

Abstract
The accumulation of smooth muscle and endothelial cells is essential for remodeling and repair of injured blood vessel walls. Bone marrow–derived progenitor cells have been implicated in vascular repair and remodeling; however, the mechanisms underlying their recruitment to the site of injury remain elusive. Here, using real-time in vivo fluorescence microscopy, we show that platelets provide the critical signal that recruits CD34+ bone marrow cells and c-Kit+ Sca-1+ Lin− bone marrow–derived progenitor cells to sites of vascular injury. Correspondingly, specific inhibition of platelet adhesion virtually abrogated the accumulation of both CD34+ and c-Kit+ Sca-1+ Lin− bone marrow–derived progenitor cells at sites of endothelial disruption. Binding of bone marrow cells to platelets involves both P-selectin and GPIIb integrin on platelets. Unexpectedly, we found that activated platelets secrete the chemokine SDF-1α, thereby supporting further primary adhesion and migration of progenitor cells. These findings establish the platelet as a major player in the initiation of vascular remodeling, a process of fundamental importance for vascular repair and pathological remodeling after vascular injury.
Proliferation of endothelial cells (ECs) and vascular smooth muscle cells (SMCs) is essential for cardiovascular development (1), but also contributes to the repair and remodeling of the injured vessel wall (2). The major trigger of vascular injury is atherosclerosis, which is today the most important cause of morbidity in the Western world. Spontaneous rupture of the atherosclerotic plaque and vascular interventions for atherothrombotic disease (e.g., balloon angioplasty) induce EC damage. Disruption of the endothelial integrity initiates proliferation of ECs, which promotes reendothelialization of the vascular lesion (3, 4), but also triggers local accumulation of SMCs, leading to intimal hyperplasia and occlusion of the diseased vessel (2).
At first, neointimal ECs and SMCs were believed to originate exclusively from adjacent cells within the vessel wall that migrate to the site of injury and start to proliferate (3). However, recently BM-derived progenitor cells (BM-PCs) have been implicated in remodeling and repair of the injured vessel wall (4–9). Indeed, BM-PCs appear to give rise to substantial numbers of neointimal ECs and SMCs after endothelial denudation (4–9). However, the signals that target BM-PCs to foci of vascular injury have not been understood thus far.
The first response to vascular injury is platelet adhesion to the exposed subendothelium (10–13). Here, we show that platelet adhesion not only triggers vascular thrombosis, leading to myocardial infarction and ischemic stroke, but also represents the critical step for the targeting of BM-PCs to sites of endothelial disruption. Using real-time in vivo double fluorescence microscopy of the mouse carotid artery, we demonstrate that CD34+ and c-Kit+ Sca-1+ Lin− (KSL) BM-PCs directly adhere to platelets after vascular injury in a process that involves platelet P-selectin and GPIIb integrin. Once activated, platelets secrete the chemokine SDF-1α, which supports primary adhesion of PCs on the surface of arterial thrombi in vivo. BM-PCs recruited to platelet aggregates give rise to neointimal cells, indicating that accumulation of BM-PCs in arterial thrombi may contribute to vascular repair and, eventually, to pathological remodeling. Together, the present results identify a central role of platelets for the targeting of BM-PCs to the arterial intima, a process of fundamental importance for vascular repair and pathological remodeling after vascular injury.
RESULTS
BM-PCs are recruited rapidly to sites of endothelial denudation
Because primitive long-term repopulating mouse hematopoietic PCs are enriched among CD34+ BM cells (14), we first addressed the accumulation of mouse CD34+ BM-PCs to foci of vascular injury. We injured the carotid artery of C57BL/6J mice (12) and infused CD34+ cells tagged with dichlorofluorescein (DCF). We used intravital video fluorescence microscopy (IVM) to directly visualize and quantify the dynamic process of BM-PC accumulation. In the absence of vascular injury, CD34+ BM-PCs rolled along short distances of the intact vessel wall; however, firm adhesion was not detected. In contrast, numerous CD34+ BM-PCs were recruited to the vascular wall within 5 min after endothelial denudation, reaching a plateau 60 min after vascular injury (375 ± 83 adherent cells/mm2 60 min after injury, P < 0.05 vs. baseline; Fig. 1, a and b).
Figure 1.

Recruitment of BM-PCs to the injured carotid artery. (a) CD34+ BM-PC adhesion before and after carotid injury was monitored by in vivo microscopy. *, P < 0.05 vs. baseline (pre). (b) The microphotographs show representative in vivo fluorescence microscopy images of CD34+ BM-PCs at distinct time points. Bars, 50 μm. (c) Purification of c-Kit+ Sca-1+ Lin− PCs. Lin− cells were stained with propidium iodide (PI), FITC-conjugated anti–Sca-1 (Ly 6A/E) antibody, and PE-conjugated anti–c-Kit (CD117) antibody. Viable (PI−) c-Kit+ Sca-1+ cells were sorted. After sorting, the purity of c-Kit+ Sca-1+ Lin− cells was >98%. (d) Assessment of c-Kit+ Sca-1+ Lin− BM-PC adhesion before and after carotid injury by in vivo microscopy. *, P < 0.05 vs. no injury. (e) The microphotographs show representative in vivo fluorescence microscopy images of c-Kit+ Sca-1+ Lin− BM-PCs before and after vascular injury. Bars represent 50 μm. Arrows in b and e indicate nonadherent BM-PCs, arrowheads indicate adherent BM-PCs. Data are means ± SEM.
CD34 expression on mouse BM-PCs has been demonstrated to vary with the activation status of the precursor cells (15) and considerable amounts of mouse long-term reconstituting BM-PCs reside in the CD34−/low cell fraction (16). Hence, we next analyzed the recruitment of phenotypically defined mouse hematopoietic BM-PCs (16). We isolated KSL cells by fluorescence-activated cell sorting (purity of KSL >98%; Fig. 1 c). DCF-tagged KSL cells were infused i.v. and visualized by intravital microscopy before and after carotid injury. Like CD34+ cells, KSL BM-PCs did not firmly adhere to the intact vessel wall. However, after endothelial disruption, KSL cells readily attached to the site of injury (1,110 ± 428 adherent KSL cells/mm2 30 min after injury; P < 0.05 vs. uninjured vessel wall; Fig. 1, d and e). This demonstrates for the first time in vivo that BM-PCs are recruited rapidly to sites of vascular injury.
BM-PCs do not adhere directly to subendothelial matrix proteins under high arterial shear
Next, we elucidated the determinants that initiate BM-PC recruitment to the subendothelium. Adhesion of CD34+ or KSL BM-PCs was monitored in a parallel plate flow chamber at a wall shear rate of 1,000 s−1. To our surprise, neither vitronectin, nor fibronectin, fibrinogen, or collagen, the major constituents of extracellular matrices exposed at sites of vascular injury, promoted considerable BM-PC adhesion under flow (Fig. 2 a). Correspondingly, flow cytometric examination of mouse CD34+ or KSL BM-PCs failed to demonstrate expression of adhesion molecules that would allow adhesion of flowing cells to the subendothelial matrix under arterial shear conditions, such as GPIb-V-IX or GPVI (Fig. 2 b). However, as reported previously by others, α4 integrin (CD49d) and P-selectin glycoprotein ligand (PSGL)-1 were the major adhesion receptors present on the surface of CD34+ and KSL BM-PCs (17, 18) (Fig. 2 b). PSGL-1 and α4 integrin mediate PC homing to the endothelial lining of BM microvessels; however, they are not sufficient to promote cellular adhesion to subendothelial matrices. Collectively, the latter results imply that BM-PCs are not able to adhere directly to the exposed extracellular matrix after vascular injury.
Figure 2.

BM-PCs do not adhere directly to subendothelial matrix proteins under arterial shear conditions. (a) The adhesion of BM-PCs (2 × 104/ml KSL cells or CD34+ cells) to coverslips coated with vitronectin (Vn; Becton Dickinson), collagen (Col; Becton Dickinson), fibrinogen (Fb; Sigma-Aldrich), fibronectin (Fn; Sigma-Aldrich), or to surface adherent platelets was assessed in a transparent flow chamber at a wall shear rate of 1,000 s−1 as described elsewhere (reference 22). The number of adherent BM-PCs is given per mm2 surface. *, P < 0.05 vs. collagen. (b) To determine the adhesion receptors present on the surface of BM-PCs, KSL or CD34+ cells were incubated with fluorophore-labeled anti-αIIb, anti-α4, anti–PSGL-1, anti-α2β1, anti-GPIbα, or anti-GPVI mAb, or irrelevant isotype-matched control antibody and directly analyzed on a FACSCalibur. Data are given as difference between the mean fluorescence intensity [arbitrary units] obtained with the specific antibodies and the signal obtained with the corresponding irrelevant isotype-matched control antibody. Data are means ± SEM.
Platelets interact with BM-PCs in vitro and at sites of vascular injury in vivo
One of the first responses to vascular injury is platelet adhesion and aggregation at the site of endothelial denudation (Fig. 3, a and b) (11, 12, 19, 20). To evaluate the potential role of platelets for the recruitment of PCs to the injured vessel wall, we investigated whether BM-PCs are able to interact with platelets. Using flow cytometry, we observed that resting and particularly thrombin-activated platelets bind to BM-derived CD34+ and KSL cells in vitro (Fig. 3 c). Scanning electron microscopy revealed that platelets interact directly with BM-PCs (Fig. 3 d).
Figure 3.

Platelets contribute to the recruitment of BM-PCs to the injured carotid artery. (a) Interactions of rhodamine-6G–tagged platelets before and after carotid injury were investigated by in vivo microscopy. *, P < 0.05 vs. baseline (pre). Representative microscopic images are presented in b. Arrows indicate adherent platelets. (c) BM-PCs were incubated with PBS, thrombin, or resting platelets or platelets activated with thrombin. Flow cytometry was used to detect the binding of CD41+ platelets to BM-PCs. Data are given as increase in mean fluorescence intensity compared with vehicle (PBS)-treated BM-PCs. *, P < 0.05 vs. resting. (d) CD34+ BM-PCs were cultivated in the presence of platelets. Scanning electron microscopy revealed that platelets (arrows) bind directly to the surface of CD34+ BM-PCs. Bar, 5 μm. (e) The carotid artery of C57BL/6J mice was injured, and differentially tagged KSL cells (DCF, green; left) and platelets (rhodamine-6G chloride, red; middle) were visualized by real-time double fluorescence microscopy. The overlay (right) shows that BM-derived c-Kit+ Sca-1+ Lin− cells bind exclusively to adherent platelets (arrows). Bars, 50 μm. Data are means ± SEM.
To identify whether similar interactions also occur under physiological shear conditions, mouse platelets were allowed to adhere to collagen-coated coverslips and the adhesion of CD34+ or KSL BM-PCs was monitored in a parallel plate flow chamber at a wall shear rate of 1,000 s−1 as described in previous paragraphs (Fig. 2 a). BM-PCs did not attach to collagen-coated coverslips in the absence of platelets (Fig. 2 a, Col). However, when the collagen matrix was covered with platelets, BM-PCs readily adhered in the presence of arterial shear conditions (Fig. 2 a, Plts).
To find out whether similar interactions between BM-PCs and platelets are present in vivo, we injured the carotid artery of mice. Differentially tagged KSL cells (DCF) and platelets (rhodamine-6G) were infused i.v. Platelet–KSL interactions at the site of the vascular lesion were monitored by real-time double fluorescence IVM. Although platelet adhesion occurred within seconds after endothelial disruption, KSL BM-PCs started to adhere to the injured vessel wall 5–10 min after induction of vascular injury. Interestingly, we observed that KSL BM-PCs bound exclusively to adherent platelets, whereas no adherent KSL cells were detected at areas devoid of platelet adhesion (Fig. 3 e).
Platelet adhesion is critically involved in BM-PC recruitment after vascular injury
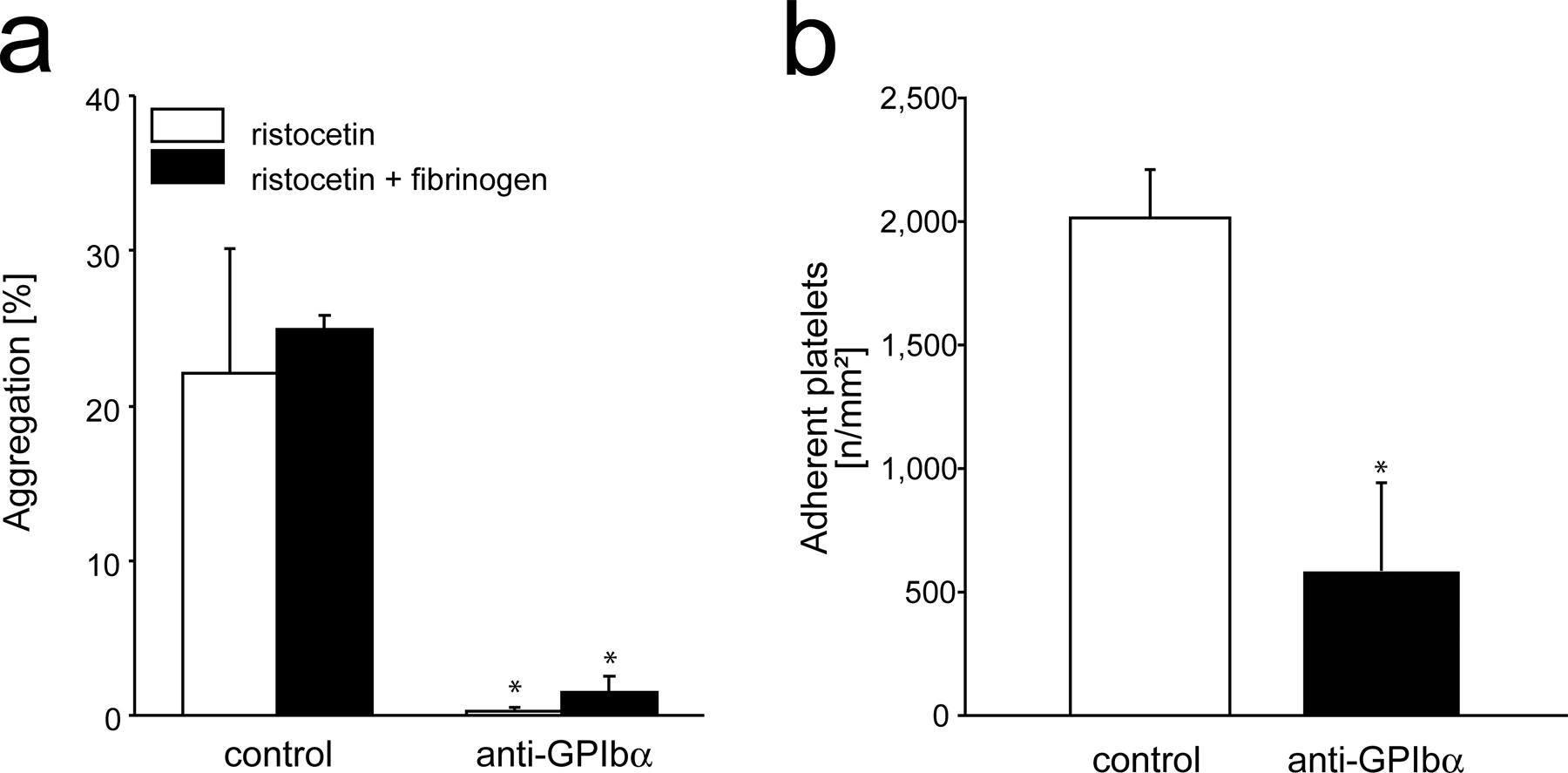
Because these findings were very suggestive, we subsequently addressed whether the presence of adherent platelets might actually be mandatory for the recruitment of BM-PCs. Platelet adhesion to the subendothelium involves the platelet vWF receptor GPIbα and the major platelet collagen receptor GPVI (10–12, 21). Correspondingly, inhibition of platelet GPIbα or GPVI by function-blocking mAb almost completely prevents platelet adhesion at sites of vascular injury (12). Because the platelet adhesion receptors GPIbα or GPVI are not expressed by BM-PCs (Fig. 2 b), inhibition of GPIbα or GPVI provides a powerful tool to selectively address the role of platelet adhesion for BM-PC recruitment at the injured vessel wall. Hence, to evaluate the functional importance of platelets for BM-PC accumulation, we treated mice with 2 mg/kg function-blocking anti-GPIbα mAb or isotype-matched control IgG. Thereafter, carotid injury was induced and the adhesion of DCF-tagged CD34+ (Fig. 4 a) or KSL BM-PCs (Fig. 4 b) was monitored in vivo by IVM. As reported previously, inhibition of GPIbα virtually abrogated platelet aggregation in response to ristocetin in vitro and reduced platelet adhesion to the injured vessel wall in vivo (Fig. S1, a and b, available at http://www.jem.org/cgi/content/full/jem.20051772/DC1). However, to our surprise, interference with GPIbα–vWF interactions not only prevented platelet adhesion but also virtually completely blocked recruitment of CD34+ (Fig. 4 a) and KSL BM-PCs (Fig. 4 b) in response to carotid denudation, indicating that platelet adhesion is crucial for BM-PC recruitment to the injured vessel wall.
Figure 4.

Platelet adhesion contributes to the recruitment of BM-PCs to the injured carotid. (a) Carotid injury was induced in wild-type mice treated with function-blocking anti-GPIbα, GPVI-Fc, anti–PSGL-1, anti-CD62P mAbs, or isotype-matched control IgG. In a separate group of animals, carotid injury was induced in mice lacking P-selectin. Adhesion of CD34+ BM-PCs was monitored by in vivo fluorescence microscopy. *, P < 0.05 vs. control IgG-treated WT mice. (b) Carotid injury was induced in wild-type mice treated with function-blocking anti-GPIbα or anti–PSGL-1 mAbs. Adhesion of KSL BM-PCs was monitored by in vivo fluorescence microscopy. *, P < 0.05 vs. control IgG-treated mice. (c) CD34+ BM-PCs were incubated with thrombin-activated platelets in the absence or presence of function-blocking anti–PSGL-1 or anti-CD11b mAb. CD41 expression on the surface of CD34+ BM-PCs was determined to assess platelet binding. Data are means ± SEM.
A separate group of C57BL/6J wild-type mice was treated with soluble GPVI-Fc, which acts as a competitive inhibitor of platelet GPVI in vivo and virtually abrogates platelet accumulation in response to vascular injury (22). In a similar manner, as reported in the previous paragraph for inhibition of GPIbα, blockade of GPVI abrogated the recruitment of precursor cells in response to carotid denudation (Fig. 4 a). These data demonstrate for the first time that platelet adhesion is strictly required for targeting of CD34+ and KSL BM-PCs to foci of vascular injury in vivo.
Platelets promote BM-PC recruitment after vascular injury by expression of P-selectin
Upon adhesion, platelets become activated and expose P-selectin. CD34+ and KSL BM-PCs express PSGL-1 (Fig. 2 b), the major ligand of P-selectin, and roll on recombinant P-selectin in vitro (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20051772/DC1). Hence, we next evaluated the significance of P-selectin and PSGL-1 for BM-PC–platelet interactions. We incubated activated platelets with CD34+ BM-PCs in the presence of function-blocking anti–mouse PSGL-1 mAb 4RA10 (23). Notably, platelet binding to BM-PCs was greatly reduced after inhibition of PSGL-1 in vitro, as determined by flow cytometry (Fig. 4 c). To further substantiate the role of P-selectin and PSGL-1 for BM-PC recruitment in vivo, CD34+ or KSL BM-PCs were transfused into mice pretreated with mAbs directed against P-selectin or PSGL-1. Inhibition of P-selectin or PSGL-1 significantly reduced the recruitment of BM-derived CD34+ (Fig. 4 a) or KSL BM-PCs (Fig. 4 b) after carotid injury. Similarly, BM-PC accumulation to the injured vessel walls was significantly reduced in P-selectin–deficient mice (Fig. 4 a).
Platelets mediate BM-PC recruitment by release of SDF-1α
Recently, the CXC chemokine SDF-1α has been implicated in attracting blood-borne PCs to the vascular intima (24, 25). Hence, we asked whether SDF-1α might contribute to PC accumulation on arterial thrombi at sites of vascular injury. First, we evaluated the distribution of SDF-1α in the vessel wall in response to endothelial denudation. Although SDF-1α was not detected in intact carotid arteries, endothelial disruption triggered a substantial increase in vascular SDF-1α expression (Fig. 5 a). SMCs were a major source of SDF-1α at 4 and 24 h after endothelial disruption (Fig. 5 a and not depicted). This is consistent with previous reports (25–27), showing that medial SMCs express SDF-1α very early after vascular damage. However, 30 min after vascular injury, SDF-1α expression in SMCs was negligible (Fig. 5 a), indicating that SMC-derived SDF-1α is unlikely to contribute to BM-PC accumulation on arterial thrombi during the first minutes after endothelial disruption (Fig. 1, a and d).
Figure 5.

Platelets express and secrete SDF-1α and recruit endogenous PCs to sites of vascular injury. (a) Paraffin-embedded sections of a mouse carotid artery injured as described (panel a and see Materials and methods) and a fresh human coronary thrombus (b) were stained for SDF-1α. Sections incubated with irrelevant isotype-matched IgG served as controls. The intense red (a) or brown (b) staining indicates that SDF-1α is expressed in both mouse and human intravascular thrombi. In a, L, EC, M, and A indicate lumen, endothelial cells, media, and adventitia, respectively. Arrows in b indicate mononuclear cells. Bars, 50 (a) or 20 μm (b). (c) Triple immunofluorescence staining of an injured carotid artery shows that SDF-1α expression in thrombi is largely confined to CD41-expressing platelets. Mononuclear cells recruited to the thrombus are indicated by blue (DAPI). Bars, 25 μm. Sections incubated with the secondary but not the primary antibody served as controls (right). (d) We used ELISA to determine SDF-1α in mouse platelets or whole blood. The mouse melanoma cell line B16-D5 served as control. SDF-1α protein expression is given in nanograms per 100 μg total protein. *, P < 0.05 vs. whole blood. (e) Thrombin-activated human platelets surface express SDF-1α, as indicated by flow cytometry (for details, see Materials and methods). The diagram (left) summarizes three independent experiments and a representative histogram is shown (right). (f) ELISA also demonstrates that platelet activation triggers release of SDF-1α, as indicated by an increase in SDF-1α protein concentration in the supernatants of α-thrombin–activated platelets. *, P < 0.05 vs. resting. (g) Confocal laser scanning microscopy demonstrates that SDF-1α is present in the cytosol of resting platelets, whereas it becomes surface expressed after platelet activation. (top left) Resting platelets at high magnification. SDF-1α is present in the platelet cytoplasm as identified by the red staining (arrowheads); CD41 expression is indicated by green. Platelet activation by 20 μM ADP (top, middle and right) or 0.2 U/ml thrombin (bottom) induced surface mobilization of SDF-1α (green; arrowheads). The platelet cytoskeleton is highlighted by the red staining (phalloidin). Thrombin-activated platelets (bottom right) were stained with irrelevant control mAb (green) and counter-stained with phalloidin (red). Bars, 10 μm (top, middle; bottom, left and right) or 5 μm (top, left and right; bottom, middle). Data are means ± SEM.
Importantly though, medial SMCs were not the only source of SDF-1α at sites of vascular damage. To our surprise, a robust staining for SDF-1α was also present in arterial thrombi attached to the injured vessel wall. SDF-1α expression within arterial thrombi was found as early as 30 min after vascular injury and could still be detected at 4 and 24 h (Fig. 5 a and not depicted) after injury. SDF-1α expression was not restricted to mouse thrombi, but could also be observed during thrombus formation in the human vasculature. Correspondingly, arterial thrombi isolated from ruptured atherosclerotic plaques of human coronary arteries showed substantial SDF-1α protein expression (Fig. 5 b). Together, these data suggest that thrombi that develop at the site of endothelial denudation are the initial source of SDF-1α (Fig. 5 a), whereas SMCs account for SDF-1α release at later stages (25–27).
To identify the source of SDF-1α within arterial thrombi, we subsequently performed immunohistochemistry of injured mouse carotid arteries. Unexpectedly, we found that SDF-1α expression in thrombi is largely confined to the surface of aggregated platelets (Fig. 5 c). To further substantiate SDF-1α expression by platelets, we analyzed lysates of purified platelets (purity >99%) for the presence of SDF-1α using ELISA. Indeed, purified platelets contained substantial amounts of the CXC chemokine SDF-1α (Fig. 5 d). Upon platelet activation, SDF-1α can be detected on the platelet surface (Fig. 5 e) and is released into the platelet supernatant (Fig. 5). Correspondingly, confocal scanning microscopy demonstrated that SDF-1α is present in the cytosol of resting platelets, whereas it becomes surface expressed after platelet activation (Fig. 5 g).
To define the source of platelet SDF-1α, we examined the BM of mice. Interestingly, we found that, apart from stromal cells, megakaryocytes stain positive for SDF-1α, albeit to a lesser extent (Fig. 6 a). To further substantiate SDF-1α expression by the megakaryocytic lineage, we determined SDF-1α mRNA and protein expression in culture-derived megakaryocytes (28). By RT-PCR (Fig. 6 b) and using ELISA (Fig. 6 c), we show that megakaryocytes contain SDF-1α mRNA and protein. Notably, SDF-1α is targeted to α-granules in the megakaryocytic/platelet lineage (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20051772/DC1). Together, this suggests that platelets obtain SDF-1α basically from their PCs.
Figure 6.

Megakaryocytes express SDF-1α. (a) Paraffin-embedded sections of mouse femura were stained with anti–SDF-1α. SDF-1α was expressed not only in stromal cells (arrows) but also in megakaryocytes (arrowheads). Bars, 25 μm. (b) Megakaryocytic and platelet SDF-1α mRNA expression was determined using RT-PCR. The mRNA expression of GPIIb integrin and β-actin served as controls. The primer sequences and their corresponding product sizes and annealing temperatures are in Table S1. (c) ELISA was used to determine SDF-1α in mouse megakaryocytes or mouse BM. SDF-1α protein expression is given in nanograms per 100 μg total protein. Data are means ± SEM.
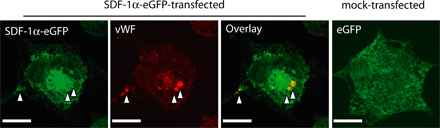
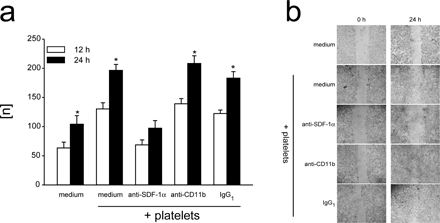
To test the biological significance of platelet SDF-1α release, subconfluent monolayers of adherent embryonic endothelial PCs (T17B) (29) were wounded and subsequently incubated in the absence or presence of platelets. Notably, platelets induced considerable migration of PCs into the wounded area. Function-blocking anti–SDF-1α mAb, but not control IgG, virtually abolished platelet-induced PC migration, indicating that platelet-derived SDF-1α is functionally active (Fig. S4, a and b, available at http://www.jem.org/cgi/content/full/jem.20051772/DC1).
Next, we addressed the biological relevance of platelet SDF-1α for the recruitment of BM-PCs into arterial thrombi in vivo. We injured the carotid artery of C57BL/6J mice to induce arterial thrombosis. Next, the animals were treated with function-blocking anti–SDF-1α mAb, and DCF-tagged KSL BM-PCs were infused. Adhesion of KSL PCs on the thrombus surface was visualized in vivo by IVM as described in previous paragraphs. Inhibition of SDF-1α by function-blocking mAb significantly attenuated KSL accumulation within the growing platelet-rich thrombus (267 ± 33 adherent KSL cells/mm2 30 min after vascular injury in the presence of anti–SDF-1α mAb; *, P < 0.05 vs. control IgG-treated mice).
Platelet GPIIb contributes to BM-PC recruitment in vivo
During adhesion, platelets become activated and start to aggregate in a process mediated by the platelet integrin GPIIb-IIIa (11). To find out whether GPIIb-dependent platelet aggregation is essential for BM-PC accumulation in vivo, we injured the carotid artery of GPIIb-deficient mice and simultaneously visualized the recruitment of differentially tagged GPIIb−/− platelets and wild-type CD34+ or KSL BM-PCs by IVM. As reported previously, platelet aggregation and thrombus formation after vascular injury are abolished in mice lacking GPIIb integrin (13). Remarkably, the defect in GPIIb-dependent platelet aggregation translated into a profound and significant reduction in CD34+ BM-PC accumulation compared with wild-type animals (P < 0.05; Fig. 7 a). Likewise, adhesion of KSL PCs was significantly attenuated in mice lacking GPIIb integrin (128 ± 16 or 964 ± 393 adherent KSL cells/mm2 15 min after vascular injury in GPIIb-deficient or wild-type mice, respectively).
Figure 7.

GPIIb-dependent platelet aggregation promotes BM-PC recruitment during arterial thrombosis in vivo and contributes to SDF-1α release in vitro. (a) To find out whether platelet GPIIb contributes to BM-PC recruitment in vivo, carotid injury was induced in mice lacking GPIIb. Adhesion of CD34+ BM-PCs was monitored by in vivo fluorescence microscopy. *, P < 0.05 vs. wild type. (b) Platelets were incubated with medium, thrombin, 100 μg/ml soluble fibrinogen, 5 μg/ml mAb anti-fibrinogen, or a combination of both for 30 min. Platelet SDF-1α release was determined by ELISA as described in Materials and methods. Similar experiments were performed with anti-GPIIb mAb 7E3 (whole IgG, 5 μg/ml) in the presence or absence of 5 μg/ml mAb goat anti–mouse IgG. *, P < 0.05 vs. resting platelets. (c) The carotid artery of wild-type (WT) or GPIIb-deficient mice was injured as described in Materials and methods. Paraffin-embedded sections were stained for SDF-1α. L, M, and A indicate lumen, media, and adventitia, respectively. Arrows identify SDF-1α deposited at the site of vascular injury. Bars, 20 μm. Data are means ± SEM.
During aggregation, clustering of GPIIb-IIIa transduces inward signals that may affect important platelet functions including the release of chemokines (30, 31). Hence, we next asked whether, beyond promoting platelet aggregation, GPIIb-IIIa might contribute to platelet SDF-1α exposure. Platelets were incubated with soluble fibrinogen in the presence or absence of bivalent mAb antifibrinogen (IgG1), which enhances receptor cross-linking (Fig. 7 b). Interestingly, SDF-1α release was significantly increased when GPIIb-IIIa was clustered by combined fibrinogen/antifibrinogen incubation. Similarly, mouse anti-GPIIb mAb 7E3 significantly increased SDF-1α release in the presence of anti–mouse mAb (IgG) that binds mAb 7E3. This indicates that engagement of GPIIb during platelet aggregation by itself initiates platelet SDF-1α release. Correspondingly, the expression of SDF-1α at the site of injury was substantially reduced in mice lacking GPIIb as compared with wild-type animals (Fig. 7 c). This shows that GPIIb considerably contributes to the local delivery and release of SDF-1α during arterial thrombosis by promoting the accumulation of SDF-1α–bearing platelets and by triggering the release of SDF-1α from platelet storage granules during platelet aggregation.
Platelets recruit endogenous PCs to the injured vessel wall in vivo
To further substantiate the biological significance of platelet–PC interactions, we finally determined whether platelets might also contribute to the targeting of circulating endogenous PCs to sites of vascular lesions. We mechanically injured the carotid artery of C57BL/6J, P-selectin–, or GPIIb-deficient recipient mice or C57BL/6J mice treated with anti–SDF-1α mAb. The injured and the uninjured contralateral carotid arteries were harvested 24 h after injury. To detect the recruitment of endogenous PCs to the site of vascular injury, we determined c-Kit and Sca-1 mRNA expression. In wild-type mice, carotid injury induced considerable PC recruitment, as indicated by an approximately five- to sixfold increase of c-Kit and Sca-1 mRNA expression in the injured vessel compared with the uninjured contralateral control carotid artery (Fig. 8 a). Correspondingly, on day 1 after vascular injury, the carotid arteries of wild-type mice showed thrombi containing numerous nucleated cells that stained positive for c-Kit upon immunohistochemical observation (Fig. 8 b). In contrast, the recruitment of c-Kit+ cells (unpublished data) and the concomitant increase in c-Kit and Sca-1 mRNA expression after vascular injury was substantially attenuated in P-selectin– or GPIIb-deficient mice, whereas it was completely absent in anti–SDF-1α mAb-treated animals (Fig. 8 c).
Figure 8.

Platelets recruit endogenous PCs to sites of vascular injury. (a) Carotid arteries of C57BL/6J mice were injured as described in Materials and methods. 24 h thereafter, the injured (right) and the uninjured (left, control) carotid arteries were excised and Sca-1 and c-Kit mRNA expression were determined as described in Materials and methods. Isolated mouse CD34+ BM-PCs and the mouse heart EC line MHEC5-T served as positive and negative controls, respectively. The constitutively expressed β-actin transcript was amplified as an internal control to compare relative abundance of PCR products. One representative PCR gel (out of three) is presented. (b) A mouse carotid artery 24 h after vascular injury. Immunohistochemistry demonstrates that c-Kit+ cells are recruited to the luminal aspect of arterial thrombi in vivo (arrowhead). The arrow shows a c-Kit negative cell. Bars, 50 μm (left) and 15 μm (right). (c) Injured carotid arteries of P-selectin–, GPIIb-deficient mice or in anti–SDF-1α mAb-treated wild-type mice were analyzed for c-Kit and Sca-1 mRNA expression. The relative expression of each mRNA was normalized to the expression of β-actin for semiquantification and is presented as fold-increase (mean values) of the injured carotid artery compared with the uninjured control. (d) GFP+ KSL BM-PCs were isolated from the BM of GFP transgenic mice and injected into GFP− recipient animals before mechanical injury of the carotid artery. 5 d later, the injured carotid arteries were excised and GFP fluorescence was assessed on cryostat sections. The corresponding hematoxylin and eosin stain of the identical section is shown on the right. Arrows identify GFP+ cells within the neointima. Bars, 25 μm.
Having shown that platelets recruit BM-PCs to sites of vascular injury, we were next interested whether BM-PCs targeted to thrombi are a source of neointimal cells. To address this, we isolated c-Kit+ Sca-1+ Lin− GFP+ BM-PCs from the BM of GFP transgenic mice and injected 105 GFP-positive BM-PCs into GFP− recipient animals. We mechanically injured the carotid artery and killed the animals 5 d thereafter. The injured carotid arteries showed platelet-rich thrombi at the site of endothelial denudation. The thrombi contained abundant numbers of nucleated cells, indicating the onset of thrombus organization and neointima formation. Numerous GFP+ cells were detected within the organized thrombus 5 d after vascular injury (Fig. 8 d). Together, these findings clearly indicate that BM-PCs are recruited into platelet-rich thrombi and give rise to neointimal cells in vivo, a process that might potentially contribute to vascular repair and remodeling.
DISCUSSION
Platelets are crucial for hemostasis and limit blood loss after vascular injury. Here, we have identified a novel and totally unexpected role for platelets in the targeting of primitive BM-PCs to the vessel wall. We directly visualize BM-PCs in vivo using IVM and demonstrate that platelets are absolutely mandatory to recruit BM-PCs to foci of vessel injury. Correspondingly, specific abrogation of platelet adhesion by inhibition of GPVI or GPIbα, both of which are not expressed by BM-PCs, virtually abolished the accumulation of PCs at sites of endothelial disruption. We provide evidence that platelets attract BM-PCs to the injured vessel wall in a multi-step process involving the following: (a) surface exposure of platelet P-selectin; (b) GPIIb-dependent platelet aggregate formation; and, highly unexpected, (c) the release of the CXC chemokine SDF-1α by activated platelets (Fig. 9). Together, the present study identifies the mechanism underlying the selective recruitment of circulating PCs to the vascular intima, a process that plays a fundamental role for the repair and regeneration of vascular tissue after vessel injury, but also during cardiovascular development (1, 2).
Figure 9.

Adherent platelets recruit BM-PCs to injured vessel wall. Within minutes after vessel injury, platelets adhere to the exposed subendothelium in a process involving GPVI and GPIbα-IX. Adherent/activated platelets surface express P-selectin and release SDF-1α after engagement of GPIIb integrin, thereby initiating BM-PC recruitment within minutes after vessel injury. Within the ensuing hours and days after endothelial disruption, apoptotic SMCs appear to account for the long-term SDF-1α release (reference 25). Hence, SDF-1α delivered by platelets early on and by SMCs at later stages act in concert to promote the entry of BM-PCs at sites of vascular damage.
Platelet P-selectin and SDF-1α act in concert to recruit PCs to the injured vessel wall in vivo
P-selectin expressed on the platelet surface has previously been involved in the accumulation of mature leukocytes, including monocytes, neutrophils, and lymphocytes (32, 33). Here, we provide direct in vivo evidence that platelet P-selectin is also central for the targeting of primitive precursor cells to foci of vascular injury. Correspondingly, inhibition or loss of P-selectin or its ligand PSGL-1 drastically reduced BM-PC accumulation on the surface of adherent/aggregated platelets in vivo. Notably, platelet aggregation and arterial thrombus formation were not affected by lack or inhibition of P-selectin, indicating that the decrease of BM-PC accumulation was not the result of a loss of platelet accumulation at the site of vessel injury.
Although P-selectin is required for initial tethering/rolling of BM-PCs, the subsequent firm arrest of BM-PCs is regulated by chemokines. In particular, the CXC chemokine SDF-1α, which is critical for mobilization and homing of BM-PCs (34), is required to arrest PCs under flow in vitro (35). Only recently, SDF-1α has been involved in the process of vascular remodeling and intimal hyperplasia (24) by inducing recruitment of PCs to the vascular wall. Originally, SMCs cells have been identified as the major resource of SDF-1α in both atherosclerotic lesions (27) and after vascular injury (25, 26). Here, we have identified another important source of SDF-1α at sites of vascular lesions. Highly unexpected, we found that, apart from SMCs, platelets and their megakaryocytic precursors abundantly express SDF-1α. Activated platelets rapidly release SDF-1α from internal stores, which is functionally active and promotes the migration of primitive PCs in vitro. Thus, in addition to SDF-1α expressed by vascular SMCs, platelet recruitment during arterial thrombosis is associated with a substantial and rapid rise in the local concentrations of SDF-1α released into the microenvironment of sites of vascular injury.
SDF-1α deposited by activated platelets and SDF-1α released by injured SMCs (25) appear to act in a sequential manner. Correspondingly, we show here that, 30 min after vascular injury, the platelet-rich thrombus is the major source of SDF-1α at the site of vascular injury, whereas 4 h after vascular injury, SDF-1α is abundantly expressed in both SMCs and the thrombus. This is consistent with a recent report showing that SDF-1α expression in vascular SMCs occurs within 6–24 h after vascular injury, whereas it is negligible within the first hours after endothelial disruption (25). We show here that inhibition of SDF-1α significantly reduces BM-PC adhesion to the growing thrombus as early as 5–30 min after vascular injury. Hence, it is tempting to speculate that platelets constitute the initial and presumably more short-lived source of SDF-1α that first directs BM-PCs to the site of vessel damage. In contrast, SMCs appear to account for the long-term SDF-1α release over the ensuing days and weeks after vessel injury required to sustain the process of vascular remodeling and repair (24, 25). The proposed temporal relationship of SDF-1α release by platelets and SMCs, and their relative contributions to the recruitment of BM-PCs at sites of vessel injury is illustrated in Fig. 9.
GPIIb-dependent platelet aggregation modulates the recruitment of PCs to the injured vessel wall
We report here that GPIIb-dependent platelet aggregation not only initiates arterial thrombosis (13), but also plays a fundamental role for the recruitment of primitive PCs at sites of vascular injury. GPIIb may directly promote adhesion of BM-PCs into the growing thrombus by forming a link between platelets and PCs using a GPIIb-dependent bridging mechanism previously reported to occur during platelet–EC interactions (36). Moreover, GPIIb might also operate indirectly in that cross-linking of platelets through GPIIb forms a structure in which PCs get trapped. Finally, we report here that engagement of GPIIb during platelet aggregation initiates platelet SDF-1α release. Hence, distinct GPIIb-dependent pathways are likely to act sequentially to promote BM-PC recruitment at sites of vessel injury.
BM-PCs recruited to arterial thrombi give rise to neointimal cells
There is increasing evidence that KSL or CD34+ BM-PCs give rise to ECs and SMCs in the vascular (neo-) intima in response to vessel injury (4, 6–8). Here, we demonstrate that GFP+ BM-PCs recruited to platelet-rich thrombi grow into neointimal cells. Notably, it has been reported recently that platelet-derived growth factors, including PDGF-BB and VEGF, may modulate PC differentiation in vitro (7, 37). We show here that, apart from growth factors, activated platelets release the CXC chemokine SDF-1α. Beyond its role for PC recruitment to sites of vessel injury, SDF-1α has been demonstrated to support BM-PC survival and proliferation (38). In addition, SDF-1α appears to be involved in the differentiation of c-Kit+ cells into endothelial precursors (39). Although we did not define the progeny of KSL or CD34+ BM-PCs recruited to thrombi in vivo, it is tempting to speculate, based on the findings outlined here, that platelets establish a microenvironment permissive for BM-PC proliferation and early differentiation after vascular injury in vivo.
To conclude, PCs normally circulate within the blood in a “surveillance mode,” encountering only brief contacts with the vascular endothelium. Requirement of tissue renewal or tissue repair is communicated to circulating PCs by adherent/aggregated platelets. We propose that platelet-dependent BM-PC recruitment is essential for vascular repair and local wound healing and/or tissue regeneration at sites of vessel damage. However, it may also contribute to undesirable PC recruitment and proliferation in the process of neointima formation after vascular injury and during atheroprogression (4, 6, 9).
MATERIALS AND METHODS
Animals.
C57BL/6J mice were purchased from Charles River Laboratories. Transgenic C57BL/6J mice ubiquitously expressing enhanced GFP under the control of the chicken β-actin promoter and CMV enhancer were obtained from The Jackson Laboratory. P-selectin–deficient mice were generated and provided by A.L. Beaudet (Baylor College of Medicine, Houston, TX). GPIIb (αIIb integrin)-deficient mice were generated as described previously (40). All experimental procedures performed on animals met the requirements of the German legislation on protection of animals and were approved by the Government of Bavaria/Germany.
Monoclonal antibodies.
4RA10 (function-blocking anti–mouse PSGL-1) were raised as described previously (22, 23, 41). Rat anti–mouse GPIbα mAb was obtained from Emfret Analytics. Function-blocking anti–mouse/human SDF-1α (clone 79014) was obtained from R&D Systems. Anti-α2β1 mAb (clone: BMA2.1) was obtained from Chemicon. mAb 7E3 (gift from B. Coller, The Rockefeller University, New York, NY) inhibits fibrinogen binding to GPIIb-IIIa.
Separation and labeling of CD34+ BM-PCs, KSL BM-PCs, and platelets.
For separation of BM-PCs, mice (C57BL/6J or GFP+) were killed by an overdose of pentobarbital, and both femura were harvested from each animal. Single cell suspensions of the BM were obtained by flushing the femura with PBS using a 21-gauge needle. CD34+ BM-PCs were separated as described previously (42). Primitive KSL BM-PCs were purified according to a previously published protocol (6) (for details, see supplemental Materials and methods, available at http://www.jem.org/cgi/content/full/jem.20051772/DC1). For IVM, CD34+ or KSL BM-PCs were fluorescently labeled with 5-carboxyfluorescein diacetate succinimidyl ester (DCF). For each experiment, 107 fluorescent CD34+ BM-PCs or 1.5 × 105 fluorescent KSL cells were infused intravenously. Mouse platelets were isolated from whole blood as described previously (12, 19). For IVM, platelets were tagged with rhodamine-6G or DCF as reported previously (12, 19).
BM-PC adhesion to purified proteins under flow conditions.
BM-PCs were isolated as described in the previous paragraph. BM-PC (2 × 104/ml) adhesion to coverslips coated with BSA, vitronectin (Becton Dickinson), fibrinogen (Sigma-Aldrich), collagen (Becton Dickinson), or recombinant mouse P-selectin, generated as described previously (23, 41), was assessed in a transparent flow chamber at a wall shear rate of 1,000 s−1 as described elsewhere (22). In a separate set of experiments, platelets (4 × 108/ml) were allowed to adhere to collagen-coated coverslips before perfusion with BM-PCs. Where indicated, function-blocking anti–PSGL-1 mAb (5 μg/ml) was added to the BM-PC suspension.
Characterization of BM-PC adhesion receptors and determination of platelet-binding by flow cytometry.
BM-PCs (10 × 103 CD34+ or KSL cells/ml) resuspended in PBS were incubated with fluorophore-labeled anti-CD41 (αIIb integrin, clone: MWReg 30), anti-CD49d (α4 integrin, clone: 9C10), anti-CD162 (PSGL-1, clone: 2PH1) (all from Becton Dickinson), anti-GPIbα (Xia.B2, Emfret) or anti-GPVI mAb (2G3, generated as described (22)), or irrelevant isotype-matched control antibody for 30 min at room temperature and directly analyzed on a FACSCalibur (Becton Dickinson). In a separate set of experiments, CD34+ or KSL BM-PCs (2 × 104/ml) were coincubated with 108/ml resting mouse platelets or platelets activated with mouse thrombin (2 U/ml, Sigma-Aldrich) for 15 min. Where indicated function-blocking anti-CD11b (M1/70, BD Biosciences) or anti-PSGL-1 mAb (4RA10, 5 μg/ml) was added to the cell suspension. PBS-incubated BM-PCs in the absence or presence of thrombin served as controls. Thereafter, the samples were incubated with FITC-labeled anti–mouse CD41 (MW Reg30) for 30 min to detect platelets attached to BM-PCs at room temperature and directly analyzed.
Assessment of BM-PCs and platelet adhesion and BM-PC–platelet interaction in response to carotid injury.
Platelet and BM-PC (CD34+ or c-Kit+ Sca-1+ Lin−) adhesion dynamics before and after vascular injury were monitored in vivo by use of IVM. Carotid injury was induced in anesthetized C57BL6/J, P-selectin–, or GPIIb-deficient mice as described previously (12, 20). Before and after vascular injury, differentially fluorescent-tagged cells (platelets and BM-PCs) were visualized using a Zeiss Axiotech microscope (20× water immersion objective, W 20×/0.5, Zeiss) with a 100W HBO mercury lamp for epi-illumination as reported previously (12, 20). Where indicated, C57BL6/J mice were pretreated with 2 mg/kg body weight RB40.34 (anti–mouse P-selectin), 4RA10 (anti–mouse PSGL-1), rat anti–mouse GPIbα, or anti–mouse/human SDF-1α (clone 79014) 5 min before intravital microscopy (n = 4–6 per group). BM-PC adhesion to the carotid artery after infusion of irrelevant isotype-matched IgG served as control (n = 10).
For direct visualization of platelet–KSL cell interactions at the site of vascular lesion differentially fluorescent-tagged cells (rhodamine-tagged platelets and DCF-tagged KSL BM-PCs) were infused i.v. KSL binding to adherent platelets was analyzed 5 min after induction of carotid injury by real-time double fluorescence IVM using a BX51WI microscope (Olympus), equipped with an Olympus MT20 monochromator.
Assessment of SDF-1α expression by immunohistochemistry.
To determine the presence of SDF-1α in megakaryocytes and aggregated platelets, paraffin-embedded sections of mouse femura, injured mouse carotid arteries (WT or GPIIb-deficient, 0.5, 4, and 24 h after endothelial denudation), and a fresh thrombus isolated from an occluded human coronary artery were cut into 2-μm sections and incubated with anti–mouse SDF-1α (clone 79018; R&D Systems), or anti–human SDF-1α (polyclonal goat; R&D Systems), respectively, and stained using APAAP chemMATE or DAB chemMATE detection kits (both obtained from DakoCytomation). For immunofluorescence microscopy, cryostat sections of injured mouse carotid arteries (24 h after endothelial denudation) were incubated with anti–mouse CD41 mAb (MW Reg30; BD Biosciences), anti–mouse SDF-1α mAb (clone 79014; R&D Systems), Alexa488-labeled goat anti–rat IgG, Alexa594-labeled goat anti–mouse IgG (both obtained from Invitrogen), and DAPI (D1306; Invitrogen). Fluorescence images were obtained using a Leica fluorescence microscope.
Determination of platelet and megakaryocytic SDF-1α protein and mRNA expression.
Platelet SDF-1α release was determined in vitro by enzyme-linked immunosorbent assay (R&D Systems). In brief, mouse SDF-1α standard lysates obtained from isolated resting mouse platelets, whole blood, or mouse BM cells were incubated in SDF-1α mAb precoated microplates and reacted with a polyclonal anti–SDF-1α–horseradish peroxidase antibody (all obtained from R&D Systems). For detection a tetramethyl benzidine peroxidase substrate was used. The mouse melanoma cell line D5 served as a control. Absorbance was read at 450 nm and the background was corrected. SDF-1α concentrations were calculated with standards and are adjusted to 100 μg total protein.
To determine SDF-1α expression by the megakaryocytic lineage, we generated megakaryocytes from mouse CD34+ cells as reported previously (28) (for details see supplemental Materials and methods). Megakaryocytic SDF-1α expression was assessed by ELISA as described in the previous paragraphs. In addition, megakaryocytic SDF-1α mRNA expression was determined using RT-PCR. The mRNA expression of GPIIb integrin and β-actin served as controls. The primer sequences and their corresponding product sizes and annealing temperatures are shown in the Table S1 (available at http://www.jem.org/cgi/content/full/jem.20051772/DC1).
Assessment of SDF-1α release upon platelet activation.
To determine the effect of platelet activation on SDF-1α release, human platelets (0.5 × 109 cells/ml) were incubated with 2 U/ml α-thrombin or PBS for 30 min. To determine the effects of GPIIb receptor cross-linking, human platelets were incubated with 100 μg/ml of soluble fibrinogen (Sigma-Aldrich) in the presence or absence of 5 μg/ml of bivalent mAb antifibrinogen (2C2-G7; BD Biosciences), or with 5 μg/ml of mouse anti-GPIIb mAb 7E3 in the presence or absence of 5 μg/ml of anti–mouse IgG mAb (DakoCytomation). SDF-1α release into the supernatants of resting or activated platelets was determined by ELISA as described in previous paragraphs.
To analyze the surface exposure of SDF-1α in response to platelet activation, resting or thrombin-activated human platelets were incubated with mouse anti–SDF-1α mAb (R&D Systems) or irrelevant isotype-matched control antibody for 30 min at room temperature, followed by staining with FITC-labeled rabbit anti–mouse IgG antibody (Becton Dickinson). The samples were directly analyzed on a FACSCalibur (Becton Dickinson).
To define the distribution of SDF-1α in resting and activated platelets, human platelets (2 × 108 cells/ml) were stimulated with 2 U/ml α-thrombin, 5 μM ADP, or PBS for 60 min. The platelets were fixed and stained with FITC-labeled anti-CD41 (P2; Beckman Coulter), monoclonal (R&D Systems) or polyclonal anti–SDF-1α antibody (goat; R&D Systems), Alexa Fluor 488–tagged goat anti–mouse, Alexa 568–tagged rabbit anti–goat, Alexa Fluor 488–tagged rabbit anti–goat (all obtained from Invitrogen), or Rhodamine-Phalloidin (R415; Invitrogen) as indicated. Confocal immunofluorescence analysis was performed using a LSM510 META confocal laser microscope equipped with the LSM510 META software (Carl Zeiss MicroImaging, Inc.).
Incorporation of BM-PCs into the neointima.
GFP+ CD34+ BM-PCs or KSL GFP+ CD34+ BM-PCs were isolated from the BM of GFP transgenic mice as described in previous paragraphs and injected into C57BL/6J recipient animals (n = 5; 105 cells per experiment). We mechanically injured the carotid artery. After 5 d, the injured carotid arteries were excised. GFP fluorescence was detected on cryostat sections by standard fluorescence microscopy.
Recruitment of endogenous PCs to the injured carotid artery.
To determine recruitment of endogenous circulating PCs, carotid arteries of C57BL/6J (n = 3), P-selectin–, (n = 4) or GPIIb-deficient mice (n = 2) or in anti–SDF-1α mAb-treated wild-type mice (n = 2) were injured as described in previous paragraphs and c-Kit and Sca-1 mRNA expression was assessed by RT-PCR (see supplemental Materials and methods). The constitutively expressed β-actin transcript was amplified as an internal control to compare relative abundance of PCR products, and the relative expression of each mRNA was normalized to the expression of β-actin for semiquantification. To detect c-Kit positive paraffin-embedded sections of injured mouse, carotid arteries (24 h after endothelial denudation) were incubated with anti–mouse c-Kit mAb (2B8; BD Biosciences) and stained using APAAP chemMATE mouse detection kit (DakoCytomation).
Statistical analysis.
Comparisons between group means were performed using one-way analysis of variance on Ranks. Data represent mean ± SEM. A value of P < 0.05 was regarded as significant.
Online supplemental material.
Online supplemental Materials and methods provides additional methodological information in the following categories: separation of BM-PCs; effects of anti-GPIbα mAb on platelet function; transfection of DAMI cells; scanning electron microscopy; generation of mouse megakaryocytes; in vitro PC migration; and recruitment of endogenous PCs. Table S1 contains primer sequences, corresponding product sizes, and annealing temperatures. Fig. S1 shows the effects of anti- GPIbα mAb on platelet function. Fig. S2 addresses BM-PC binding to recombinant P-selectin. Fig. S3 illustrates the subcellular localization of SDF-1α in megakaryocytes. Fig. S4 investigates the effect of platelet SDF-1α on PC migration. All online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20051772/DC1.
Supplemental Material
Acknowledgments
We are indebted to K. Langenbrink and H. Wehnes for their excellent technical assistance. Histomorphometry was performed with the skillful help of R. Hegenloh with the support of Dr. R. Brandl (Br 1583/1-2).
The study was supported by grants from the Bayerische Forschungsstiftung and the Deutsche Forschungsgemeinschaft DFG (Ma 2186/3-1 to S. Massberg) and the Wellcome Trust (to N.R. Emambokus, J. Frampton). S. Massberg is a Heisenberg fellow of the DFG (Ma 2186/4-1). K. Schürzinger and P. Goyal were graduate students supported by the DFG Graduate Student Program GRK 438.
The authors have no conflicting financial interests.
Abbreviations used: BM-PC, BM-derived PC; DCF, dichlorofluorescein; EC, endothelial cell; IVM, intravital video fluorescence microscopy; KSL, c-Kit+ Sca-1+ Lin−; PC, progenitor cell; PSGL, P-selectin glycoprotein ligand; SDF-1α, stromal cell–derived factor 1α; SMC, smooth muscle cell.
S. Massberg's present address is CBR Institute for Biomedical Research and Department of Pathology, Harvard Medical School, Boston, MA 02115.
References
- 1.Carmeliet, P. 2000. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 6:389–395. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz, S.M. 1997. Smooth muscle migration in atherosclerosis and restenosis. J. Clin. Invest. 100:S87–S89. [PubMed] [Google Scholar]
- 3.Carmeliet, P., L. Moons, J.M. Stassen, M. De Mol, A. Bouche, J.J. van den Oord, M. Kockx, and D. Collen. 1997. Vascular wound healing and neointima formation induced by perivascular electric injury in mice. Am. J. Pathol. 150:761–776. [PMC free article] [PubMed] [Google Scholar]
- 4.Werner, N., J. Priller, U. Laufs, M. Endres, M. Bohm, U. Dirnagl, and G. Nickenig. 2002. Bone marrow-derived progenitor cells modulate vascular reendothelialization and neointimal formation: effect of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition. Arterioscler. Thromb. Vasc. Biol. 22:1567–1572. [DOI] [PubMed] [Google Scholar]
- 5.Jiang, S., L. Walker, M. Afentoulis, D.A. Anderson, L. Jauron-Mills, C.L. Corless, and W.H. Fleming. 2004. Transplanted human bone marrow contributes to vascular endothelium. Proc. Natl. Acad. Sci. USA. 101:16891–16896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sata, M., A. Saiura, A. Kunisato, A. Tojo, S. Okada, T. Tokuhisa, H. Hirai, M. Makuuchi, Y. Hirata, and R. Nagai. 2002. Hematopoietic stem cells differentiate into vascular cells that participate in the pathogenesis of atherosclerosis. Nat. Med. 8:403–409. [DOI] [PubMed] [Google Scholar]
- 7.Simper, D., P.G. Stalboerger, C.J. Panetta, S. Wang, and N.M. Caplice. 2002. Smooth muscle progenitor cells in human blood. Circulation. 106:1199–1204. [DOI] [PubMed] [Google Scholar]
- 8.Shimizu, K., S. Sugiyama, M. Aikawa, Y. Fukumoto, E. Rabkin, P. Libby, and R.N. Mitchell. 2001. Host bone-marrow cells are a source of donor intimal smooth-muscle-like cells in murine aortic transplant arteriopathy. Nat. Med. 7:738–741. [DOI] [PubMed] [Google Scholar]
- 9.Sata, M., K. Tanaka, and R. Nagai. 2003. Origin of smooth muscle progenitor cells: different conclusions from different models. Circulation. 107:e106–e107. [DOI] [PubMed] [Google Scholar]
- 10.Chen, J., and J.A. Lopez. 2005. Interactions of platelets with subendothelium and endothelium. Microcirculation. 12:235–246. [DOI] [PubMed] [Google Scholar]
- 11.Ruggeri, Z.M. 1997. Mechanisms initiating platelet thrombus formation. Thromb. Haemost. 78:611–616. [PubMed] [Google Scholar]
- 12.Massberg, S., M. Gawaz, S. Gruner, V. Schulte, I. Konrad, D. Zohlnhofer, U. Heinzmann, and B. Nieswandt. 2003. A crucial role of glycoprotein VI for platelet recruitment to the injured arterial wall in vivo. J. Exp. Med. 197:41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Massberg, S., K. Schurzinger, M. Lorenz, I. Konrad, C. Schulz, N. Plesnila, E. Kennerknecht, M. Rudelius, S. Sauer, S. Braun, et al. 2005. Platelet adhesion via glycoprotein IIb integrin is critical for atheroprogression and focal cerebral ischemia. An in vivo study in mice lacking glycoprotein IIb. Circulation. 112:1180–1188. [DOI] [PubMed] [Google Scholar]
- 14.Morel, F., A. Galy, B. Chen, and S.J. Szilvassy. 1998. Equal distribution of competitive long-term repopulating stem cells in the CD34+ and CD34− fractions of Thy-1low Lin−/low Sca-1+ bone marrow cells. Exp. Hematol. 26:440–448. [PubMed] [Google Scholar]
- 15.Sato, T., J.H. Laver, and M. Ogawa. 1999. Reversible expression of CD34 by murine hematopoietic stem cells. Blood. 94:2548–2554. [PubMed] [Google Scholar]
- 16.Osawa, M., K. Hanada, H. Hamada, and H. Nakauchi. 1996. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science. 273:242–245. [DOI] [PubMed] [Google Scholar]
- 17.Mazo, I.B., J.C. Gutierrez-Ramos, P.S. Frenette, R.O. Hynes, D.D. Wagner, and U.H. von Andrian. 1998. Hematopoietic progenitor cell rolling in bone marrow microvessels: parallel contributions by endothelial selectins and vascular cell adhesion molecule 1. J. Exp. Med. 188:465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frenette, P.S., S. Subbarao, I.B. Mazo, U.H. von Andrian, and D.D. Wagner. 1998. Endothelial selectins and vascular cell adhesion molecule-1 promote hematopoietic progenitor homing to bone marrow. Proc. Natl. Acad. Sci. USA. 95:14423–14428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Massberg, S., K. Brand, S. Gruner, S. Page, E. Muller, I. Muller, W. Bergmeier, T. Richter, M. Lorenz, I. Konrad, et al. 2002. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J. Exp. Med. 196:887–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moers, A., B. Nieswandt, S. Massberg, N. Wettschureck, S. Gruner, I. Konrad, V. Schulte, B. Aktas, M.P. Gratacap, M.I. Simon, et al. 2003. G(13) is an essential mediator of platelet activation in hemostasis and thrombosis. Nat. Med. 9:1418–1422. [DOI] [PubMed] [Google Scholar]
- 21.Cruz, M.A., J. Chen, J.L. Whitelock, L.D. Morales, and J.A. Lopez. 2005. The platelet glycoprotein Ib-von Willebrand factor interaction activates the collagen receptor α2β1 to bind collagen: activation-dependent conformational change of the α2-I domain. Blood. 105:1986–1991. [DOI] [PubMed] [Google Scholar]
- 22.Massberg, S., I. Konrad, A. Bultmann, C. Schulz, G. Munch, M. Peluso, M. Lorenz, S. Schneider, F. Besta, I. Muller, et al. 2004. Soluble glycoprotein VI dimer inhibits platelet adhesion and aggregation to the injured vessel wall in vivo. FASEB J. 18:397–399. [DOI] [PubMed] [Google Scholar]
- 23.Pendl, G.G., C. Robert, M. Steinert, R. Thanos, R. Eytner, E. Borges, M.K. Wild, J.B. Lowe, R.C. Fuhlbrigge, T.S. Kupper, et al. 2002. Immature mouse dendritic cells enter inflamed tissue, a process that requires E- and P-selectin, but not P-selectin glycoprotein ligand 1. Blood. 99:946–956. [DOI] [PubMed] [Google Scholar]
- 24.Schober, A., S. Knarren, M. Lietz, E.A. Lin, and C. Weber. 2003. Crucial role of stromal cell-derived factor-1α in neointima formation after vascular injury in apolipoprotein E-deficient mice. Circulation. 108:2491–2497. [DOI] [PubMed] [Google Scholar]
- 25.Zernecke, A., A. Schober, I. Bot, P. von Hundelshausen, E.A. Liehn, B. Mopps, M. Mericskay, P. Gierschik, E.A. Biessen, and C. Weber. 2005. SDF-1α/CXCR4 axis is instrumental in neointimal hyperplasia and recruitment of smooth muscle progenitor cells. Circ. Res. 96:784–791. [DOI] [PubMed] [Google Scholar]
- 26.Nuhrenberg, T.G., R. Voisard, F. Fahlisch, M. Rudelius, J. Braun, J. Gschwend, M. Kountides, T. Herter, R. Baur, V. Hombach, et al. 2005. Rapamycin attenuates vascular wall inflammation and progenitor cell promoters after angioplasty. FASEB J. 19:246–248. [DOI] [PubMed] [Google Scholar]
- 27.Abi-Younes, S., A. Sauty, F. Mach, G.K. Sukhova, P. Libby, and A.D. Luster. 2000. The stromal cell-derived factor-1 chemokine is a potent platelet agonist highly expressed in atherosclerotic plaques. Circ. Res. 86:131–138. [DOI] [PubMed] [Google Scholar]
- 28.Ungerer, M., M. Peluso, A. Gillitzer, S. Massberg, U. Heinzmann, C. Schulz, G. Munch, and M. Gawaz. 2004. Generation of functional culture-derived platelets from CD34+ progenitor cells to study transgenes in the platelet environment. Circ. Res. 95:e36–e44. [DOI] [PubMed] [Google Scholar]
- 29.Hatzopoulos, A.K., J. Folkman, E. Vasile, G.K. Eiselen, and R.D. Rosenberg. 1998. Isolation and characterization of endothelial progenitor cells from mouse embryos. Development. 125:1457–1468. [DOI] [PubMed] [Google Scholar]
- 30.Shattil, S.J., and M.H. Ginsberg. 1997. Integrin signaling in vascular biology. J. Clin. Invest. 100:S91–S95. [PubMed] [Google Scholar]
- 31.May, A.E., T. Kalsch, S. Massberg, Y. Herouy, R. Schmidt, and M. Gawaz. 2002. Engagement of glycoprotein IIb/IIIa (α(IIb)β3) on platelets upregulates CD40L and triggers CD40L-dependent matrix degradation by endothelial cells. Circulation. 106:2111–2117. [DOI] [PubMed] [Google Scholar]
- 32.Wagner, D.D., and P.C. Burger. 2003. Platelets in inflammation and thrombosis. Arterioscler. Thromb. Vasc. Biol. 23:2131–2137. [DOI] [PubMed] [Google Scholar]
- 33.Diacovo, T.G., K.D. Puri, R.A. Warnock, T.A. Springer, and U.H. von Andrian. 1996. Platelet-mediated lymphocyte delivery to high endothelial venules. Science. 273:252–255. [DOI] [PubMed] [Google Scholar]
- 34.Lapidot, T., and I. Petit. 2002. Current understanding of stem cell mobilization: the roles of chemokines, proteolytic enzymes, adhesion molecules, cytokines, and stromal cells. Exp. Hematol. 30:973–981. [DOI] [PubMed] [Google Scholar]
- 35.Peled, A., V. Grabovsky, L. Habler, J. Sandbank, F. Arenzana-Seisdedos, I. Petit, H. Ben Hur, T. Lapidot, and R. Alon. 1999. The chemokine SDF-1 stimulates integrin-mediated arrest of CD34(+) cells on vascular endothelium under shear flow. J. Clin. Invest. 104:1199–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bombeli, T., B.R. Schwartz, and J.M. Harlan. 1998. Adhesion of activated platelets to endothelial cells: evidence for a GPIIbIIIa-dependent bridging mechanism and novel roles for endothelial intercellular adhesion molecule 1 (ICAM-1), αvβ3 integrin, and GPIbα. J. Exp. Med. 187:329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu, Y., Z. Zhang, E. Torsney, A.R. Afzal, F. Davison, B. Metzler, and Q. Xu. 2004. Abundant progenitor cells in the adventitia contribute to atherosclerosis of vein grafts in ApoE-deficient mice. J. Clin. Invest. 113:1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ponomaryov, T., A. Peled, I. Petit, R.S. Taichman, L. Habler, J. Sandbank, F. Arenzana-Seisdedos, A. Magerus, A. Caruz, N. Fujii, et al. 2000. Induction of the chemokine stromal-derived factor-1 following DNA damage improves human stem cell function. J. Clin. Invest. 106:1331–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Falco, E., D. Porcelli, A.R. Torella, S. Straino, M.G. Iachininoto, A. Orlandi, S. Truffa, P. Biglioli, M. Napolitano, M.C. Capogrossi, and M. Pesce. 2004. SDF-1 involvement in endothelial phenotype and ischemia-induced recruitment of bone marrow progenitor cells. Blood. 104:3472–3482. [DOI] [PubMed] [Google Scholar]
- 40.Emambokus, N.R., and J. Frampton. 2003. The glycoprotein IIb molecule is expressed on early murine hematopoietic progenitors and regulates their numbers in sites of hematopoiesis. Immunity. 19:33–45. [DOI] [PubMed] [Google Scholar]
- 41.Bosse, R., and D. Vestweber. 1994. Only simultaneous blocking of the L- and P-selectin completely inhibits neutrophil migration into mouse peritoneum. Eur. J. Immunol. 24:3019–3024. [DOI] [PubMed] [Google Scholar]
- 42.Dimmeler, S., A. Aicher, M. Vasa, C. Mildner-Rihm, K. Adler, M. Tiemann, H. Rutten, S. Fichtlscherer, H. Martin, and A.M. Zeiher. 2001. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J. Clin. Invest. 108:391–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}