

Abstract
To generate the forces needed for motility, the plasma membranes of nonmuscle cells adopt an activated state that dynamically reorganizes the actin cytoskeleton. By usurping components from focal contacts and the actin cytoskeleton, the intracellular pathogens Shigella flexneri and Listeria monocytogenes use molecular mimicry to create their own actin-based motors. We raised an antibody (designated FS-1) against the FEFPPPPTDE sequence of Listeria ActA, and this antibody: (a) localized at the trailing end of motile intracellular Shigella, (b) inhibited intracellular locomotion upon microinjection of Shigella-infected cells, and (c) cross-reacted with the proteolytically derived 90-kD human vinculin head fragment that contains the Vinc-1 oligoproline sequence, PDFPPPPPDL. Antibody FS-1 reacted only weakly with full-length vinculin, suggesting that the Vinc-1 sequence in full-length vinculin may be masked by its tail region and that this sequence is unmasked by proteolysis. Immunofluoresence staining with a monoclonal antibody against the head region of vinculin (Vin 11-5) localized to the back of motile bacteria (an identical staining pattern observed with the anti-ActA FS-1 antibody), indicating that motile bacteria attract a form of vinculin containing an unmasked Vinc-1 oligoproline sequence. Microinjection of submicromolar concentrations of a synthetic Vinc-1 peptide arrested Shigella intracellular motility, underscoring the functional importance of this sequence. Western blots revealed that Shigella infection induces vinculin proteolysis in PtK2 cells and generates p90 head fragment over the same 1–3 h time frame when intracellular bacteria move within the host cell cytoplasm. We also discovered that microinjected p90, but not full-length vinculin, accelerates rates of pathogen motility by a factor of 3 ± 0.4 in Shigella-infected PtK2 cells. These experiments suggest that vinculin p90 is a rate-limiting component in actin-based Shigella motility, and that supplementing cells with p90 stimulates rocket tail growth. Earlier findings demonstrated that vinculin p90 binds to IcsA (Suzuki, T.A., S. Saga, and C. Sasakawa. 1996. J. Biol. Chem. 271:21878– 21885) and to vasodilator-stimulated phosphoprotein (VASP) (Brindle, N.P.J., M.R. Hold, J.E. Davies, C.J. Price, and D.R. Critchley. 1996. Biochem. J. 318:753– 757). We now offer a working model in which proteolysis unmasks vinculin's ActA-like oligoproline sequence. Unmasking of this site serves as a molecular switch that initiates assembly of an actin-based motility complex containing VASP and profilin.
The microbial pathogens Listeria monocytogenes, Shigella flexneri, rickettsia, and vaccinia have evolved functionally related mechanisms to harness the forces of actin polymerization for propelling themselves within the host cell cytoplasm and for promoting their cell-to-cell spread (Bernardini et al., 1989; Dabiri et al., 1990; Heinzen et al., 1993; Cudmore et al., 1995). Listeria requires the proline-rich surface protein ActA to initiate host cell actin assembly (Domann et al., 1992; Kocks et al., 1992), whereas Shigella uses another unrelated cell wall protein called IcsA (Bernardini et al., 1989; Goldberg et al., 1993). Listeria and Shigella move through the cytoplasm of PtK2 host cells at speeds as rapid as 0.4 μm/s (Dabiri et al., 1990; Zeile et al., 1996). Upon reaching the periphery of the host cell, these bacteria induce the formation of filopods, and these membrane projections can be ingested by adjacent cells, allowing these microorganisms to maximize their infectivity. As Listeria move through the cytoplasm, each of their trailing poles promotes actin filament assembly into rocket tails (Tilney and Portnoy, 1989; Dabiri et al., 1990); actin monomers add to the tails at the bacteria–actin interface, and such localized actin assembly provides the force for intracellular movement (Sanger et al., 1992; Peskin et al., 1993). The host cell components required for this actin-based motor appear to include constituents of focal contacts, among them actin filaments (Tilney and Portnoy, 1989; Dabiri et al., 1990), α-actinin (Dabiri et al., 1990; Dold et al., 1994), profilin (Theriot et al., 1994), and the vasodilator-stimulated phosphoprotein (VASP)1 (Chakraborty et al., 1995). The cell wall protein ActA is the only known bacterial component required for Listeria intracellular motility (Domann et al., 1992; Kocks et al., 1992). ActA contains four oligoproline repeats of the type FEFPPPPTDE that are essential for binding VASP (Chakraborty et al., 1995; Pistor et al., 1995). The consensus sequence (D/E)FPPPPX(D/E)(D/E) is characterized by a stretch of four prolines flanked NH2-terminally by aromatic and acidic residues and COOH-terminally by acidic residues. These features define a new class of docking sequences designated as actin-based motility-1 (ABM-1) sequences (Purich and Southwick, 1997). This sequence binds VASP, which in turn contains its own set of GPPPPP repeats for profilin binding (Reinhard et al., 1995a ). The latter sequence is representative of another class of oligoproline sequences (ABM-2 sequences) for profilin binding (Purich and Southwick, 1997).
Shigella flexneri also form actin rocket tails while moving within the host's cytoplasm (Bernardini et al., 1989), and VASP colocalizes with intracellular Shigella (Chakraborty et al., 1995). While the bacterial surface protein IcsA is necessary for Shigella's actin-based motility (Bernardini et al., 1989; Goldberg et al., 1993; Goldberg and Theriot, 1995), IcsA bears no obvious structural homology to Listeria ActA and lacks ABM-1 sequences for VASP binding. Nevertheless, microinjection of the ActA ABM-1 peptide FEFPPPPTDE into Shigella-infected cells blocks bacterial motility over the same concentration range found to inhibit Listeria movement (Zeile et al., 1996), indicating that Shigella may recruit a host cell adapter protein to supply ABM-1 sequence(s) in place of ActA. Shigella infection has been shown to deplete vinculin from the focal contacts of host cells (Kadurugamuwa et al., 1991), and IcsA is known to bind vinculin and to concentrate vinculin to the back of intracellular bacteria (Suzuki et al., 1996). Using an antibody directed against the FEFPPPPTDE sequence of the ActA protein, we have discovered that one or more cross-reactive proteins concentrate focally at the rearward pole of motile intracellular Shigella. We have identified the 90-kD vinculin head fragment, which contains an ABM-1 sequence at its COOH terminus, as the major cross-reactive protein. Our data suggest that Shigella infection results in the proteolysis of intact 120-kD vinculin, thereby generating a p90 polypeptide that specifically binds to IcsA and concentrates on the bacterial surface. Microinjection of the p90 polypeptide, but not intact vinculin, into Shigella-infected PtK2 cells accelerates intracellular motility of the microbes by a factor of three, thus illustrating that the p90 fragment is functionally engaged in the locomotory process. Our findings indicate that the 90-kD head fragment of vinculin can serve as the ActA homolog required for Shigella actin-based motility, and vinculin proteolysis is likely to serve as a molecular switch that unmasks this protein's ABM-1 oligoproline sequence to bind VASP on the bacterial surface and to promote the assembly of an actin-based motor.
Materials and Methods
Materials
PtK2 kangaroo rat kidney cells were grown and infected with S. flexneri strain M90T, serotype 5, or L. monocytogenes 10403S, virulent strain serotype 1, as previously described (Dabiri et al., 1990; Zeile et al., 1996). Outdated platelet-rich plasma was obtained from the local blood bank and processed within 24 h of the storage expiration date. Thermolysin, poly- l-proline (mol wtaverage = 5,600; degree of polymerization = 57 residues), aprotinin, leupeptin, pepstatin A, PMSF, and DTT were obtained from Sigma Chemical Co. (St. Louis, MO). NBD-bodipy-phalloidin was purchased from Molecular Probes, Inc. (Eugene, OR). ActA peptide (CFEFPPPPTDE) and vinculin Vinc-1 peptide (PDFPPPPPDL) were synthesized and HPLC purified in the University of Florida Protein Sequencing Core Lab.
Antibodies
Anti-vinculin 11-5 mouse monoclonal antibody (Vin 11-5) was obtained from Sigma Chemical Co. The rabbit serum containing anti-VASP polyclonal antibody was a gift of U. Walter (Medical University Clinic, Wurzburg, Germany). Polyclonal rabbit anti–ActA-peptide antibody was raised by immunization with a peptide (CFEFPPPPTDE), corresponding to the ActA's second oligoproline repeat (Southwick and Purich, 1994) and coupled to keyhole limpet hemocyanin (Cocalico Biologicals, Reamstown, PA). Monospecific IgG was then isolated by immuno-affinity chromatography on CFEFPPPPTDE peptide coupled to cyanogen bromide-activated Sepharose 4B (Laine et al., 1988; Laine and Esser, 1989). Anti– actin-related protein (ARP3) antibody was provided by M. Welch and T. Mitchison (University of California, San Francisco, CA). Commercial secondary antibodies conjugated to rhodamine, fluorescein, alkaline phosphatase, or horseradish peroxidase were used without further purification.
Proteins
Human platelet vinculin was obtained from soluble platelet fraction by a modification of the method of Rosenfeld et al. (1985). Briefly, a vinculin-enriched fraction was obtained from the 0–35% ammonium sulfate fractionation. The pellet was resuspended in and dialyzed against 20 mM Tris/ acetate, 20 mM NaCl, 0.5 mM EDTA, 0.5 mM EGTA, 0.5 mM PMSF, 15 mM β-mercaptoethanol, pH 7.6. After centrifugation at 100,000 g for 1 h, the protein solution was loaded onto a 1.6 × 30 cm Q-Sepharose (Pharmacia Biotechnology Inc., Piscataway, NJ) column equilibrated with the same buffer and eluted with a 20–500 mM NaCl gradient. The first column peak (issuing from the column over the 50–100 mM salt range) was highly enriched in vinculin, as evaluated by SDS-PAGE and Western blotting. Vinculin was further purified from the pooled fractions of this peak by a gel filtration column (Superose-12, 10/30; Pharmacia Biotechnology Inc.).
The 90-kD vinculin head fragment was proteolytically cleaved from the 30-kD tail region of purified vinculin using thermolysin (1:250 wt/wt) in phosphate-buffered saline, pH 7.2, for 20 min at 37°C (Kilic and Ball, 1991), whereupon the solution was adjusted to pH 5.5 with 0.5 M acetic acid. The head fragment was purified from the tail fragment by Mono S chromatography (Pharmacia Biotechnology Inc.). The head fragment failed to remain adsorbed to the column, whereas the p30 tail fragment eluted at ∼200 mM NaCl.
Recombinant human platelet profilin was expressed and purified as described elsewhere (Fedorov et al., 1994; Kang et al., 1997).
Microinjection Experiments
Individual PtK2 cells were microinjected with the antibody, peptides, or protein, and velocities were measured as previously described (Southwick and Purich, 1994, 1995; Zeile et al., 1996). Differences in mean velocity were compared using the nonparametric Wilcoxon test. Immediately before microinjection, stock samples (10–12 μM) of full-length vinculin and p90 in 20 mM Hepes, 1 mM EDTA, 1 mM DMSF, 1 mM DTT, pH 7.4, were diluted with phosphate-buffered saline, pH 7.2, to a final concentration of 1.2 μM.
Identification of Vinculin p90 Polypeptide
Soluble and membrane fractions from ∼7 × 1012 human platelets were prepared as described in Waldmann et al. (1986). The platelet membrane fractions were extracted and passed through a 1 ml profilin affinity column as previously described by Reinhard et al. (1995a). The 90-kD polypeptide was identified as the major ABM-1–containing protein by cross-reaction with FS-1 anti-ActA antibody. This protein eluted in the flowthrough fraction that was subjected to two-dimensional IEF, pH range 4–7, and SDS-PAGE. After electrotransfer to polyvinylidene difluoride (PVDF) membrane, reaction with the FS-1 antibody was demonstrated using the alkaline phosphatase method. This protein was then subjected to proteolysis and automated Edman microsequencing.
Western Blot Analysis
Proteins transferred to the membrane were incubated in Tris-buffered saline (100 mM Tris/HCl, pH 7.5, 0.9% NaCl) containing 5% instant milk during the blocking and antibody binding steps. Secondary antibodies used included goat anti–rabbit IgG and rabbit anti–mouse IgG conjugated to horseradish peroxidase. Bound secondary antibody was visualized using the enhanced chemiluminescence method (ECL) (Pierce Chemical Co., Rockford, IL).
Vinculin Proteolysis After Shigella Infection of PtK2 Cells
To quantitate the p90 content of PtK2 cells, 150-mm culture dishes containing semiconfluent PtK2 cells were infected with Shigella as described above. At various times (1, 2, and 3 h) after initiating infection, cells were harvested into a cocktail of protease inhibitors (2 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, 1 mM PMSF, 1 mM EGTA, 1 mM EDTA, and 1 mM DTT in 0.1% Triton X-100). The samples were concentrated with a Centriprep-10 membrane concentrator (Amicon Corp., Danvers, MA). Samples were standardized with respect to protein concentration, and then suspended in 2× SDS sample buffer, boiled for 5 min, and subjected to SDS-PAGE. Uninfected cells and cells exposed to Escherichia coli were extracted and processed in an identical manner.
Indirect Immunofluorescence Microscopy
PtK2 cells were infected with Shigella, fixed in 3.7% formalin in a standard salt solution, permeabilized with 0.2% (vol/vol) Triton X-100, and stained as previously described (Sanger et al., 1983).
Results
Anti–ActA-peptide Immunofluorescence Microscopy Localizes a Cross-reactive Protein at the Back of Moving Bacteria
VASP was recently shown to concentrate on the surface of intracellular Shigella (Chakraborty et al., 1995). To identify the host cell ActA-like protein that binds VASP, we used an antibody designated FS-1, raised against the ABM-1 FEFPPPPTDE sequence of Listeria ActA (Southwick and Purich, 1994). This rabbit polyclonal antibody specifically reacted with the ActA surface protein in Listeria cell wall extracts and bound to the surface of both intra- and extracellular Listeria, as demonstrated by immunofluorescence microscopy. The same antibody failed to cross-react with Shigella or with cell-free extracts of Shigella grown in bacterial culture (data not shown). However, immunofluorescence micrographs of Shigella-infected PtK2 cells demonstrated the presence of a cross-reactive protein or proteins on intracellular bacteria (Fig. 1, A–C). Motile bacteria possessing F-actin rocket tails were identified by bodipy phalloidin staining (Fig. 1 A, rhodamine channel) and by phase contrast microscopy (Fig. 1 B). The same bacteria were also stained with the anti-ActA peptide antibody FS-1 (Fig. 1 C, fluorescein channel). Of 56 bacteria with actin filament rocket tails, 54 demonstrated focal staining with the FS-1 anti-ActA antibody. As previously reported (Goldberg et al., 1993; Suzuki et al., 1995; D'Hauteville et al., 1996), the IcsA surface protein is shed from the bacterial surface into the actin tails, and the cuplike staining pattern observed in Fig. 1 C reflects the same distribution that is observed with IcsA (i.e., the greatest concentration was found immediately behind the motile bacteria). These observations suggested that the intracellular Shigella bacterium does attract a mammalian ActA homolog to its surface, as suggested by our earlier inhibition experiments with ABM-1 and ABM-2 oligoproline peptides (Zeile et al., 1996).
Figure 1.
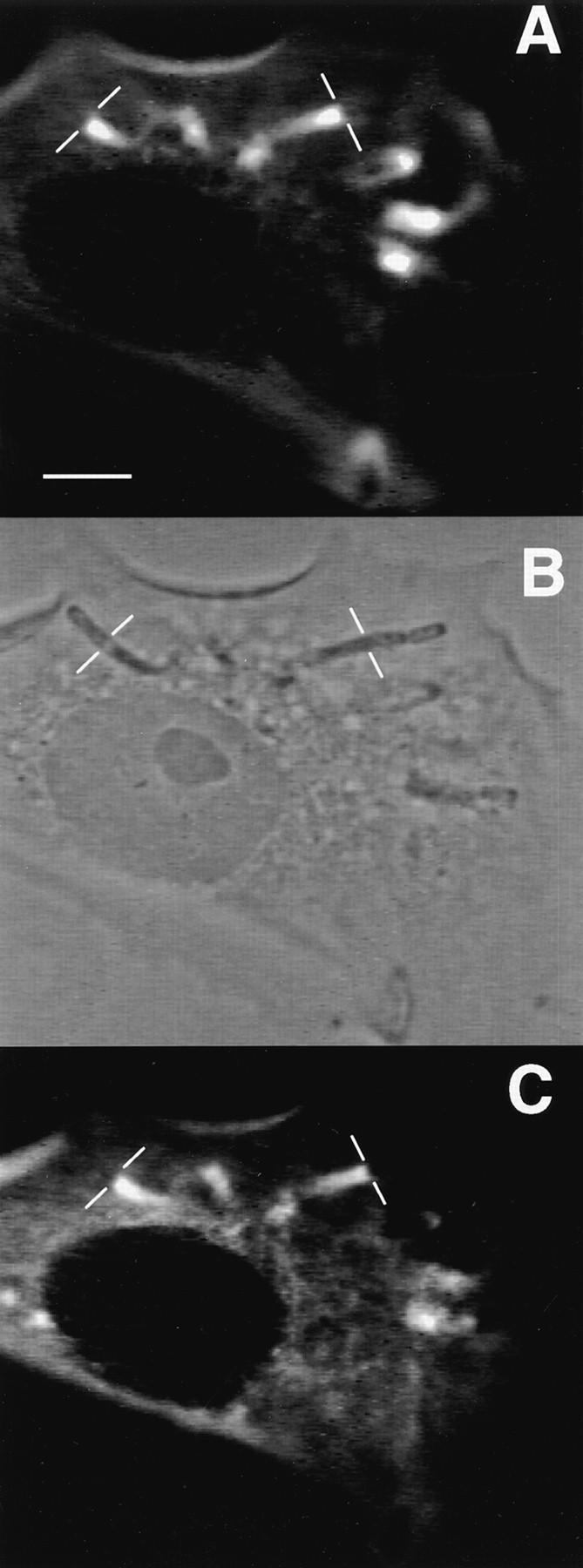
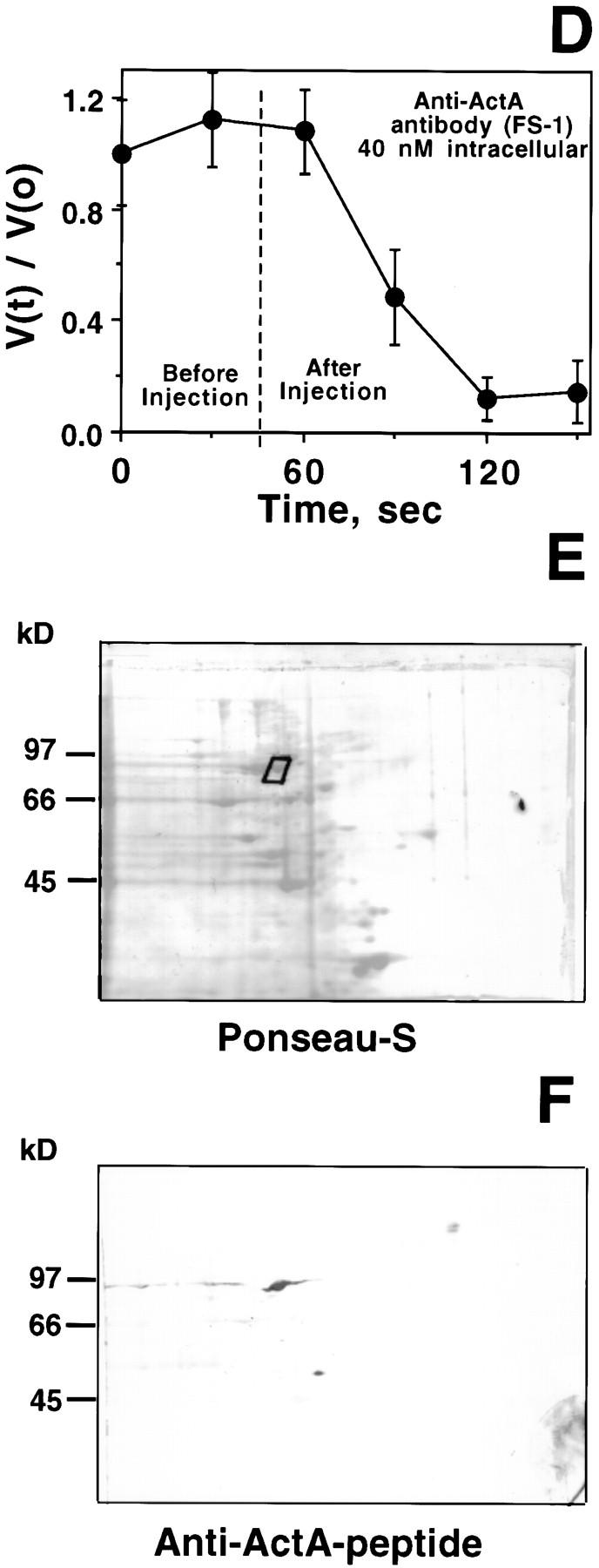
Characteristics of anti–ActA-peptide antibody: immunolocalization, inhibition of motility, and identification of human platelet p90 polypeptide. (A) Fluorescence image of Shigella-infected PtK2 using bodipy-phalloidin to label polymerized actin. The thin white bars demarcate the interface between the bacterium and the trailing actin rocket tail. (B) Phase-contrast image of the same field shown in A. (C) Indirect immunofluorescence image of the same cells using the FS-1 antibody raised against the FEFPPPPTDE sequence in Listeria ActA protein. (D) Speed measurements of Shigella in PtK2 cells before and after microinjection of FS-1 anti– ActA-peptide antibody. The dashed line indicates the time of microinjection of antibody (40 nM calculated intracellular concentration; needle concentration 0.4 μM). Bars represent the SEM of 13 different bacteria at each time point. To compare different bacteria, values were graphed as the ratio V(t)/V(0), where V(t) is the velocity at each time point and V(0) is the initial velocity at t = 0 s. Comparisons of actual pre- and postinjection speeds also demonstrated a highly significant inhibition of Shigella motility after introduction of the FS-1 antibody (mean preinjection speed: 0.11 ± 0.01 μm/s SEM n = 46, vs. postinjection: 0.02 ± 0.01 μm/s n = 48, P < 0.0001). This same concentration of antibody also significantly inhibited Listeria intracellular motility (mean preinjection velocity 0.11 ± 0.01 μm/s n = 14, vs. 0.02 ± 0.01 μm/s n = 18, P < 0.0001). (E) Ponseau S staining of an electroblot of a two-dimensional IEF/SDS electrophoresis gel. The boxed area shows the major spot identified by FS-1 antibody (raised against ActA peptide). (F) Same electroblot stained with the FS-1 antibody. Two major cross-reactive polypeptides are identified: the 90-kD polypeptide selected for microsequencing, and a 53-kD polypeptide. Bar, 10 μm.
We also found that microinjection of the FS-1 antibody (at intracellular concentrations as low as 40 nM) into Listeria-infected PtK2 cells rapidly halted bacterial locomotion, suggesting that the antibody binds to an ActA region that is critical for actin-based Listeria motility. The same intracellular concentration of the anti-ActA antibody also rapidly blocked Shigella motility in PtK2 cells (Fig. 1 D). Microinjection of the same concentration of FS-1 antibody preincubated with twice the molar concentration of the Listeria ActA ABM-1 peptide (FEFPPPPTDE) failed to block bacterial intracellular movement (data not shown), thereby excluding nonspecific inhibition by IgG.
The ActA-binding protein VASP is abundant in platelets (Reinhard et al., 1992), and we expected that one or more ActA homolog(s) might be present in higher abundance in platelets relative to PtK2 cells. Accordingly, we used the FS-1 antibody to identify ActA homolog(s) in platelet membrane extracts that were subjected to IEF and subsequent SDS-PAGE. After electrotransfer of proteins from the two-dimensional gel, an immunoblot revealed the presence of a major cross-reactive 90-kD polypeptide with an approximate isoelectric point of 6.0– 6.3 (Fig. 1, E and F). A second, less-abundant 53-kD protein (with a slightly more acidic isoelectric point) reacted more weakly with the FS-1 antibody.
Identification of the 90-kD ActA Homolog as the Vinculin Head Fragment
The 90-kD polypeptide (designated hereafter as p90) recognized by FS-1 anti-ActA peptide antibody was collected by excising the Ponseau S–stained protein from the two-dimensional electrophoresis blot. Upon our finding that the NH2 terminus was blocked, we treated the p90 species with Lys-C protease. Subsequent gas phase microsequencing yielded five different peptides (281-GXLRDPSAXPGDAG; 315-ERREILGTXK; 417-IAELXDDPK; 607-LLAVAATAPPTDAPNREEVF; 815-SFLDSGYRI LGA-826), and all corresponded exactly to the numbered positions within human vinculin (Weller et al., 1990). That all five matched vinculin also indicated that no other protein was a major constituent of the p90 spot excised from the PVDF membrane. Furthermore, Kilic and Ball (1991) reported that thermolysin digestion of intact human vinculin generated 90-kD head and 30-kD tail fragments; and all of our sequenced peptides are present in the 90-kD head fragment (Fig. 2). Therefore, the FS-1 antibody cross-reacted with p90 head fragment of vinculin. The identity of the 53-kD cross-reactive polypeptide could not be determined by the same technique, because multiple polypeptides migrated within the same region (Fig. 1, E and F). It is possible that this polypeptide represents a further proteolytic degradation product of vinculin p90, and attempts to further purify the 53-kD polypeptide are presently underway.
Figure 2.

Structural organization of human vinculin. Full-length vinculin has a rigid 90-kD head region and a 30-kD tail region defined by a proteolytic cleavage site. As shown, the tail region is folded to indicate the latent binding site for F-actin as proposed by Johnson and Craig (1995). Below is a linearized representation of human vinculin indicating the reported binding regions and the proteolytic cleavage site that generates the p90 head and p30 tail fragments. The head fragment binds monoclonal anti-vinculin 11-5 antibody (Kilic and Ball, 1991), talin (Price et al., 1989), and α-actinin (Wachsstock et al., 1987). The location of the IcsA binding site on vinculin's head fragment has not been determined. The tail fragment contains the sites for binding F-actin (Johnson and Craig, 1995) and paxillin (Turner et al., 1990). Human vinculin also has an ABM-1 sequence (residues 840–849) named Vinc-1 located at the COOH terminus of the p90 fragment generated from vinculin by limited digestion with thermolysin, and included for comparison is the second ABM-1 repeat of Listeria ActA (residues 269–278). Also shown are the residues (including the conservative substitution of D for E) shared by the two sequences. The p30 tail contains two other oligoproline sequences (Vinc-2 and Vinc-3) that fail to fulfill the consensus features of ABM-1 homology sequences.
As an additional test of the FS-1 antibody's specificity for the 90-kD vinculin fragment, Western blots of thermolysin-cleaved and intact vinculin were performed. Johnson and Craig (1994, 1995) reported that vinculin's tail region is folded over and many binding sites for known actin regulatory proteins, as well as internal epitopes, are masked by interactions between the head and tail protein (Fig. 2). Consistent with this model was our finding that the anti– ActA-peptide antibody only weakly cross-reacted with full-length vinculin, yet reacted strongly with the 90-kD head fragment (Fig. 3). The COOH-terminal region of the p90 vinculin domain also contains an ABM-1 oligoproline sequence (PDFPPPPPDL, designated Vinc-1) that bears sequence homology to the ActA peptide used to generate the FS-1 antibody. Two other oligoproline sequences (named Vinc-2 and Vinc-3) are also found in the vinculin p30 tail (Fig. 2). These proline-rich sequences are distinctly different from Vinc-1, because they have intervening basic amino acids that were not recognized by the FS-1 antibody, even on Western blots. Our observations indicate that the full-length vinculin refolds to mask its ABM-1 site, after transfer to the PVDF membrane and exposure to physiologic buffer.
Figure 3.

Anti–ActA-peptide identification of the p90 fragment from digested human vinculin. Full-length human vinculin (lanes 2 and 4) and thermolysin- digested (lanes 1 and 3) were separated by SDS-PAGE and transferred to PVDF membrane. Lanes 1 and 2 are the Coomassie blue stain of the membrane, whereas lanes 3 and 4 are a set of duplicate samples reacted against the FS-1 anti– ActA-peptide antibody. Note the p90 fragment of the digested human vinculin (lane 3) is strongly recognized by the antibody, whereas, the full-length molecule shows very faint recognition (lane 4). Lane M contains molecular weight markers.
Vinculin's Head Domain Localizes to the Surface of Intracellular Shigella
To learn whether the vinculin head fragment serves as a mammalian ActA homolog involved in Shigella motility, bacteria migrating in PtK2 cells were subjected to immunofluorescence microscopy using a monoclonal antibody (Vin 11-5) directed against vinculin's head region (Fig. 2). Motile bacteria were identified by bodipy-phalloidin staining (rhodamine channel), and the vinculin head fragment was localized (Fig. 4) by using Vin 11-5 antibody staining (fluorescein channel). As previously observed with the FS-1 antibody, nearly all motile bacteria (identified by the presence of actin filament rocket tails) likewise exhibited focal staining with this anti-vinculin head region antibody (i.e., 53 of 54 bacteria with actin rocket tails demonstrated focal staining with Vin 11-5 antibody). This observation, combined with our results with the FS-1 antibody, strongly suggests that motile bacteria attract a form of vinculin containing the unmasked Vinc-1 oligoproline sequence. As had been observed with our FS-1 antibody, the monoclonal Vin 11-5 antibody did not cross-react with any Shigella proteins (data not shown). Vin 11-5 antibody specifically cross-reacted with intact vinculin and the p90 head region on Western blot analysis of PtK2 cell extracts (Fig. 6).
Figure 4.
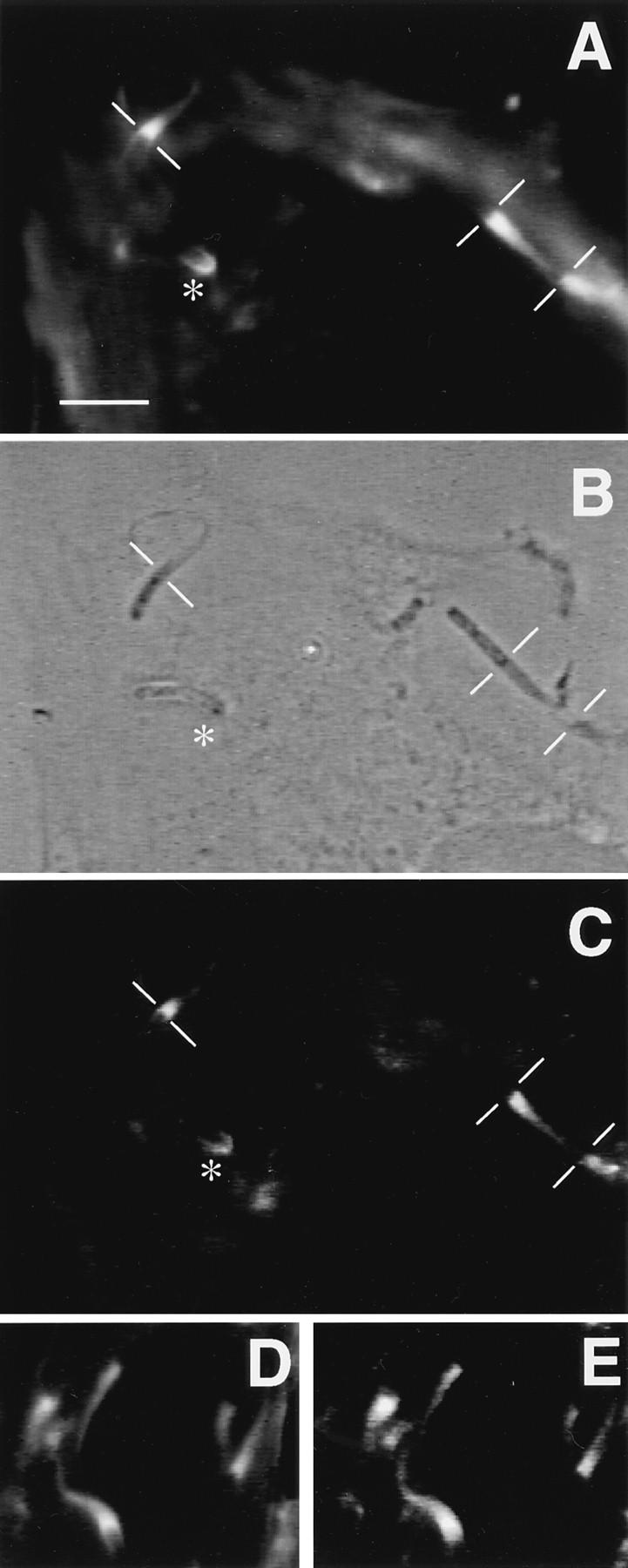
Immunofluorescence microscopy of Shigella-infected PtK2 cells using anti-vinculin antibody. (A) Fluorescence image of Shigella-infected PtK2 cells using bodipy-phalloidin to label polymerized actin. The thin white lines demarcate the junction between the bacterium and the actin rocket tail. The asterisk identifies a bacterium that has a small focal cluster of F-actin. (B) Phase-contrast image of the same field as shown in A. (C) Indirect immunofluorescence micrograph of the same cells using the anti-vinculin 11-5 antibody (monoclonal antibody directed against the head fragment). Note that this anti-vinculin antibody localizes to the F-actin tails and to the focal F-actin cluster at one end of the bacterium identified by the asterisk. (D) An additional fluorescence image of Shigella-infected PtK2 cells using bodipy-phalloidin. (E) Same cell visualized by indirect immunofluorescence using the vin11-5 antibody. Bar, 10 μm.
Figure 6.

Shigella infection induces the production of the 90-kD vinculin head fragment. (A) Western blots of PtK2 extracts from uninfected and infected cells using anti-vinculin clone 11-5. Left lanes, Coomassie blue–stained samples; right lanes, ECL-developed Western blots. A 120-kD cross-reactive polypeptide (full-length vinculin) is evident in both extracts. However the 90-kD head fragment is detected only in the infected cell extract. Cells were infected for 3 h before generation of the extract. (B) Time course of p90 formation after Shigella infection. Note the appearance of the p90 band within 1 h of infection. Far right lane shows extract from PtK2 cells infected for 3 h with a similar number of E. coli.
To assess the potential functional significance of the ABM-1 sequence in vinculin p90 in Shigella motility, we then examined the inhibitory properties of the synthetic microinjected Vinc-1, a synthetic peptide based on the vinculin ABM-1 oligoproline sequence PDFPPPPPDL. As noted above, this sequence is located at the COOH terminus of the p90 head fragment and shows strong homology to the oligoproline sequences of Listeria ActA (Fig. 2). Zeile et al. (1996) observed that microinjection of the ActA ABM-1 peptide FEFPPPPTDE arrested intracellular Shigella motility, and the Vinc-1 peptide likewise inhibited intracellular bacterial movement in PtK2 cells. Complete inhibition was observed at an estimated intracellular concentration of 800 nM Vinc-1 peptide (mean velocity preinjection 0.11 ± 0.05 μm/s, mean and standard deviation, n = 44 versus 0.00 μm/s, n = 44 postinjection) (Fig. 5). Further examination revealed that a 10-fold higher concentration of Vinc-1 peptide was required (Fig. 5, inset) to produce the same level of inhibition observed with the ActA ABM-1 peptide (i.e., half-maximal inhibition was observed at 0.5 μM for Vinc-1 versus 0.05 μM for the ActA ABM-1 peptide). Introduction of poly-l-proline (intracellular concentration, 1 μM) failed to inhibit motility, thereby excluding any nonspecific inhibitory effect (Zeile et al., 1996).
Figure 5.

Effect of Vinc-1 peptide on Shigella speed. Representative experiment showing Shigella flexneri speed measurements before and after introduction of the Vinc-1 peptide. Arrow indicates time of microinjection of 800 nM Vinc-1 peptide, calculated as the intracellular concentration. This experiment is representative of multiple determinations (mean preinjection velocities: 0.11 ± 0.01, SEM, n = 44, vs. postinjection: 0.00 ± 0.01 n = 44, P < 0.0001). Inset demonstrates the concentration dependence of Vinc-1 peptide inhibition. The relative values V(t)/V(0) were determined as described in Fig. 1 D. Bars represent the (SEM) for 24–44 observations.
The Vinculin Head Fragment Is Generated after Shigella Infection
Although present in outdated platelets, the p90 vinculin head fragment was not detected in cell-free extracts of uninfected PtK2 cells. Using the high sensitivity ECL detection method, Western blot analysis indicated that relatively high concentrations of full-length, 120-kD vinculin were present in PtK2 cells extracts (Fig. 6 A), and even prolonged exposure of the autoradiograms failed to demonstrate any vinculin p90. However, vinculin p90 was observed in extracts of PtK2 cells that had been infected with Shigella for 3 h (Fig. 6 A). The time course of p90 generation upon Shigella infection was also examined (Fig. 6 B). The vinculin p90 polypeptide was formed and persisted throughout the period over which Shigella are typically observed to move within the cytoplasm of PtK2 cells (i.e., 1–3 h after the initiation of infection). Densitometry scans of our autoradiograms revealed that the p90 proteolytic fragment represented 5.6–7.7% of the total vinculin in Shigella-infected PtK2 cells. To exclude the possibility that proteolysis was being stimulated by extracellular bacteria, PtK2 cells were exposed to a similar number of E. coli, and then incubated for 3 h. While closely related to S. flexneri, E. coli lacks the 220-kb virulence plasmid that permits Shigella to enter host cells, and therefore remains extracellular. Infection with E. coli failed to generate the p90 fragment in PtK2 cells (Fig. 6 B). Comparisons of Coomassie blue–stained extracts from Shigella-infected and -uninfected bacteria also demonstrated that there were no noticeable changes in the abundance of other proteins in this molecular weight range, either before or after Shigella infection (Fig. 6 A, left 2 lanes). Western blots of uninfected and infected cell extracts, using an anti–α-actinin antibody, likewise revealed no differences in α-actinin proteolysis (data not shown) in uninfected versus infected cells. These observations indicate that the generation of vinculin p90 did not simply arise from generalized proteolysis in infected PtK2 cells. In addition, phase-contrast microscopy revealed no discernible differences in morphology of uninfected and infected PtK2 cells, again indicating that over the time course of the infection, the host cells maintained their normal cytoarchitecture.
Microinjected p90 Head Fragment, but Not Intact Vinculin, Markedly Accelerates Shigella Intracellular Motility
Based on our Western analysis of Shigella-infected PtK2 cells, we hypothesized that cells containing slowly moving bacteria may be suboptimal with respect to the intracellular concentrations of the vinculin p90. To test this prediction, purified platelet p90 (needle concentration = 1.2 μM; estimated intracellular concentration = 0.12 μM) was introduced into PtK2 cells by microinjection. Cells were chosen that contained Shigella moving at slow speeds (i.e., those with rates of 0.02–0.06 μm/s). Within 30 s after microinjection, all moving bacteria began to increase their rates of locomotion (Fig. 7), and within 60 s, they reached velocities that were greater than three times their preinjection rates (Fig. 7 B, V(t)/V(0)). To our knowledge, this is the first observation of stimulated Shigella locomotion by the addition of a fragment of vinculin. In some cases, even previously stationary bacteria began to move at rapid rates (Fig. 7 A, middle curve).
Figure 7.

Effects of the p90 head fragment and intact vinculin on speed of Shigella motility in PtK2 cells. (A) Individual Shigella bacteria migrating in PtK2 cells before and after microinjection of the p90 head fragment or intact vinculin (estimated intracellular concentrations 0.12 μM, needle concentrations 1.2 μM). Dashed line represents the time of microinjection. Two examples of bacteria exposed to the p90 fragment are shown. In one instance the bacterium was stationary before microinjection (lower ○). After introduction of p90, the bacterium began rapidly accelerating to a speed of 0.2 μm/sec. (B) Relative velocity values before and after microinjection of the same concentrations of the p90 and vinculin depicted in A. V(t)/V(0) values were determined as described in the Fig. 1 D. Bars represent the standard error of n = 12 bacteria. All motile bacteria in each microinjected cell were analyzed before and after microinjection.
The difference between the mean preinjection (0.05 ± 0.01 μm/s, SEM, n = 31) and the mean postinjection velocity (0.17 ± 0.01 μm/s, n = 56) was highly significant (P < 0.001). Introduction of identical concentrations of full-length vinculin p120 failed to cause any significant change in bacterial migration speeds (Fig. 7, A and B), and we obtained mean preinjection speeds of 0.04 ± 0.01 μm/s, SEM, n = 24 versus postinjection speeds of 0.04 ± 0.01 μm/s, n = 28. Introduction of a twofold greater concentration of p120 vinculin (needle concentration 2.2 μM, estimated intracellular concentration 0.22 μM) also failed to accelerate Shigella intracellular motility.
Discussion
Because locomoting Shigella and Listeria mimic the activated state of the leading edge of nonmuscle cells, these pathogens are powerful tools for dissecting the molecular machinery of actin-based motility. Transposon mutation studies identified the cell wall protein IcsA as a necessary component for Shigella-induced actin assembly (Bernardini et al., 1989), and expression of only IcsA on the surface of E. coli stimulates actin-based motility in Xenopus oocyte extracts (Goldberg and Theriot, 1995). Therefore, IcsA is required for the assembly of actin filaments to propel Shigella through the cytoplasm. The IcsA protein, however, exhibits no homology to the ActA protein, which was found to be a critical component for Listeria actin-based motility. In particular, IcsA contains none of the ABM-1 VASP-binding sites. Nonetheless, like its counterpart Listeria, Chakraborty et al. (1995) demonstrated that Shigella attracts VASP to its surface, and we have confirmed focal binding of VASP to the trailing pole of migrating bacteria by using anti-hVASP antibody (Laine, R.O., W. Zeile, F. Southwick, and D.L. Purich, unpublished observations). VASP also has a tandem series of three GPPPPP (or ABM-2) sequences for profilin binding, and microinjection of the synthetic GPPPPP peptide blocks Shigella actin-based motility (Zeile et al., 1996). Moreover, we have confirmed that this octadecapeptide GPPPPP-triplet binds to profilin using fluorescence spectroscopy (Kang et al., 1997). These findings indicate that VASP and profilin are key components in the Shigella actin-based motor.
How then is VASP attracted to the surface of Shigella? We and others have proposed that the IcsA protein must bind an ActA-like adapter protein containing one or more ABM-1 sequences to bind VASP. Vinculin is known to bind on the surface of intracellularly motile Shigella (Kadurugamuwa et al., 1991; Suzuki et al., 1996), and IcsA is capable of binding the head fragment of vinculin (Suzuki et al., 1996). However, vinculin's role in generating actin rocket tails needed for Shigella motility in host cells remained to be elucidated. Suzuki et al. (1996) proposed that vinculin might serve to bind F-actin and thereby promote cross-linking of actin filaments. Our experiments strongly support a role for the p90 head fragment as an ActA homolog that attracts VASP. Vinculin p90 fulfills the following essential features of an ActA homolog: (a) an ability to bind to IcsA on the surface of Shigella, (b) possession of an ActA-like ABM-sequence PDFPPPPPDL for VASP binding (Brindle et al., 1996), (c) the capacity to be generated by vinculin proteolysis in Shigella-infected cells, and (d) the ability to accelerate actin-based motility of Shigella when introduced into infected cells. These properties suggest a mechanism for assembly of an actin-based motor (Fig. 8). First, proteolysis generates the 90-kD head, allowing vinculin's newly exposed VASP-binding sites to link VASP to the surface of Shigella. Second, IcsA has recently been shown to have a considerably higher affinity for the p90 head fragment than for intact vinculin (Suzuki et al., 1996) suggesting that vinculin proteolysis may also unmask vinculin's IcsA binding site. Therefore, the p90 head fragment would be expected to be preferentially attracted to Shigella's surface. Third, binding of p90 to the surface of Shigella could then attract VASP to the same polarized location, followed by the concentration of profilin via the 20–24 ABM-2 sites found on the VASP tetramer. Brindle et al. (1996) used GST fusion protein fragments of vinculin (residues 836–940) to demonstrate binding to VASP, and this region of vinculin contains the ABM-1 sequence. The oligoproline sequences present in the tail region failed to bind VASP (Brindle et al., 1996).
Figure 8.
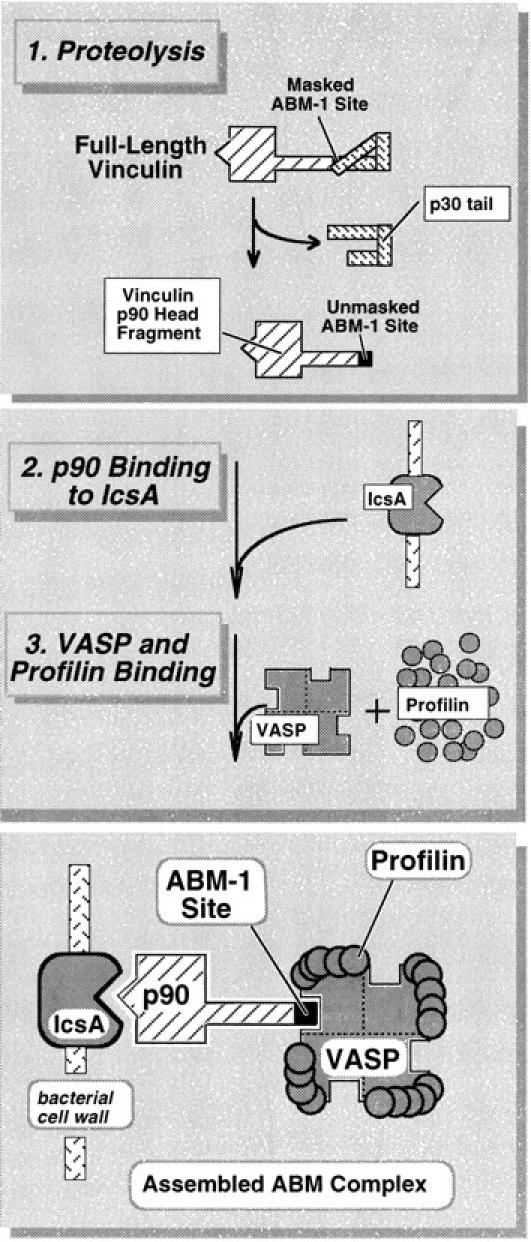
A working model for vinculin proteolysis and the assembly of the Shigella actin-based motility complex.
Very recently, Sechi et al. (1997) convincingly demonstrated that most actin filaments within bacterial rocket tails are organized into long, cross-linked arrays. Their findings disagree with those of Tilney and Portnoy (1989) and Tilney et al. (1992a ,b) who observed much shorter actin filaments in Listeria rocket tails. Based on their findings, Sechi et al. (1997) proposed the existence of a polymerization zone on the bacterial surface that accelerates filament assembly without increasing spontaneous nucleation or the capture of new filaments. Kang et al. (1997) have likewise proposed a cluster model for concentrating VASP and profilin into narrow zone on the trailing pole of motile bacteria. They suggest that profilin tethered within this zone may reach submillimolar concentrations. Recently, Perelroizen et al. (1996) suggested that profilin promotes assembly by increasing the efficiency of actin monomer addition to the barbed ends of growing filaments, and they conclude that this process need not involve profilin-catalyzed exchange of actin-bound nucleotide. Our scheme (Fig. 8) now offers a mechanism for using vinculin p90 as an adapter molecule for concentrating VASP and profilin in a similar polymerization zone on the surface of Shigella. In a sense, the IcsA–p90 complex replaces the function of ActA, but further investigation is warranted to determine how other actin regulatory proteins bind to ActA and the IcsA–p90 complex.
The potent inhibitory action of a synthetic Vinc-1 peptide on Shigella intracellular motility also supports the conclusion that this ABM-1 site is functionally engaged in the motile process. Zeile et al. (1996) obtained apparent inhibitory constants (i.e., the concentration needed to inhibit motility by 50%) of 0.05 and 6 μM for the Listeria ABM-1 peptide (Phe-Glu-Phe-Pro-Pro-Pro-Pro-Thr-Asp-Glu), and the ABM-2 peptide (Gly-Pro-Pro-Pro-Pro Pro), respectively. In the present study, we obtained an approximate inhibitory constant of 0.5 μM for the vinculin ABM-1 peptide (Asp-Phe-Pro-Pro-Pro-Pro-Pro-Asp-Leu) (Fig. 5). This indicates that VASP binds to the Vinc-1 ABM-1 peptide about 10 times more weakly than the corresponding ActA sequence. This may explain why Listeria form rocket tails having a greater F-actin content than Shigella rocket tails. It is very unlikely that our ABM-1 peptides exert their inhibitory actions by binding to Src SH3 domains. The latter interact with two classes of oligoproline ligands, designated as the PLR1 and RLP2 sequences: APPPLPRR, and RALPPLP, respectively (Feng et al., 1994). Positively charged guanidinium groups on PLR1 and RLP2 interact with an essential carboxyl group located on SH3 domains, and this charge neutralization is an essential binding interaction. The negatively charged ABM-1 sequences most likely interact with one or more cationic sidechains on VASP. Given the diametrically opposite charge interactions for binding of SH3 domains to their ligands and VASP to ABM-1 peptides, any interaction between ABM-1 peptides and Src SH3 domains is doubtful.
While our studies indicate that the vinculin p90 is a component of the Shigella actin-based motor, other homologous proteins may also participate. In particular, zyxin possesses several ABM-1 sequences (Reinhard et al., 1995b ; Purich and Southwick, 1997), but zyxin is about 10– 30 times less abundant than vinculin in nonmuscle cells (Beckerle, 1986). Our findings demonstrate that vinculin p90 is clearly present at the trailing pole of motile Shigella, and zyxin may also play an analogous role as an adapter. In view of a recent study suggesting that Shigella motility is unimpaired in the vinculin-deficient cell line 5.51 (Goldberg, 1997), one might propose that zyxin is fulfilling this role. However, Western blot analysis of 5.51 cell extracts using the Vin 11-5 monoclonal antibody (directed against the head region of vinculin) reveal several truncated vinculin head polypeptides. Furthermore, analysis of vinculin messenger mRNA in 5.51 cells detects a message spanning the entire p90 head region of vinculin (Southwick, F., E. Adamson, D. Purich, unpublished findings). In fact the nM p90 concentrations required for Shigella motility (Fig. 7), may fall below the detection limits of Western blot analysis.
Platelet extract experiments have recently identified a complex involving the ARPs as another potential component of the actin motor (Welch et al., 1997). This complex may be attracted by the high concentrations of profilin (Machevsky et al., 1994) concentrated by both VASP and another VASP-like protein, Mena (Gertler et al., 1996), at the back of Listeria. Similarly, Shigella's ability to attract VASP raises the possibility that the ARP complex may be a component of Shigella's actin-based motor; however, preliminary experiments using an anti-ARP3 antibody failed to disclose the presence of ARP3 on the surface of Shigella migrating in the cytoplasm of PtK2 cells (data not shown).
Identification of the p90 vinculin head fragment on the surface of intracellular Shigella also raises questions about the abundance of this proteolytic fragment in host cells. Western blot analysis fails to detect the p90 proteolytic fragment in uninfected PtK2 cells, but this head fragment is known to accumulate in aging platelets (Reid et al., 1993). The latter finding probably accounts for the observation that zyxin is the only VASP-binding protein in fresh platelets (Reinhard et al., 1995b ). Intracellular infection by Shigella appears to be required to generate the vinculin p90 fragment in PtK2 cells, and our studies suggest proteolysis may serve as one molecular switch that unmasks the vinculin ABM-1 site. The coincident appearance of p90 over the same time frame as Shigella motility, as well as the absence of generalized proteolysis, point to vinculin proteolysis as a likely key step in the generation of Shigella's actin-based motor. Furthermore, the profound functional consequences of generating the p90 vinculin fragment are dramatically illustrated by microinjecting this product into infected PtK2 cells. Introduction of the p90 head fragment, but not full-length vinculin, consistently resulted in the acceleration of Shigella intracellular speeds by a factor of three. These experiments suggest that the p90 vinculin head fragment is a rate-limiting component of Shigella's actin-based motor and that supplementing the cell with this product allows Shigella to stimulate the formation of actin filaments. The inability of intact vinculin to enhance intracellular bacterial motility is also consistent with our working model (Fig. 8), which suggests that the unmasking of vinculin's ABM-1 site is critical for generating Shigella's actin-based motor.
Uninfected cells are apt to engage multiple pathways for unmasking VASP binding sites on vinculin and related actin cytoskeletal components. Our FS-1 antibody recognizes vinculin's ABM-1 binding site for VASP, yet fails to stain focal contacts. By contrast, anti-vinculin antibody recognizes different epitopes on the head region and localizes to focal contacts (data not shown). These observations suggest that vinculin's binding sites for VASP in resting cells are likely to be fully masked. In response to host cell regulatory signals, either vinculin proteolysis or some other nonproteolytic route(s) for unmasking the ABM-1 site may serve as molecular switches for activating or remodeling localized actin assembly. For example, Weekes et al. (1996) showed that acidic phospholipids inhibit the intramolecular association between the NH2-terminal head region and the COOH-terminal tail. The conformational flexibility of vinculin and its ability to become incorporated into focal contacts may also be regulated by phosphorylation. The vinculin tail region contains several serine and threonine residues that are appropriately flanked by residues typically recognized by protein kinases A and C and other kinases involved in signal transduction. These structural characteristics make vinculin an ideal candidate for integrating metabolic signals that may lead to the reorganization of the actin cytoskeleton. Our experiments with Shigella extend the concept that changes in vinculin structure may serve as a critical molecular switch for activating the assembly of actin filaments needed to generate the forces for cell motility.
Acknowledgments
We thank T. Stossel for valuable discussions concerning actin-based motility mechanisms. We are also indebted to J. Pohl (Emory University Microsequencing Laboratory, Atlanta, GA) for carrying out the p90 sequencing and P. Shen for her technical assistance.
Abbreviations used in this paper
- ABM-1
actin-based motility–1
- ARP
actin-related protein
- ECL
enhanced chemiluminescence
- PVDF
polyvinylidene difluoride
- VASP
vasodilator-stimulated protein
Footnotes
Address all correspondence to Frederick S. Southwick, Division of Infectious Diseases, Box 100277, University of Florida College of Medicine, Gainesville, FL 32610. Tel.: (352) 392-2928. Fax: (352) 392-6481.
This work was funded by National Institutes of Health grant RO1AI24276 and RO1AI23262.
References
- Beckerle MC. Identification of a new protein localized at sites of cell– substrate adhesion. J Cell Biol. 1986;103:1679–1687. doi: 10.1083/jcb.103.5.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardini ML, Mounier J, d'Hauteville H, Coquis-Rondon M, Sansonetti PJ. Identification of IcsA, a plasmid locus of Shigella flexnerithat governs bacterial intra- and intercellular spread through interaction with F-actin. Proc Natl Acad Sci USA. 1989;86:3867–3871. doi: 10.1073/pnas.86.10.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindle NPJ, Hold MR, Davies JE, Price CJ, Critchley DR. The focal-adhesion vasodilator-stimulated phosphoprotein (VASP) binds to the proline-rich domain in vinculin. Biochem J. 1996;318:753–757. doi: 10.1042/bj3180753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty T, Ebel F, Domann E, Niebuhr K, Gerstel B, Pistor S, Temm-Grove J, Jockusch BM, Reinhard M, Walter U, et al. A focal adhesion factor directly linking intracellularly motile Listeria monocytogenes and Listeria ivanoviito the actin-based cytoskeleton of mammalian cells. EMBO (Eur Mol Biol Organ) J. 1995;14:1314–1321. doi: 10.1002/j.1460-2075.1995.tb07117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cudmore S, Cossart P, Griffiths G, Way M. Actin-based motility of vaccinia virus. Nature (Lond) 1995;378:636–638. doi: 10.1038/378636a0. [DOI] [PubMed] [Google Scholar]
- Dabiri GA, Sanger JM, Portnoy DA, Southwick FS. Listeria monocytogenesmoves rapidly through the host cell cytoplasm by inducing directional actin assembly. Proc Natl Acad Sci USA. 1990;87:6068–6072. doi: 10.1073/pnas.87.16.6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Hauteville H, Lagelouse RD, Nato F, Sansonetti PJ. Lack of Cleavage of IcsA in Shigella flexnericauses aberrant movement and allows demonstration of a cross-reactive eukaryotic protein. Infect Immun. 1996;64:511–517. doi: 10.1128/iai.64.2.511-517.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dold F, Sanger JM, Sanger JW. Intact alpha-actinin molecules are needed for both the assembly of actin into the tails and locomotion of Listeria monocytogenes. . Cell Motil Cytoskeleton. 1994;28:97–107. doi: 10.1002/cm.970280202. [DOI] [PubMed] [Google Scholar]
- Domann E, Wehland J, Rohde M, Pistor S, Hartl M, Goebel W, Leimeister-Wachter M, Wuenscher M, Chakraborty T. A novel bacterial virulence gene in Listeria monocytogenesrequired for host cell microfilament interaction with homology to the proline-rich region of vinculin. EMBO (Eur Mol Biol Organ) J. 1992;11:1981–1990. doi: 10.1002/j.1460-2075.1992.tb05252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov AA, Pollard TD, Almo SC. Purification, characterization and crystallization of human platelet profilin expressed in Escherichia coli. . J Mol Biol. 1994;241:480–482. doi: 10.1006/jmbi.1994.1522. [DOI] [PubMed] [Google Scholar]
- Feng S, Chen JK, Yu H, Simon JA, Schreiber SL. Two binding orientations for peptides to the Src SH3 domain: development of a general model for SH3-ligand interactions. Science (Wash DC) 1994;266:1241–1247. doi: 10.1126/science.7526465. [DOI] [PubMed] [Google Scholar]
- Gertler F B, Niebuhr K, Reinhard M, Wehland J, Soriano P. Mena, a relative of VASP and Drosophilaenabled, is implicated in the control of microfilament dynamics. Cell. 1996;87:227–239. doi: 10.1016/s0092-8674(00)81341-0. [DOI] [PubMed] [Google Scholar]
- Goldberg MB. Shigellaactin-based motility in the absence of vinculin. Cell Motil Cytoskeleton. 1997;37:44–53. doi: 10.1002/(SICI)1097-0169(1997)37:1<44::AID-CM5>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Goldberg MB, Theriot JA. Shigella flexnerisurface protein IcsA is sufficient to direct actin-based motility. Proc Natl Acad Sci USA. 1995;92:6572–6576. doi: 10.1073/pnas.92.14.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MB, Barzu O, Parsot C, Sansonetti PJ. Unipolar localization and ATPase activity of IcsA, a Shigella flexneriprotein involved in intracellular movement. J Bacteriol. 1993;175:2189–2196. doi: 10.1128/jb.175.8.2189-2196.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzen RA, Hayes SF, Peacock MG, Hackstadt T. Directional actin polymerization associated with spotted fever group rickettsia infection of vero cells. Infect Immun. 1993;61:1926–1935. doi: 10.1128/iai.61.5.1926-1935.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RP, Craig SW. An intramolecular association between the head and tail regions of vinculin. J Biol Chem. 1994;269:12611–12619. [PubMed] [Google Scholar]
- Johnson RP, Craig SW. F-actin binding site masked by intramolecular association of vinculin head and tail regions. Nature (Lond) 1995;373:261–264. doi: 10.1038/373261a0. [DOI] [PubMed] [Google Scholar]
- Kadurugamuwa JL, Rohde M, Wehland J, Timmis KN. Intercellular spread of Shigella flexnerithrough a monolayer mediated by membranous protrusions and associated with reorganization of the cytoskeletal protein vinculin. Infect Immun. 1991;59:3463–3471. doi: 10.1128/iai.59.10.3463-3471.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang F, Laine RO, Bubb MR, Southwick FS, Purich DL. Profilin interacts with the Gly-Pro-Pro-Pro-Pro-Pro sequences of vasodilator-stimulated phosphoprotein (VASP): implications for actin-based Listeriamotility. Biochemistry. 1997;36:8384–8392. doi: 10.1021/bi970065n. [DOI] [PubMed] [Google Scholar]
- Kilic G, Ball EH. Partial cleavage mapping of the cytoskeletal protein vinculin. J Biol Chem. 1991;266:8734–8740. [PubMed] [Google Scholar]
- Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, Cossart P. L. monocytogenes-induced actin assembly requires actAgene product, a surface protein. Cell. 1992;68:521–531. doi: 10.1016/0092-8674(92)90188-i. [DOI] [PubMed] [Google Scholar]
- Laine RO, Esser AF. Detection of refolding conformers of complement protein C9 during insertion into membranes. Nature (Lond) 1989;341:63–65. doi: 10.1038/341063a0. [DOI] [PubMed] [Google Scholar]
- Laine RO, Morgan BP, Esser AF. Comparison between complement and melittin hemolysis: anti-melittin antibodies inhibit complement lysis. Biochemistry. 1988;27:5308–5314. doi: 10.1021/bi00414a054. [DOI] [PubMed] [Google Scholar]
- Machevsky LM, Atkinson SJ, Ampe C, Vandekerckhove J, Pollard TD. Purification of a cortical complex containing two unconventional actins from Acanthamoebaby affinity chromatography on profilin-agarose. J Cell Biol. 1994;127:107–115. doi: 10.1083/jcb.127.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelroizen I, Didry D, Christensen H, Chua NH, Carlier MF. Role of nucleotide exchange and hydrolysis in the function of profilin in action assembly. J Biol Chem. 1996;271:12302–12309. doi: 10.1074/jbc.271.21.12302. [DOI] [PubMed] [Google Scholar]
- Peskin CS, Odell GM, Oster GF. Cellular motions and thermal fluctuations: the Brownian ratchet. Biophys J. 1993;65:316–324. doi: 10.1016/S0006-3495(93)81035-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistor S, Chakraborty T, Walter U, Wehland J. The bacterial actin nucleator protein ActA of Listeria monocytogenescontains multiple binding sites for host microfilament proteins. Curr Biol. 1995;5:517–525. doi: 10.1016/s0960-9822(95)00104-7. [DOI] [PubMed] [Google Scholar]
- Price D, Jones P, Davison MD, Patel B, Bendori R, Geiger B, Critchley DR. Primary sequence and domain structure of chicken vinculin. Biochem J. 1989;259:453–461. doi: 10.1042/bj2590453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purich DL, Southwick FS. ABM-1 and ABM-2 homology sequences: oligoproline regions in ListeriaActA surface protein and human VASP define consensus docking sites for actin-based motility. Biochem Biophys Res Commun. 1997;231:686–691. doi: 10.1006/bbrc.1997.6158. [DOI] [PubMed] [Google Scholar]
- Reid DM, Jones CE, Luo C, Shulman NR. Immunoglobulins from normal sera bind platelet vinculin and talin and their proteolytic fragments. Blood. 1993;81:745–751. [PubMed] [Google Scholar]
- Reinhard M, Halbrugge M, Scheer U, Wiegand C, Jockusch BM, Walter U. The 46/50-kD phosphoprotein VASP purified from human platelets is a novel protein associated with actin filaments and focal contacts. EMBO (Eur Mol Biol Organ) J. 1992;11:2063–2070. doi: 10.1002/j.1460-2075.1992.tb05264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard M, Giehl K, Abel K, Haffner C, Jarchau T, Hoppe V, Jockusch BM, Walter U. The proline-rich focal adhesion and microfilament protein VASP is a ligand for profilins. EMBO (Eur Mol Biol Organ) J. 1995a;14:1583–1589. doi: 10.1002/j.1460-2075.1995.tb07146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard M, Jouvenal K, Tripier D, Walter U. Identification, purification, and characterization of a zyxin-related protein that binds the focal adhesion and microfilament protein VASP (vasodilator-stimulated phosphoprotein) Proc Natl Acad Sci USA. 1995b;92:7956–7960. doi: 10.1073/pnas.92.17.7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld GC, Hou DC, Dingus J, Meza I, Bryan J. Isolation and partial characterization of human platelet vinculin. J Cell Biol. 1985;100:669–676. doi: 10.1083/jcb.100.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger JW, Sanger JM, Jockusch BM. Differences in the stress fibers between fibroblasts and epithelial cells. J Cell Biol. 1983;96:961–969. doi: 10.1083/jcb.96.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger JM, Sanger JW, Southwick FS. Host cell actin assembly is necessary and likely to provide the propulsive force for intracellular movement of Listeria monocytogenes. . Infect Immun. 1992;60:3609–3619. doi: 10.1128/iai.60.9.3609-3619.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechi AS, Wehland J, Small JV. The isolated comet tail pseudopodium of Listeria monocytogenes:a tail of two actin filament populations, long and axial and short and random. J Cell Biol. 1997;137:155–167. doi: 10.1083/jcb.137.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwick FS, Purich DL. Arrest of Listeriamovement in host cells by a bacterial actA analogue: implications for actin-based motility. Proc Natl Acad Sci USA. 1994;91:5168–5172. doi: 10.1073/pnas.91.11.5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwick FS, Purich DL. Inhibition of Listerialocomotion by mosquito oostatic factor, a natural oligoproline peptide uncoupler of profilin action. Infect Immun. 1995;63:182–190. doi: 10.1128/iai.63.1.182-190.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Lett M-C, Sasakawa C. Extracellular transport of VirG protein in Shigella. . J Biol Chem. 1995;270:3874–3880. doi: 10.1074/jbc.270.52.30874. [DOI] [PubMed] [Google Scholar]
- Suzuki TA, Saga S, Sasakawa C. Functional analysis of ShigellaVirG domains essential for interaction with vinculin and actin-based motility. J Biol Chem. 1996;271:21878–21885. doi: 10.1074/jbc.271.36.21878. [DOI] [PubMed] [Google Scholar]
- Theriot JA, Rosenblatt J, Portnoy DA, Goldschmidt-Clermont RJ, Mitchison TJ. Involvement of profilin in the actin-based motility of L. monocytogenesin cells and in cell-free extracts. Cell. 1994;76:505–515. doi: 10.1016/0092-8674(94)90114-7. [DOI] [PubMed] [Google Scholar]
- Tilney LB, Portnoy DA. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocyto-genes. . J Cell Biol. 1989;109:1597–1608. doi: 10.1083/jcb.109.4.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney LG, DeRosier DJ, Tilney MS. How Listeriaexploits host cell actin to form its own cytoskeleton. I. Formation of a tail and how that tail might be involved in movement. J Cell Biol. 1992a;118:71–82. doi: 10.1083/jcb.118.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney LG, DeRosier DJ, Weber A, Tilney MS. How Listeriaexploits host cell actin to form its own cytoskeleton. II. Nucleation, actin filament polarity, filament assembly, and evidence for a pointed end capper. J Cell Biol. 1992b;118:83–93. doi: 10.1083/jcb.118.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CE, Glenney JR, Jr, Burridge K. Paxillin: a new vinculin-binding protein present in focal adhesions. J Cell Biol. 1990;111:1059–1068. doi: 10.1083/jcb.111.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachsstock DH, Wilkins JA, Lin S. Specific interaction of vinculin with alpha-actinin. Biochem Biophys Res Commun. 1987;146:554–560. doi: 10.1016/0006-291x(87)90564-x. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Bauer S, Gobl C, Hofmann F, Jakobs KH, Walter U. Demonstration of cGMP-dependent protein kinase and cGMP-dependent phosphorylation in cell-free extracts of platelets. Eur J Biochem. 1986;138:203–210. doi: 10.1111/j.1432-1033.1986.tb09739.x. [DOI] [PubMed] [Google Scholar]
- Weekes J, Barry ST, Critchley DR. Acidic phospholipids inhibit the intramolecular association between the N- and C-terminal regions of vinculin, exposing actin-binding and protein kinase C phosphorylation sites. Biochem J. 1996;314:827–832. doi: 10.1042/bj3140827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch MD, Iwamatsu A, Mitchison TJ. Actin polymerization is induced by ARP 2/3 protein complex at the surface of Listeria monocytogenes. . Nature (Lond) 1997;385:265–268. doi: 10.1038/385265a0. [DOI] [PubMed] [Google Scholar]
- Weller PA, Ogryzko EP, Corben EB, Zhidkova NI, Patel B, Price GJ, Spurr NK, Koteliansky VE, Critchley DR. Complete sequence of human vinculin and assignment of the gene to chromosome 10. Proc Natl Acad Sci USA. 1990;87:5667–5671. doi: 10.1073/pnas.87.15.5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeile W, Purich DL, Southwick FS. Recognition of two classes of oligoproline sequences in profilin-mediated acceleration of actin-based Shigellamotility. J Cell Biol. 1996;133:49–59. doi: 10.1083/jcb.133.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]