

Abstract
Acentric, autonomously replicating extrachromosomal structures called double-minute chromosomes (DMs) frequently mediate oncogene amplification in human tumors. We show that DMs can be removed from the nucleus by a novel micronucleation mechanism that is initiated by budding of the nuclear membrane during S phase. DMs containing c-myc oncogenes in a colon cancer cell line localized to and replicated at the nuclear periphery. Replication inhibitors increased micronucleation; cell synchronization and bromodeoxyuridine–pulse labeling demonstrated de novo formation of buds and micronuclei during S phase. The frequencies of S-phase nuclear budding and micronucleation were increased dramatically in normal human cells by inactivating p53, suggesting that an S-phase function of p53 minimizes the probability of producing the broken chromosome fragments that induce budding and micronucleation. These data have implications for understanding the behavior of acentric DNA in interphase nuclei and for developing chemotherapeutic strategies based on this new mechanism for DM elimination.
The accumulation of structural and numerical chromosome abnormalities in eukaryotic cells is limited by coordinating biosynthetic and repair processes with cell cycle checkpoints (Hartwell and Kastan, 1994). Mutations in genes involved in these transactions occur commonly during cancer progression and can greatly elevate the frequencies of base alterations or large-scale chromosome rearrangements. For example, defects in cell cycle–control pathways involving the p53 tumor suppressor gene create a permissive environment in which cells with aneuploidy, chromosome translocations, and gene amplification arise at high frequency in response to stresses created by antimetabolites or oncogene overexpression (Livingstone et al., 1992; Yin et al., 1992; Denko et al., 1994).
The types of aberrant chromosomal structures generated in cells with defective repair and cell cycle control functions are likely to be constrained by nuclear structure. For example, chromosomes with very long arms tend to generate nuclear projections variously referred to as “blebs” or “buds” (Ruddle, 1962; Lo and Fraccaro, 1974; Toledo et al., 1992; Pedeutour et al., 1994). A recent study in peas demonstrated that excessive DNA within a single chromosome arm generated a nuclear projection that was cut when the cell division plate formed after telophase (Schubert and Oud, 1997). Sequences enclosed in such projections are often detected in micronuclei, suggesting that projections can be precursors of micronuclei (Toledo et al., 1992; Pedeutour et al., 1994), and that the chromosomal sequences they contain can be lost from the nucleus. These data indicate that a maximum allowable size exists for each chromosome arm within the nuclei of specific cell types.
Circular, autonomously replicating DNA fragments such as double-minute chromosomes (DMs)1 are also frequently generated in cancer cells (Barker, 1982; Cowell, 1982; Benner et al., 1991). These structures encode proteins that provide survival advantages in vivo, or resistance to a variety of chemotherapeutic agents in vitro (Alitalo and Shwab, 1986; Wahl, 1989; Von Hoff et al., 1992; Brison, 1993; Shimizu et al., 1994; Eckhardt et al., 1994). DMs replicate using cellular replication origins (Carroll et al., 1993), but lacking centromeres, they do not segregate by the same mechanisms used by chromosomes. Consequently, DMs are lost spontaneously in the absence of selection. Drugs such as hydroxyurea (HU) significantly increase the loss rate of DMs in human and rodent cell lines (Snapka and Varshavsky, 1983; Von Hoff et al., 1991; Von Hoff et al., 1992; Eckhardt et al., 1994; Canute et al., 1996). DM elimination results in increased drug sensitivity, reduced tumorigenicity, or differentiation, depending on the proteins expressed by DM-encoded genes (Snapka and Varshavsky, 1983; Snapka, 1992; Von Hoff et al., 1992; Eckhardt et al., 1994; Shimizu et al., 1994). Identifying the mechanisms by which DMs are eliminated could enable the development of new and more selective chemotherapeutic strategies, since DMs are uniquely found in cancer cells, and chromosome loss should not be induced by such treatments.
Like abnormally long chromosome arms, DMs have also been reported to be preferentially incorporated within micronuclei that are removed from the cell (Von Hoff et al., 1992; Shimizu et al., 1996). It is clear that small size alone does not guarantee selective enclosure of DNA fragments within micronuclei because a centric minichromosome the size of a typical DM is effectively excluded from micronuclei (Shimizu et al., 1996). This observation is consistent with the classical mechanism of micronucleus formation that involves the enclosure of lagging acentric chromosome fragments as nuclear membranes reform at the end of mitosis (Heddle and Carrano, 1977; Heddle et al., 1983). Thus, one would expect postmitotic enclosure of DMs within micronuclei since they typically lack functional centromeres (Levan et al., 1976). However, DMs appear to associate with chromosomes or nucleoli, which may enable most of them to evade such a postmitotic mechanism. The ability of DMs to “hitchhike” by association with mitotic chromosomes or nucleoli provides one explanation of why few micronuclei were detected at the midbody in a cell line containing numerous DMs (Levan and Levan, 1978), and their surprisingly efficient partitioning to daughter cells in some cell lines (Levan and Levan, 1978; Hamkalo et al., 1985). However, the interphase behavior of normal chromosomes and DMs may differ because DMs lack the centromeres and/or telomeres that position chromosomes in restricted territories and produce a choreographed set of chromosome movements during S phase (DeBoni and Mintz, 1986; Cremer et al., 1993). It has neither been determined whether acentric DM–DNA occupies positions different from chromosomes in interphase, nor whether this could enable their removal from the nucleus by a budding process like that observed for abnormally long chromosomes (Ruddle, 1962; Jackson and Clement, 1974; Lo and Fraccaro, 1974; Miele et al., 1989; Toledo et al., 1992).
DMs provide an excellent model for analyzing the nuclear behavior of multimegabase replicons lacking a centromere and telomeres. We show that DMs can preferentially localize the nuclear periphery, whereas chromosomally amplified sequences occupy a more central position. Although the micronuclei that entrap acentric chromosome fragments have typically been viewed to be generated postmitotically, we provide evidence for a novel micronucleation mechanism that involves the formation of nuclear projections which we refer to as buds. Buds form during S phase and appear to selectively associate with DMs replicating near or at the nuclear periphery. Since micronuclei are indicators of DNA damage and are produced at much higher frequencies in tumor cells than in normal cells, we investigated whether micronucleation frequency is increased in cells with defects in cellular responses to DNA damage. We show that loss of p53 function increases the frequency of micronucleation and enables buds and micronuclei to be produced under conditions expected to lead to chromosome breakage.
Materials and Methods
Cell Culture
Human COLO 320DM (CCL 220) and COLO 320HSR (CCL 220.1) neuroendocrine tumor cells were obtained from American Type Culture Collection (Rockville, MD) and then single cell subclones were obtained by limiting dilution (Von Hoff et al., 1988). The locations of amplified c-myc genes to DMs or HSRs were confirmed by fluorescence in situ hybridization (FISH) using c-myc cosmid DNA. The cells were grown in RPMI 1640 medium supplemented with 10% FBS. The WS1 human embryonic skin fibroblast, obtained from American Type Culture Collection (CRL 1502), was cultured in DME supplemented with 10% heat-inactivated, dialyzed FBS, and 1× MEM nonessential amino acids. WS1-neo and WS1-E6 were kind gifts of S. Linke (National Institutes of Health, Bethesda, MD), and were generated by infecting WS1 with retroviral vectors expressing genes encoding either neomycin resistance or both neomycin resistance and the E6 protein from human papilloma virus 16, respectively (Linke et al., 1996). RPE-h (normal human retinal pigmented epithelial cells) and its neo and E6 derivatives were also kindly provided by S. Linke and the parental cells were obtained from Cell Genesys, Inc. (Foster City, CA). Epithelial cells were cultured in the same way as WS1.
Chemicals
Aphidicolin, 5-bromo-2′-deoxyuridine (BrdU), coumarin (1, 2-benzopyrone), deferoxamine mesylate (desferrioxamine mesylate), DMSO, hydroxyurea, nicotinamide, thymidine, and nocodazole (methyl-[5-{2-thienylcarbonyl}-1H-benzimidazol-2-YL]carbamate) were obtained from Sigma Chemical Co. (St. Louis, MO). Guanazole (3,5-diamino-1,2,4-triazole) was from Aldrich Chemical Co. (Milwaukee, WI). PALA (N-phosphonacetyl-l-aspartate) was provided by the Drug Biosynthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment, National Cancer Institute (Bethesda, MD).
Cell Cycle Analysis
Cell cycle distribution was analyzed using flow cytometry as described previously (Yin et al., 1992; Di Leonardo et al., 1994). Cells treated with the indicated concentrations of drugs for the indicated times were labeled with 10 μM BrdU for 30 min. The cells were collected, fixed with 70% ethanol, treated with 0.1 N HCl containing 0.5% Triton X-100 (Mallinckrodt, Paris, KY), and then followed by boiling for 10 min and rapid cooling to denature the DNA. The nuclei were then incubated with FITC-conjugated anti-BrdU antibodies (Boehringer Mannheim Biochemicals, Indianapolis, IN) and counterstained with 2 μg/ml of propidium iodide (PI) containing RNase (200 μg/ml). Samples were analyzed using a Becton Dickinson FACScanTM (Sparks, MD). 10,000 events were collected for each sample. Data were analyzed using Sun Display as described previously (Yin et al., 1992; Di Leonardo et al., 1994).
Quantification of Micronuclei
Micronuclei containing DM sequences in COLO 320DM cells (see Figs. 2 and 3) were detected by preparing chromosome spreads using standard hypotonic swelling conditions (Lawce and Brown, 1991), followed by hybridization with a biotinylated c-myc cosmid probe as described previously (Shimizu et al., 1996). Total micronuclei (see Fig. 3) were determined by staining chromosome spreads with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma Chemical Co.; 1 μg/ml in VectaShield, Vector Labs, Inc., Burlingame, CA). The adherent cells (WS1, RPE-h, and their derivatives) were grown on coverslips, fixed with cold acetone (−20°C for 5 min) followed by cold methanol (−20°C for 5 min), rehydrated with PBS, and then stained with DAPI (1 μg/ml in VectaShield). The numbers of total or DM-enriched micronuclei were scored using 60 or 100× objectives and a fluorescence microscope equipped with appropriate epifluorescence filters (model Zeiss WL; Carl Zeiss, Inc., Thornwood, NY). The results are expressed as Frequency of Micronuclei (%) relative to the number of interphase nuclei scored (⩾1,000 for each point).
Figure 2.



Formation of micronuclei and nuclear buds in a synchronized culture of COLO 320DM cells. COLO 320DM cells were synchronized at the G1/S boundary using a two-step procedure as described in Materials and Methods. The culture was divided into two portions and then released in the absence of any drug (A, C, and E), or the presence of 0.4 μg/ml of nocodazole (B, D, and F). [3H]thymidine incorporation (closed circles) and the fraction of mitotic cells (open circles) were determined to monitor the synchronous progression through S and M phases, respectively (A and B). The numbers of total micronuclei (closed circles) and total nuclear buds (open circles) were determined in slides stained with DAPI (C and D). The numbers of DM+ micronuclei (closed circles) or DM+ nuclear buds (open circles) were determined in the slides hybridized with the purified micronuclei probe (E and F). These values are expressed as the frequency relative to the number of interphase nuclei scored (more than 1,000 for each point). The degree of synchronization was further evaluated by pulse labeling the cells for 30 min with BrdU. At the times indicated, the cells were harvested and the incorporated BrdU was detected by anti-BrdU antibody and rhodamin-conjugated secondary antibody, and then examined by confocal microscopy. The distribution of BrdU labeling inside each nucleus was classified into 6 patterns (G); i.e., Pattern 0, no labeling; 1, only at the internal euchromatin; 2, spread of the labeling to periphery; 3, onset of labeling of peripheral heterochromatin; 4, almost exclusive labeling of peripheral heterochromatin; 5, labeling of peripheral and internal heterochromatin; and 6, exclusive labeling of internal heterochromatin. At each time point after release from aphidicolin block, the frequencies of each pattern scored from 100 nuclei were plotted (H).
Figure 3.
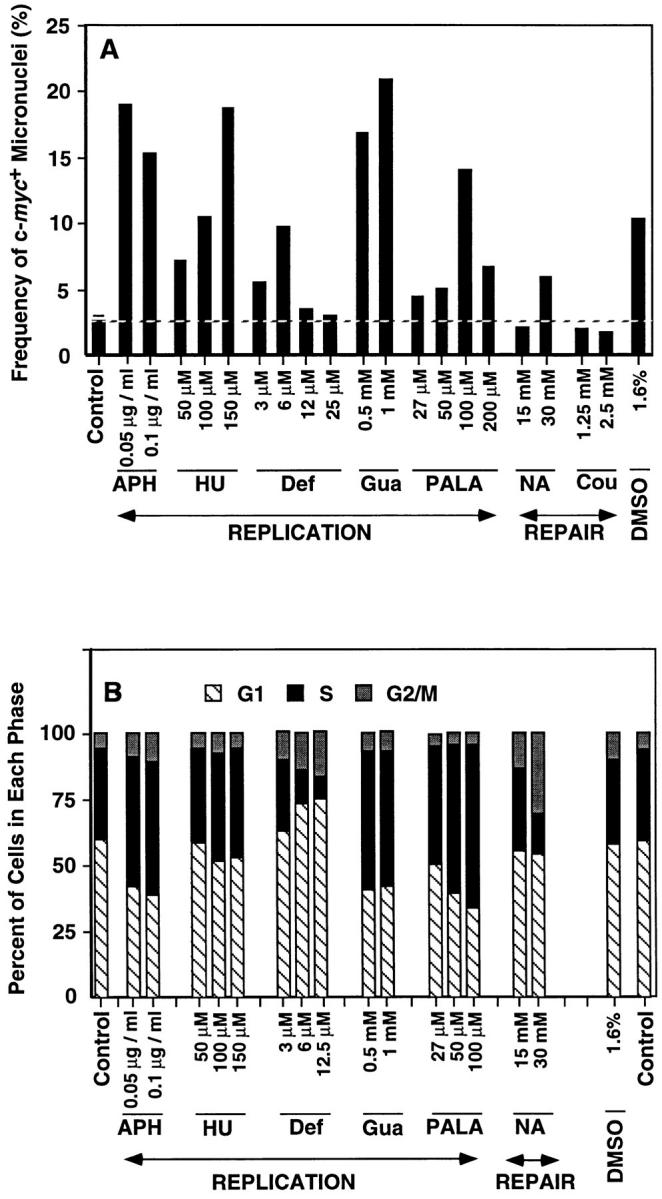
Effects of various drugs on induction of micronuclei and cell cycle distribution. COLO 320DM cells were treated for 3 d with inhibitors of DNA replication (APH, aphidicolin; Def, deferoxamine; Gua, guanazole; HU, hydroxyurea; and PALA), repair (Cou, coumarin; NA, nicotinamide) or the membrane-active agent DMSO at the concentrations indicated. (A) The treated cells were fixed with methanol/acetic acid and then hybridized with c-myc cosmid probe. The numbers of micronuclei that were stained brightly with c-myc probe were scored and expressed as frequency of c-myc + micronuclei (%) relative to the number of interphase nuclei scored (more than 1,000 for each point). (B) The cells were pulse labeled with BrdU (for 30 min) at the end of the drug treatment and then analyzed by flow cytometry as described under Materials and Methods to determine the fraction of cells in G1, S, and G2/M.
Cell Synchronization
Synchronization was performed as described previously (Stein et al., 1994). Rapidly growing COLO 320DM cells were first arrested in early S phase using excess thymidine (2 mM) for 17 h. The cells were then washed with growth medium, released into growth medium containing 25 μM 2′-deoxycytidine for 12 h (to reverse thymidine toxicity), and then incubated in 2.5 μg/ml aphidicolin for 17 h to arrest cells as they entered S phase. The arrested cells were washed with growth medium and then either released into medium lacking drug, or into medium containing nocodazole (0.4 μg/ml). Cell cycle progression was monitored using incorporation of [3H]thymidine (Stein et al., 1994). To monitor the progression through mitosis, a portion (1 ml) of culture was fixed by paraformaldehyde (PFA; 2%) and then stained with DAPI. The frequency of cells in mitosis was scored using fluorescence microscopy. WS1-E6 cells were synchronized by seeding them at low density in 15-cm dishes with or without coverslips (18 × 18 mm). The day after subculture, the medium was removed and then replaced with medium containing 0.1% FCS, and then followed by the culture for an additional 48 h. Cells arrested at G0 by serum deprivation were released into growth medium containing 5 μg/ml aphidicolin for 15 h to arrest them at the beginning of S phase. Cells were released into S phase by replacing the medium with fresh growth medium lacking drug. Progression into S phase was monitored by the incorporation of [3H]thymidine (Stein et al., 1994). The number of cells that were in S phase just before release from synchrony was monitored by BrdU–pulse labeling (30 min) followed by confocal examination of the labeling pattern as described in the following section (see Simultaneous Determination of DM Location and DNA Replication Using FISH and BrdU Incorporation). Concurrently, coverslips were removed, fixed with acetone and methanol, stained by DAPI, and then the frequency of micronuclei, nuclear budding, and mitotic cells were scored as described above.
Terminal Deoxynucleotidyl Transferase-mediated dUTP-Biotin Nick-end Labeling Assay
Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick-end labeling (TUNEL) assay was done according to a previously published procedure (Gavrieli et al., 1992) modified as described below. In brief, COLO 320DM cells were fixed in 2% PFA (10 min at room temperature) and then centrifuged onto a glass slide using a cytospin (Salk Institute Shop) apparatus. The cells were further fixed in cold methanol (−20°C, 5 min) followed by cold acetone (−20°C, 5 min). The slides were rehydrated in PBS and then equilibrated in reaction buffer (200 mM sodium cacodylate, 1 mM MgCl2, 1 mM β-mercaptoethanol, pH 7.2) for 15 min at room temperature. The end-labeling reaction was done by incubating the slides with the reaction buffer containing 10 μM biotin-dUTP (Boehringer Mannheim GmbH, Mannheim, Germany), and 0.3 U/μl of terminal deoxynucleotidyl transferase (Toyobo Co., Osaka, Japan), for 60 min at 37°C. The slides were washed extensively, blocked with 20% FCS, and then the incorporated biotin was detected using FITC–conjugated streptavidin as in the protocol for FISH (see below). The slides were treated with RNase A (100 μg/ml, 37°C, for 20 min), counterstained with PI, and then observed under the conditions used for FISH.
Probe Preparation and FISH
Preparation of probe from purified micronuclei was as described (Shimizu et al., 1996), except that DNA in the purified micronuclei was directly used for biotin labeling by using the BioPrime DNA Labeling System (Life Technologies Inc., Gaithersburg, MD). C-myc cosmid DNA probe was prepared as described (Shimizu et al., 1996). FISH using standard methanol/acetic acid-fixed nuclei was performed as described previously (Shimizu et al., 1996). Assessments of DM localization by confocal microscopy required the following procedure to preserve the spherical shape of the nuclei. This protocol, based on that developed for human lymphocytes (Ferguson and Ward, 1992; Vourc'h et al., 1993), could not be applied directly to COLO 320DM due to severe nuclear aggregation. The modified procedure involves pelleting 10 ml of COLO 320DM cells by centrifugation at 260 g for 5 min, followed by complete removal of the supernatant. The cells were gently suspended in 50 μl of growth medium and then 10 ml of prewarmed (37°C) 75 mM KCl, 2 mM CaCl2 was added slowly. The suspension was centrifuged immediately as described above and the supernatant was removed completely. The cell pellet was loosened gently, suspended in 1 ml of 75 mM KCl and 2 mM CaCl2 at 4°C, and then followed by addition of 1 ml of 75 mM KCl, 2 mM CaCl2, 0.5% Triton X-100 at 4°C. The suspension was kept on ice for 10 min, then Dounce homogenized (loose fitting pestle, 5 times, at 4°C; Fisher, Pittsburgh, PA). 1.5 vol of 5% PFA in PBS was added to the suspension and then incubated for 10 min at room temperature with occasional gentle shaking. After incubation, a 1:10 vol of 1 M Tris-HCl, pH 7.4, containing 1% BSA was added and further incubated for 10 min at room temperature with gentle shaking. The fixed nuclei were washed twice with PBS containing 1% FCS and then stored at 4°C up to 1 wk. Before FISH hybridization, the fixed nuclei were sedimented by cytospin onto poly l-lysine–coated glass slides (Matsunami Glass Ind., Ltd., Osaka, Japan). Slides were treated with RNase A (Sigma Chemical Co., 100 μg/ml in 2× SSC, 37°C for 60 min), washed once with 2× SSC for 3 min, and then followed by blocking with 3% BSA in PBS for 30 min at 37°C. Slides were incubated in 50% formamide dissolved in 2× SSC for 30 min at room temperature to enable buffer equilibration, followed by addition of the hybridization mixture containing labeled probe (prepared as for standard FISH [Shimizu et al., 1996]). The sample was covered by a coverslip, sealed completely with rubber cement, denatured at 85°C, and then hybridized using overnight incubation at 37°C. Washing and the detection of the hybridized probe were performed as described previously (Shimizu et al., 1996). In some cases, intact cells were fixed directly and then hybridized. For this purpose, the cells were cytocentrifuged onto poly l-lysine–coated slides, fixed with ethanol/acetic acid (19:1) for 3 min at −20°C, rehydrated with PBS, and then treated with 4% formaldehyde in PBS for 10 min at 4°C. The slides were then washed extensively with PBS and hybridized as described above for isolated nuclei. Images were obtained using a Bio-Rad MRC600 confocal system (Hercules, CA) on a Zeiss Axiovert 135 microscope (see Fig. 6). Most images were obtained using a 63× objective (Apochromat, 1.40, oil, Carl Zeiss, Inc.), and zoom factor two. The acquired digital images were expressed as pseudocolors and then merged using Adobe Photoshop (Adobe Systems Inc., Mountain View, CA).
Figure 6.

Analyses of DM–DNA replication and apoptosis in COLO 320DM cells. (A and B) A culture of rapidly growing COLO 320DM cells was pulse labeled with 10 μM BrdU for 1 h. The nuclei were isolated, fixed with PFA, hybridized with the biotinylated DM painting probe, and then detected using FITC-conjugated streptavidin as in Fig. 4. Sites at which BrdU was incorporated were detected with an anti-BrdU mouse monoclonal antibody followed by rhodamine-conjugated anti–mouse immunoglobulin. The double-labeled nuclei were examined using a confocal laser scanning microscope. The images of BrdU, FISH, and the merged images (red, BrdU; green, FISH) are shown for two representative fields. Nuclear buds that selectively entrap DMs are indicated by arrows, and the cells that were not in S phase during the pulse label (BrdU−) are indicated by arrowheads. (C) Analysis of apoptosis in COLO 320DM cells was done using the TUNEL method as described in Materials and Methods. This representative photograph is from one experiment in which COLO 320DM cells were treated with 100 μM HU for 3 d. The arrow points to a bud in a cell that is not undergoing apoptosis, whereas the arrowhead points to an apoptotic cell in the same field.
Localizing DMs in Interphase Nuclei
The locations of DMs in confocal nuclear sections were determined by measuring the distance from each of the hybridized signals to the center of the nucleus using the corresponding nuclear diameter as a unit length. Coplanar PI (DNA) and FITC (hybridized signal) images intercepting the center of the nucleus were obtained from randomly chosen nuclei. The digital images were merged using COMOS software (Bio-Rad Laboratories). The threshold value for each FITC signal was lowered until each signal, representing the domain of one or more DMs, became a single dot to enable accurate distance measurements. The distances from this dot to the center of the nucleus and the nuclear diameter were determined. The location of each signal in the nucleus was expressed by dividing the former number by the latter number. According to this expression, the center of the nucleus is 0, and the outer edge of the nuclear membrane is 1. At the same time, the intensity of each signal was measured using an arbitrary unit scale. These values were determined for every signal in each of the nuclear sections using a minimum of 100 randomly chosen nuclei for each sample. This procedure gives rise to a distribution of DM signals in each two-dimensional focal plane. The heights and widths of each nucleus were found to be approximately equal, indicating that the fixation procedure preserved a spherical nuclear morphology. We assume that the distribution of signals within each nuclear vol should correspond to the number of signals we detected at the corresponding radius in two-dimensional space. Therefore, we corrected the number of signals at each radial location to represent the number that should be present in the spherical volume corresponding to that radius.
Simultaneous Determination of DM Location and DNA Replication Using FISH and BrdU Incorporation
Rapidly growing COLO 320DM cultures were pulse labeled using 10 μM BrdU (Sigma Chemical Co.) for 1 h and then followed by immediate cell collection. The pulse-labeling period was 30 min for the experiment presented in Fig. 2, G and H. The isolation of nuclei, fixation by PFA, and FISH using purified micronuclei probe were done as described above. After FISH, BrdU incorporation was determined by incubating the slides with anti-BrdU mouse monoclonal antibody (PharMingen, San Diego, CA) at a final concentration of 10 μg/ml in PBS containing 0.1% BSA. After a 60-min incubation at 37°C, the slides were washed three times with PBS for 5 min each. The slides were then treated with rhodamine-labeled anti–mouse Ig (Boehringer Mannheim Biochemicals) at a final concentration of 10 μg/ml in PBS containing 0.1% BSA. The slides were incubated for 60 min at 37°C and then washed with PBS three times at 5 min each. The nuclei were viewed without counter staining, using an MRC 1024 (Bio-Rad Laboratories) confocal system equipped to Axiovert 135M microscope (Carl Zeiss, Inc.), and then the acquired digital images were processed as described above.
Results
Nuclear Budding during Interphase Can Selectively Entrap DMs
The classic mechanism by which acentric chromosome fragments such as DMs are lost from cells involves their enclosure within reforming nuclear membranes subsequent to telophase (for review see Heddle et al., 1991). However, there is one report that nuclear anomalies resembling micronuclei can be generated in interphase subsequent to γ irradiation (Duncan et al., 1985). We used a cell line with amplified c-myc genes to assess the relative contributions of postmitotic and interphase mechanisms to DM micronucleation.
A FISH analysis of COLO 320DM, a colon cancer cell line of neuroendocrine origin, is shown in Fig. 1. A biotinylated FISH probe specific for the c-myc amplicon in COLO 320 cells was obtained from micronuclei purified from COLO 320DM cells (Shimizu et al., 1996). FISH analysis with this probe showed that >95% of the cells in the population contained only DMs, and the remainder contained DMs along with one intrachromosomally amplified region (Fig. 1 A, arrow). Consistent with a previous report (Levan and Levan, 1978), the DMs in the prometaphase spread (Fig. 1 A) do not appear to be distributed randomly since many localize to the periphery of the prometaphase ring. Peripheral nuclear localization was also observed in interphase nuclei using confocal microscopy (see below).
Figure 1.

Nuclear budding selectively entraps DMs. Exponentially growing COLO 320DM cells were examined by a conventional FISH procedure that includes hypotonic treatment and fixation with methanol/acetic acid (3:1) (A–E). Alternatively, cells grown on slides were directly fixed with ethanol/ acetic acid (19:1) followed by formaldehyde fixation (F and G). The slides were treated with RNase and hybridized with biotinylated DNA from purified micronuclei, except E, which was hybridized with biotinylated c-myc cosmid DNA. The hybridized probe was detected by FITC-conjugated streptavidin and then the DNA was counterstained with PI. (A) A prometaphase figure that shows both the specificity of the probe for DM painting and documents the peripheral location of DMs around the prometaphase chromosomes. One chromosomally integrated HSR region is indicated by an arrow. These DMs are selectively incorporated into the nuclear buds formed in the interphase nuclei (B–G, arrows). DM capture by buds appears to be very selective because the three dense PI-positive signals obvious in the nuclear bud (D) label intensely with the FITC–FISH probe (D′). (D′′) The merged image.
Analyses of exponentially growing cultures of COLO 320DM cells by a conventional FISH procedure revealed that 2.8 ± 0.9% of interphase nuclei have projections or buds. These buds can be classified into two types by FISH analysis using the probe described above. The first class (56 ± 12.2% of the nuclear buds) contained highly concentrated DM sequences (Fig. 1, B–D show representative buds that contain DMs; C shows two micronuclei and one bud). We also observed a second class of buds that were stained with PI, but did not hybridize with the DM probe. This suggests that DNA that does not hybridize with the DM probe was contained in these buds. (see Discussion). However, the selectivity for acentromeric sequences such as DMs appears to be very high. This is indicated by the nuclear bud shown in Fig. 1 D that contains three DNA clusters that stain with PI (the red signal represents DNA staining as these samples were first treated extensively with RNAse) and that hybridize intensely with the micronuclear DNA–FISH probe (Fig 1 D′; merge shown in D′′). The selective inclusion of DMs into buds was also readily apparent when we used a FISH probe derived from a cosmid containing the c-myc gene. (Fig. 1 E), PFA-fixed nuclei isolated by a hypotonic method (see Fig. 6), or an isotonic method (data not shown). Furthermore, the selective inclusion of DM sequences into nuclear buds was also apparent in intact cells fixed directly by ethanol/acetic acid (19:1) followed by formaldehyde (Fig. 1, F and G). The apparent preferential inclusion of DM sequences in buds is reminiscent of our previous finding that DMs are highly enriched in micronuclei and that chromosomes or minichromosomes with functional centromeres are excluded from micronuclei (Von Hoff et al., 1992; Shimizu et al., 1996). These data suggest that buds are precursors of micronuclei.
Nuclear Buds and Micronuclei Are Formed during S Phase
The nuclei with buds exhibit a morphology typical of an interphase cell, not one in mitosis. Therefore, we determined the kinetics of formation of micronuclei and nuclear buds to ascertain whether these structures can be generated during S phase. COLO 320DM cells were synchronized at the G1/S boundary using a two-step procedure involving treatment with high thymidine concentration to arrest cells during S phase, release for 12 h to enable progression through and exit from S phase, and then incubation with the DNA polymerase inhibitor aphidicolin to arrest cells at the beginning of the next S phase (Stein et al., 1994). Removal of aphidicolin resulted in rapid entry into S phase with a peak of [3H]thymidine uptake at 4 and a peak of mitosis at 10 h, respectively (Fig. 2 A). The cells entered a second, less synchronous cycle ⩽19 h after release. The synchronization level of the first S phase was further quantified by BrdU–pulse labeling followed by confocal microscopic examination of the nuclear labeling patterns. The labeling pattern progressed as reported previously (O'Keefe et al., 1992), with six readily distinguishable patterns (Fig. 2 G, Patterns 0–6). Examination of the arrested cells (t = 0) revealed that 96.1% of the cells did not incorporate label (Fig. 2 H, Pattern 0) but the remaining 3.9% of cells exhibited the pattern expected for early S phase (Fig. 2 G, Pattern 1). The small but significant fraction of cells in early S phase most likely reflects the leakiness of the synchronization procedure. After the release from the aphidicolin block, these patterns progressed sequentially as shown in Fig. 2, G and H. At the peak of [3H]thymidine uptake (4 h after release), 8.3% of cells still did not incorporate BrdU, suggesting that some cells may have been arrested irreversibly by aphidicolin.
The frequencies of micronuclei and buds were ascertained using the DNA specific dye DAPI (Fig. 2, C and D). We determined whether these structures contain amplified sequences by hybridizing all samples with the DM-painting probe obtained from micronuclei. The frequency of nuclei with buds at the G1/S boundary (i.e., t = 0) was nearly zero, increased dramatically as the cells progressed through early S phase (t = 0–5 h), declined in later S phase, and then gradually increased as the cells entered and progressed through a second S phase (Fig. 2 C). FISH analysis demonstrated that the buds enriched in DM sequences (DM + buds) showed the same time course (Fig. 2 E). The frequency of buds peaked concurrent with a BrdU labeling pattern characteristic of the onset of replication of peripheral heterochromatin (Fig. 2, G and H, Patterns 3). Importantly, the number of total micronuclei, or those with DMs, increased and declined in register with the number of nuclear buds (Fig. 2 C), suggesting that buds are precursors of micronuclei. We recently found that micronuclei containing DM sequences are released into the growth medium. The extracellular micronuclei were distinct from apoptotic bodies since they did not have condensed chromatin, the DNA was not degraded extensively, and the nuclear lamina were still intact (Shimizu, N., and G.M. Wahl, manuscript in preparation). Therefore, it is reasonable to propose that the decrease of micronuclei in late S phase reflects their release from the cells.
Whereas the frequency of nuclei with buds was low at the G1/S boundary, micronuclei were readily apparent (Fig. 2 C). One likely possibility is that these micronuclei were generated during the telophase of a previous cell cycle. We modified the synchronization strategy to include nocodazole to block cells in prometaphase after release from the aphidicolin block. This protocol restricts the analysis to micronuclei and buds generated within a single S phase, and to prevent buds or micronuclei produced from cells arrested before prometaphase of the previous cycle from entering the analysis (Cassimeris and Salmon, 1991). The treated cells entered and progressed through S phase at approximately the same rate as those not exposed to nocodazole (Fig. 2, compare A and B), and the mitotic index increased significantly by 8 h after release. The effectiveness of nocodazole treatment is also indicated by the inability of the drug-treated cells to progress into a second S phase over the time course used. Importantly, the number of nuclei with buds was again nearly zero at the G1/S boundary, and both the number of buds and micronuclei increased after release into nocodazole-containing medium. These data are consistent with the interpretation that nuclear buds and micronuclei can arise de novo during S-phase progression.
The kinetic and nuclear morphologic analyses of budding and micronucleation indicate that both events can occur during S phase. Since slowing replication fork progression may lead to DNA breakage (Eki et al., 1987; Linke et al., 1996), and micronuclei preferentially capture acentric fragments (Von Hoff et al., 1992; Shimizu et al., 1996), we determined whether replication inhibitors increase S phase micronucleation efficiency. The drugs tested included inhibitors of ribonucleotide reductase (HU, deferoxamine, and guanazole), an inhibitor (PALA) of the carbamyl phosphate synthatase, dihydro-orotase, aspartate transcarbamylase (CAD) enzyme complex that catalyzes the first three steps of de novo pyrimidine biosynthesis, and aphidicolin. DNA synthesis inhibitors produced substantial increases in micronucleation, and this generally correlated with an increased fraction of cells in S phase (Fig. 3, A and B). A sharp decrease in micronucleation efficiency was observed for deferoxamine and PALA when these drugs were used at concentrations that severely inhibited S phase (Fig. 3 B and data not shown). These data indicate that micronucleation can result from inhibitors that retard replication fork progression and lengthen S phase.
We analyzed the effects of inhibitors that do not affect DNA synthesis to ascertain whether micronucleation can result from interfering with other DNA transactions such as DNA repair, or by interfering with membrane structure. Two inhibitors of poly(ADP-ribose) polymerase (nicotinamide and coumarin) were tested since ADP ribosylation has been implicated in DNA repair (Satoh and Lindahl, 1992), and inhibiting repair could increase the probability of generating acentric chromosome fragments. We tested the effects of DMSO, a membrane-active polar compound previously reported to reduce DM copy number in some tumor cell lines (Shima et al., 1989; Eckhardt et al., 1994). DMSO increased micronucleation without lengthening S phase (Fig. 3, A and B). Coumarin had no effect on micronucleation, whereas nicotinamide produced a small increase under conditions that apparently increased the amount of damage in the cells since there was a significant increase in the G2 fraction (Fig. 3, A and B). These data are consistent with the view that micronucleation efficiency can be increased by at least two mechanisms, one of which presumably involves perturbing replication fork progression, and another of which may involve events occurring outside of S phase.
Peripheral Nuclear Localization of DMs Correlates with Their Elimination by Budding
Insight into the mechanisms underlying the selective inclusion of DMs into nuclear buds and the formation of these structures in interphase was obtained by confocal microscopy. PFA fixation of nuclei was used for optimal preservation of nuclear morphology (Manuelidis and Borden, 1988).
Confocal sections from three representative nuclei isolated from rapidly growing untreated COLO 320DM cells are shown in Fig. 4, A–C. FISH revealed preferential localization of most DM sequences to the nuclear periphery, as indicated by the significant hybridization intensity, clustering, and number of DM signals at the extreme edge of each nucleus. Note the substantial deviation from a random distribution of DM sequences at the nuclear periphery (quantified in Fig. 5 A by measuring DM positions relative to the center of each nucleus in 100 interphase nuclei). By contrast, sequences amplified within chromosomes in the closely related cell line COLO 320HSR showed a nearly random distribution throughout the nucleus (Fig. 5 C). HU treatment preferentially depleted DMs from the nuclear periphery (Fig. 4, D–F and Fig. 5 B, quantification) and then reduced the DM content per cell by approximately threefold as determined by competitive PCR amplification (for method see Shimizu et al., 1996; data not shown). Taken together with the data reported above, these results indicate that DM sequences located at the nuclear periphery are preferentially incorporated into nuclear buds, which are then removed from the nucleus through the formation of micronuclei.
Figure 4.

Localization of DMs in interphase COLO 320DM nuclei. PFA-fixed isolated nuclei from a culture of COLO 320DM cells were hybridized with a DM painting probe and then counterstained with PI. Optical sections near the center of each nucleus were obtained using confocal laser scanning microscopy. Each of three representative images of nuclei from the rapidly growing culture (A–C) and from a culture treated with 100 μM HU for 3 d (D–F) are shown. In untreated cultures, DMs preferentially located just beneath the nuclear membrane as indicated by the arrows. Very few peripheral DMs were detected in the nuclei from the HU-treated culture, and most of the signals localized well within the nucleus as shown.
Figure 5.

Quantitative analysis of the nuclear positions of DMs and chromosomally amplified sequences. The nuclei from a rapidly growing culture of COLO 320DM cells (A), COLO 320DM treated with 100 μM HU for 3 d (B), and rapidly growing COLO 320 cells with chromosomally amplified c-myc sequences (COLO 320HSR) (C) were hybridized with a probe for the c-myc amplicon. Sections intercepting the center of each nucleus were obtained as in Fig. 4. For each section, the position and intensity of each hybridized signal was measured. Each analysis represents measurements on 100 randomly chosen nuclei. The abscissa depicts the fractional distance from the nuclear center (0, center; 1, periphery), and the ordinate is the number of signals detected at each position in 100 nuclei. The theoretical random distribution curves based on signals per nuclear vol at each position are shown in each graph as explained in greater detail in Materials and Methods.
Incorporation of Replicating DM Sequences into Nuclear Buds
The correlations between S phase progression, nuclear budding, and micronucleation reported above led us to investigate whether DM sequences undergoing replication are targeted for inclusion into buds. This possibility was examined by pulse labeling COLO 320DM cells with BrdU, and then hybridizing the isolated nuclei with the DM–FISH probe. Subsequent reaction with an anti-BrdU antibody and fluorescein-labeled secondary antibody enabled simultaneous detection of nuclei, buds, and micronuclei containing DMs that were undergoing DNA replication during the brief labeling interval. FISH analysis of two representative confocal sections (Fig. 6, A–A′′ and B–B′′) shows that nuclear buds in these cells (arrows) contain highly concentrated DM sequences. The nuclei, nuclear buds, and peripheral regions of each nucleus incorporated BrdU, indicating that these buds were formed in nuclei that were synthesizing DNA at the time of bud formation. Table I shows that BrdU+, DM+ buds (type 1, 1′) represent 48% of the total population of DM-containing buds. Some nuclei incorporated BrdU, but the buds they produced were not labeled (type 2, 2′; 35%). We infer that some of these buds were also generated during S phase (as opposed to during the previous cycle), and that they did not reveal BrdU incorporation because their DNA was not undergoing replication during the brief BrdU incubation period. Samples in which neither nuclei nor buds labeled with BrdU may represent examples where buds were generated outside of S phase (type 3; 17%). Alternatively, the presence of such buds (type 3) may indicate that these structures persist after DNA replication has finished, and may be sources of some of the micronuclei observed to form during mitosis. These data reveal a strong correlation between DMs undergoing replication and their inclusion in buds and micronuclei, and they lead to a conservative estimate of ⩾50% of the micronuclei produced during each cell cycle being generated during S phase.
Table I.
Quantification of Budding and Micronucleation
| BrdU labeling in: | Frequency of DM+buds that belong to each type | |||||
|---|---|---|---|---|---|---|
| Type | DM+buds | Nuclei | ||||
| 1 | + | + | 26/60 (43%) | |||
| 1′ | ± | + | 3/60 (5%) | |||
| 2 | − | + | 9/60 (15%) | |||
| 2′ | − | ± | 12/60 (20%) | |||
| 3 | − | − | 10/60 (17%) | |||
Nuclei from COLO 320DM were pulse labeled with BrdU and then analyzed for BrdU incorporation and DMs as described in Fig. 6. Nuclei were observed using epifluorescence microscopy. 60 nuclei with DM+buds were classified according to whether they labeled with BrdU (classes 1, 1′), whether the nuclei to which they were attached labeled with BrdU (classes 2, 2′), or whether neither bud nor nucleus labeled with BrdU (class 3). The number of nuclei belonging to each type were scored and expressed as nuclei in that class/total number scored.
The S phase micronucleation process described here superficially resembles the induction of “nuclear anomalies” by an apoptotic mechanism following γ irradiation (Duncan et al., 1985) or colchicine treatment (Duncan et al., 1984). However, the absence of highly condensed DNA in the nuclei-producing buds suggests that they were not undergoing apoptosis at the time of bud formation. To determine whether budding and micronucleation are separable from apoptosis, we determined whether buds contain the condensed, fragmented DNA that typifies apoptotic cells. Fragmented DNA was detected using the TUNEL assay in which terminal transferase is used to add BrdU to the 3′-OH groups generated by apoptotic DNA fragmentation (Gavrieli et al., 1992). The cells were also stained with PI to visualize all nuclei and buds. An example of the data obtained from such an analysis of COLO 320DM cells is shown in Fig. 6, C–C′′. The TUNEL assay shows one cell with a lobular nucleus that exhibits a strong TUNEL reaction and typifies the fragmented, condensed DNA observed in apoptotic nuclei (Cohen, 1993). The PI staining in the middle panel reveals a cell producing a nuclear bud that does not stain by the TUNEL assay and does not exhibit the pycnotic structure of the apoptotic nucleus. Synchronization experiments showed that whereas ∼5% of cells generated buds at the peak of S phase (e.g., refer to Fig. 2 C), only 0.5–1% were TUNEL positive. Other data (see Discussion) provide additional evidence that S phase budding and micronucleation do not require activation of an apoptotic program.
Micronucleation Occurs Infrequently in Normal Cells, and Is Increased upon p53 Inactivation
Micronucleation occurs at significantly lower rates in normal cells than tumor cell lines (Roser et al., 1989; Bondy et al., 1993). Since micronucleation can be induced by chromosome breakage (Heddle and Carrano, 1977), the observed increase in micronucleation in tumor cells might result from mutations that increase the probability of DNA breakage. The tumor suppressor p53 controls G1 arrest responses activated by DNA breakage and rNTP depletion induced by PALA treatment, and DNA breakage can occur in p53-deficient cell lines that enter S phase during PALA treatment (Livingstone et al., 1992; Yin et al., 1992; Linke et al., 1996). As reported above, PALA also induces S-phase micronucleation in COLO 320 cells. These data led us to investigate whether p53 inactivation in normal diploid fibroblasts results in increased S-phase budding and micronucleation.
Human WS1 normal diploid fibroblasts, and two nearly isogenic derivatives generated by retroviral transduction of the neomycin phosphotransferase gene (WS1-neo) or an oncogenic derivative of the human papilloma virus E6 gene (WS1-E6) were used. The E6 gene product facilitates p53 degradation by a ubiquitin dependent pathway (Scheffner et al., 1990; Crook et al., 1991). Cell cycle checkpoint controls that regulate entry into S phase in the presence of DNA damage or limiting rNTP concentrations appear to be inactivated to equivalent degrees in human cells expressing mutant p53, oncogenic E6 protein, and mouse embryo fibroblasts with homozygous p53 knock out (Kastan et al., 1992; Kuerbitz et al., 1992; Livingstone et al., 1992; Yin et al., 1992; White et al., 1994; Linke et al., 1996; Linke et al., 1997). Importantly, we previously showed that the frequency of γ radiation induced micronucleation is higher in p53−/− MEFs than in wild-type MEFs (Huang et al., 1996). It is likely, therefore, that effects on micronucleation observed upon expression of oncogenic E6 protein relate to inactivation of p53 rather than to other proteins that may be affected by E6.
The data shown in Fig. 7 demonstrate that E6 gene expression increases the micronucleation rate of WS1 cells. WS1 cells exhibit a low micronucleation rate that is not increased by HU or PALA (Fig. 7 A). Consistent with our previous studies (Linke et al., 1996), PALA induced a G1 cell cycle arrest, whereas HU did not significantly affect the percentage of WS1 cells in S phase at the concentration used (Fig. 7 B). By contrast, E6 expression increased the micronucleation efficiency of these fibroblasts growing under normal conditions, and both HU and PALA produced a substantial further increase in micronucleation rate (Fig. 7 A), which correlated with a significant increase in the number of cells in S phase (Fig. 7 B). The importance of an E6 target, which we infer to be p53, in limiting micronucleation is evident in other cell types since similar results were obtained using RPE-h and their E6 expressing derivatives (data not shown).
Figure 7.

p53 deficiency increases S-phase budding and micronucleation in normal human diploid fibroblasts. Wild-type (wt) WS1 human diploid fibroblasts, or transformants expressing the neomycin resistance gene (neo) or both neo and the human papilloma virus E6 protein (E6) were generated by retroviral transduction. These cells were cultured in the absence or the presence of HU or PALA at the indicated concentrations for 3 d. (A) Cells grown on coverslips and treated as above, fixed, and then stained by DAPI. The number of micronuclei were scored and expressed as the frequency of micronuclei (%) relative to the number of interphase nuclei scored (more than 1,000 for each point). (B) Cell cycle effects of these drugs were examined as in Fig. 2 B. (C and D) The culture of WS1-neo (open and closed triangles) or WS1-E6 (open and closed circles) were synchronized at the G1/S boundary by the procedure described in Materials and Methods, and were released into growth medium. (C) Progression through S or M phase was monitored by the incorporation of [3H]thymidine (closed symbols) or the fraction of mitotic cells determined by microscopic detection of metaphase figures (open symbols). (D) The cells on the coverslips were fixed, stained with DAPI, and then the frequencies of micronuclei relative to the number of interphase nuclei were scored. >500 nuclei were counted for each point and the results are expressed as mean ± SD for WS1-neo and WS1-E6 cells (three independent determinations for each strain; error bars for WS1-neo data points were smaller than the sizes of the symbols). Nuclear budding frequencies were determined on the same slides as used to determine the micronucleation index (more than 1,000 nuclei for each point).
The elongation of S phase and induction of micronuclei by HU and PALA in both COLO 320DM and WS1-E6 cells led us to assess whether budding in S phase is the predominant mechanism of micronucleation in WS1-E6 cells. WS1-neo and WS1-E6 cells were arrested in G0 by serum deprivation and then released in the presence of aphidicolin to arrest the cells at the G1/S boundary (Fig. 7 C). Synchronization by serum depletion did not increase micronucleation rate (data not shown) and then the micronucleation and budding frequencies did not increase in S phase in WS1-neo cells (Fig. 7 D). By contrast, removal of aphidicolin from WS1-E6 cells resulted in significant increases in the frequencies of both nuclear budding and micronucleation as the cells progressed through S phase (Fig. 7 D). Since DNA damage does not induce apoptosis in either WS1 or WS1-E6 cells (Di Leonardo et al., 1994; Linke et al., 1996; Linke et al., 1997), the increased S-phase micronucleation observed in these cells occurs independent of an apoptotic program.
Discussion
Loss of cell cycle checkpoints during cancer progression creates a permissive environment for the initiation and propagation of chromosomal rearrangements such as amplification of cellular protooncogenes. The persistence or elimination of these structures within the nucleus can, respectively, promote or inhibit cancer progression. Interestingly, in human tumors analyzed at biopsy, oncogene amplification occurs most frequently in acentric chromosomal fragments such as DMs (Benner et al., 1991), and micronucleation represents a major pathway for the elimination of such structures (Von Hoff et al., 1992; Shimizu et al., 1996). Before this report, micronucleation had been considered to result from imperfect segregation of acentric chromosomal fragments or fragments of overly long chromosomes during karyokinesis (Heddle and Carrano, 1977; Heddle et al., 1983). The results presented here, by contrast, reveal that acentric DMs are sorted to the nuclear periphery during S phase, and are then selectively eliminated from the nucleus by micronucleation in advance of karyokinesis.
S Phase Budding and Micronucleation May Be the Predominant Mechanism for DM Elimination
We infer that S-phase micronucleation is a general characteristic of human cells with an aberrant p53 pathway since this process was observed in a human tumor cell line, and p53-deficient normal human fibroblasts and epithelial cells. The cell synchrony and release experiments provide direct evidence that budding begins as cells enter S phase. The experiment summarized in Table I shows that BrdU+ micronuclei account for almost 50% of the total micronuclei generated during a single S-phase. It is reasonable to infer that a fraction of the 35% of BrdU− micronuclei derived from BrdU+ nuclei did not incorporate the label because their DNA did not replicate during the brief BrdU pulse used. Such micronuclei would, therefore, also have originated during S phase, but we have no means of providing a precise estimate of this fraction. The available data do, however, demonstrate that S-phase micronucleation mediated by budding is at least as common as the classic postmitotic process, and may be the predominant mechanism for removing DMs from human cancer cells.
The parallel increases in budding and micronucleation frequencies as cells enter and proceed through S phase, and the absence of a significant temporal lag between the two, is consistent with a precursor–product relationship in which a micronucleus is produced shortly after a bud is generated. The decrease in budding and micronucleation during the latter part of S phase is also consistent with a tight linkage to the replication program, and suggests that once micronuclei are generated during S phase, some may be released from the cell, whereas others may fuse to nuclei or be degraded intracellularly. Among these possibilities, we have direct experimental evidence that some micronuclei may be expelled from the cell through the cytoplasmic membrane since we have observed extracellular micronuclei containing three membranes and amplified c-myc genes (Fig. 8; Shimizu, N., and G.M. Wahl, unpublished data). We are currently testing whether such micronuclei are bound by a cytoplasmic membrane, and whether they can fuse to the same or other cells in the population (Fig. 8).
Figure 8.

Model for the formation of micronuclei. Models for DM elimination by budding and micronucleation in S-phase and postmitotic micronucleation are shown. In the S-phase budding mechanism, DMs are preferentially located at the periphery of the interphase nucleus and then selectively encapsulated into nuclear buds that then pinch off to form micronuclei during DNA replication (a–d). This process is an alternative to the classical postmitotic mechanism of generating micronuclei depicted in f–h. One possible fate of these micronuclei is that they refuse to the main nucleus (i to j and d to e). At present, this is a speculation as there is no direct experimental evidence to support it. On the other hand, we do have direct evidence that micronuclei can be released into the culture supernatant as extracellular micronuclei, suggesting that they may be extruded through the cell membrane (j; Shimizu, N., and G.M. Wahl, unpublished data). Decrease in DM content can be achieved by either expulsion of micronuclei containing DMs from the cell, or by degradation of DM–DNA within intracellular micronuclei. In either case, loss of DM sequences from the nucleus results in reversion of the tumor phenotype, differentiation, or apoptosis (k; Von Hoff et al., 1992; Eckhardt et al., 1994; Shimizu et al., 1994).
The baseline frequency of micronuclei observed in cultures at the beginning of S phase in which budding was not yet evident suggests that some micronuclei were generated in a previous cycle and persisted for an extended time, which would be consistent with previous reports of long lived micronuclei (Heddle et al., 1983). It is conceivable, therefore, that there are two classes of micronuclei that differ from each other by their stability, perhaps because of structural distinctions deriving from their mechanisms of formation. Consistent with the inference of multiple types of micronuclei, we found that micronuclei can differ in their lamin and nuclear pore contents (Shimizu, N., and G. Wahl, unpublished observations). Experiments in progress are designed to elucidate whether such differences correlate with stability and alternative mechanisms of formation.
S-phase Budding and Micronucleation Do Not Require Previous Engagement of an Apoptotic Program, but Can Result in Apoptosis
Apoptosis can generate nuclear blebs (Dini et al., 1996) and has been inferred to produce “nuclear anomalies” that resemble micronuclei (Duncan and Heddle, 1984; Duncan et al., 1985). However, our analyses show that nuclei that produced buds were not pycnotic and fragmented like apoptotic nuclei. Buds and micronuclei generated in COLO 320DM cells within a single S phase were not TUNEL positive, indicating that they did not contain fragmented DNA. Whereas apoptotic cells did arise in HU-treated COLO 320DM cultures, this required prolonged incubation, and occurred after a substantial fraction of the amplified c-myc genes had been removed. Furthermore, cells undergoing budding and micronucleation survived for several days, which is not expected if an apoptotic program were activated before micronucleation. A time course experiment similar to that described above (e.g., Fig. 2) revealed that budding increases during a single S phase whereas the fraction of cells undergoing apoptosis remained roughly constant, and that most of those generating buds were not TUNEL positive (data not shown). Finally, though we have not observed apoptosis in normal fibroblasts (Di Leonardo et al., 1994), and loss of p53 function typically makes cells more resistant to apoptosis induced by growth conditions that can lead to DNA damage (White, 1994), S-phase budding and micronucleation were induced in normal diploid fibroblasts upon expression of oncogenic papillomavirus E6 protein, presumably due to elimination of p53 function. These observations lead us to propose that buds and micronuclei in COLO 320DM and WS1-E6 cells are produced by a mechanism that does not require prior engagement of the apoptotic program.
Mechanisms of Budding and Micronucleation
The molecular basis for inclusion of DMs in buds and micronuclei, and exclusion of similarly sized centric fragments from such structures (Shimizu et al., 1996), remains to be elucidated. We offer two potential explanations. The first relates to replication of DMs at an inappropriate nuclear location. Previous studies showed that heterochromatic DNA, such as that comprising inactive X chromosome–Barr bodies, typically replicates late in S phase at the nuclear periphery and is often enclosed within micronuclei, whereas the active X replicates earlier in S phase at a more internal position and is not subject to micronucleation (Dyer et al., 1989; Tucker et al., 1996). DMs are euchromatic and tend to replicate early in S phase, typically at approximately the same time as the native chromosomal locus (Carroll et al., 1991, 1993). However, as shown here, they can localize to and replicate at the nuclear periphery. Since chromosomes occupy specific territories within interphase nuclei (Cremer et al., 1993), the independence of DMs from chromosomes may prevent them from occupying the correct nuclear position and could explain their localization to the periphery. Peripheral localization may be a default position reflecting the inability of DMs to undergo the nuclear movements choreographed by centromeres and/or telomeres during S phase (DeBoni and Mintz, 1986; Ferguson and Ward, 1992; Vourc'h et al., 1993). Replication at such peripheral locations, or perhaps inability to move away from such sites into protected inner nuclear regions following replication, may then precipitate bud formation. A second explanation is that some chromosomal sequences may bind proteins that target the protein and associated nucleic acid to a particular nuclear location, such as the periphery. In the absence of a centromere/ telomere, this protein–nucleic acid complex may be destined to form a nuclear bud until membrane fusion produces a micronucleus. This view is supported by recent studies in Tetrahymena demonstrating that cis-acting heterochromatic regions bound by the chromodomain protein Pdd1p are targeted to the nuclear periphery when the DNA is fragmented during macronuclear development (Madireddi et al., 1996). The peripheral, Pdd1p-associated acentric chromosomal fragments are then removed from the nucleus via micronucleation. The striking similarities between the observations in Tetrahymena and those reported here raise the intriguing possibility of an evolutionarity conserved process that distinguishes intact chromosomes from chromosome fragments or other acentric DNA such as DNA viruses to facilitate removal of the latter from the nucleus.
S-Phase Micronucleation Suggests That Loss of p53 Function Affects the Probability of Chromosome Breakage
Treatment of normal fibroblasts with HU or PALA did not induce S-phase micronucleation, whereas identical treatment of isogenic p53–deficient normal fibroblasts increased S-phase micronucleation frequency significantly. These data, along with previous observations, lead us to propose that this micronucleation increase most likely reflects a higher probability of DNA breakage occurring in the p53-deficient cells when they attempt DNA replication under adverse conditions. In support of this idea, micronucleation is an indicator of chromosome breakage, and has long been used as an assay for clastogens (Heddle et al., 1991). Inhibition of replication fork progression induces chromosome breakage in bacteria, yeast, and mammals (Eki et al., 1987; Kuzminov, 1995; Michel et al., 1997). Furthermore, replication inhibitors including PALA, methotrexate, and aphidicolin can induce chromosome breakage through expression of fragile sites (Kuo et al., 1994; Coquelle et al., 1997). Although the relationship between fragile site induction and p53 function has not been tested, the cell lines used for such studies were competent for gene amplification and consequently should have had a defective p53 pathway (Livingstone et al., 1992; Yin et al., 1992). Consistent with a breakage-moderating function of p53, we previously reported that PALA generates chromosome damage in cells with defective, but not normal, p53 function (Linke et al., 1996). Taken together, the data lead us to propose that p53 minimizes the frequency at which structural chromosomal alterations are induced during exposure to suboptimal growth conditions by at least two mechanisms. First, as shown previously, p53 mediates a G1 arrest in response to low rNTP pools, which prevents cells from entering S phase with inadequate precursors for DNA replication (Linke et al., 1996). Second, the data presented here suggest that p53 minimizes S-phase DNA breakage in cells that attempt DNA replication during diverse metabolic challenges. The mechanisms underlying the proposed S-phase function of p53 are currently under investigation.
Micronucleation and Cancer Treatment
Drugs that enhance S-phase budding should prove valuable for chemotherapy as they affect a process (micronucleation) and cytogenetic aberration (DMs) that are restricted to cancer cells. However, their efficacy is limited by the extent to which DM removal prevents further cell growth, or enhances sensitivity to other therapeutic strategies. In COLO 320DM cells, induction of the S-phase budding mechanism can decrease the number of DMs sufficiently to reduce plating efficiency in soft agar and tumorigenicity in vivo (Von Hoff et al., 1992). Interestingly, COLO 320HSR cells, which have approximately the same number of c-myc genes amplified within chromosomes, did not exhibit such phenotypic changes upon HU treatment and did not decrease c-myc copy number, even though they produced approximately the same number of micronuclei as COLO 320DM cells (Von Hoff et al., 1992). The same HU treatment conditions also induced apoptosis more rapidly and in a higher fraction of COLO 320DM than COLO 320HSR cells (data not shown). Similarly, HU treatment of HL-60 DM, but not HL-60 HSR cells, reduced c-myc copy number, induced differentiation, and later, apoptosis (Eckhardt et al., 1994; Shimizu et al., 1994). These results suggest that reduced tumorigenicity may result from induction of an apoptotic program when a sufficient number of extrachromosomally amplified sequences encoding oncogenes are removed from the cell. The recognition of a mechanism for the segregation and elimination of amplified sequences and other acentric DNA macromolecules, and the recognition that some agents can enhance this process, present important opportunities to expand and refine chemotherapeutic strategies, and to gain insight into the cis-acting elements and trans-acting factors that determine nuclear DNA localization.
Abbreviations used in this paper
- BrdU
5-bromo-2′-deoxyuridine
- DAPI
4′-6-diamidino-2-phenylindole
- DM
double-minute chromosome(s)
- FISH
fluorescence in situ hybridization
- HU
hydroxyurea
- PALA
N-phosphoracetyl-l-aspartate
- PFA
paraformaldehyde
- PI
propidium iodide
- RPE-h
normal human retinal pigmented epithelial cells
Footnotes
We would like to acknowledge S.P. Linke (National Cancer Institute, Bethesda, MD) for kindly providing cell lines, D. Peterson (Salk Institute, La Jolla, CA) for his kind help on the operation of confocal microscopy that appeared in Fig. 6, and D. Von Hoff and K. Davidson (both from Institute of Drug Development, University of Texas Health Science Center, San Antonio, TX) for allowing us to cite their unpublished results. We thank T. Shimura and N. Kumon (both from Hiroshima University, Higashi-Hiroshima, Japan) for their technical help. T. Paulson, T. Kanda, S. O'Gorman, S. Pfaff, G. Karpen, O. Vafa, and L.-c. Huang (all from Salk Institute except Paulson [Fred Hutchinson Cancer Research Center, Seattle, WA] and Huang [UBI, Menlo Park, CA]) provided helpful discussions concerning experimental procedures and topics presented in this manuscript.
This work was in part supported by a grant from the United States Department of Army, (grant number DAMD17-94-J4359) and the International Collaboration Grant from Japanese Ministry of Education (grant number 07044271), and the G. Harold and Leila Y. Mathers Charitable Foundation.
Address all correspondence to Geoffrey Wahl, Gene Expression Laboratory, The Salk Institute, 10010 North Torrey Pines Road, La Jolla, CA 92037. Tel: (619) 453-4100. Fax: (619) 552-8285. E-mail: wahl@salk.edu
References
- Alitalo K, Shwab M. Oncogene amplification in tumor cells. Adv Cancer Res. 1986;47:235–281. doi: 10.1016/s0065-230x(08)60201-8. [DOI] [PubMed] [Google Scholar]
- Barker PE. Double minutes in human tumor cells. Cancer Genet Cytogenet. 1982;5:81–94. doi: 10.1016/0165-4608(82)90043-7. [DOI] [PubMed] [Google Scholar]
- Benner SE, Wahl GM, Von Hoff DD. Double minute chromosomes and homogeneously staining regions in tumors taken directly from patients versus in human tumor cell lines. Anti-Cancer Drugs. 1991;2:11–25. doi: 10.1097/00001813-199102000-00002. [DOI] [PubMed] [Google Scholar]
- Bondy ML, Spitz MR, Halabi S, Fueger JJ, Schantz SP, Sample D, Hsu TC. Association between family history of cancer and mutagen sensitivity in upper aerodigestive tract cancer patients. Cancer Epidemiol Biomarkers Prev. 1993;2:103–106. [PubMed] [Google Scholar]
- Brison O. Gene amplification and tumor progression. Biochim Biophys Acta. 1993;1155:25–41. doi: 10.1016/0304-419x(93)90020-d. [DOI] [PubMed] [Google Scholar]
- Canute GW, Longo SL, Longo JA, Winfield JA, Nevaldine BH, Hahn PJ. Hydroxyurea accelerates the loss of epidermal growth factor receptor genes amplified as double-minute chromosomes in human glioblastoma multiforme. Neurosurgery. 1996;39:976–983. doi: 10.1097/00006123-199611000-00019. [DOI] [PubMed] [Google Scholar]
- Carroll SM, Trotter J, Wahl GM. Replication timing control can be maintained in extrachromosomally amplified genes. Mol Cell Biol. 1991;11:4779–4785. doi: 10.1128/mcb.11.9.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SM, DeRose ML, Kolman JL, Nonet GH, Kelly RE, Wahl GM. Localization of a bidirectional DNA replication origin in the native locus and in episomally amplified murine adenosine deaminase loci. Mol Cell Biol. 1993;13:2971–2981. doi: 10.1128/mcb.13.5.2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassimeris, L., and E.D. Salmon. 1991. Kinetochore microtubules shorten by loss of subunits at the kinetochores of prometaphase chromosomes. J. Cell Sci. 151–158. [DOI] [PubMed]
- Cohen S. Apoptosis. Immunol Today. 1993;14:126–130. doi: 10.1016/0167-5699(93)90214-6. [DOI] [PubMed] [Google Scholar]
- Coquelle A, Pipiras E, Toledo F, Buttin G, Debatisse M. Expression of fragile sites triggers intrachromosomal mammalian gene amplification and sets boundaries to early amplicons. Cell. 1997;89:215–225. doi: 10.1016/s0092-8674(00)80201-9. [DOI] [PubMed] [Google Scholar]
- Cowell JK. Double minutes and homogenously staining regions: gene amplification in mammalian cells. Annu Rev Gen. 1982;16:21–59. doi: 10.1146/annurev.ge.16.120182.000321. [DOI] [PubMed] [Google Scholar]
- Cremer T, Kurz A, Zirbel R, Dietzel S, Rinke B, Schrock E, Speicher MR, Mathieu U, Jauch A, Emmerich P. Role of chromosome territories in the functional compartmentalization of the cell nucleus. Cold Spring Harbor Symp Quant Biol. 1993;58:777–792. doi: 10.1101/sqb.1993.058.01.085. [DOI] [PubMed] [Google Scholar]
- Crook T, Tidy JA, Vousden KH. Degradation of p53 can be targeted by HPV E6 sequences distinct from those required for p53 binding and trans-activation. Cell. 1991;67:547–556. doi: 10.1016/0092-8674(91)90529-8. [DOI] [PubMed] [Google Scholar]
- DeBoni U, Mintz AH. Curvilinear, three-dimensional motion of chromatin domains and nucleoli in neuronal interphase nuclei. Science. 1986;234:863–866. doi: 10.1126/science.3775367. [DOI] [PubMed] [Google Scholar]
- Denko NC, Giaccia AJ, Stringer JR, Stambrook PJ. The human Ha-ras oncogene induces genomic instability in murine fibroblasts within one cell cycle. Proc Natl Acad Sci USA. 1994;91:5124–5128. doi: 10.1073/pnas.91.11.5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Leonardo A, Linke SP, Clarkin K, Wahl GM. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev. 1994;8:2540–2551. doi: 10.1101/gad.8.21.2540. [DOI] [PubMed] [Google Scholar]
- Dini L, Coppola S, Ruzittu MT, Ghibelli L. Multiple pathways for apoptotic nuclear fragmentation. Exp Cell Res. 1996;223:340–347. doi: 10.1006/excr.1996.0089. [DOI] [PubMed] [Google Scholar]
- Duncan AM, Heddle JA. The frequency and distribution of apoptosis induced by three non-carcinogenic agents in mouse colonic crypts. Cancer Lett. 1984;23:307–311. doi: 10.1016/0304-3835(84)90098-3. [DOI] [PubMed] [Google Scholar]
- Duncan AM, Heddle JA, Blakey DH. Mechanism of induction of nuclear anomalies by gamma-radiation in the colonic epithelium of the mouse. Cancer Res. 1985;45:250–252. [PubMed] [Google Scholar]
- Dyer KA, Canfield TK, Gartler SM. Molecular cytological differentiation of active from inactive X domains in interphase: implications for X chromosome inactivation. Cytogenet Cell Genet. 1989;50:116–120. doi: 10.1159/000132736. [DOI] [PubMed] [Google Scholar]
- Eckhardt SG, Dai A, Davidson KK, Forseth BJ, Wahl GM, Von Hoff DD. Induction of differentiation in HL60 cells by the reduction of extrachromosomally amplified c-myc. Proc Natl Acad Sci USA. 1994;91:6674–6678. doi: 10.1073/pnas.91.14.6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eki T, Enomoto T, Murakami Y, Hanaoka F, Yamada M. Characterization of chromosome aberrations induced by incubation at a restrictive temperature in the mouse temperature-sensitive mutant tsFT20 strain containing heat-labile DNA polymerase a. Cancer Res. 1987;47:5162–5170. [PubMed] [Google Scholar]
- Ferguson M, Ward DC. Cell cycle dependent chromosomal movement in pre-mitotic human T-lymphocyte nuclei. Chromosoma. 1992;101:557–565. doi: 10.1007/BF00660315. [DOI] [PubMed] [Google Scholar]
- Gaubatz JW. Extrachromosomal circular DNA and genomic sequence plasticity in eukaryotic cells. Mutat Res. 1990;237:271–292. doi: 10.1016/0921-8734(90)90009-g. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben SS. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamkalo BA, Farnham PJ, Johnston R, Schimke RT. Ultrastructural features of min chromosomes in a methotrexate-resistant mouse 3T3 cell line. Proc Natl Acad Sci USA. 1985;82:1026–1030. doi: 10.1073/pnas.82.4.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Kastan MB. Cell cycle control and cancer. Science. 1994;266:1821–1828. doi: 10.1126/science.7997877. [DOI] [PubMed] [Google Scholar]
- Heddle JA, Carrano AV. The DNA content of micronuclei induced in mouse bone marrow by γ-irradiation: evidence that micronuclei arise from acentric chromosomal fragments. Mutat Res. 1977;44:63–69. doi: 10.1016/0027-5107(77)90115-4. [DOI] [PubMed] [Google Scholar]
- Heddle JA, Cimino MC, Hayashi M, Romagna F, Shelby MD, Tucker JD, Vanparys P, MacGregor JT. Micronuclei as an index of cytogenetic damage: past, present, and future. Environ Mol Mutagen. 1991;18:277–291. doi: 10.1002/em.2850180414. [DOI] [PubMed] [Google Scholar]
- Heddle JA, Hite M, Kirkhart B, Mavournin K, MacGregor JT, Newell GW, Salamone MF. The induction of micronuclei as a measure of genotoxicity. A report of the U.S. Environmental Protection Agency Gene-Tox Program. Mutat Res. 1983;123:61–118. doi: 10.1016/0165-1110(83)90047-7. [DOI] [PubMed] [Google Scholar]
- Huang L-c, Clarkin KC, Wahl GM. p53 dependent cell cycle arrests are preserved in DNA-activated protein kinase deficient mouse fibroblasts. Cancer Res. 1996;56:2940–2944. [PubMed] [Google Scholar]
- Jackson JF, Clement EG. Letter: nuclear projections and chromosome abnormalities. Lancet. 1974;2:1270–1271. doi: 10.1016/s0140-6736(74)90791-0. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Zhan Q, El-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- Kuerbitz SJ, Plunkett BS, Walsh WV, Kastan MB. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc Natl Acad Sci USA. 1992;89:7491–7495. doi: 10.1073/pnas.89.16.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo MT, Vyas RC, Jiang LX, Hittelman WN. Chromosome breakage at a major fragile site associated with P-glycoprotein gene amplification in multidrug-resistant CHO cells. Mol Cell Biol. 1994;14:5202–5211. doi: 10.1128/mcb.14.8.5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzminov A. Instability of inhibited replication forks in E. coli. . Bioessays. 1995;17:733–741. doi: 10.1002/bies.950170810. [DOI] [PubMed] [Google Scholar]
- Lawce, H.J., and G. Brown. 1991. Harvesting, Slide Making, and Chromosome Elongation Techniques. M.J. Barch, editor. Raven Press, Ltd. New York. 31–66.
- Levan A, Levan G. Have double minutes functioning centromeres? . Hereditas. 1978;88:81–92. doi: 10.1111/j.1601-5223.1978.tb01606.x. [DOI] [PubMed] [Google Scholar]
- Levan G, Mandahl N, Bregula U, Klein G, Levan A. Double minute chromosomes are not centromeric regions of the host chromosomes. Hereditas. 1976;83:83–90. doi: 10.1111/j.1601-5223.1976.tb01573.x. [DOI] [PubMed] [Google Scholar]
- Linke SP, Clarkin KC, Di Leonardo A, Tsou A, Wahl GM. A reversible p53-dependent G0/G1 cell cycle arrest induced by ribonucleotide depletion in the absence of detectable DNA damage. Genes Dev. 1996;10:934–947. doi: 10.1101/gad.10.8.934. [DOI] [PubMed] [Google Scholar]
- Linke SP, Clarkin KC, Wahl GM. p53 mediates permanent arrest over multiple cell cycles in response to gamma irradiation. Cancer Res. 1997;57:1171–1179. [PubMed] [Google Scholar]
- Livingstone LR, White A, Sprouse J, Livanos E, Jacks T, Tlsty TD. Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell. 1992;70:923–935. doi: 10.1016/0092-8674(92)90243-6. [DOI] [PubMed] [Google Scholar]
- Lo CF, Fraccaro M. Letter: nuclear projections in tumour cells. Lancet. 1974;2:847. doi: 10.1016/s0140-6736(74)91119-2. [DOI] [PubMed] [Google Scholar]
- Madireddi MT, Coyne RS, Smothers JF, Mickey KM, Yao MC, Allis CD. Pdd1p, a novel chromodomain-containing protein, links heterochromatin assembly and DNA elimination in Tetrahymena. Cell. 1996;87:75–84. doi: 10.1016/s0092-8674(00)81324-0. [DOI] [PubMed] [Google Scholar]
- Manuelidis L, Borden J. Reproducible compartmentalization of individual chromosome domains in human CNS cells revealed by in situ hybridization and three-dimensional reconstruction. Chromosoma (Berl) 1988;96:397–410. doi: 10.1007/BF00303033. [DOI] [PubMed] [Google Scholar]
- Michel B, Ehrlich SD, Uzest M. DNA double-strand breaks caused by replication arrest. EMBO (Eur Mol Biol Organ) J. 1997;16:430–438. doi: 10.1093/emboj/16.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miele M, Bonatti S, Menichini P, Ottaggio L, Abbondandolo A. The presence of amplified regions affects the stability of chromosomes in drug-resistant Chinese hamster cells. Mutat Res. 1989;219:171–178. doi: 10.1016/0921-8734(89)90012-x. [DOI] [PubMed] [Google Scholar]
- O'Keefe RT, Henderson SC, Spector DL. Dynamic organization of DNA replication in mammalian cell nuclei: Spatially and temporally defined replication of chromosome-specific α-satellite DNA sequences. J Cell Biol. 1992;116:1095–1110. doi: 10.1083/jcb.116.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedeutour F, Suijkerbuijk RF, Forus A, Van de Klundert GJ, Coindre WJM, Nicolo G, Collin F, VanHaelst U, Huffermann K, et al. Complex composition and co-amplification of SAS and MDM2 in ring and giant rod marker chromosomes in well-differentiated liposarcoma. Genes Chromosom Cancer. 1994;10:85–94. doi: 10.1002/gcc.2870100203. [DOI] [PubMed] [Google Scholar]
- Roser M, Bohm A, Oldigs M, Weichenthal M, Reimers U, Schmidt PU, Breitbart EW, Rudiger HW. Ultraviolet-induced formation of micronuclei and sister chromatid exchange in cultured fibroblasts of patients with cutaneous malignant melanoma. Cancer Genet Cytogenet. 1989;41:129–137. doi: 10.1016/0165-4608(89)90117-9. [DOI] [PubMed] [Google Scholar]
- Ruddle FH. Nuclear bleb: a stable interphase marker in established line of cells in vitro. J Natl Cancer Inst. 1962;28:1247–1251. [PubMed] [Google Scholar]
- Satoh MS, Lindahl T. Role of poly (ADP-ribose) formation in DNA repair. Nature. 1992;356:356–358. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- Schubert I, Oud JL. There is an upper limit of chromosome size for normal development of an organism. Cell. 1997;88:515–520. doi: 10.1016/s0092-8674(00)81891-7. [DOI] [PubMed] [Google Scholar]
- Shima H, Nakayasu M, Aonuma S, Sugimura T, Nagao M. Loss of the MYC gene amplified in human HL-60 cells after treatment with inhibitors of poly (ADP-ribose) polymerase or with dimethyl sulfoxide. Proc Natl Acad Sci USA. 1989;86:7442–7445. doi: 10.1073/pnas.86.19.7442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu N, Kanda T, Wahl GM. Selective capture of acentric fragments by micronuclei provides a rapid method for purifying extrachromosomally amplified DNA. Nat Genet. 1996;12:65–71. doi: 10.1038/ng0196-65. [DOI] [PubMed] [Google Scholar]
- Shimizu N, Nakamura H, Kadota T, Kitajima K, Oda T, Hirano T, Utiyama H. Loss of amplified c-mycgenes in the spontaneously differentiated HL-60 cells. Cancer Res. 1994;54:3561–3567. [PubMed] [Google Scholar]
- Snapka RM. Gene amplification as a target for cancer chemotherapy. Oncol Res. 1992;4:145–150. [PubMed] [Google Scholar]
- Snapka RM, Varshavsky A. Loss of unstably amplified dihydrofolate reductase genes from mouse cells is greatly accelerated by hydroxyurea. Proc Natl Acad Sci USA. 1983;80:7533–7537. doi: 10.1073/pnas.80.24.7533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, S.J., J.L. Stein, J.B. Lian, T.J. Last, T. Owen, and L. McCabe. 1994. Synchronization of Normal Diploid and Transformed Mammalian Cells. J.E. Celis, editor. Academic Press, San Diego, CA. 282–287.
- Toledo F, Le RD, Buttin G, Debatisse M. Co-amplified markers alternate in megabase long chromosomal inverted repeats and cluster independently in interphase nuclei at early steps of mammalian gene amplification. EMBO (Eur Mol Biol Organ) J. 1992;11:2665–2673. doi: 10.1002/j.1460-2075.1992.tb05332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker JD, Nath J, Hando JC. Activation status of the X-chromosome in human micronucleated lymphocytes. Human Genet. 1996;97:471–475. doi: 10.1007/BF02267069. [DOI] [PubMed] [Google Scholar]
- Von Hoff D, Needham-VanDevanter D, Yucel J, Windle B, Wahl G. Amplified human MYC oncogenes localized to replicating submicroscopic circular DNA molecules. Proc Natl Acad Sci USA. 1988;85:4804–4808. doi: 10.1073/pnas.85.13.4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff DD, McGill JR, Forseth BJ, Davidson KK, Bradley TP, Van Devanter DR, Wahl GM. Elimination of extrachromosomally amplified MYC genes from human tumor cells reduces their tumorigenicity. Proc Natl Acad Sci USA. 1992;89:8165–8169. doi: 10.1073/pnas.89.17.8165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff DD, Waddelow T, Forseth B, Davidson K, Scott J, Wahl GM. Hydroxyurea accelerates the loss of extrachromosomally amplified genes from tumor cells. Cancer Res. 1991;51:6273–6279. [PubMed] [Google Scholar]
- Vourc'h C, Taruscio D, Boyle AL, Ward DC. Cell cycle-dependent distribution of telomeres, centromeres, and chromosome-specific subsatellite domains in the interphase nucleus of mouse lymphocytes. Exp Cell Res. 1993;205:142–151. doi: 10.1006/excr.1993.1068. [DOI] [PubMed] [Google Scholar]
- Wahl GM. The importance of circular DNA in mammalian gene amplification. Cancer Res. 1989;49:1333–1340. [PubMed] [Google Scholar]
- White AE, Livanos EM, Tlsty TD. Differential disruption of genomic integrity and cell cycle regulation in normal human fibroblasts by the HPV oncoproteins. Genes Dev. 1994;8:666–677. doi: 10.1101/gad.8.6.666. [DOI] [PubMed] [Google Scholar]
- White E. Tumour biology. p53, guardian of Rb. Nature. 1994;371:21–22. doi: 10.1038/371021a0. [DOI] [PubMed] [Google Scholar]
- Yin Y, Tainsky MA, Bischoff FZ, Strong LC, Wahl GM. Wild-type p53 restores cell cycle control and inhibits gene amplification in cells with mutant p53 alleles. Cell. 1992;70:937–948. doi: 10.1016/0092-8674(92)90244-7. [DOI] [PubMed] [Google Scholar]