon astrocytic activation. Targeting mechanisms that counterregulate the spinal consequences of persistent peripheral inflammation provide a novel endogenous mechanism by which chronic pain may be controlled.
Chronic pain secondary to injury and inflammation is a prevalent, persistent, and debilitating problem. Although early work dissected the role of sensitization of peripheral nerves in painful conditions, it is now appreciated that facilitation of spinal processing is also a critical component in states of inflammatory pain. Work with these systems has revealed an important role for spinal nonneuronal cells, such as astrocytes and microglia, in the regulation of nociception (for review see reference 1). In addition, it has been widely emphasized that peripheral injury leads to rapid expression and release of proinflammatory factors (e.g., TNF-α, IL-1β, and prostaglandins [PGs]) at the injury site, which reduce the threshold for activation and increase the responsiveness of peripheral nociceptor terminals. There is increasing evidence that there is a comparable release of such mediators in the spinal dorsal horn where these factors mediate facilitation of pain processing, further amplifying the nociceptive signaling conveyed to the brain (for review see reference 1). Hence, many factors that are involved in the initiation of inflammation at the peripheral site also drive spinal sensitization.
Insights from recent work point to the formation of antiinflammatory lipid mediators in response to inflammation (for review see reference 2). In this regard, lipoxins represent a unique class of lipid mediators that can function as “braking signals” in inflammation. Lipoxin A4 (LXA4) and LXB4 are positional isomers generated from arachidonic acid via the phospholipase A2–lipoxygenase pathway during cell–cell interactions. LXA4 binds with high affinity to a G protein–coupled receptor termed LXA4 receptor (ALXR) (3–6), also known as FPRL1 and FPR2. Activation of ALXR stops recruitment of neutrophils and facilitates resolution of inflammation by stimulating monocytes and macrophages to perform phagocytosis without releasing cytokines or chemokines. LXA4 also attenuates NF-κB activation and blocks phosphorylation of p38 and extracellular signal-regulated kinase (ERK; for review see reference 2).
It is noteworthy that many of the factors that are attenuated by the lipoxin–ALXR interaction are involved in pain processing, both at the site of injury and in the spinal cord. Another intriguing link between lipoxins and pain is the action of aspirin, which, through acetylation of cyclooxygenase-2, redirects the activity of cyclooxygenase-2 from generating intermediates of PGs and thromboxane from arachidonic acid to generating intermediates of a series of aspirin-triggered lipoxins (ATLs). ATLs display the same antiinflammatory activities as the native lipoxins but are more resistant to metabolic inactivation (for review see reference 2). It is thus possible that the analgesic action of aspirin involves not only the inhibition of PG generation but also the formation of antiinflammatory lipoxin analogues. We reasoned that processing of pain signals at the site of inflammation, but more importantly at the spinal level, is regulated by factors such as lipoxins that have resolving and antiinflammatory endogenous actions in peripheral tissues, and we accordingly performed a series of studies focused on the role of lipoxins in inflammatory pain.
RESULTS AND DISCUSSION
Painful inflammatory conditions are associated with sensitization of specialized sensory neurons that compose the nociceptive (pain) pathway, leading to enhanced pain sensations in response to both noxious and nonnoxious stimuli (termed hyperalgesia and allodynia, respectively). Lipoxins have antiinflammatory actions in vivo when administered to the site of inflammation or systemically by the i.v. or oral route (for review see reference 2). To investigate whether lipoxins block pain associated with inflammation, LXA4, LXB4, and the more stable ATL analogue (ATLa) and an analogue of LXB4, (8,9)-acetylenic LXB4 (8,9-aLXB4), were administered i.v. to rats 2 min before injection of carrageenan to the hind paw. Hyperalgesia was assessed by measuring the response latency to a thermal stimulus. Typically, intraplantar injection of carrageenan results in a transient inflammation, apparent as an increase in paw volume and reddening of the skin, and hyperalgesia with an onset at 2 h that is resolved after 24 h (7, 8). Intravenous injection of 10 μg/kg LXA4 (28 nmol/kg), 10 μg/kg ATLa (24 nmol/kg) and 10 μg/kg LXB4 (28 nmol/kg), but not 10 μg/kg 8,9-aLXB4 (29 nmol/kg), had antihyperalgesic effects (Fig. 1, A and B). The withdrawal latencies were significantly longer for the inflamed paw in the LXA4-, ATLa-, and LXB4-injected animals, as compared with the vehicle-treated animals (Fig. 1, A and B), indicating that lipoxins can alter pain processing. These results were also processed for calculation of the hyperalgesic index (HI), and i.v. LXA4, ATLa, and LXB4 significantly reduced the HI for the 0–4-h time span (Fig. 1 C). Assessment of paw volume by measuring paw height with calipers applied across the highest point of the dorsum and plantar aspects of the paw revealed a reduction in carrageenan-evoked paw edema (Fig. 1 D), suggesting a local antiinflammatory action of lipoxins when this delivery route is used.
Figure 1.

Intravenous administration of lipoxins reduces inflammation-evoked hyperalgesia and edema. Paw withdrawal latency (PWL) is plotted versus time for the ipsilateral (ip; injected) and contralateral (c; uninjected) hind paw showing that i.v. injection of LXA4, ATLa (A and C), and aLXB4 (B and C), but not 8,9-aLXB4 (B and C), before injection of carrageenan reduces thermal hyperalgesia. HI (C) is calculated for 0–4 h. Paw thickness (D) was measured by calipers at different time points after induction of inflammation. Each time point and bar represents the mean ± SEM (n = 4–6). *, P < 0.05 as compared with vehicle (ip) measurements.
Peripheral inflammation triggers release of factors such as PGs not only at the site of inflammation but also in the spinal cord (9). It is important to note that PGs not only have proinflammatory actions but can also drive the initiation of the resolution phase via PGE2 and PGD2 actions on PMN that include the induction of lipoxygenases that enable the formation of lipoxins (10). This led us to investigate whether spinal (intrathecal [i.t.])-administrated lipoxins have an antihyperalgesic effect. Peripheral afferents innervating the lower hind limbs, including the paws, terminate at the lumbar level of the spinal cord. To enable local delivery of lipoxins, injection catheters were implanted so that the tip of the catheter was placed at the level of the lumbar spinal cord. Although i.t. injection of the vehicle (saline) had no effect on carrageenan-induced hyperalgesia, i.t. LXA4 (0.3 nmol) attenuated the carrageenan-evoked hyperalgesia in a dose-dependent fashion (Fig. 2, A and C), and equimolar dosing of ATLa had a similar effect (Fig. 2 C). On the contrary, i.t. LXA4 did not alter normal nociceptive thresholds, as indicated by the lack of change in withdrawal latency of the contralateral (uninflamed) paw (Fig. 2 A). Intrathecal injection of the positional isomer LXB4 (0.3 nmol) also caused antihyperalgesia, whereas a higher dose of 8,9-aLXB4 (10 nmol) was required for similar effects (Fig. 2, B and C). None of the lipoxins altered carrageenan-induced paw edema (Fig. 2 D). The therapeutic potential of lipoxins was tested by i.t. injection of LXA4 at time points when the carrageenan-induced hyperalgesia was established. Although a single dose of LXA4 (0.3 nmol) showed a modest effect, two consecutive injections resulted in a statistically significant reversal of the hyperalgesia (Fig. 2, E and F). To examine whether the antihyperalgesic effect of i.t.-delivered lipoxins is caused by redistribution, the same dose of LXA4 that was effective when injected i.t. was administered i.v. Intravenous administration of 0.3 nmol LXA4 did not reduce carrageenan-evoked thermal hyperalgesia or peripheral inflammation (Fig. 1 A), and it is thus unlikely that the effect observed with spinal administration of lipoxins is the result of redistribution from the spinal compartment to the site of inflammation. Because the trihydroxytetraene structure of native lipoxins is sensitive to metabolic inactivation by dehydrogenation, LXA4 was administered via continuous i.t. infusion that resulted in a prolonged antihyperalgesic effect compared with i.t. bolus injection (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20061826/DC1). These results suggest that lipoxins have antinociceptive action not only after systemic delivery but also when delivered spinally, and that the antihyperalgesiceffect exerted at the spinal level is mediated via a mechanism that is distinct from regulation of peripheral inflammation and edema.
Figure 2.

Intrathecal injection of lipoxins reduces inflammation-evoked hyperalgesia. Paw withdrawal latency (PWL) is plotted versus time for the left ipsilateral (ip) and contralateral (c) hind paw, showing that i.t. pretreatment (−2 min) with equimolar doses of LXA4 (A and C), LXB4, and ATLa and a higher dose of 8,9-aLXB4 (B and C), as well as posttreatment with LXA4 (injection at 120 min or at 120 min followed by a second injection at 190 min; E and F), reduces inflammation-evoked hyperalgesia. HI is calculated for 0–4 h (C) and 2–6 h (F). None of the i.t.-delivered lipoxins altered paw thickness (D). Each time point and bar represents the mean ± SEM (n = 6–8). *, P < 0.05 as compared with vehicle (ip) measurements.
Lipoxins act on ALXR in peripheral tissues, and this receptor has been cloned from both immune and nonimmune cell types, including PMNs, monocytes, T cells, intestinal enterocytes, and synovial fibroblasts (for review see reference 2). The antihyperalgesic action observed after i.t. administration of LXA4, LXB4, and ATLa indicates that this class of lipid mediators also acts on specific targets in the spinal cord. Accordingly, we examined whether the lipoxin receptor is expressed at the spinal cord of naive rodents. At this point in time there is no commercially available antibody against the rat ALXR. Based on the fact that rat ALXR shares 74 and 84% homology with human and mouse ALXR, respectively, the immunoreactivity of an antibody against human ALXR was examined in rodent tissues. Rat and mouse spinal cord homogenates were subjected to gel electrophoresis and Western blotting. ALXR immunoreactive bands, running at 70 kD (Fig. 3 A), were detected on membranes probed with this antibody, indicating that it recognizes rat, as well as human and mouse, ALXR and that the ALXR is constitutively expressed in naive rat and mouse spinal cords (Fig. 3 A).
Figure 3.

ALXR is expressed on astrocytes in the naive rat spinal cord. (A) Representative Western blots depicting ALXR expression in mouse and rat spinal cords 0–24 h after injection of carrageenan to the paw. Rat spleen (from rats 8 h after carrageenan injection) was used as positive control. Immunohistological processing of spinal cord sections from the naive rat show that ALXR is homogenously expressed throughout the dorsal horn of the spinal cord (B), colocalizing with the astrocyte marker GFAP (C–E, green; F, enlarged image) but not with the microglia marker OX-42 (K, green) or the neuronal marker NeuN (L, green). ALXR immunoreactivity was detected with both fluorescence (G) and avidin–biotin–3,3-diaminobenzidine (H) detection in spleens from carrageenan-injected rats (F). ALXR antibody replaced with goat serum IgG was used as a negative control (I). ALXR immunoreactivity was absent in dorsal root ganglia (DRG; J). Bars, 50 μm.
ALXR expression in intestinal enterocytes increases in response to cytokine (IL-13 and IFN-γ) and LPS stimulation in vitro (for review see reference 2). To assess whether inflammation in peripheral tissues and persistent afferent input alter the expression of spinal ALXR, spinal cords were collected at different time points after intraplantar injection of carrageenan. Lumbar spinal cords harvested from rats 6, 12, and 24 h after injection of carrageenan showed protein expression levels of ALXR very similar to those observed in naive animals (6 h, 115 ± 8%; 12 h, 125 ± 11%; and 24 h, 119 ± 13% as compared with naive animals; n = 3–5 per group; Fig. 3 A). These results emphasize that the spinal expression of this receptor does not change in the face of peripheral inflammation. To understand the interplay between lipoxins, ALXR, and spinal pain processing, it is important to determine in which spinal cell type ALXR is expressed. To address this question, lumbar spinal cords from naive rats were sectioned and incubated with ALXR antibody. ALXR immunoreactivity was observed in stellate-like cells throughout the spinal cord, homogenously distributed in the dorsal (Fig. 3 B) and ventral horns (not depicted). Double labeling of spinal cord sections revealed that ALXR expression colocalized with the astrocyte marker glial fibrillary acidic protein (GFAP; Fig. 3, C–F) but not with the microglia marker OX-42 or the neuronal marker NeuN (Fig. 3, K and L). In line with our present results, earlier in vitro studies demonstrated ALXR (FPRL1) expression in astrocytoma cells (11). As a positive control, a rat spleen (8 h after paw carrageenan injection) was probed for ALXR, and positive cells were observed with both fluorescence (Fig. 3 G) and avidin-biotin detection (Fig. 3 H). The ALXR immunoreactivity was abolished in the goat-serum control (Fig. 3 I). ALXR expression was not detected in dorsal root ganglia (Fig. 3 J).
The finding that spinal astrocytes express ALXR is intriguing, as the number of reports indicating that spinal nonneuronal cells play an important role in spinal facilitation of pain processing is rapidly increasing. It has been shown that both nerve injury and peripheral inflammation lead to activation of spinal dorsal horn astrocytes and microglia (12). Spinal delivery of inhibitors or modulators of astrocyte function block initiation and maintenance of persistent pain states (13–16), supporting an important role for these cells in spinal sensitization. Astrocytes respond to changes in their environment by releasing, for example, cytokines, chemokines, and nitric oxide. Thus, it is possible that lipoxins, by acting on astrocyte ALXR, dampen this response by counterregulating the production of proinflammatory factors. Based on reports demonstrating that ERK and c-Jun N-terminal kinase (JNK) mitogen-activated protein kinase (MAPK) are activated (phosphorylated) in spinal astrocytes in models of persistent pain (14, 17), we sought to examine the effect of lipoxins on ERK and JNK phosphorylation in astrocytes. For this purpose, primary cultures of astrocytes (>95% astrocytes) were established from postnatal rat spinal cord, and ALXR expression was verified (Fig. 4 A). The astrocyte cultures were subjected to 250 μM of stable ATP, 50 ng/ml TNF-α, 10 ng/ml IL-1β, and 10 μM substance P (SP), factors indicated to act on glia and play a role in spinal pain processing, for 15 min. This study showed that ATP drives activation of both ERK and JNK, whereas TNF-α stimulation activated only JNK. SP and IL-1β did not evoke phosphorylation of these MAPKs at the chosen concentration and time point. Strikingly, ATP-evoked ERK and JNK phosphorylation, but not TNF-α–evoked JNK phosphorylation, was reduced in the presence of 10 nM ATLa (30-min pretreatment). This observation is in line with studies in fibroblasts and T cells where it has been shown that ERK activation is attenuated in the presence of lipoxins (for review see reference 2). Of importance, ERK activity in spinal astrocytes is thought to be important for the maintenance of chronic pain, and inhibition of spinal ERK, at the time point when it is activated in astrocytes, attenuates neuropathic pain (17). In addition to ERK, this work has identified the JNK family as an intracellular mechanism through which lipoxins may exert their antiinflammatory and antihyperalgesic actions in spinal tissues. Importantly, it has been reported that persistent activation of JNK in spinal astrocytes appears critical for the maintenance of neuropathic pain (14). In the current study, increased JNK activation was observed in spinal astrocytes after peripheral inflammation, and the carrageenan-evoked JNK phosphorylation was reduced in the presence of i.t. LXA4 (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20061826/DC1). The antihyperalgesic effects observed after i.t. injection of lipoxins in our model of inflammatory pain, in conjunction with the prevention of JNK activation in vivo and JNK and ERK phosphorylation in primary astrocytes, suggest that ALXR activation mediates antinociception through prevention of intracellular MAPK signaling in spinal astrocytes. This is a striking finding, as it provides support for the existence of a mechanism through which spinal nonneuronal cells can participate in the regulation of sensory neuronal activity, not only through sensitization, but also by normalization, of pain signaling.
Figure 4.

ATLa prevents ATP-evoked ERK and JNK phosphorylation in primary astrocyte cultures. (A) Representative images demonstrating that ALXR colocalizes with the astrocyte marker GFAP in cultured primary astrocytes. Bar, 50 μm. (B) Western blots probed for phosphorylated ERK and JNK in samples from primary astrocytes stimulated with ATP, SP, IL-1β, and TNF-α for 15 min. Incubation with 10 nM ATLa, starting 30 min before TNF-α stimulation, had no effect on JNK phosphorylation (C), whereas ATLa prevented both ERK and JNK phosphorylation evoked by ATP (D and E). Each bar represents the mean ± SEM (n = 4–5). *, P < 0.05 as compared with control; #, P < 0.05 as compared with PBS + ATP.
Earlier results also suggested that lipoxins play an important role in pain processing by regulating the communication between the immune and sensory nervous system. The neuropeptide nociceptin (orphanin FQ) elicits leukocyte recruitment in vitro and in vivo (18). It is hypothesized that nociceptin, released from afferent nerve terminals, leads to the recruitment of inflammatory cells that contribute to the facilitation of pain processing. Along these lines, i.v. injection of ATLa blocked nociceptin-evoked PMN infiltration and may thereby reduce inflammation-evoked pain (18). In the present study, systemic administration of lipoxins reduced signs of inflammation (i.e., hyperalgesia and edema); even though not examined here, it is reasonable to consider that this is mediated in part by lipoxin reduction in PMN infiltration. Lipoxins also act on monocytes and macrophages, stimulating these cells to facilitate resolution of inflammation by increased monocyte infiltration and phagocytosis of apoptotic PMNs in a nonphlogistic fashion. It is likely that lipoxins can also attenuate sensitization and activation of sensory neurons through their antiinflammatory and proresolving properties, limiting the local release of inflammatory, pronociceptive factors.
A considerable amount of effort has been focused on blocking signals that drive chronic pain either by reducing the generation of a single factor (e.g., PGE2 or NO) or by blocking factors from acting on their specific receptors (e.g., SP, glutamate, or TNF-α). Although these strategies have proven to reduce nociception, it is becoming increasingly appreciated that targeting systems that affect a broad spectrum of factors involved in pain processing may be more advantageous. The present results suggest that using agonists that activate pathways for antiinflammatory proresolving actions is an effective approach for regulation of nociceptive signaling in, but not limited to, painful inflammatory conditions. Of interest, reduced levels of the antiinflammatory cytokines IL-4 and IL-10 was recently reported in patients with chronic pain (19), and IL-4 induces both ALXR and the enzymes in LX biosynthesis (for review see reference 2). In accordance, studies showing that systemic as well as spinal delivery of IL-4 and IL-10, as a protein or through gene transfer, reduces hypersensitivity in experimental models of pain (20, 21) support the importance of a counterregulatory system in the control of pain signaling. Though not examined in this study, it is possible that there is an elevation of lipoxins in the spinal cord during normal resolution of pain and, conversely, that a reduced capacity to produce peripheral and/or spinal lipoxins may lead to persistent pain.
In summary, our findings suggest that lipoxins attenuate nociception not only at the site of inflammation but also by participating in the regulation of spinal pain processing through actions on astrocyte-expressed lipoxin receptors. To date, aside from the initial studies noted here, the role of lipoxins in pain regulation has not been investigated. Accordingly, these results provide insights into a novel lipid cascade regulating peripheral and spinal sensitization and hyperalgesia and promise to shed light onto the role of nonneuronal cells in spinal nociceptive processing. Moreover, they point to novel therapeutic targets for antihyperalgesic agents in chronic pain states.
MATERIALS AND METHODS
Animals.
All experiments were performed according to protocols approved by the Institutional Animal Care Committee of the University of California, San Diego. 250–300 g male Sprague Dawley rats (Harlan Industries) were housed individually on a 12-h light/dark cycle with free access to food and water. Intrathecal injection catheters were implanted through an incision in the atlanto-occipital membrane and advanced to the lumbar level under isoflurane anesthesia. All agents were delivered in 10 μl saline followed by 10-μl saline flush. For continuous infusion, the catheter was attached to an s.c. osmotic minipump (ALZET). For these studies, synthetic LXA4, LXB4, and 8,9-LXB4 were purchased from D. Clissold (CASCADE Limited), and ATLa was a gift from W. Gilford, J. Parkinson, and H.D. Perez at Berlex Biosciences.
Astrocyte cultures.
Astrocyte cultures were prepared from spinal cords of 1–2-d-old rat pups and plated in DMEM containing 10% FBS and 1% penicillin/streptomycin in 75-cm2 flasks. At days 13 and 14, the flasks were mechanically shaken for 2 h to remove any remaining oligodendrocytes and microglia. On day 15, the astrocyte cultures were trypsinized and replated into six-well dishes. The cells were used for stimulation experiments with SP, TNF-α, Bz-ATP (Sigma-Aldrich), and IL-1β (R&D Systems) when confluent.
Carrageenan-induced hyperalgesia.
100 μl carrageenan 2% in saline (λ; Sigma-Aldrich) was injected s.c. into the plantar surface of the left hind paw under light isoflurane anesthesia. The rats were placed in plexiglass cubicles on a glass surface maintained at 25°C for assessment of thermal hyperalgesia (8). A thermal nociceptive stimulus was generated by a projection light bulb positioned below the glass surface. Paw withdrawal latency was defined as the time until the rat showed a brisk withdrawal response to the heat generated by the light projected to the paw. The time was determined by a timer activated by the light source and stopped by photodiode motion sensors. The HI represents the area beneath the time-course curve after stimulation in which the percent reduction from baseline latency is plotted against time such that the numeric value increases with increased hyperalgesia. The resulting calculated number is the percent change × hours. The formula for calculating the percent change is (baseline latency − postdrug latency) × 100(baseline latency)−1, where latency is expressed in seconds.
Western blotting.
The lumbar part of the spinal cord and cultured astrocytes were homogenized and subjected to PAGE. Membranes were incubated with antibodies against ALXR (1:2,000; Biologicals), phosphorylated JNK (P-JNK), total JNK, phosphorylated (P-ERK), and total ERK (1:10,000; Cell Signaling). The signal was detected with chemiluminescent reagents (SuperSignal; Pierce Chemical Co.). The membranes were reblotted with GAPDH or β-actin (1:50,000; Sigma-Aldrich). The intensity of immunoreactive bands was quantified using ImageQuant software (Molecular Dynamics), pooled for P-ERK and P-JNK, respectively, normalized against β-actin, and presented as the percentage of control.
Immunohistochemistry.
Animals were perfused with saline followed by 4% paraformaldehyde. Spinal lumbar enlargement was removed, postfixed, and cryoprotected. Nonspecific binding was blocked, and cultured astrocytes and spinal floating sections (30 μm) were incubated with ALXR antibody (1:500) overnight at 4°C. Alexa Fluor–conjugated secondary antibodies (Invitrogen) were used for detection. The slides were double stained with GFAP (1:1,000; Chemicon). Images were captured using a multiphoton laser point scanning confocal microscopy system (Radiance 2100/AGR-3Q; Bio-Rad Laboratories) operated by Lasersharp 2000 software (Bio-Rad Laboratories).
Statistics.
One-way analysis of variance was performed to analyze HI, paw thickness, and densitometric calculations from Western blot studies. Multiple post hoc comparisons used the Bonferroni correction. P < 0.05 was considered significant.
Online supplemental material.
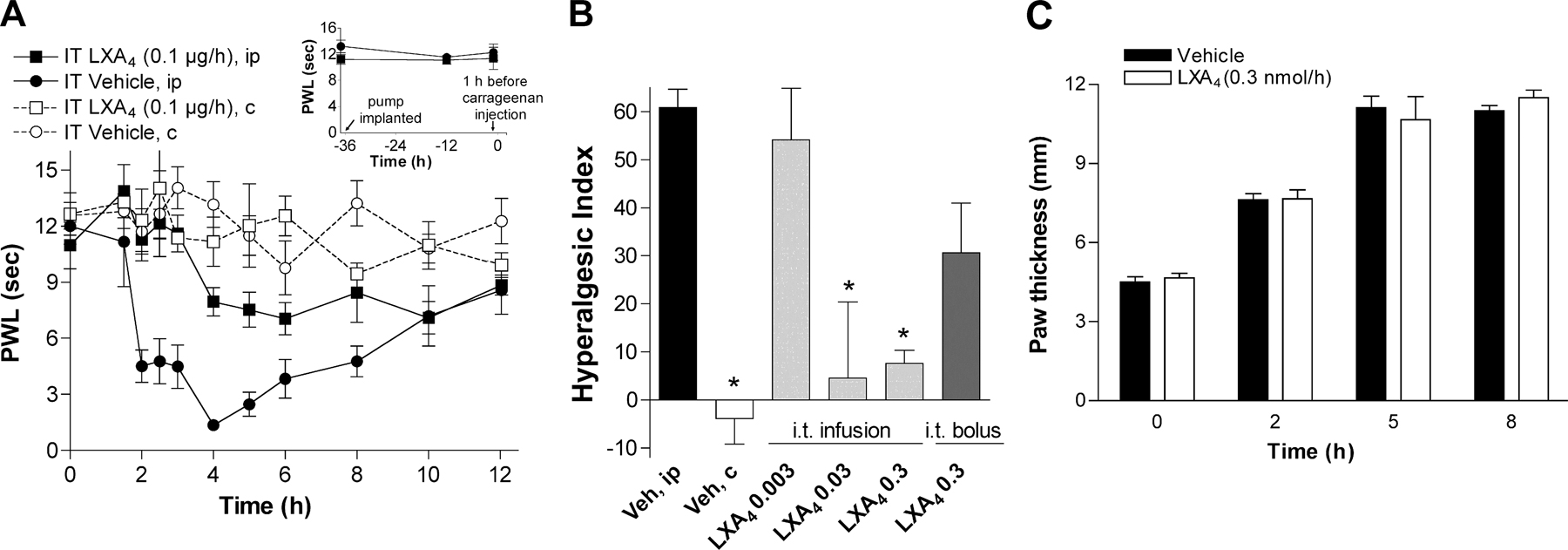
Fig. S1 shows that continuous i.t. infusion of LXA4 resulted in a prolonged antihyperalgesic effect compared with i.t. bolus injection of LXA4. Intrathecal catheters were implanted and connected to osmotic minipumps (pump rate = 1 μl/h) prefilled with 0.01–0.1 μg/μl LXA4 solution (0.03–0.3 nmol/h). The pumps and i.t catheters were implanted and connected 36 h before the carrageenan injection. It takes 10–14 h before the infusate reaches the spinal fluid; hence, this administration regimen provide a 20–24 h lipoxin pretreatment. Hyperalgesia was assessed by measuring time to response to thermal stimuli. Paw volume was assessed by paw height measured with calipers applied across the highest point of the dorsum and plantar aspects of the paw.
Fig. S2 demonstrates that phosphorylation of spinal JNK, evoked by peripheral inflammation, was reduced in the presence of i.t. LXA4. Rats received i.t. injections of vehicle (saline) or 0.3 nmol LXA4 2 min before and 120 min after carrageenan injection to the paw. The dorsal ipsilateral lumbar enlargement of spinal cords was harvested 4 h after injection of carrageenan, homogenized, and subjected to PAGE. Membranes were incubated with antibodies against P-JNK, total JNK (1:10,000; Cell Signaling), and β-actin (1:50,000; Sigma-Aldrich). The signal was detected with chemiluminescent reagents. Intensity of immunoreactive bands were quantified using ImageQuant software. The signal for P-JNK bands was pooled, normalized against total JNK, and presented as the percentage of control.
For immunohistochemistry, animals were perfused with saline followed by 4% paraformaldehyde. Spinal lumbar enlargement was removed, postfixed, and cryoprotected. Nonspecific binding was blocked, and the 30-μm floating sections were incubated with monoclonal P-JNK antibody (1:100; Cell Signaling) overnight at room temperature. Alexa Fluor–conjugated secondary antibodies (Invitrogen) were used for detection. The slides were double stained with polyclonal GFAP antibody (1:500; Chemicon). Images were captured using a confocal microscopy system operated by Lasersharp 2000 software.
Supplemental Material
Acknowledgments
We thank Dr. T.L. Yaksh for support and helpful discussions.
Parts of this work were supported by National Institutes of Health grants P50DE016191 and GM38765 (both to C.N. Serhan), and by an Arthritis Foundation fellowship (to C.I. Svensson). M. Zattoni is a student supported by T.L. Yaksh and National Institutes of Health grant NS16541.
C.I. Svensson and M. Zattoni have no conflicting financial interests. Brigham and Women's Hospital is assigned patents on lipoxins and ATL stable analogues that are licensed for clinical development; C.N. Serhan is the inventor. These programs and their clinical development are the subject of consultant agreements.
References
- 1.McMahon, S.B., W.B. Cafferty, and F. Marchand. 2005. Immune and glial cell factors as pain mediators and modulators. Exp. Neurol. 192:444–462. [DOI] [PubMed] [Google Scholar]
- 2.Serhan, C.N. 2005. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot. Essent. Fatty Acids. 73:141–162. [DOI] [PubMed] [Google Scholar]
- 3.Fiore, S., J.F. Maddox, H.D. Perez, and C.N. Serhan. 1994. Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. J. Exp. Med. 180:253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fiore, S., S.W. Ryeom, P.F. Weller, and C.N. Serhan. 1992. Lipoxin recognition sites. Specific binding of labeled lipoxin A4 with human neutrophils. J. Biol. Chem. 267:16168–16176. [PubMed] [Google Scholar]
- 5.Takano, T., S. Fiore, J.F. Maddox, H.R. Brady, N.A. Petasis, and C.N. Serhan. 1997. Aspirin-triggered 15-epi-lipoxin A4 (LXA4) and LXA4 stable analogues are potent inhibitors of acute inflammation: evidence for antiinflammatory receptors. J. Exp. Med. 185:1693–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiang, N., I.M. Fierro, K. Gronert, and C.N. Serhan. 2000. Activation of lipoxin A4 receptors by aspirin-triggered lipoxins and select peptides evokes ligand-specific responses in inflammation. J. Exp. Med. 191:1197–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menezes-de-Lima, O., Jr., C.A. Kassuya, A.F. Nascimento, M.G. Henriques, and J.B. Calixto. 2006. Lipoxin A4 inhibits acute edema in mice: implications for the anti-edematogenic mechanism induced by aspirin. Prostaglandins Other Lipid Mediat. 80:123–135. [DOI] [PubMed] [Google Scholar]
- 8.Hua, X.Y., C.I. Svensson, T. Matsui, B. Fitzsimmons, T.L. Yaksh, and M. Webb. 2005. Intrathecal minocycline attenuates peripheral inflammation-induced hyperalgesia by inhibiting p38 MAPK in spinal microglia. Eur. J. Neurosci. 22:2431–2440. [DOI] [PubMed] [Google Scholar]
- 9.Yang, L.C., M. Marsala, and T.L. Yaksh. 1996. Characterization of time course of spinal amino acids, citrulline and PGE2 release after carrageenan/kaolin-induced knee joint inflammation: a chronic microdialysis study. Pain. 67:345–354. [DOI] [PubMed] [Google Scholar]
- 10.Levy, B.D., C.B. Clish, B. Schmidt, K. Gronert, and C.N. Serhan. 2001. Lipid mediator class switching during acute inflammation: signals in resolution. Nat. Immunol. 2:612–619. [DOI] [PubMed] [Google Scholar]
- 11.Le, Y., J. Hu, W. Gong, W. Shen, B. Li, N.M. Dunlop, D.O. Halverson, D.G. Blair, and J.M. Wang. 2000. Expression of functional formyl peptide receptors by human astrocytoma cell lines. J. Neuroimmunol. 111:102–108. [DOI] [PubMed] [Google Scholar]
- 12.Sweitzer, S.M., R.W. Colburn, M. Rutkowski, and J.A. DeLeo. 1999. Acute peripheral inflammation induces moderate glial activation and spinal IL-1beta expression that correlates with pain behavior in the rat. Brain Res. 829:209–221. [DOI] [PubMed] [Google Scholar]
- 13.Sweitzer, S.M., P. Schubert, and J.A. DeLeo. 2001. Propentofylline, a glial modulating agent, exhibits antiallodynic properties in a rat model of neuropathic pain. J. Pharmacol. Exp. Ther. 297:1210–1217. [PubMed] [Google Scholar]
- 14.Zhuang, Z.Y., Y.R. Wen, D.R. Zhang, T. Borsello, C. Bonny, G.R. Strichartz, I. Decosterd, and R.R. Ji. 2006. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J. Neurosci. 26:3551–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Obata, H., J.C. Eisenach, H. Hussain, T. Bynum, and M. Vincler. 2006. Spinal glial activation contributes to postoperative mechanical hypersensitivity in the rat. J. Pain. 7:816–822. [DOI] [PubMed] [Google Scholar]
- 16.Watkins, L.R., D. Martin, P. Ulrich, K.J. Tracey, and S.F. Maier. 1997. Evidence for the involvement of spinal cord glia in subcutaneous formalin induced hyperalgesia in the rat. Pain. 71:225–235. [DOI] [PubMed] [Google Scholar]
- 17.Zhuang, Z.Y., P. Gerner, C.J. Woolf, and R.R. Ji. 2005. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 114:149–159. [DOI] [PubMed] [Google Scholar]
- 18.Serhan, C.N., I.M. Fierro, N. Chiang, and M. Pouliot. 2001. Cutting edge: nociceptin stimulates neutrophil chemotaxis and recruitment: inhibition by aspirin-triggered-15-epi-lipoxin A4. J. Immunol. 166:3650–3654. [DOI] [PubMed] [Google Scholar]
- 19.Uceyler, N., R. Valenza, M. Stock, R. Schedel, G. Sprotte, and C. Sommer. 2006. Reduced levels of antiinflammatory cytokines in patients with chronic widespread pain. Arthritis Rheum. 54:2656–2664. [DOI] [PubMed] [Google Scholar]
- 20.Milligan, E.D., S.J. Langer, E.M. Sloane, L. He, J. Wieseler-Frank, K. O'Connor, D. Martin, J.R. Forsayeth, S.F. Maier, K. Johnson, et al. 2005. Controlling pathological pain by adenovirally driven spinal production of the anti-inflammatory cytokine, interleukin-10. Eur. J. Neurosci. 21:2136–2148. [DOI] [PubMed] [Google Scholar]
- 21.Vale, M.L., J.B. Marques, C.A. Moreira, F.A. Rocha, S.H. Ferreira, S. Poole, F.Q. Cunha, and R.A. Ribeiro. 2003. Antinociceptive effects of interleukin-4, -10, and -13 on the writhing response in mice and zymosan-induced knee joint incapacitation in rats. J. Pharmacol. Exp. Ther. 304:102–108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}