

Abstract
Respiratory syncytial virus (RSV) infection is widely spread and is a major cause of bronchiolitis in infants and high-risk adults, often leading to hospitalization. RSV infection leads to obstruction and inflammation of the airways and induction of innate and acquired immune responses. Because dendritic cells (DCs) are essential in the elicitation of these immune responses, we investigated the presence and the role of dendritic cell subtypes upon RSV infection in the lung. Here, we report that RSV infection increased the number of both conventional and plasmacytoid dendritic cells in the lung and the lung-draining lymph nodes. In particular, the increase in plasmacytoid dendritic cell numbers was sustained and lasted until 30 d after infection. Depletion of plasmacytoid dendritic cells resulted in decreased RSV clearance. In addition, depletion of plasmacytoid dendritic cells resulted in an exacerbation of all manifestations of immune-mediated pathology caused by RSV infection. In conclusion, this study demonstrates that both conventional and plasmacytoid dendritic cells are attracted to the site of RSV infection. It is demonstrated that plasmacytoid dendritic cells play a protective role during RSV infection by modulation of local immune responses.
Respiratory syncytial virus (RSV) is one of the most common causes of viral bronchiolitis and a major cause for hospitalization in infants, elderly and high risk adults (1). RSV infection is characterized by epithelial cell necrosis, a lymphocytic infiltrate in the lung and edema leading to obstruction of the smaller airways by cellular debris and mucus (2). In addition to the acute effects of RSV infection on the immune system, RSV also causes long-term changes in the immunological environment in the lung. This is of particular concern in light of the observation that RSV infection in early infancy was correlated with development of allergic and asthmatic symptoms later in life (3). However, the immune response to RSV is still complex and puzzling. Infection with RSV leads to poor development of immunity and recurrent infections are common, exemplified by the failure of an RSV vaccine in the 1960s (4). Therefore, a better understanding of the immunological responses elicited during RSV infection may contribute to the development of new treatment strategies or novel vaccines.
Murine models of RSV infection have been effectively used to study innate and acquired immune responses. RSV infection of BALB/c mice elicits a vigorous inflammatory response in the lung leading to airway hyperresponsiveness, CD4+ and CD8+ T cell influx, and increased production of mucus (5, 6). Crucial in the elicitation of acquired immune responses in the lung is the DC. During priming of the immune response in the lung, immature pulmonary DCs internalize antigens, mature, and emigrate the tissue to the local lymph nodes where they present the antigen to naive T cells (7). In addition, during ongoing inflammation, DCs migrate into the tissue where they maintain and enhance local immune responses (8). In humans and mice, several subtypes of DCs have been described, as characterized by surface markers and function. Generally, DCs can be distinguished into conventional DCs (cDC or “myeloid” DCs) and plasmacytoid DCs (pDCs) (9).
Not much is known about the role of different subsets of DCs in respiratory viral infections. After influenza A virus infection of mice, a strong increase in both immature and mature DCs was observed in the lung (10), whereas an increase of CD11b−/CD8α− DCs, most likely pDCs, was observed in the mediastinal lymph nodes of mice after influenza A infection (11). In a mouse RSV infection model, it was demonstrated that RSV caused a persistent increase in pulmonary DCs, which displayed a mature phenotype (12). The different subtypes of DCs were not distinguished in the latter studies. Interestingly, a recent study showed that both cDC and pDC numbers were increased in nasal washings of RSV-infected infants (13). However, so far, nothing is known about the contribution of different DC subtypes to the immunopathology in RSV infection. Therefore, in this study, we investigated the effect of RSV infection on cDC and pDC numbers in the lung and lung-draining lymph nodes. Moreover, we investigated the functional role of pDCs in RSV-mediated lung immunopathology and in RSV viral clearance.
RESULTS AND DISCUSSION
The number of cDCs and pDCs are increased in lungs of RSV-infected mice
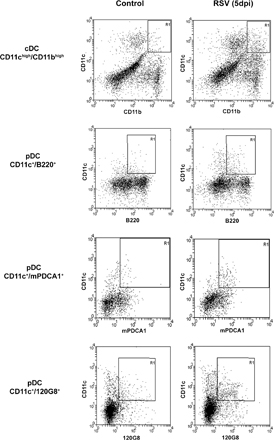
To investigate the number of cDCs and pDCs after RSV infection, lungs and lung-draining lymph nodes of mice were analyzed at different time points after infection for the presence of CD11chigh/CD11bhigh cDCs or CD11c+/B220+, CD11c+/mPDCA1+, and CD11c+/120G8+ pDCs (Fig. 1 and Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20052359/DC1). During RSV infection, the number of both cDCs and pDCs increased significantly. Interestingly, the increase in the number of pDCs in the lung was sustained until 30 days post-infection (dpi) when infected lungs still contained significantly more pDCs compared with naive mice. In the lymph nodes, both cDC and pDC numbers showed a temporary peak at 7 dpi. These data show that both cDC and pDC cell numbers are increased considerably in both lungs and lung-draining lymph nodes after RSV infection. However, only the increase in pDCs in the lung was sustained until 30 dpi.
Figure 1.

Numbers of cDCs and pDCs in the lung and the lung-draining lymph nodes are increased upon RSV infection. Before and at different time points after RSV (▪) or uvRSV (▴) infection, single cell suspensions of lungs and lung-draining lymph nodes were analyzed by flow cytometry for the presence of cDCs (CD11chigh/CD11bhigh) and pDCs (CD11c+/B220+). Results are displayed as percentage of lung cells (A), total number of cells per lung (B), percentage of lymph node cells (C), or total number of cells per lymph node (D) ± SEM. *, P < 0.05 compared with noninfected control mice, n = 4–7 mice per group.
These data suggest that the sustained number of CD11c+/MHChigh DCs, observed in studies in mice by Beyer et al. in the lung after RSV infection, may be pDCs (12). In line with these and our results is a recent study that showed that both cDC and pDC numbers were increased significantly in nasal washings of RSV-infected children (13). In this study, it was also observed that the number of pDCs was still increased after apparent resolution of RSV infection. Collectively, these data support the hypothesis that recruitment of recently attracted DCs in the lung leads to maintenance and control of the immune response in the lung as suggested by van Rijt et al. (8), even after the actual infection and inflammation in the lung is resolved. In addition, the sustained presence of pDCs in the lung after apparent resolution of RSV infection may also suggest that RSV infection of BALB/c mice can become either latent or persistent, as suggested by studies both in mice (14) and infants (15). So far, nobody has described the role of pDCs in respiratory viral infections in vivo. Previously, others have suggested that pDCs in the lung have tolerogenic functions after pulmonary allergen exposure, by inhibition of T effector cell induction or indirectly by induction of regulatory T cells in the lung-draining lymph nodes (16–18).
The 120G8 antibody effectively depletes pDCs in the lung
Unfortunately, selective targeting of cDCs is still impossible because of the surface markers it shares with other immune cells. However, depletion of pDCs has been performed in studies before, using anti-Gr-1, 120G8, 440c, or anti–mPDCA-1 antibodies (for review see reference 19), including selective depletion in the lung (17). Therefore, in the next series of experiments, mice received the 120G8 antibody 1 d before and after RSV infection. The 120G8 antibody effectively decreased the number of pDCs in lungs of RSV-infected mice, as demonstrated by a significant decrease in the number of CD11c/B220, CD11c/mPDCA1, and CD11c/Gr-1 double-positive cells on 1 dpi and 6 dpi (Table S1, available at http://www.jem.org/cgi/content/full/jem.20052359/DC1). In contrast, the number of cDCs (CD11c/CD11b double-positive cells) was not decreased in the lungs of RSV-infected mice after treatment with the 120G8 antibody. In addition, no effect of pDC depletion was seen on the number of CD4+, CD8+ T cells, DX5+ NK cells, or CD19+ B cells on 1 dpi (unpublished data). These data show that the pDC depletion was selective in our model and effectively prevents the influx of pDCs during RSV infection.
Depletion of pDCs enhances RSV-induced immunopathology in the lung
This study next investigated whether pDCs influence manifestations of RSV-induced immunopathology in the lung. RSV-infected mice developed airway hyperreactivity, as measured by a strong increase in airway resistance in response to methacholine compared with control mice on 9 dpi (Fig. 2 A). Depletion of pDCs around the time of RSV infection enhanced this airway hyperreactivity significantly. Histological examination of control mice showed that RSV infection leads to a peribronchial and perivascular inflammatory infiltrate consisting of mononuclear cells, mostly lymphocytes, and leads to goblet cell hyperplasia and mucus production in the lung (Fig. 2 B and Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20052359/DC1). Lungs of pDC-depleted mice showed enhanced inflammation and mucus production. In addition, RSV infection elicited transcription of the mucus genes GOB-5 and Muc5ac, which was significantly increased in pDC-depleted mice (Fig. S2).
Figure 2.

Depletion of pDCs enhances RSV-induced airway hyperresponsiveness, inflammation, and mucus production in the lung and decreases clearance of RSV. (A) On 9 dpi, airway responses were measured in control (cont), RSV-infected (RSV), and pDC-depleted, RSV-infected (RSV+120G8) mice after one dose of methacholine and compared with basal measurements. Data are represented as mean airway resistance in cm H2O/ml/s ± SEM. *, P < 0.01 compared with noninfected control mice; #, P < 0.05 compared with RSV infected mice, n = 5 mice per group. (B) Lungs were isolated on 9 dpi and processed and sections were stained with H&E and PAS. Shown are representative lung sections of control (cont), RSV-infected (RSV), and pDC-depleted, RSV-infected (RSV+120G8) mice. Bars, 300 μM (H&E) and 75 μM (PAS). (C) On 6 dpi, RSV titers were determined in the lungs of RSV-infected (RSV) or pDC-depleted, RSV-infected (RSV+120G8) mice by plaque assay. Data are presented as mean PFU per gram lung ± SEM, n = 4. *, P < 0.05 compared with RSV-infected mice, n = 5 mice. In addition, RSV G protein RNA transcription in the lung on 6 dpi was measured by quantitative real-time PCR (Taqman). Data are represented as mean fold increase to RSV-infected mice ± SEM. *, P < 0.05 compared with RSV-infected mice, n = 5 mice per group.
The aforementioned effect of altered immunopathology in pDC-depleted mice may be the result of either an inability to clear virus properly, leading to an increased load of RSV virus, and/or immune deviation leading to enhanced T cell–mediated responses. To determine whether pDCs play a role in clearance of RSV from the lung, RSV viral titers from lungs were measured by plaque assay and by viral G protein mRNA assay on 6 dpi (Fig. 2 C). Plasmacytoid DC depletion of RSV-infected mice significantly enhanced both viral titers and RSV G protein RNA levels in the lung. This demonstrates that pDCs enhance viral clearance during RSV infection. However, pDC depletion enhanced viral titers approximately twofold, indicating that the observed increase in pathology may not be the result of increased viral titer only.
Next, we investigated whether depletion of pDCs in the lung had an effect on the cytokine environment induced by RSV. RSV infection enhanced production of both the Th1 cytokine IFNγ and the Th2-type cytokines, IL-4, IL-5, and IL-13, by lymph node T cells after anti-CD3 restimulation compared with naive mice (Fig. 3 A). Depletion of pDCs significantly increased the production of all these cytokines. In addition, real-time PCR analysis showed that RSV infection elicited transcription of all of these cytokines in the lung (Fig. 3 B). Again, depletion of pDCs significantly enhanced the transcription of these cytokines. There are two compatible possibilities to explain these data. First, pDCs in the lung or lung-draining lymph nodes may suppress the generation of effector T cells as show by de Heer et al. after allergen exposure in the lung (17). This may lead to the observed increased T cell responses after pDC depletion. Second, IFNα, produced mainly by pDCs, may enhance the cell-mediated antiviral immune response, allowing more efficient clearance of the virus. In this way, pDC depletion may lead to an increased viral load after viral infection, which may lead to the enhanced immune responses observed.
Figure 3.

Depletion of pDCs enhances RSV-induced immunological responses in the lung. (A) Cytokine production of lymph node T cells in response to anti-CD3 stimulation. On 6 dpi, lung lymph nodes were isolated from control (Cont), RSV-infected (RSV), and pDC-depleted RSV-infected (RSV + 120G8) mice and stimulated with anti-CD3 at a concentration of 5 × 106 cells/ml. Supernatants were analyzed using ELISA. Data are represented as mean ng/ml ± SEM. *, P < 0.01 compared with noninfected control mice, #, P < 0.05 compared with RSV-infected mice, n = 4 mice per group. (B) Messenger RNA levels of cytokines and chemokines in the lungs of mice. On 6 dpi, mRNA was isolated from lungs from control (Cont), RSV-infected (RSV), and pDC-depleted, RSV-infected (RSV + 120G8) mice and analyzed using quantitative real-time PCR by Taqman. Each sample was normalized using a GAPDH control and the figure shows mean fold increase to control ± SEM. *, P < 0.05 compared with noninfected control mice; #, P < 0.05 compared with RSV-infected mice, n = 5–6 mice per group.
To address the latter possibility, we investigated the direct role of IFNα during RSV infection in control and pDC-depleted mice. After RSV infection, levels of both IFNα4 mRNA and IFNα protein in the lung were elevated significantly at 1 dpi followed by a decrease during viral replication at dpi 4, and an increase again during clearance of the virus at 7 dpi (Fig. 4 A). Depletion of pDCs by 120G8 (Fig. 4 A) completely abolished transcription and production of IFNα throughout RSV infection. The diminished IFNα production in the lung after pDC depletion is in line with earlier reports that showed that pDCs are the primary producers of IFNα during viral infection (20). Classically, IFNα generates a general antiviral environment by activation of NK cells and CD8+ T cells that both mediate lysis of infected cells. In addition, IFNα together with IL-6 enhances the differentiation of B cells into antibody-producing plasma cells (21). In mouse models of RSV infection, the protective role of IFNα was suggested by the observation that RSV infection was exacerbated in STAT1 and IFNαβγR knockout mice (22, 23). For this reason, we investigated whether local administration of IFNα would modify the immune response after RSV infection. As shown in Fig. 4 B, IFNα significantly enhanced the clearance of RSV in both control and pDC-depleted mice. This confirms earlier reports in which administration of IFNα, or poly-IC, a potent IFNα inducer in vivo, decreased RSV titers and disease scores (24). In contrast, in our experiments, IFNα administration did not change airway hyperreactivity (Fig. 4 C) and lung lymph node T cell responses (Fig. 4 D) after RSV infection. Moreover, IFNα had no effect on the elevated responses in pDC-depleted mice.
Figure 4.

Role of IFNα during RSV infection in control and pDC-depleted mice. (A) IFNα4 mRNA and IFNα protein was measured before and at different time points after RSV infection in homogenates of lungs of control (▴) or pDC-depleted (▪) mice. Results are displayed as mean increase to control or pg/ml ± SEM. *, P < 0.05 compared with control mice, n = 5 mice per group. (B) On 6 dpi, RSV titers were determined in the lungs of control (cont), RSV-infected (RSV), IFNα-treated, RSV-infected (RSV+IFNα), or DC-depleted, IFNα-treated, RSV-infected (RSV +120G8+IFNα) mice by the plaque assay. Data are presented as mean PFU per gram lung ± SEM. In addition, RSV G protein RNA transcription in the lung on 6 dpi was measured by quantitative real-time PCR (Taqman). Data are represented as mean fold increase to RSV-infected mice ± SEM. *, P < 0.05 compared with control mice; #, P < 0.05 compared with RSV-infected mice, n = 5 mice per group. (C) Airway responses were measured in control (cont), RSV-infected (RSV), IFNα-treated, RSV-infected (RSV+IFNα), or pDC-depleted, IFNα-treated, RSV-infected (RSV + 120G8+IFNα) mice after one dose of methacholine and compared with basal measurements. Data are represented as mean airway resistance in cm H2O/ml/s ± SEM. *, P < 0.01 compared with noninfected control mice; #, P < 0.05 compared with RSV-infected mice, n = 5 mice per group. (D) Lung-draining lymph nodes were isolated from control (cont), RSV-infected (RSV), IFNα-treated, RSV-infected (RSV+IFNα), and pDC-depleted, IFNα-treated, RSV-infected (RSV + 120G8+IFNα) mice and stimulated with anti-CD3 at a concentration of 5 × 106 cells/ml. Supernatants were analyzed using ELISA. Data are represented as mean ng/ml ± SEM. *, P < 0.01 compared with noninfected control mice; #, P < 0.05 compared with RSV infected mice, n = 4 mice per group.
These data would suggest that IFNα, secreted by pDCs, helps to clear RSV but does not change the adaptive immune response and airway hyperreactivity induced later in the course of RSV infection. This suggests that pDCs, next to playing a direct antiviral role by secreting IFNα, play a modulatory role in establishing T cell–mediated responses to pathogens in the lung as well. Although our experiments have not determined the direct mechanism of pDC function in this regard, we hypothesize that depletion of pDCs initially leads to increased viral antigen presentation by the immuno-stimulatory cDC population. The enhanced T cell cytokine response would likely lead to the increased pathophysiology observed in the absence of pDCs, especially as it relates to the increased mucus overexpression and airway hyperreactivity.
In summary, this study demonstrated that RSV infection of mice caused a sustained increase in the number of pDCs in the lung. Plasmacytoid DCs play a substantial protective role during RSV infection because depletion of pDCs exacerbated all the measured parameters of RSV-induced airway disease. This protective effect of pDCs is only partially mediated through IFNα, as treatment with IFN-α correlated with enhanced clearance of RSV, but had little effect on the development of immune responses in the presence or absence of pDCs. Possibly, pDCs provide additional functions that modulate the adaptive immune response in the lung. These data highlight the critical role of pDCs in pulmonary viral infection and these data may be of use in future studies for the development of new treatment strategies or novel vaccines for RSV infection.
MATERIALS AND METHODS
Animals.
Female BALB/cByJ mice, 6–8 wk of age, were obtained from The Jackson Laboratory. All mice were housed under specific pathogen-free conditions within the animal care facility at the University of Michigan. All experiments were approved by the University of Michigan Committee on the Use and Care of Animals.
RSV infection model and RSV plaque assay.
RSV subtype A, Umich/line 19 strain was derived from a clinical isolate at the University of Michigan and was propagated in Hep2 cells. Infection was allowed to proceed until syncytia were observed. Cells were frozen at −80°C and the supernatant was harvested, clarified, and aliquoted. This RSV preparation was tested negative for both endotoxin and mycoplasma. To determine viral titers in culture supernatants and lung homogenates, an immunoplaque assay was performed as described previously (25). For RSV infection, mice were anesthetized with ketamine/xylazine and intratracheally infected with ∼105 PFU of RSV. For determination of viral titers, lungs from RSV-infected mice were isolated on 6 dpi followed by dispersion and analysis by RSV plaque assay. Administration of UV-irradiated RSV does not elicit any responses in the lungs in mice, demonstrating no confounding effects of the medium used to culture RSV in vivo.
Detection of DC subtypes in lung.
Mice were infected with RSV and at 1, 5, 7, 9, 14, 21, and 30 dpi, lungs and lung-draining mediastinal lymph nodes were isolated and dispersed using 0.2% collagenase (Type IV; Sigma-Aldrich) in RPMI 1640 with 10% FCS at 37°C for 45 min. After lysis of red blood cells and blocking nonspecific binding by FcR, cells were counted and stained with anti-mPDCA1–FITC (Miltenyi Biotec), 120G8-Alexa 488 (AbCys S.A.) or anti-CD4–PerCp-Cy5.5, anti-–DX-5–PE, anti–CD19-PE, anti–CD8-PE, anti–CD11c-PE, anti-CD11c–FITC, anti-B220–FITC, anti-Gr-1–PE, anti-CD11b–FITC antibodies, or isotype controls (all antibodies were obtained from BD Biosciences). Cells were fixed with paraformaldehyde (1%, overnight) and kept in the dark at 4°C until analysis on a Cytomics FC500 flow cytometer (Beckman Coulter). Double positive cells were selected using single staining and isotype controls. Fixation of cells does not affect analysis and number of cells recovered.
Depletion of pDCs and IFNα treatment in vivo.
pDCs were depleted in vivo by i.p. injecting mice 1 d before and day 1 after RSV infection with 150 μg 120G8 antibody (provided by Dr. P. Zavodny, Schering-Plough Research Institute, Kenilworth, NJ) as described previously (17, 26). All control and RSV-infected mice received an isotype control antibody (rat IgG; Sigma-Aldrich).
In a separate set of experiments, mice were treated i.t. with 104 IU murine recombinant IFNα (PBL Biomedical Laboratories) or with control diluent alongside the RSV infection as described previously (24).
Measurement of airway responsiveness.
At day 9 after RSV infection, airway reactivity in anesthetized mice was measured as described previously (6). In brief, mice were anesthetized with sodium pentobarbital and the trachea was cannulated and ventilated using a pump ventilator. After baseline measurements, mice were injected i.v. with 2.5 μg of methacholine (Sigma-Aldrich) and the peak airway resistance was recorded.
Histological analysis.
Lungs were lavaged via the tracheal tube with 1 ml of PBS. Total cell numbers were obtained and differential cell counts were performed by counting cells on cytocentrifuged preparations stained with Diffquick. After obtaining the lung lavage, lungs were inflated and maintained in formalin for 24 h before being processed into paraffin using standard histological techniques. Lung tissue sections were stained with hematoxylin and eosin (H&E) for analysis of inflammatory cell accumulation and alcian blue/periodic acid-Schiff (PAS) for assessment of mucus production. To quantify the mucus production in the lung, PAS sections were randomized and examined and scored on a scale from 1 to 4, with 1 representing no mucus cell content.
Lymph node restimulation.
On 6 dpi, single cell suspensions of lung-draining lymph nodes were prepared by isolation of mediastinal lymph nodes, pushing cells trough a nylon mesh using a syringe and lysis of red blood cells. Cells were cultured in RPMI 1640 medium (RPMI 1640 with 10% FCS, l-glutamin and Pen/Strep) at a concentration of 5 × 106 cells/ml in the presence of either 10 μg/ml plate-bound anti-CD3 (BD Biosciences) or medium alone and incubated at 37°C. After 3 d, supernatants were collected and stored at −80°C until further analysis.
ELISA and quantitative PCR analysis.
Standardized sandwich ELISAs were performed to measure cytokine and chemokine levels in culture supernatants as described previously (6). All antibody pairs were obtained from R&D Systems. IFNα was measured in lung homogenates using a commercial ELISA kit obtained from PBL Biomedical Laboratories that has a detection limit of 13 pg/ml.
RNA was isolated from lungs using TRIzol reagent (Invitrogen), according to the instructions of the manufacturer. Each sample was reverse transcribed into cDNA and analyzed by quantitative real-time PCR using Taqman (Applied Biosytems). Raw data were normalized to GAPDH control standards in each sample.
Statistical analysis.
Significance was determined using one-way analysis of variance or single Student's t tests with 95% confidence intervals. A p-value of <0.05 was considered significant.
Online supplemental material.
Table S1 shows the efficacy of the 120G8 antibody in depleting pDCs in the lung. The 120g8 antibody decreased the number of pDCs, but not the number of cDCs in the lung after RSV infection. Fig. S1 displays the analysis of CD11chigh/CD11bhigh, CD11c+/B220+, and CD11c+/mPDCA1+ DCs as enumerated in Fig. S1. Fig. S2 shows the quantification of the inflammation and mucus production in the lung as shown in Fig. S2 B. Shown are mucus scores from the PAS stained slides, the number of lymphocytes in the lung lavage fluid, and the expression of Gob5 and Muc5ac mRNA in the lung. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20052359/DC1.
Supplemental Material
Acknowledgments
We thank Dr. D.T. Deurloo for critically reading the manuscript.
This work was supported in part by National Institutes of Health grant no. AF36302.
The authors have no conflicting financial interests.
References
- 1.Falsey, A.R., P.A. Hennessey, M.A. Formica, C. Cox, and E.E. Walsh. 2005. Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 352:1749–1759. [DOI] [PubMed] [Google Scholar]
- 2.Hall, C.B. 2001. Respiratory syncytial virus and parainfluenza virus. N. Engl. J. Med. 344:1917–1928. [DOI] [PubMed] [Google Scholar]
- 3.Sigurs, N., P.M. Gustafsson, R. Bjarnason, F. Lundberg, S. Schmidt, F. Sigurbergsson, and B. Kjellman. 2005. Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. Am. J. Respir. Crit. Care Med. 171:137–141. [DOI] [PubMed] [Google Scholar]
- 4.Kim, H.W., J.G. Canchola, C.D. Brandt, G. Pyles, R.M. Chanock, K. Jensen, and R.H. Parrott. 1969. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am. J. Epidemiol. 89:422–434. [DOI] [PubMed] [Google Scholar]
- 5.Peebles, R.S., Jr., and B.S. Graham. 2005. Pathogenesis of respiratory syncytial virus infection in the murine model. Proc. Am. Thorac. Soc. 2:110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tekkanat, K.K., H.F. Maassab, D.S. Cho, J.J. Lai, A. John, A. Berlin, M.H. Kaplan, and N.W. Lukacs. 2001. IL-13-induced airway hyperreactivity during respiratory syncytial virus infection is STAT6 dependent. J. Immunol. 166:3542–3548. [DOI] [PubMed] [Google Scholar]
- 7.Vermaelen, K., and R. Pauwels. 2005. Pulmonary dendritic cells. Am. J. Respir. Crit. Care Med. 172:530–551. [DOI] [PubMed] [Google Scholar]
- 8.van Rijt, L.S., S. Jung, A. Kleinjan, N. Vos, M. Willart, C. Duez, H.C. Hoogsteden, and B.N. Lambrecht. 2005. In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J. Exp. Med. 201:981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steinman, R.M. 2003. Some interfaces of dendritic cell biology. APMIS. 111:675–697. [DOI] [PubMed] [Google Scholar]
- 10.Dahl, M.E., K. Dabbagh, D. Liggitt, S. Kim, and D.B. Lewis. 2004. Viral-induced T helper type 1 responses enhance allergic disease by effects on lung dendritic cells. Nat. Immunol. 5:337–343. [DOI] [PubMed] [Google Scholar]
- 11.Belz, G.T., C.M. Smith, L. Kleinert, P. Reading, A. Brooks, K. Shortman, F.R. Carbone, and W.R. Heath. 2004. Distinct migrating and nonmigrating dendritic cell populations are involved in MHC class I-restricted antigen presentation after lung infection with virus. Proc. Natl. Acad. Sci. USA. 101:8670–8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beyer, M., H. Bartz, K. Horner, S. Doths, C. Koerner-Rettberg, and J. Schwarze. 2004. Sustained increases in numbers of pulmonary dendritic cells after respiratory syncytial virus infection. J. Allergy Clin. Immunol. 113:127–133. [DOI] [PubMed] [Google Scholar]
- 13.Gill, M.A., A.K. Palucka, T. Barton, F. Ghaffar, H. Jafri, J. Banchereau, and O. Ramilo. 2005. Mobilization of plasmacytoid and myeloid dendritic cells to mucosal sites in children with respiratory syncytial virus and other viral respiratory infections. J. Infect. Dis. 191:1105–1115. [DOI] [PubMed] [Google Scholar]
- 14.Schwarze, J., D.R. O'Donnell, A. Rohwedder, and P.J. Openshaw. 2004. Latency and persistence of respiratory syncytial virus despite T cell immunity. Am. J. Respir. Crit. Care Med. 169:801–805. [DOI] [PubMed] [Google Scholar]
- 15.Cubie, H.A., L.A. Duncan, L.A. Marshall, and N.M. Smith. 1997. Detection of respiratory syncytial virus nucleic acid in archival postmortem tissue from infants. Pediatr. Pathol. Lab. Med. 17:927–938. [PubMed] [Google Scholar]
- 16.Oriss, T.B., M. Ostroukhova, C. Seguin-Devaux, B. Dixon-McCarthy, D.B. Stolz, S.C. Watkins, B. Pillemer, P. Ray, and A. Ray. 2005. Dynamics of dendritic cell phenotype and interactions with CD4+ T cells in airway inflammation and tolerance. J. Immunol. 174:854–863. [DOI] [PubMed] [Google Scholar]
- 17.de Heer, H.J., H. Hammad, T. Soullie, D. Hijdra, N. Vos, M.A. Willart, H.C. Hoogsteden, and B.N. Lambrecht. 2004. Essential role of lung plasmacytoid dendritic cells in preventing asthmatic reactions to harmless inhaled antigen. J. Exp. Med. 200:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Demedts, I.K., G.G. Brusselle, K.Y. Vermaelen, and R.A. Pauwels. 2005. Identification and characterization of human pulmonary dendritic cells. Am. J. Respir. Cell Mol. Biol. 32:177–184. [DOI] [PubMed] [Google Scholar]
- 19.Colonna, M., G. Trinchieri, and Y.J. Liu. 2004. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 5:1219–1226. [DOI] [PubMed] [Google Scholar]
- 20.Cella, M., D. Jarrossay, F. Facchetti, O. Alebardi, H. Nakajima, A. Lanzavecchia, and M. Colonna. 1999. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat. Med. 5:919–923. [DOI] [PubMed] [Google Scholar]
- 21.Le Bon, A., G. Schiavoni, G. D'Agostino, I. Gresser, F. Belardelli, and D.F. Tough. 2001. Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity. 14:461–470. [DOI] [PubMed] [Google Scholar]
- 22.Hashimoto, K., J.E. Durbin, W. Zhou, R.D. Collins, S.B. Ho, J.K. Kolls, P.J. Dubin, J.R. Sheller, K. Goleniewska, J.F. O'Neal, et al. 2005. Respiratory syncytial virus infection in the absence of STAT 1 results in airway dysfunction, airway mucus, and augmented IL-17 levels. J. Allergy Clin. Immunol. 116:550–557. [DOI] [PubMed] [Google Scholar]
- 23.Johnson, T.R., S.E. Mertz, N. Gitiban, S. Hammond, R. Legallo, R.K. Durbin, and J.E. Durbin. 2005. Role for innate IFNs in determining respiratory syncytial virus immunopathology. J. Immunol. 174:7234–7241. [DOI] [PubMed] [Google Scholar]
- 24.Guerrero-Plata, A., S. Baron, J.S. Poast, P.A. Adegboyega, A. Casola, and R.P. Garofalo. 2005. Activity and regulation of α interferon in respiratory syncytial virus and human metapneumovirus experimental infections. J. Virol. 79:10190–10199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller, A.L., T.L. Bowlin, and N.W. Lukacs. 2004. Respiratory syncytial virus-induced chemokine production: linking viral replication to chemokine production in vitro and in vivo. J. Infect. Dis. 189:1419–1430. [DOI] [PubMed] [Google Scholar]
- 26.Asselin-Paturel, C., G. Brizard, J.J. Pin, F. Briere, and G. Trinchieri. 2003. Mouse strain differences in plasmacytoid dendritic cell frequency and function revealed by a novel monoclonal antibody. J. Immunol. 171:6466–6477. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}