

Summary
Mismatch repair proteins, DNA damage-recognition proteins and translesion DNA polymerases discriminate between Pt-GG adducts containing cis-diammine ligands (formed by cisplatin (CP) and carboplatin) and trans-RR-diaminocyclohexane ligands (formed by oxaliplatin (OX)) and this discrimination is thought to be important in determining differences in the efficacy, toxicity and mutagenicity of these platinum anticancer agents. We have postulated that these proteins recognize differences in conformation and/or conformational dynamics of the DNA containing the adducts. We have previously determined the NMR solution structure of OX-DNA, CP-DNA and undamaged duplex DNA in the 5'-d(CCTCAGGCCTCC)-3' sequence context and have shown the existence of several conformational differences in the vicinity of the Pt-GG adduct. In this study we have used molecular dynamics simulations to explore differences in the conformational dynamics between OX-DNA, CP-DNA and undamaged DNA in the same sequence context. Twenty-five 10 ns unrestrained fully solvated molecular dynamics simulations were performed starting from two different DNA conformations using AMBER v8.0. All twenty-five simulations reached equilibrium within 4 ns, were independent of the starting structure and were in close agreement with previous crystal and NMR structures. Our data show that the cis-diammine (CP) ligand preferentially forms hydrogen bonds on the 5' side of the Pt-GG adduct, while the trans-RR-diaminocyclohexane (OX) ligand preferentially forms hydrogen bonds on the 3' side of the adduct. In addition, our data show that these differences in hydrogen bond formation are strongly correlated with differences in conformational dynamics, specifically the fraction of time spent in different DNA conformations in the vicinity of the adduct, for CP- and OX-DNA adducts. We postulate that differential recognition of CP- and OX-GG adducts by mismatch repair proteins, DNA damage-recognition proteins and DNA polymerases may be due, in part, to differences in the fraction of time that the adducts spend in a conformation favorable for protein binding.
Keywords: Cisplatin, Oxaliplatin, Molecular Dynamics, Simulations, Conformation
Introduction
Cisplatin (CP; cis-diamminedichloroplatinum(II)) and carboplatin (CBDCA,cis-diammine-1,1-cyclobutanedicarboxylatoplatinum(II)) are widely used for treatment of testicular cancer, ovarian cancer, head and neck tumors and a variety of other solid tumors. However, many tumors are intrinsically resistant or develop acquired resistance to these chemotherapeutic agents, and tumors that are resistant to one of these two platinum compounds are usually cross-resistant to the other. The mutagenicity of CP in vivo1 is also of concern because secondary malignancies have been associated with CP chemotherapy2. Considerable effort has been made to develop third generation platinum anticancer agents that would not share these limitations. Oxaliplatin (OX; trans-R,R-1,2-diaminocyclohexaneoxalatoplatinum(II)) is one such compound, and has recently been approved for the treatment of colorectal cancer and tumors that are resistant to CP and CBDCA. While OX does have some mutagenicity3, it appears to be less mutagenic than CP4. CP and CBDCA form Pt-DNA adducts that contain the cis-diammine carrier ligands, while OX forms Pt-DNA adducts that contain the trans-RR-1,2-diaminocyclohexane carrier ligand. For simplicity, we will refer to these as CP-DNA and OX-DNA adducts throughout this paper. Other than the differences in carrier ligand, the adducts formed by CP, CBDCA and OX appear to be identical in terms of the type of adduct formed (60-65% intrastrand GG, 25-30% intrastrand AG, 5-10% intrastrand GNG and 1-3% interstrand) and the site of adduct formation5-7.
Because of their abundance, the intrastrand GG adducts are thought to be major determinants of the cytotoxic response to platinum anticancer agents. The basis for the differences in tumor range and mutagenicity of OX compared to CP and CBDCA is not known, but is thought to be determined by the ability of proteins involved in damage recognition, damage repair and/or damage tolerance to discriminate between CP and OX adducts. For example, hMSH2 and MutS bind with greater affinity to CP-GG adducts than to OX-GG adducts8 ,9, and, as might be expected from this difference in binding affinity, defects in mismatch repair result in resistance to CP and CBDCA, but not to OX8 ,10-13. Similarly, a number of damage recognition proteins and transcription factors, especially those with HMG domains, have been shown to discriminate between CP- and OX-GG adducts14 ,15. The mechanism(s) by which the binding of these proteins to Pt-DNA adducts influences the cytotoxic response is not known, but has been postulated to involve shielding of the adducts from DNA repair and tolerance mechanisms16-19, activation of signaling pathways leading to cell cycle arrest or apoptosis, and/or hijacking of transcription factors needed for DNA replication or cell division15 ,20. The binding specificity has been determined for only a few of these proteins, but where it has been studied these proteins bind to CP-GG adducts with higher affinity than to OX-GG adducts14 ,15 ,21. Finally, translesion DNA polymerases such as hpol β and hpol η have been shown to bypass OX-GG adducts with higher efficiency than CP-GG adducts19 ,22 ,22 ,23, which might contribute to the differences in CP and OX mutagenicity.
The CP- and OX-GG adducts form in the major groove and bend the DNA in the direction of the major groove. The proteins that discriminate between CP- and OX-GG adducts either bind to bent DNA or bend the DNA in the direction of the major groove after binding24-27. Because these proteins primarily interact with the minor groove, we have hypothesized that the ability of the proteins to discriminate between CP- and OX-GG adducts probably results from subtle differences in conformation or conformational dynamics in the DNA containing the two adducts rather than from physical interaction of the proteins with the carrier ligands of the adducts in the major groove. A number of structures have been reported for CP-GG and OX-GG adducts. The overall conformation of DNA containing these adducts appears to be similar, but exact comparisons have been difficult to make because the structures have been determined by different techniques (crystallography versus NMR), in different sequence contexts and with oligonucleotides of different lengths. The NMR structures obtained to date have varied with respect to the number and resolution of NMR constraints obtained and the molecular mechanics simulations used to convert the NMR constraints to final structures28-30. X-ray crystallographic structures have been reported for the CP-GG and OX-GG adducts in the same sequence context31, 32, but these structures may have been constrained by crystal packing restraints. We have recently obtained high resolution NMR solution structures of the OX-GG30, CP-GG adducts33 and undamaged DNA33 in the same mutagenic AGGC sequence context (The underlined bases indicate the position of the Pt-GG adduct). The NMR studies have identified several conformational differences between the solution structures of the two Pt-GG adducts. In this study we have used molecular dynamics (MD) simulations to extend this analysis to differences in conformational dynamics between CP-GG adducts, OX-GG adducts and undamaged DNA in the AGGC sequence context. Our data are consistent with earlier reports of greater distortion on the 5' side of Pt-GG adducts29 ,30 ,34-39. Our data are also consistent with previous reports that the cis-diammine (CP) ligand preferentially forms hydrogen bonds on the 5' side of the Pt-GG adduct, while the trans-RR-diaminocyclohexane (OX) ligand preferentially forms hydrogen bonds on the 3' side of the adduct. Finally, our data show that these differences in hydrogen bond formation are strongly correlated with differences in conformational dynamics, specifically the fraction of time spent in different DNA conformations, for CP- and OX-DNA adducts. These differences were particularly evident for propeller twist, buckle, slide and shift in the vicinity of the Pt-GG adduct. We postulate that these differences in conformational dynamics could allow differential recognition of CP- and OX-GG adducts by critical DNA-binding proteins that influence the cytotoxic response to these adducts. For example, we postulate that CP-GG adducts spend a greater percentage of time in conformations favorable for binding of mismatch repair and HMG-domain DNA-binding proteins, while OX adducts spend a greater percentage of time in conformations(s) favorable for bypass by hpol β and hpol η. Experiments are currently underway to test these hypotheses. Experiments are also underway to compare the effect of sequence context on the conformational differences between these two adducts.
Results
The MD simulations were independent of starting structure
In total, twenty-five 10 ns unrestrained fully solvated molecular dynamics simulations were performed starting from five different DNA conformations. In order to critically evaluate whether the starting structures influenced the MD simulations, two very different starting structures were used for both the CP-DNA and OX-DNA simulations. One of the starting structures for the CP- and OX-DNA simulations used the NMR structure of DNA complexed with hSRY (Protein DataBank accession code 1J46.pdb, bend angle = 54°) for the DNA backbone. The other starting structures were based on the NMR structures of the CP-DNA (Protein DataBank accession code 2NMW.pdb; bend angle = 22°) and OX-DNA (Protein DataBank accession code 1PGC.pdb; bend angle = 31°) adducts alone. Idealized B-DNA was used as the starting structure for the BDNA simulation. Five simulations each were performed with the CP-SRY, CP-NMR, OX-SRY, OX-NMR and B-DNA starting structures as described in methods. The all-atom mass-weighted root-mean-square deviations (RMSDs) referenced to the corresponding NMR structures for the CP-DNA and OX-DNA structures and the starting B-DNA structure for undamaged DNA calculated over all twenty-five trajectories were used to determine how long it took the simulations to reach equilibrium and, in turn, the range of trajectories to be used for the subsequent analyses. Plots of RMSD over time are shown for CP-DNA (Figure 1A), OX-DNA (Figure 1B) and B-DNA (Figure S1). The average RMSD plots of all ten CP-DNA simulations were within close proximity of one another within the final 6 ns. Thus, the simulations of the CP-DNA adducts converged to similar equilibrium structures even when the starting structures differed significantly in the initial degree of DNA bending and/or MD initial velocity. Similar behavior was evident for the OX-DNA adducts. Based on these plots of average RMSD over time, we concluded that the simulations reached equilibrium within 4 ns. Therefore our comparisons were made based on the final 6 ns of the trajectories. The average RMSD compared to the NMR solution structures30 ,33 was 2.08 ± 0.43 Å over all ten simulations for OX-DNA, 2.73 ± 0.53 Å over all ten simulations for CP-DNA and 2.80 ± 0.30 Å over all five simulations for B-DNA.
Figure 1. Average RMSDs for the MD simulations over time.

The RMSDs for each of the 20 simulations compared to the corresponding NMR solution structure30 ,33 for the CP-DNA (Figure 1A) and OX-DNA adducts (Figure 1B) are shown for the full 10 ns of each simulation. RMSD at time t represents the average of RMSD from time zero to time t. CP-NMR and OX-NMR represent simulations starting from NMR solution structures of the Pt-DNA 12-mer duplexes. CP-SRY and OX-SRY stand for simulations starting from the more distorted DNA•SRY structures. The five simulation trajectories starting from CP-NMR and OX-NMR structures with different initial MD velocities are represented as solid lines (circle symbol), dotted lines(square symbol), dashed lines (diamond symbol), long dashed lines (upward triangle symbol) and dot-dashed lines (left triangle symbol). The five simulation trajectories starting from the more distorted CP-SRY and OX-SRY structures with different initial MD velocities are represented as solid lines (downward triangle symbol), dotted lines (right triangle symbol), dashed lines (plus symbol), long dashed lines (cross symbol) and dot-dashed lines (asterisk symbol).
Comparison of the MD simulations with previously reported structures
For the purposes of comparison with previous structures, centroid structures (the structure from the simulation with the lowest RMSD compared to the average structure) were determined for both the CP-DNA and OX-DNA simulations. Figure 2 shows an overlay of the CP-DNA and OX-DNA centroid structures and “sausage diagrams” of each structure individually showing the overall variation in the structures during the simulations. The centroid structures of the CP-DNA and OX-DNA adducts were very similar (The all atom RMSD for the DNA portion of the structures was 1.3 Å). The “sausage diagrams” showed that the greatest variations in both the CP-DNA and OX-DNA structures occurred at the ends of the DNA molecules, but that there was relatively little fluctuation of the DNA backbone during the simulations.
Figure 2.

Average CP-DNA and OX-DNA structures were obtained using the AMBER ptraj tool. In each case, the overall average structure was computed over the equilibrated final 6 ns trajectories combined for the five independent simulations starting with the CP-SRY, CP-NMR and OX-SRY, OX-NMR respectively. Pairwise Kabsch alignment was performed for the SRY and NMR starting structures. Because the RMSDs indicated that the starting structure had little influence on the average structure (RMSD = 0.83 Å for CP-SRY versus CP-NMR and 1.03 Å for OX-SRY versus OX-NMR), the two average structures were further averaged to obtain a single average structure for the last 6 ns of all 10 simulations of the CP-DNA and OX-DNA adducts. Kabsch alignment was then performed against CP-DNA and OX-DNA average structures against every 1 ps snapshot of simulation trajectory. The CP-DNA and OX-DNA MD simulation snapshots having lowest RMS deviations with corresponding average structures were selected as the centroid structures. A. Overlay of CP-DNA and OX-DNA centroid structures. Cyan = OX-DNA; purple = CP-DNA. B and C “Sausage diagrams” of CP-DNA and OX-DNA centroid structures, respectively. Root mean square atomic fluctuations of the CP-DNA and OX-DNA phosphate backbone were obtained by averaging the backbone fluctuations over the 30000 snapshots of equilibrated final 6 ns of all 10 simulation trajectories. The atomicfluct routine in the AMBER ptraj tool was used to compute these fluctuations. The backbone thickness and color (in a scale varying from blue to red) corresponds to the relative fluctuations of CP-DNA (B) and OX-DNA (C) backbone phosphates.
The centroid structures of the CP-DNA and OX-DNA simulations were also compared with the previous crystal31,32 and NMR30,33 structures of the same adducts (Figure 3). In both cases, the simulations agreed slightly better with the NMR structures than the crystal structures. For example, the all atom RMSD comparisons of the CP-DNA centroid structure with the corresponding crystal31 and NMR33 structures were 4.2 Å and 3.1 Å, respectively. Similarly, the all atom RMSD comparisons of the OX-DNA centroid structure with the corresponding crystal32 and NMR30 structures were 4.0 Å and 3.1 Å, respectively. For the central four base pairs, the RMSDs were 3.5 Å, 2.8 Å, 3.6 Å and 2.9 Å for the CP-crystal versus CP-centroid, CP-NMR versus CP-centroid, OX-crystal versus OX-centroid and OX-NMR versus OX-centroid, repectively.
Figure 3. Comparison of CP-DNA and OX-DNA centroid structures with previously determined crystal and NMR structures.
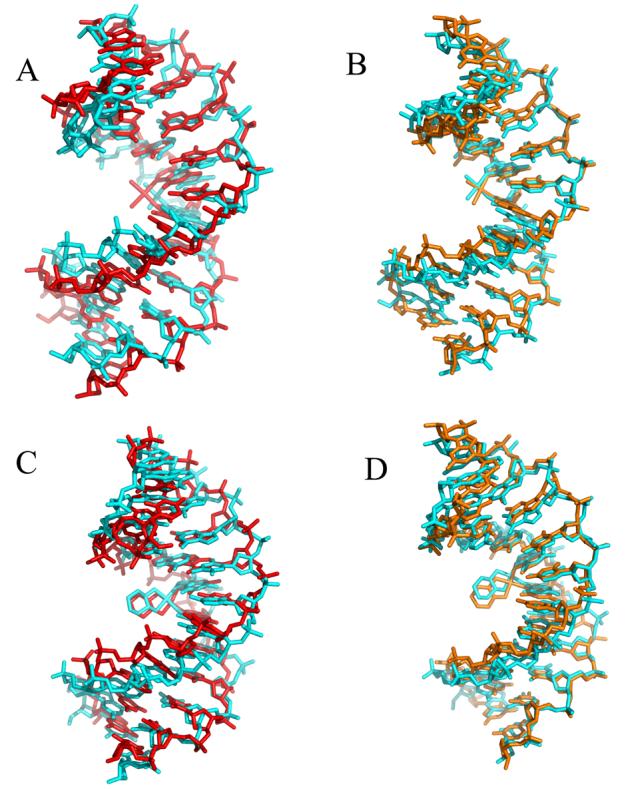
Pairwise Kabsch alignment of the centroid structure (cyan) obtained from the CP-DNA and OX-DNA simulations versus the corresponding crystal structures (red) and NMR structures (orange). The NMR solution structures of CP-DNA33 and OX-DNA30 (Protein DataBank entries 2NPW and 1PG9, respectively) in the d(CCTCAGGCCTCC)-3' sequence context were used to compare against corresponding centroid structures. The crystal structures corresponding to CP-DNA31 and OX-DNA32 were derived from the available structures in the 5'-d(CCTCTGGTCTCC)-3' sequence context (Protein DataBank entries 1AIO and 1IHH, respectively). Representative crystal structures in the 5'-d(CCTCAGGCCTCC)-3' sequence context were derived by mutating the nucleotide sequence using Tripos Sybyl. Pairwise Kabsch alignment of the CP-DNA and OX-DNA centroid structures was performed using PyMol's pair_fit tool.
In order to further assess how well the MD simulation structures agreed with our previously reported NMR structures30,33, the extent to which the MD simulations reproduced NMR-derived inter-proton distances was also evaluated. There were 171 and 160 inter-proton distance constraints derived from NMR data within the central four base pairs 5'-d(A5G6G7C8)-3' of CP-DNA and OX-DNA adducts respectively30 ,33. The statistics of the inter-proton distance violations (defined as an inter-proton distance deviating from the range of corresponding distance constraints by more than 0.5 Å, see Methods) are listed in Table 1 and the details of these distance violations are listed in Table S1 of Supplementary Data. Overall, there were 2 (< 2%) and 5 (< 3%) violations for OX-DNA and CP-DNA respectively. Most of the violations in CP-DNA simulation structures and one of the two violations in OX-DNA simulation structures involved A5 and G6 nucleotides, which were on the 5' side of the adduct and may represent the intrinsic dynamics of this portion of the molecule. Furthermore, one of the OX-DNA violations and three of the CP-DNA violations involved sugar protons, and may represent differences in sugar pucker, rather than differences in the position of the purine or pyrimidine bases. The very small number of violations compared to the total number of distance constraints indicated that our MD simulations largely reproduced the NMR data.
Table 1.
Violations of NMR constraints for the central four base pairs
| Violations | Total Constraints |
||||
|---|---|---|---|---|---|
| Base-Base Protons |
Base-Pucker Protons | Total Violations |
|||
|
CP-DNA (171) |
Interstrand | 1 | 0 | 1 | 18 |
| Intrastrand | 1 | 1 | 2 | 43 | |
|
Same Nucleotide |
0 | 2 | 2 | 110 | |
|
OX-DNA (160) |
Interstrand | 0 | 0 | 0 | 18 |
| Intrastrand | 1 | 1 | 2 | 44 | |
|
Same Nucleotide |
0 | 0 | 0 | 98 | |
Inter-proton differences averaged from the final 6 ns of the simulation trajectories were compared to the NOE derived inter-proton differences for CP-DNA and OX-DNA30,33 as described in Methods. All average inter-proton distances from the MD simulations trajectories which differed from the NOE inter-proton distance constraints by more than 0.5 Å were considered violations. The total number of inter-proton distance constraints for the central four base pairs is reported in parentheses in the first column. Restraints are reported according to category in the last column. The middle columns give the number of violations between base and base protons or between base and sugar pucker protons, and the total number of violations in each category.
The geometry of the Pt-DNA adducts was also compared with previously reported structures for the CP- and OX-DNA adducts (Table S2 in Supplementary Data). The platinum displacement from the plane of the central guanines and the platinum out of guanine plane bending angles were much closer to the previously reported values for the CP- and OX-DNA adducts than was the recent MD simulation of a CP-DNA adduct reported by Elizondro-Riojas and Kozelka40, possibly reflecting the refinement of partial charges that we derived and utilized for these simulations. Our MD simulations do not reflect the differences between CP- and OX-DNA adducts with respect to platinum displacement from the plane of the central guanines and the platinum out of guanine plane bending angles that were seen in our previous NMR solution structures of the adducts30 ,33 (Table S2). However, it is important to note that the position of the platinum is not directly determined by NMR constraints in the solution structures and that the CNS software with the partial charges reported earlier by Yao et al41 and Scheef et al42 was used to derive the lowest energy solution structures. Thus, this discrepancy between our MD simulations and the solution structures in the geometry of the platinum adduct may simply reflect differences in structure refinement, such as partial charges or differences between the AMBER and the CNS software used for calculations of the NMR structures. AMBER and CNS use different force fields and the AMBER simulation was performed under unrestrained and fully solvated condition, whereas CNS calculation used the simulated annealing protocol in vacuum and was restrained by the inter-proton distance constraints.
Principle Component Analysis of major conformational dynamics
Principle component analysis can be used to segregate large-scale correlated motions from random thermal fluctuations, thereby probing the essential dynamics of the system. Principal component analysis PCA is an orthogonal linear transformation that maps the data to a new coordinate system so that the system can be deconvoluted along these coordinates. The order of coordinates is rank-ordered according to their contribution to the motion of the system. Principle component analysis was used to analyze the trajectories from each set of simulations for major conformational motions. The first three principal components described >90% of the essential modes of dynamics for the platinum-adducted DNA complexes. As expected for DNA simulations, the first three components of conformational motions roughly corresponded to a superposition of bending (Movie S1), twisting (Movie S2) and winding (Movie S3) motions, in that order. By superimposing the major motions for CP- and OX-DNA adducts (Movies 1-3) it is evident that the overall conformational flexibility of DNA containing CP-GG and OX-GG adducts is very similar.
Hydrogen bonds
To assess the stability of the DNA duplex, the occupancy of all possible hydrogen bonds (calculated as the percentage of the time during the simulation that the hydrogen bonds existed), was measured for CP-DNA and OX-DNA adducts and B-DNA of the same sequence. The data for Watson-Crick hydrogen bond occupancy are shown in Figure 4. When compared to B-DNA, both CP-DNA and OX-DNA adducts show a significant decrease in standard Watson-Crick hydrogen bond occupancy for the A5•T20 and G6•C19 base pairs on the 5' side of the adduct, whereas the base pairs on the 3' side of the adduct are almost completely intact.
Figure 4. Hydrogen bond occupancy of the central four base pairs.

The hydrogen bond occupancy of the central four base pairs was calculated as described in Methods. The standard Watson-Crick hydrogen bond occupancy of base pairs for B-DNA (circle), CP-DNA (square) and OX-DNA (triangle) are shown.
Two hydrogen bonds with significantly high occupancy between the platinum carrier ligand and the DNA were identified and are shown in Figure 5. One is between the 3' amine hydrogen of the platinum and the oxygen atom O6 of the 3' G7 and the other is between the 5' amine hydrogen of the platinum and the nitrogen atom N7 of A5. For the CP-DNA adduct the amine hydrogens are equivalent so only four combinations of hydrogen bond formation are possible. The occupancy of each of these hydrogen bond combinations is summarized in Table 2A. Differences in global DNA conformation associated with 5' only, 5' and 3' and 3' only hydrogen bonds were minimal. However, significant conformational differences were observed in the central four base pair region and these are shown in Figure S2. The CP-DNA adduct spends 40.2% of its time in conformations that allow formation of the 5' hydrogen bond only and 13.3% of its time in conformations that allow formation of the 3' hydrogen bond only. However, the CP-DNA adduct also spends a significant amount of time (34.0%) in conformations that allow formation of both the 5' and 3' hydrogen bonds. The total occupancy of the 5' and 3' hydrogen bonds for the CP-DNA adduct is 74.2% and 47.2%, respectively. Thus, for the CP-DNA adduct the occupancy is greater for the hydrogen bond on the 5' side of the adduct, suggesting that the CP-DNA adduct is preferentially oriented towards the 5' side.
Figure 5. Hydrogen bonds between platinum carrier ligands and DNA.
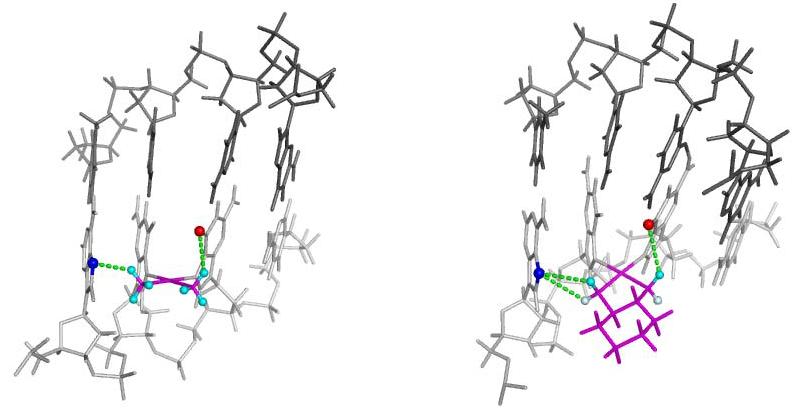
Structures illustrating observed hydrogen bond formation between Pt-amine hydrogens and the surrounding DNA bases are shown for both the CP-DNA adduct (left) and the OX-DNA adduct (right). The DNA backbone is shown in light gray for the strand containing the Pt-DNA adduct and darker gray for the complementary strand. The Pt adduct including the carrier ligand is shown in maroon. For the CP adduct, all amine hydrogens are in equilibrium and are shown in cyan. For the OX adduct the amine hydrogens are not in equilibrium. The equatorial hydrogen is shown in cyan and the axial hydrogen is shown in light blue. The dashed lines illustrate the potential hydrogen bonds between the platinum amine groups and the DNA bases.
Table 2.
Occupancy of Hydrogen Bonds Between Platinum Amine Hydrogens and Nearby Bases on the DNA
| Pattern of H Bond Formation | % Occupancy of H Bond | |
|---|---|---|
| 5' Side | 3' Side | |
| A: CP-DNA Adduct | ||
| Pt-amine-H • A5 | Pt-amine-H • G7 | |
| + | − | 40.2% |
| + | + | 34.0% |
| − | + | 13.3% |
| − | − | 12.5% |
| B: OX-DNA Adduct | ||
| Pt-amine-axial-H • A5 | Pt-amine-equitorial-H • G7 | |
| + | − | 7.7% |
| + | + | 3.5% |
| Pt-amine-equitorial-H • A5 | Pt-amine-equitorial-H • G7 | |
| + | − | 6.0% |
| + | + | 40.9% |
| No Pt-amine-H • A5 | Pt-amine-equitorial-H • G7 | |
| − | + | 34.3% |
| − | − | 7.6% |
The percentage occupancy of hydrogen bonds between Pt-amine hydrogens and near-by DNA bases (see Figure 5) was calculated as described in Methods.
For the OX-DNA adduct the situation is somewhat more complicated because the axial and equatorial amine hydrogens are not equivalent (Figure 5). Thus, on the 5' side of the adduct, the hydrogen bond between the axial hydrogen and N7 of A5 is not equivalent to the hydrogen bond between the equatorial hydrogen and N7 of A5. However, on the 3' side of the adduct only the equatorial hydrogen is in sufficient proximity to the O6 of G7 to form a hydrogen bond. Thus, for the OX-DNA adducts there are six possible combinations of hydrogen bond formation. The occupancy of each is summarized in Table 2B. As seen for the CP-DNA adduct, differences in global DNA conformations associated with these hydrogen patterns were minimal. The differences in DNA conformation in the central four base pair region associated with 5' equatorial only, both 5' equatorial and 3' equatorial and 3' equatorial only hydrogen bonds are shown in Figure S3. The OX-DNA adduct spends 34.2% of its time in conformations that allow formation of the 3' hydrogen bond only and 13.7% of its time in conformations that allow formation of 5' hydrogen bonds only. However, it also spends a significant amount of time (40.9%) in conformations that allow simultaneous formation of both a hydrogen bond between the equatorial hydrogen on the 5' side and N7 of A5 and a hydrogen bond between the equatorial hydrogen on the 3' side and O6 of G7. The total occupancy of the 5' and 3' hydrogen bonds for the OX-DNA adduct is 58.1% and 78.7%, respectively. Thus, for the OX-DNA adduct the occupancy is greater for the hydrogen bond on the 3' side of the adduct, suggesting that the OX-DNA adduct is preferentially oriented towards the 3' side.
DNA conformational dynamics
While the overall centroid structures of the CP-DNA and OX-DNA simulations were very similar, we observed significant differences between the two simulations in terms of DNA conformational dynamics. To determine the effect of CP- and OX-GG adducts on the conformational dynamics of DNA, the frequency distributions (fraction of the time spent in each conformation) from the trajectories of the CP-DNA, OX-DNA and undamaged DNA simulations were calculated using the program CURVES v5.3. From previous comparisons of CP- and OX-DNA adducts, it appeared that they were most likely to differ in terms of overall bend angle and the DNA helical parameters of the central four base pair region30-33. The overall bend angle was calculated from the CURVES output using MADBEND. No significant differences in the frequency distribution of bend angles was observed for the CP- and OX-DNA adduct simulations (data not shown). The frequency distributions of DNA helical parameters for the central four base pairs were taken directly from the CURVES output and were analyzed for statistical significance by the Kolmogorov-Smirnov test43 (Table S3). The Kologorov-Smirnov test determines how significantly two distributions differ from each other, without making any assumption regarding the distribution of data (non-parametric and distribution-free). Cases in which the frequency distributions of DNA helical parameters were significantly different from undamaged DNA for both CP- and OX-DNA adducts are indicated in bold in Table S3. The distribution of frequency values for those helical parameters between both types of Pt-DNA adducts and undamaged B-DNA are shown in Figure 6. When comparing both Pt-DNA adducts to B-DNA, there were several striking differences identified, including buckle and propeller twist for the A5·T20 base pair, shear and propeller twist for the G6·C19 base pair, buckle for the G7·C18 base pair; roll at the G6-G7 base pair step, and slide, tilt and roll at the G7-C8 base pair step. These differences indicated that the conformational dynamics profile of B-DNA was altered by the platinum adducts and are consistent with the previously reported conformational distortions imposed on B-DNA by Pt-GG adducts28 ,29 ,31 ,32 ,44. At the G6-G7 base pair step, the profiles of frequency distribution of roll were almost identical for CP-DNA and OX-DNA adducts, which was consistent with the GG dihedral angles for these two adducts.
Figure 6. Frequency distributions of representative DNA duplex helical parameters for the central four base pairs: Differences between Pt-DNA adducts and undamaged DNA.

Frequency distributions of each DNA duplex helical parameter for the central four base pairs were calculated from the simulation trajectories by CURVES v5.3 as described in Methods. The frequency distribution histograms were calculated from the structures obtained at every picosecond over the final 6 nanosecond of each equilibrated MD simulation. Thus, the histograms for CP-DNA and OX-DNA adducts were derived from 60,000 structures, while the histograms for undamaged DNA were derived from 30,000 structures. Selected frequency distributions that show differences between Pt-DNA adducts and undamaged DNA are shown. B-DNA (dotted lines with diamond symbols); CP-DNA (solid lines with circle symbols); OX-DNA (long dashed lines with square symbols) (The symbols do not represent the data ponts; rather they are shown to help distinguish the curves).
The significance of differences in the frequency distributions of DNA helical parameters between CP- and OX-DNA adducts was also analyzed by the Kolmogorov-Smirnov test43 (Table S4). The distributions of those helical parameters showing the greatest difference between CP- and OX-DNA adducts are shown in Figure 7, along with the distribution pattern of undamaged B-DNA for comparison. When comparing CP-DNA adducts to OX-DNA adducts, there were some noticeable differences. For base-base helical parameters, differences were observed for propeller twist for the G6·C19 and G7·C18 base pairs and buckle for the C8·G17 base pair. For base-pair-step helical parameters, differences were observed for slide at the A5-G6, G6-G7 and G7-C8 base pair steps and shift at the G7-C8 base pair step. All of the above differences suggest that CP-DNA and OX-DNA adducts differ in conformational dynamics.
Figure 7. Frequency distributions of representative DNA duplex helical parameters for the central four base pairs: Differences between CP-DNA and OX-DNA adducts.

Frequency distributions of each DNA duplex helical parameter for the central four base pairs were calculated from the simulation trajectories by CURVES v5.3 as described in Methods. The frequency distribution histograms were calculated from the structures obtained at every picosecond over the final 6 nanosecond of each equilibrated MD simulation. Thus, the histograms for CP-DNA and OX-DNA adducts were derived from 60,000 structures, while the histograms for undamaged DNA were derived from 30,000 structures. Selected frequency distributions that show differences between CP-DNA and OX-DNA adducts are shown. B-DNA (dotted lines with diamond symbols), CP-DNA (solid lines with circle symbols); OX-DNA (long dashed lines with square symbols) (The symbols do not represent the data points; rather they are shown to help distinguish the curves.) The vertical dash-dot lines show the value of the corresponding DNA helical parameter on the 3' side of the adduct in the crystal structure of the HMG-CP-DNA adduct26.
Correlation between platinum amine hydrogen bond formation and DNA conformational dynamics
As described above the % occupancy for hydrogen bonds between the Pt-amine hydrogens and nearby bases was greater on the 5′ side for the CP-DNA adduct and the 3′ side for the OX-DNA adduct. The data described above indicated that the CP- and OX-DNA adducts primarily influenced the conformational dynamics of the central four base pairs. Thus, to determine whether the conformational dynamics of CP- and OX-DNA adducts might be influenced by the formation of these hydrogen bonds, the trajectory data for DNA helical parameters for the central 4 base pairs were separated according to the patterns of hydrogen bond formation (Figures 8 and 9). The resulting frequency distributions were analyzed for significance by the Kolmogorov-Smirnov test43 (Tables S5 and S6) and cases in which the frequency distribution of DNA helical parameters associated with 5′ only hydrogen bonds were significantly different than the DNA helical parameters associated with 3′ only hydrogen bonds are indicated in bold Tables S5 and S6. The large number of examples in which the frequency distribution of DNA helical parameters is associated with the pattern of hydrogen bond formation (5′ only versus 3′ only) suggests that the conformational dynamics of many of the DNA helical parameters in the central four base pair region are strongly correlated with the pattern of hydrogen bond formation.
Figure 8. Effect of hydrogen bonding patterns on frequency distributions of selected DNA duplex helical parameters for the central four base pairs of CP-DNA adducts.

The effect of hydrogen bond patterns between the Pt-amine hydrogens and adjacent DNA bases on the frequency distributions of each DNA duplex helical parameter for the central four base pairs of the CP-DNA adduct were determined as described in Methods. The frequency distribution histograms were obtained from all of the CP-DNA structures containing a particular hydrogen bond pattern (Table 2). Thus, the histograms for 5' only, 5' plus 3' and 3' only hydrogen bonds were obtained from 24120, 20400 and 7980 structures, respectively. Selected frequency distributions that show difference in the distribution of DNA helical parameters when the hydrogen bond exists on the 5' side compared to when the hydrogen bond exists on the 3' side of the CP-DNA adduct are shown. 3' Hydrogen bond only (solid lines with square symbols); 5' hydrogen bond only (dotted lines with diamond symbols); hydrogen bonds on both 3' and 5' sides (long-dashed lines with upward triangle symbols) (The symbols do not represent the data points; rather they are shown to help distinguish the curves.) The frequency distributions for no hydrogen bonds on either the 3' side or 5' side are not shown for ease of viewing.
Figure 9. Effect of hydrogen bonding patterns on frequency distributions of selected DNA duplex helical parameters for the central four base pairs of OX-DNA adducts.

The effect of hydrogen bond patterns between the Pt-amine hydrogens and on adjacent DNA bases on the frequency distributions of each DNA duplex helical parameter for the central four base pairs of the OX-DNA adduct were determined as described in Methods. The frequency distribution histograms were obtained from all of the OX-DNA structures containing a particular hydrogen bond pattern (Table 2). Thus, the histograms for 5' axial only, 5' equatorial only, 5' axial plus 3' equatorial and 3′ equatorial only hydrogen bonds were obtained from 3600, 4620, 24540 and 20500 structures, respectively. Selected frequency distributions that show difference in the distribution of DNA helical parameters when the hydrogen bond exists on the 5′ side compared to when the hydrogen bond exists on the 3′ side of the OX-DNA adduct are shown. 3′ Equatorial hydrogen bond only (solid lines with square symbols); 5′ axial hydrogen bond only (dot-dashed lines with circle symbols); 5′ equatorial hydrogen bond only (dotted lines with diamond symbols); hydrogen bonds with both 3′ equatorial and 5′ equatorial hydrogens (long-dashed lines with upward triangle symbols) (The symbols do not represent the data points; rather they are shown to help distinguish the curves. The frequency distributions for no hydrogen bonds on either the 3′ side or 5′ side and hydrogen bond formation for both the 3′ equatorial and 5′ axial hydrogens are not shown for ease of viewing.
Selected examples for the CP-DNA adduct are shown in Figure 8. For most of the DNA helical parameters shown there is a clear difference in the distribution of DNA helical parameters when the hydrogen bond exists on the 5′ side compared to when the hydrogen bond exists on the 3′ side. In the case of the CP-DNA adduct, the two major hydrogen bond patterns are hydrogen bond on 5′ side only (40.2%) and hydrogen bonds on both the 5′ and 3′ side (34.0%). Furthermore, the frequency distributions of DNA helical parameters associated with these two hydrogen bond patterns are generally either almost identical (A5-G6 slide) or very similar (G6·C19 propeller twist, G7·C18 propeller twist, C8·G17 buckle, G6-G7 slide) G7-C8 shift, G7-C8 slide). These data suggest that DNA conformations in which the hydrogen bonds are seen on both the 5′ and 3′ side may represent conformational transitions between the 5′ only and 3′ only hydrogen bond conformations.
For the OX-DNA adduct (Figure 9 and Table S6) there also appear to be clear differences in the distribution of DNA helical parameters when the hydrogen bond exists on the 5′ side only (either for the axial hydrogen or the equatorial hydrogen) compared to when the hydrogen bond exists on the 3′ side only. In the case of the OX-DNA adduct, the two major hydrogen bond patterns are a hydrogen bond with the equatorial hydrogen on 3′ side only (34.3%) and hydrogen bonds with both the equatorial hydrogen on the 5′ and the equatorial hydrogen on the 3′ side (40.9%). As with the CP-DNA adduct, the frequency distributions of DNA helical parameters associated with these two hydrogen bond patterns are generally either almost identical (G6-G7 twist, G7-C8 shift, G7-C8 slide) or very similar (G6·C19 propeller twist, G7·C18 propeller twist, C8·G17 buckle, G6-G7 slide). These data again suggest that DNA conformations in which the hydrogen bonds are seen on both the 5′ and 3′ side may represent conformational transitions between the 5′ only and 3′ only hydrogen bond conformations.
Finally, when one compares the data in Figures 8 and 9 with the data in Figure 7 it becomes apparent that most of the conformational differences associated with hydrogen bond formation in the CP- and OX-DNA adducts are highly correlated with the conformational differences seen between CP- and OX-DNA adducts in Figure 7. For example, when one looks at propeller twist at the G6·C19 base pair, the two major distribution patterns associated with 5′ hydrogen bond formation (5′ only and both 5′ and 3′) of the CP-DNA adduct (representing 74.2% of the total hydrogen bond occupancy) are centered at -18° to -16 ° (Figure 8), while the two major distribution patterns associated with 3′ hydrogen bond formation (3′ only and equatorial 5′ plus 3′) for the OX-DNA adduct (representing 75.2% of the total hydrogen bond occupancy) are centered at −29° and −21° (Figure 9). This precisely accounts for the difference in frequency distribution patterns seen between CP- and OX-DNA adducts (−18° for CP and −25° for OX) for G6•C19 propeller twist shift in Figure 7. Similar comparisons can be made for G7•C18 propeller twist, C8•G17 buckle, A5-G6 slide, G6-G7 slide, and G7-C8 slide.
However, these patterns are not universal. For G7-C8 shift, the differences in distribution patterns between CP- and OX-DNA adducts in Figure 7 correlate with the major distribution patterns for CP- and OX-DNA adducts in Figures 86 and 9, but the differences between 5' only and 3' only hydrogen bond conformational distributions are not significant for either CP- or OX-DNA adducts (Tables S5 and S6). At the opposite extreme, the 5' only and 3' only hydrogen bond conformational distributions are significantly different for G6-G7 twist (Tables S56 and S6)), but the major distribution patterns (5' only and both 5' and 3' for CP and 3' only and both 5' axial and 3' for OX) are very similar (22-26° for CP and 28° for OX). Thus, the distribution patterns for CP- and OX-DNA adducts for G6-G7 twist are not significantly different (Table S4).
Discussion
Accuracy of the MD simulations
Partial atomic charges for the platinum atom and the surrounding atoms of Pt-GG adducts are not available in the standard AMBER force field and must, therefore be developed ab initio. The previously available charges for CP-GG adducts were adopted by Yao et al41 from the ab initio calculations of the [Pt(NH3)3(Ade)]2+ complex by Kozelka et al45 over 13 years ago. The same atomic charge parameters were used by Scheef et al42 for simulations of the OX-GG adduct, with the sole exception that the charge on the amine nitrogen was modified to reflect its attachment to the cyclohexane ring. Although the partial charge parameters on the CP-GG adduct have been updated by Elizondo-Riojas and Kozelka40, those charge parameters were not actually reported. Because the partial charge parameters for the CP-GG adduct were not derived empirically for a Pt-GG adduct and the OX-GG partial charge parameters had never been determined empirically, we have developed new partial charge parameters for both CP-GG and OX-GG adducts. The new partial charges on the platinum atom were similar to the previous published values40 ,41. However, the new partial charges on the surrounding nitrogen atoms were significantly different. Therefore, it was important to validate the accuracy of our new partial charges in MD simulations.
Our MD simulations converged to a common set of structures within the first 4 ns that were independent of the starting structure and MD initial velocities and these structures reproduced the crystal and NMR solution structures of the same adducts in DNA by several criteria. First, the simulation structures in the final 6 ns had an average RMSD of < 3 Å with respect to our NMR solution structures (Figure 1). Second, the centroid structures had RMSDs of around 4.1 Å compared to the corresponding crystal structures31,32 and around 3.1 Å compared to the corresponding NMR structures30,33. Third, for the central 4 base pair region there were less than 3% violations of NMR-derived inter-proton distance constraints for the CP-GG and OX-GG adducts and the majority of these violations involved sugar pucker protons. Finally, both CP-DNA and OX-DNA adducts in our MD simulations showed significantly increased roll angle at the G6•G7 base pair step and the GG dihedral angle with respect to the undamaged B-DNA, which is consistent with both the crystal and NMR solution structures of the Pt-DNA adducts29-33. By all of these criteria, the MD simulations were excellent representations of both the crystal and NMR solution structures of the same adducts. In addition, these MD simulations provided a better estimation of the α angles and displacements of the platinum atom out of the 5' and 3' guanine planes than the previous MD simulation of the CP-GG adduct by Elizondo-Riojas and Kozelka40.
The DNA duplex is more distorted on the 5' side of the adduct than on the 3' side
We found a significantly greater decrease of occupancy of standard Watson-Crick hydrogen bonds for the 5' A5•T20 and 5' G6·C19 base pairs than for the 3' G7·C18 base pair for both CP-GG and OX-GG adducts, which suggests that the Pt-GG adduct is more distorted on the 5' side of the adduct than on the 3' side of the adduct. This is consistent with a number of biological and structural studies that have indicated that Pt-GG adducts are more distorted on the 5' side of the adduct than on the 3' side of the adduct. For example, several authors have reported that the majority of misinsertion mutations occur opposite the 5'G of CP-GG adducts34-36, and we and others have shown that hpol η has significantly greater difficulty extending the DNA chain past the 5'G than the 3'G of either CP-GG or OX-GG adducts37-39. With respect to structural studies, Marzilli et al29 have reported a faster exchange rate between the water and imino protons of the 5'G of CP-GG adducts and we have shown a faster exchange rate between water and imino protons of the 5'G of both the OX-GG30 and CP-GG adducts33. Marzilli et al.29 also reported an unusually large positive shift and slide at 5' X/G base pair step, and Elizondo-Riojas and Kozelka40, in their MD simulations, have shown greater mobility of 5' G·C base pair with respect to the base pair 5' to the Pt-GG adduct. All of these previous studies have indicated that the 5' G·C base pair was more flexible and/or more distorted than the 3' G·C base pair.
Orientation of CP-DNA and OX-DNA adducts
In the crystal structure of CP-DNA adduct, a hydrogen bond was reported between the hydrogen atom of the 5' oriented ammine NH3 ligand and an oxygen atom of the phosphate group on the 5' side of the Pt-GG adduct31. In contrast, in the crystal structure of OX-DNA adduct, a hydrogen bond was found between the 3' oriented amine NH2 group of the diaminocycloheaxane ligand and the oxygen atom O6 of the 3'G32. Thus it has been suggested that the cis-diammine carrier ligand of cisplatin was oriented more towards the 5' side of the adduct and the (trans-R,R)1,2-diaminocyclohexane carrier ligand of oxaliplatin was oriented more towards the 3' side of the adduct. However, neither hydrogen bond was observed in NMR solution structures of the CP- and OX-GG adducts28,30,33. In this study, we assessed the occupancy of these hydrogen bonds for both CP-DNA and OX-DNA adducts and found that the occupancy of the hydrogen bond between the 5'-oriented NH3 and the phosphate oxygen reported in the crystal structure of CP-DNA adduct was less than 1% for both CP-DNA and OX-DNA adducts (Figure S4). In their work based on MD simulations of CP-DNA adduct, Elizondo-Riojas and Kozelka40 have also reported that the formation of this hydrogen bond is a very rare event, unless one considers the possibility of hydrogen bonds bridged by one or two water molecules. The fact that this hydrogen bond is not observed at any significant level in either NMR solution structures or MD simulations suggests that the formation of such a hydrogen bond is a rare event and that the crystal structure of CP-DNA adduct happened to capture this rare conformation, possibly due to crystal packing.
However, we did observe significant occupancy of the hydrogen bond formed between the hydrogen atom of the 5' ammine NH3 ligand of CP or the 5' amine NH2 group of diaminocyclohexane ligand of OX and the nitrogen atom N7 of A5. The occupancy of this hydrogen bond was higher for CP-DNA adduct (74.2%) than OX-DNA adduct (58.1%). We also observed significant occupancy of the hydrogen bond formed between the hydrogen atom of the 3' ammine NH3 ligand of CP or the 3' amine NH2 group of diaminocyclohexane ligand of OX and the oxygen atom O6 of the 3' G7, which had been previously reported in the crystal structure of the OX-DNA adduct, but not the CP-DNA adduct. The occupancy of this hydrogen bond was higher for OX-DNA adduct (78.7%) than CP-DNA adduct (47.2%). The differences in occupancy of hydrogen bonds between the ammine/amine hydrogen atoms of the CP/OX and the bases on the 5' and 3' side of the Pt-GG adducts suggest a difference in the orientation of Pt carrier ligands with respect to the DNA between CP- and OX-GG adducts. The cis-diammine carrier ligand of CP appears to be oriented more towards the 5' side of the adduct and the (trans-R,R)1,2-diaminocyclohexane carrier ligand of OX appears to be oriented more towards the 3' side of the adduct. Thus our data are consistent with the observations reported by Lippard and colleagues31 ,32 with respect to the preferential orientation of carrier ligands of CP-DNA and OX-DNA adducts. This preferential formation of hydrogen bonds on the 5' side of the CP-DNA adducts and on the 3' side of OX-DNA adducts is highly correlated with differences in conformation and conformational dynamics of DNA containing CP- and OX-GG adducts. We postulate that the hydrogen bond formation between the Pt-amine hydrogens and adjacent bases drives some of the important conformational differences between CP- and OX-DNA adducts.
These experiments do not address the possible effect of sequence context on the orientation of CP- and OX-GG adducts. Our MD simulations were done in the AGGC sequence context, while the crystal structures reported by Lippard and colleagues31 ,32 were in the TGGT sequence context, which suggests that the relative orientation of CP- and OX-GG adducts are similar in those sequence contexts. However, in the MD simulations of the CP-GG adduct in the CGGA sequence context by Elizondo-Riojas and Kozelka40, a hydrogen bond was observed on the 3' side of the adduct and between 3' NH3 ligand of the CP-GG adduct and N7 of the A on the 3' side of the adduct. This observation may help explain previous reports that an A on the 3' side of Pt-GG adducts has a significant affect on the binding affinity and binding specificity of DNA damage recognition proteins for CP-GG adducts. Experiments are currently underway to determine the effect of sequence context on the orientation and conformational dynamics of CP- and OX-GG adducts.
Differences in conformational dynamics between CP-DNA and OX-DNA adducts
From the comparison of frequency distribution of DNA duplex helical parameters, we observed several differences which appear to reflect the distinct conformational dynamics between CP-DNA and OX-DNA adducts. While the total range of DNA conformations explored by CP- and OX-GG adducts is very similar, the fraction of time spent in these conformation by CP- and OX-GG adducts differed with respect to several DNA helical parameters in the vicinity of the Pt-GG adducts. We postulate that these differences in conformational dynamics could allow differential recognition of CP- and OX-GG adducts by critical DNA-binding proteins that influence the cytotoxic response to these adducts. While the differences in conformational dynamics between CP- and OX-DNA adducts are relatively small, they are fully consistent with the 1.5 to 2 fold differences in recognition of the adducts by most of the DNA-binding proteins studied to date.
We postulate that CP-GG adducts spend a greater percentage of time in conformations favorable for binding of mismatch repair and HMG-domain DNA-binding proteins, while OX adducts spend a greater percentage of time in conformations(s) favorable for bypass by hpol β and hpol η. For example, the HMG domain of both HMG-A1 and HMG-B1 has been shown to bind with higher efficiency to CP-GG adducts than to OX-GG adducts14 and the crystal structure of the HMG-CP-DNA complex has been reported26. Since HMG binds to the CP-DNA adduct on the 3'side of the adduct only26, the DNA helical parameters on the 3' side of the HMG-CP-DNA complex are shown as vertical dashed lines in Figure 7 for comparison with the frequency distributions of helical parameters for the CP-GG and OX-GG adducts. For propeller twist of the G7•C18 base pair, buckle of the C8•G17 base pair and shift and slide at the G7-C8 base pair step, the CP-GG adduct appears to spend a greater percentage of its time in a conformation that is similar to the conformation of the HMG-CP-GG complex than the OX-GG adduct. If any of these conformations are characteristic of the conformation of the protein-DNA complex in the initial recognition step, they might facilitate the recognition of the CP-GG adduct by the HMG domain. The conformational dynamics differences at the G7-C8 base pair step might be particularly critical because both the affinity of the HMG domain for the CP-GG adduct14 ,46 ,47 and the ability of the HMG domain to discriminate between CP-GG and OX-GG adducts14 is highly dependent on the base to the 3' side of the adduct.
There are a number of important limitations inherent in such comparisons. We are comparing a crystal structure of the HMG-CP-GG DNA complex with unrestrained simulations of the CP-GG and OX-GG DNA adducts; the final, stable protein DNA complex likely has a different conformation from the initial protein-DNA recognition complex; and the sequence context of the CP-GG adduct in the HMG-CP-GG complex was different from the one in the CP-GG and OX-GG simulations. To better characterize those conformational differences that are important for differential protein recognition, experiments are underway to simulate the CP-GG and OX-GG adducts in the same AGGC sequence context in complex with the HMG domains of one or more proteins that discriminate between the adducts.
Methods
Starting structures
All five starting structures contained 12-mer DNA duplex in the same sequence context of 5'-d(CCTCAGGCCTCC)-3' for the strand containing the platinum adduct. For the DNA portion of the structures, the NMR solution structures of the OX-DNA adduct30 (Protein DataBank accession code 1PGC.pdb) and the CP-DNA adduct33 (Protein DataBank accession code 2NPW.pdb) solved in our lab were used as two of the starting structures. For the CP- and OX-DNA adducts, the DNA portion of the NMR solution structure of DNA complexed with hSRY (Protein DataBank accession code 1J46.pdb) was also used as a starting structure for the DNA backbone. The original DNA sequence from 1J46.pdb was mutated to the same DNA sequence used for our NMR solution structures. Since the DNA backbone of the hSRY-DNA complex was more bent than the DNA in the NMR solution structures of the Pt-DNA adducts by themselves, it provided a test of whether our MD simulations were capable of driving significantly distinct starting structures to non-distinguishable structures when simulations reached equilibrium. For the platinum adducts on the DNA backbone derived from the hSRY•DNA complex, the oxaliplatin adduct was obtained from our NMR solution structure of OX-GG30 (1PGC.pdb) and the cisplatin adduct was obtained from the NMR solution structure of the CP-GG adduct reported by Marzilli et al29 (1KSB.pdb). The Pt-GG adducts were superimposed on the DNA from the hSRY•DNA complex, with removal of the two Gs of the adduct producing the starting structures of the CP- and OX-DNA adducts. For undamaged DNA, ideal B-DNA was used as the starting structure. The molecular modeling package InsightII (www.accelrys.com) was used to perform all manual structural preparation and building, including the undamaged DNA and the DNA sequence change for the CP- and OX-DNA adducts.
Force field parameterization
The atomic partial charges for CP-GG and OX-GG adducts are not defined in the standard AMBER force field library. However, they are required and crucial in MD simulations. In order to determine the atomic partial charges of CP-GG and OX-GG adducts, the 9-methyl-guanine derivatives cis-[Pt(NH3)2(9-Me-Guo)2]2+ (CP-meG2) and [Pt(trans-RR-1,2-diaminocyclohexane)(9-Me-Guo)2]2+ (OX-meG2) were used to simplify the calculation. These derivatives were manually built from our NMR solution structures using Insight II. The atomic partial charges were determined using the Mulliken method implemented within Gaussian03 based on either the structure geometry optimized by Gaussian03 or the NMR structure modified to the 9-methyl-guanine derivative. The density functional method B3LYP implemented within Gaussian03 was utilized; the LanL2DZ basis set was used for the platinum atom and 6-31Gd basis set was used for the rest of the atoms. The Mulliken method was found to be insensitive to the geometry of the structure and the resulting atomic partial charges were consistent in both geometry optimized and non-optimized structures.
The partial charge of the platinum atom was close to the previously published values, while the partial charges of four nitrogen atoms surrounding the platinum atom were significantly different from the previously published values. The partial charges of the rest of the atoms were within the theoretical range and comparable with their counterparts in the standard AMBER force field. The Mulliken charges based on the structure of the original 9-methyl-guanine derivative modified from our NMR solution structure were used without further geometry optimization as the new atomic partial charges for Pt-GG adducts (Table S7, Supplemental Data) and incorporated into our force field parameters. The atomic partial charges of chemically equivalent atoms (such as the pseudo-equatorial hydrogen atoms of the ammine group and the corresponding atoms in the two guanine bases) were averaged and the small charge discrepancy due to the structural difference between 9-methyl-guanine and deoxyguanine was distributed to the sugar according to the standard charge transfer technique used by Yao et al.41. Besides the atomic partial charges, other force field parameters of the Pt-GG adducts were referenced from AMBER parm99 force field parameters or from previous work by Yao et al.41 and Scheeff et al.42. The MD simulation protocol is described in Supplementary Materials.
MD simulations and trajectory analysis
Altogether twenty-five 10 ns unrestrained and fully solvated MD simulations were carried out by using SANDER module of AMBER v8.0. There were five replicas for each of the five starting structures described above. The five replicas differed only in the MD initial velocity assigned when the system was heated for the last time. The atomic coordinates of the structures were saved every 1 ps. Both the 5' and 3' terminal base pairs were excluded from analysis because in several simulations the terminal bases were not base-paired and in some extreme cases they were stacked with each other. In contrast with previous MD simulations of Pt-DNA adducts, we did not use artificial constraints to hold the terminal base pairs together. Therefore only the central ten base pairs were considered in trajectory analysis. The PTRAJ module of AMBER v8.0 was employed for trajectory analysis.
Principle component analysis
The ptraj program from the AMBER v8 simulation software was used to perform principle component analysis (PCA). The final 6 ns of equilibrated MD simulations of each of the 20 trajectories of CP-DNA and OX-DNA were subject to PCA analysis. The first three principal components described >90% of the essential modes of dynamics for the platinum-adducted DNA complexes. The dynamics of the first three principle components were visualized using the PyMOL molecular graphics tool (DeLano Scientific, http://www.pymol.org).
Hydrogen bond occupancy
A distance of less than 3.5 Å and an angle of greater than 120° between the potential hydrogen bond donor and acceptor were used as the criteria for a hydrogen bond formation. The occupancy of one hydrogen bond was defined as the percentage of frames in which the hydrogen bond existed. The hydrogen bond occupancy of one base pair was defined as the average occupancy of hydrogen bonds existing within this base pair. For example, the hydrogen bond occupancy of normal G·C base pair was the average occupancy of the three standard Watson-Crick hydrogen bonds. However, there were hydrogen bonds formed between non-complementary bases in damaged DNA adducts because of the distortion and misalignment of bases. When calculating the hydrogen bond occupancy of base pairs, the occupancy of such non-complementary hydrogen bonds of at least 5% was necessary for consideration and the occupancy was split evenly between two base pairs.
Inter-proton distance constraint comparison
A set of inter-proton distance constraints have been derived from NMR data and used to compute the NMR solution structures for the OX-DNA and CP-DNA30 ,33 by CNS (Crystallography & NMR System) program. In the CNS calculations, a distance violation is defined as an inter-proton distance deviating from the range of corresponding distance constraints by more than 0.5 Å. The same criterion was applied here when we tried to evaluate how well our MD simulation structures agreed with the NMR data. In addition, we classified these constraints into several categories based on the locations of two protons. The two protons may be from the same nucleoside, from two nucleosides within the same strand (intrastrand) or from two nucleosides on different strands (interstrand). For each proton, it may be a base proton or a sugar pucker proton. The H1' proton was treated as a base proton because its position is largely independent of sugar pucker. All the other protons of the sugar were considered as sugar pucker protons because their positions were strongly influenced by the sugar pucker conformation. Only the distance constrains of the central four base pairs 5'-d(A5G6G7C8)-3' were considered in this study.
DNA helical parameter analysis of trajectories
Five independent simulations with randomized initial velocities were performed for each of the following starting conformations: CP-SRY (the CP-GG adduct, 1KSB.pdp, superimposed on the DNA structure of the hSRY-DNA complex, 1J46.pdb), OX-SRY (the OX-GG adduct, 1PGC.pdb, superimposed on the DNA structure of the hSRY-DNA complex, 1J46.pdb), CP-NMR (the NMR structure of the CP-DNA 2NPW.pdb), OX-NMR (the NMR structure of the OX-DNA adduct, 1PGC.pdb) and B-DNA (constructed with Insight II (www.acelrys.com)). The ptraj tool from AMBER-849 was used to extract the equilibrated conformations between 4 ns and 10 ns of simulation time, recording snapshots at every 1 ps time-interval of the five independent runs of AMBER simulation trajectories. Using ptraj. these trajectory snapshots were saved in the Protein DataBank (PDB) format, resulting in a total of (10000 − 4000) × 5 snapshots, i.e. 30,000 snapshots for each of CP-SRY, OX-SRY, CP-NMR, OX-NMR and B-DNA simulations.
Each nucleotide type was converted from AMBER format to PDB format, and the resulting snapshots were subjected to CURVES analysis. The following CURVES parameters were extracted: global inter base-pair parameters: shift, slide, rise, tilt, roll and twist; global base-base parameters: shear, stretch, stagger, buckle, propeller and opening. Histograms were then constructed for percent occupancy versus discrete units of each DNA helical parameter. Initially, for both CP-DNA and OX-DNA adducts histograms were constructed for the CURVES parameters obtained with each starting structure (NMR and SRY) individually to determine whether the starting structure affected the distribution of CURVES parameters. Except for slight differences observed for shift at the A5•G6 base pair step, the CURVES parameters were not influenced by starting structure (data not shown). For CP-DNA and OX-DNA simulations, the CURVES trajectories for both the SRY and NMR starting structures were combined to get better statistics of fluctuations in the CURVES parameters (60,000 snapshots each). Percentage occupancy distributions of the DNA helical parameters were calculated by normalizing the frequency distributions to 100%.
The Kolmogorov-Smirnov test43 was performed for each helical parameter to calculate the P value for differences between CP-DNA vs. OX-DNA adducts (Table S5) and between B-DNA and both CP-DNA and OX-DNA adducts (Table S4). The Kologorov-Smirnov test determines how significantly two distributions differ from each other, without making any assumption regarding the distribution of data (non-parametric and distribution-free). For ease of comparison P values were reported on a negative logarithmic scale, −log(P) such that larger values imply greater statistical difference (Tables S4 & S5). In order to determine which of these −log(P) values were significant, the Kolomogorov-Smirnov test was also performed for the same helical parameters comparing 5 individual simulations of the same structure (i.e. 5 for CP-DNA, 5 for OX-DNA and 5 for B-DNA). The highest −log(P) value obtained from comparisons of the same structure was considered to set the threshold of significance. Only −log(P) values for the comparisons between CP-DNA and OX-DNA or between B-DNA and both CP-DNA and OX-DNA that were greater than the −log(P) value for comparisons of the same structures were considered significant.
Correlation of patterns of hydrogen bond formation with DNA helical parameters
A trajectory-wide binary profile of all combinations of the presence and absence of the following hydrogen bonds was generated using the hydrogen bond occupancy procedure described above: CP-DNA: 5' side: 5' Pt-amine hydrogen to 5N7; 3' side: 3' Pt-amine hydrogen to 7O6 and OX-DNA: 5' side: 5' Pt-amine equatorial hydrogen to 5N7; 5' Pt-amine axial hydrogen to 5N7; 3' side: 3' Pt-amine equatorial hydrogen to 7O6. DNA helical parameters of the corresponding trajectory snapshots were recorded using the helical parameter analysis procedure described above. Histograms were then constructed for percent occupancy versus discrete units of each DNA helical parameter for each of these hydrogen-bond combinations. The Kolmogorov-Smirnov test43 was performed for each helical parameter to calculate P values for the differences between 5' hydrogen bond only and 3' hydrogen bond only for both the CP-DNA (Table S6) and the OX-DNA adducts (Table S7). The significance of these P values was determined as described above.
Supplementary Material
Acknowledgements
This work was supported by USPHS grant CA84480.
Abbreviations Used
- CP
cisplatin; cis-diamminedichloroplatinum(II)
- CBDCA
carboplatin; cis-diammine-1,1-cyclobutanedicarboxylatoplatinum(II)
- OX
oxaliplatin; trans-R,R-diaminocyclohexaneoxalatoplatinum(II)
- MD
molecular dynamics
- NMR
nuclear magnetic resonance
- HMG
high mobility group
- RMSD
root mean square deviation
- hpol β
human DNA polymerase beta
- hpol η
human DNA polymerase eta
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Greene MH. Is Cisplatin a Human Carcinogen. J. Nat. Cancer Inst. 1992;84:306–312. doi: 10.1093/jnci/84.5.306. [DOI] [PubMed] [Google Scholar]
- 2.Travis LB, Curtis RE, Storm H, Hall P, Holowaty E, Van Leeuwen FE, Kohler BA, Pukkala E, Lynch CF, Andersson M, Bergfeldt K, Clarke EA, Wiklund T, Stoter G, Gospodarowicz M, Sturgeon J, Fraumeni JF, Jr., Boice JD., Jr. Risk of second malignant neoplasms among long-term survivors of testicular cancer. J. Natl. Cancer Inst. 1997;89:1429–1439. doi: 10.1093/jnci/89.19.1429. [DOI] [PubMed] [Google Scholar]
- 3.Silva MJ, Costa P, Dias A, Valente M, Louro H, Boavida MG. Comparative analysis of the mutagenic activity of oxaliplatin and cisplatin in the Hprt gene of CHO cells. Environ. Mol. Mutagen. 2005;46:104–115. doi: 10.1002/em.20138. [DOI] [PubMed] [Google Scholar]
- 4.Bassett E, King NM, Bryant MF, Hector S, Pendyala L, Chaney SG, Cordeiro-Stone M. The role of DNA polymerase η in translesion synthesis past platinum-DNA adducts in human fibroblasts. Cancer Res. 2004;64:6469–6475. doi: 10.1158/0008-5472.CAN-04-1328. [DOI] [PubMed] [Google Scholar]
- 5.Page JD, Husain I, Sancar A, Chaney SG. Effect of the diaminocyclohexane carrier ligand on platinum adduct formation, repair, and lethality. Biochemistry. 1990;29:1016–1024. doi: 10.1021/bi00456a026. [DOI] [PubMed] [Google Scholar]
- 6.Jennerwein MM, Eastman A, Khokhar AR. Characterization of adducts produced in DNA by isomeric 1,2-diaminocyclohexaneplatinum(II) complexes. Chem. -Biol. Interact. 1989;70:39–49. doi: 10.1016/0009-2797(89)90061-6. [DOI] [PubMed] [Google Scholar]
- 7.Woynarowski JM, Chapman WG, Napier C, Herzig MCS, Juniewicz P. Sequence- and region-specificity of oxaliplatin adducts in naked and cellular DNA. Molecular. Pharmacology. 1998;54:770–777. doi: 10.1124/mol.54.5.770. [DOI] [PubMed] [Google Scholar]
- 8.Fink D, Nebel S, Aebi S, Zheng H, Cenni B, Nehme A, Christen RD, Howell SB. The role of mismatch repair in platinum drug resistance. Cancer Res. 1996;56:4881–4886. [PubMed] [Google Scholar]
- 9.Zdraveski ZZ, Mello JA, Farinelli CK, Essigmann JM, Marinus MG. MutS preferentially recognizes cisplatin- over oxaliplatin-modified DNA. J. Biol. Chem. 2002;277:1255–1260. doi: 10.1074/jbc.M105382200. [DOI] [PubMed] [Google Scholar]
- 10.Aebi S, Kurdihaidar B, Gordon R, Cenni B, Zheng H, Fink D, Christen RD, Boland CR, Koi M, Fishel R, Howell SB. Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res. 1996;56:3087–3090. [PubMed] [Google Scholar]
- 11.Fink D, Zheng H, Nebel S, Norris PS, Aebi S, Lin T-P, Nehme A, Christen RD, Haas M, MacLeod CL, Howell SB. In vitro and in vivo resistance to cisplatin in cells that have lost DNA mismatch repair. Cancer Research. 1997;57:1841–1845. [PubMed] [Google Scholar]
- 12.Vaisman A, Varchenko M, Umar A, Kunkel TA, Risinger JI, Barrett JC, Hamilton TC, Chaney SG. The role of hMLH1, hMSH3, and hMSH6 defects in cisplatin and oxaliplatin resistance: Correlation with replicative bypass of platinum-DNA adducts. Cancer Research. 1998;58:3579–3585. [PubMed] [Google Scholar]
- 13.Brown R, Hirst GL, Gallagher WM, Mcilwrath AJ, Margison GP, Vanderzee AGJ, Anthoney DA. hMLH1 expression and cellular responses of ovarian tumour cells to treatment with cytotoxic anticancer agents. Oncogene. 1997;15:45–52. doi: 10.1038/sj.onc.1201167. [DOI] [PubMed] [Google Scholar]
- 14.Wei M, Cohen SM, Silverman AP, Lippard SJ. Effects of spectator ligands on the specific recognition of intrastrand platinum-DNA cross-links by high mobility group box and TATA-binding proteins. J. Biol. Chem. 2001;276:38774–38780. doi: 10.1074/jbc.M106374200. [DOI] [PubMed] [Google Scholar]
- 15.Zhai X, Beckmann H, Jantzen H-M, Essigmann JM. Cisplatin-DNA adducts inhibit ribosomal RNA synthesis by hijacking the transcription factor human upstream binding factor. Biochemistry. 1998;37:16307–16315. doi: 10.1021/bi981708h. [DOI] [PubMed] [Google Scholar]
- 16.Huang JC, Zamble DB, Reardon JT, Lippard SJ, Sancar A. HMG-domain proteins specifically inhibit the repair of the major DNA adduct of the anticancer drug cisplatin by human excision nuclease. Proc. Natl. Acad. Sci. USA. 1994;91:10394–10398. doi: 10.1073/pnas.91.22.10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mcanulty MM, Lippard SJ. The HMG-domain protein Ixr1 blocks excision repair of cisplatin- DNA adducts in yeast. Mutat. Res-DNA. Repair. 1996;362:75–86. doi: 10.1016/0921-8777(95)00037-2. [DOI] [PubMed] [Google Scholar]
- 18.Hoffmann JS, Locker D, Viliani G, Leng M. HMG1 protein inhibits the translesion synthesis of the major DNA cisplatin adduct by cell extracts. J. Mol. Biol. 1997;270:539–543. doi: 10.1006/jmbi.1997.1143. [DOI] [PubMed] [Google Scholar]
- 19.Vaisman A, Lim SE, Patrick SM, Copeland WC, Hinkle DC, Turchi JJ, Chaney SG. Effect of DNA Polymerases and High Mobility Group Protein 1 on the Carrier Ligand Specificity for Translesion Synthesis past Platinum-DNA Adducts. Biochemistry. 1999;38:11026–11039. doi: 10.1021/bi9909187. [DOI] [PubMed] [Google Scholar]
- 20.Treiber DK, Zhai XQ, Jantzen HM, Essigmann JM. Cisplatin-DNA adducts are molecular decoys for the ribosomal RNA transcription factor hUBF (human upstream binding factor) Proc. Natl. Acad. Sci. USA. 1994;91:5672–5676. doi: 10.1073/pnas.91.12.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coin F, Frit P, Viollet B, Salles B, Egly JM. TATA binding protein discriminates between different lesions on DNA, resulting in a transcription decrease. Mol. Cell. Biol. 1998;18:3907–3914. doi: 10.1128/mcb.18.7.3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vaisman A, Masutani C, Hanaoka F, Chaney SG. Efficient translesion replication past oxaliplatin and cisplatin GpG adducts by human DNA polymerase eta. Biochemistry. 2000;39:4575–4580. doi: 10.1021/bi000130k. [DOI] [PubMed] [Google Scholar]
- 23.Vaisman A, Chaney SG. The efficiency and fidelity of translesion synthesis past cisplatin and oxaliplatin GpG adducts by human DNA polymerase beta. Journal of Biological Chemistry. 2000;275:13017–13025. doi: 10.1074/jbc.275.17.13017. [DOI] [PubMed] [Google Scholar]
- 24.Lamers MH, Perrakis A, Enzlin JH, Winterwerp HHK, de Wind N, Sixma TK. The crystal structure of DNA mismatch repair protein MutS binding to a G center dot T mismatch. Nature. 2000;407:711–717. doi: 10.1038/35037523. [DOI] [PubMed] [Google Scholar]
- 25.Obmolova G, Ban C, Hsieh P, Yang W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature. 2000;407:703–710. doi: 10.1038/35037509. [DOI] [PubMed] [Google Scholar]
- 26.Ohndorf UM, Rould MA, He Q, Pabo CO, Lippard SJ. Basis for recognition of cisplatin-modified DNA by high-mobility-group proteins. Nature. 1999;399:708–712. doi: 10.1038/21460. [DOI] [PubMed] [Google Scholar]
- 27.Sawaya MR, Prasad R, Wilson SH, Kraut J, Pelletier H. Crystal structures of human DNA polymerase beta complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry. 1997;36:11205–11215. doi: 10.1021/bi9703812. [DOI] [PubMed] [Google Scholar]
- 28.Gelasco A, Lippard SJ. NMR solution structure of a DNA dodecamer duplex containing a cis-diammineplatinum(II) dGpG intrastrand cross-link, the major adduct of the anticancer drug cisplatin. Biochemistry. 1998;37:9230–9239. doi: 10.1021/bi973176v. [DOI] [PubMed] [Google Scholar]
- 29.Marzilli LG, Saad JS, Kuklenyik Z, Keating KA, Xu Y. Relationship of solution and protein-bound structures of DNA duplexes with the major intrastrand cross-link lesions formed on cisplatin binding to DNA. J. Am. Chem. Soc. 2001;123:2764–2770. doi: 10.1021/ja0007915. [DOI] [PubMed] [Google Scholar]
- 30.Wu Y, Pradhan P, Havener J, Boysen G, Swenberg JA, Campbell SL, Chaney SG. NMR solution structure of an oxaliplatin 1,2-d(GG) intrastrand cross-link in a DNA dodecamer duplex. J. Mol. Biol. 2004;341:1251–1269. doi: 10.1016/j.jmb.2004.06.066. [DOI] [PubMed] [Google Scholar]
- 31.Takahara PM, Frederick CA, Lippard SJ. Crystal structure of the anticancer drug cisplatin bound to duplex DNA. J. Am. Chem. Soc. 1996;118:12309–12321. [Google Scholar]
- 32.Spingler B, Whittington DA, Lippard SJ. 2.4 A crystal structure of an oxaliplatin 1,2-d(GpG) intrastrand cross-link in a DNA dodecamer duplex. Inorg. Chem. 2001;40:5596–5602. doi: 10.1021/ic010790t. [DOI] [PubMed] [Google Scholar]
- 33.Wu Y, Bhattacharyya D, King CL, Baskerville-Abraham I, Huh S-H, Boysen G, Swenberg JA, Temple B, Campbell SL, Chaney SG. Solution structures of a DNA dodecamer duplex with and without a cisplatin 1,2-d(GG) intrastrand cross-link: Comparison with the same DNA duplex containing an oxaliplatin 1,2-d(GG) intrastrand cross-link. Biochemistry. 2007 doi: 10.1021/bi062291f. ASAP web release. 5-12-2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bradley LJN, Yarema KJ, Lippard SJ, Essigmann JM. Mutagenicity and Genotoxicity of the Major DNA Adduct of the Antitumor Drug cis-Diamminedichloroplatinum(II) Biochemistry. 1993;32:982–988. doi: 10.1021/bi00054a031. [DOI] [PubMed] [Google Scholar]
- 35.Bubley GJ, Ashburner BP, Teicher BA. Spectrum of cis-diamminedichloroplatinum(II)-induced mutations in a shuttle vector propagated in human cells. Mol. Carcinogen. 1991;4:397–406. doi: 10.1002/mc.2940040512. [DOI] [PubMed] [Google Scholar]
- 36.Burnouf D, Daune M, Fuchs RPP. Spectrum of cisplatin-induced mutations in Escherichia coli. Proc. Natl. Acad. Sci. USA. 1987;84:3758–3762. doi: 10.1073/pnas.84.11.3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bassett E, Vaisman A, Havener JM, Masutani C, Hanaoka F, Chaney SG. Efficiency of extension of mismatched primer termini across from cisplatin and oxaliplatin adducts by human DNA polymerases beta and eta in vitro. Biochemistry. 2003;42:14197–14206. doi: 10.1021/bi035359p. [DOI] [PubMed] [Google Scholar]
- 38.Masutani C, Kusumoto R, Iwai S, Hanaoka F. Mechanisms of accurate translesion synthesis by human DNA polymerase eta. EMBO Journal. 2000;19:3100–3109. doi: 10.1093/emboj/19.12.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Washington MT, Johnson RE, Prakash S, Prakash L. Mismatch extension ability of yeast and human DNA polymerase η. J. Biol. Chem. 2001;276:2263–2266. doi: 10.1074/jbc.M009049200. [DOI] [PubMed] [Google Scholar]
- 40.Elizondo-Riojas MA, Kozelka J. Unrestrained 5 ns molecular dynamics simulation of a cisplatin-DNA 1,2-GG adduct provides a rationale for the NMR features and reveals increased conformational flexibility at the platinum binding site. J. Mol. Biol. 2001;314:1227–1243. doi: 10.1006/jmbi.2000.5216. [DOI] [PubMed] [Google Scholar]
- 41.Yao S, Plastaras JP, Marzilli LG. A molecular mechanics AMBER-type force field for modeling platinum complexes of guanine derivatives. Inorg. Chem. 1994;33:6061–6077. [Google Scholar]
- 42.Scheeff ED, Briggs JM, Howell SB. Molecular modeling of the intrastrand guanine-guanine DNA adducts produced by cisplatin and oxaliplatin. Mol. Pharmacol. 1999;56:633–643. [PubMed] [Google Scholar]
- 43.Press WH, Flannery BP, Teukolsky SA, Vetterling WT. Numerical Recipes in C: The Art of Scientific Computing. Cambridge University Press; 1992. [Google Scholar]
- 44.Yang D, van Bloom SSGE, Reedijk J, van Bloom JH, Wang AHJ. Structure and isomerization of an intrastrand cisplatin-cross-linked octamer DNA duplex by NMR analysis. Biochemistry. 1995;34:12912–12920. doi: 10.1021/bi00039a054. [DOI] [PubMed] [Google Scholar]
- 45.Kozelka J, Savinelli R, Berthier G, Flament JP, Lavery R. Force field for platinum binding to adenine. J. Comput. Chem. 1993;14:45–53. [Google Scholar]
- 46.Cohen SM, Mikata Y, He Q, Lippard SJ. HMG-Domain protein recognition of cisplatin 1,2-intrastrand d(GpG) cross-links in purine-rich sequence contexts. Biochemistry. 2000;39:11771–11776. doi: 10.1021/bi001352l. [DOI] [PubMed] [Google Scholar]
- 47.Dunham SU, Lippard SJ. DNA sequence context and protein composition modulate HMG- domain protein recognition of ciplatin-modified DNA. Biochemistry. 1997;36:11428–11436. doi: 10.1021/bi9709452. [DOI] [PubMed] [Google Scholar]
- 48.Cheatham TE, III, Cieplak P, Kollman PA. A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J Biomol.Struct.Dyn. 1999;16(4):845–862. doi: 10.1080/07391102.1999.10508297. [DOI] [PubMed] [Google Scholar]
- 49.Case DA, Darden TA, Cheatham TE, III, Simmerling CJ, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Scafmeister C, Caldwell JW, Rosa WS, Kollman PA. AMBER 8. University of California; San Francisco: 2004. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.