

Abstract
Bcl-2 family members either promote or repress programmed cell death. Bax, a death-promoting member, is a pore-forming, mitochondria-associated protein whose mechanism of action is still unknown. During apoptosis, cytochrome C is released from the mitochondria into the cytosol where it binds to APAF-1, a mammalian homologue of Ced-4, and participates in the activation of caspases. The release of cytochrome C has been postulated to be a consequence of the opening of the mitochondrial permeability transition pore (PTP). We now report that Bax is sufficient to trigger the release of cytochrome C from isolated mitochondria. This pathway is distinct from the previously described calcium-inducible, cyclosporin A–sensitive PTP. Rather, the cytochrome C release induced by Bax is facilitated by Mg2+ and cannot be blocked by PTP inhibitors. These results strongly suggest the existence of two distinct mechanisms leading to cytochrome C release: one stimulated by calcium and inhibited by cyclosporin A, the other Bax dependent, Mg2+ sensitive but cyclosporin insensitive.
Keywords: apoptosis, mitochondria, Bax, cytochrome C, permeability transition
The Bcl-2 gene family includes members which can act as either inhibitors or activators of the apoptosis process (Jacobson, 1997; Kroemer, 1997a ; Reed, 1997). These proteins can form both homodimers as well as selective pairs of heterodimers, and their ability to control cell death can be interdependent although it has been shown that antiapoptotic and proapoptotic members can also function independently (Knudson and Korsmeyer, 1997). Although the function of Bcl-2 proteins is not clearly understood, it has been demonstrated that some of them, including Bcl-x, Bcl-2, and Bax, can form ion channels in synthetic lipid membranes (Antonsson et al., 1997; Minn et al., 1997; Schendel et al., 1997). Based on the crystal structure of Bcl-XL, two central hydrophobic α helices (α5 and α6) reminiscent of the α helices which constitute the pore-forming unit of colicins and diphtheria toxins (Muchmore et al., 1996) are assumed to participate in the pore-forming channel. The channels formed by Bcl-2 family proteins can be described as multiconductance (conductances larger than 1 nS have been reported for both Bax and Bcl-2), pH-sensitive, voltage-gated channels with a rather poor ion selectivity. Bcl-2 and Bcl-x channels are rather cation selective, whereas Bax is anion selective at a physiological pH (Antonsson et al., 1997; Minn et al., 1997; Schendel et al., 1997; Schlesinger et al., 1997). Differences in the amino acid composition of the α5 and α6 helices could explain the distinct electrophysiological properties of the channels formed by the pro- and antiapoptotic proteins (Schlesinger et al., 1997).
Some Bcl-2 family members are associated with intracellular membranes including the nuclear envelope, the endoplasmic reticulum, and the outer mitochondrial membrane. Membrane attachment is presumably due to a hydrophobic amino acid sequence present at the COOH terminus. Interestingly, Bax is specifically attached to mitochondrial membranes (Zha et al., 1996; Hsu et al., 1997; Wolter et al., 1997; Rosse et al., 1998) which may indicate a role in the regulation of mitochondrial function during apoptosis.
Mitochondria have been shown to play a key function in the events leading to caspase activation in cell-free systems (Newmeyer et al., 1994) as well as in many cell types undergoing apoptosis (Kroemer et al., 1997; Bossy-Wetzel et al., 1998; Rosse et al., 1998). Induction of apoptosis is, in some cells, associated with a loss of cytochrome C (Kroemer, 1997b ; Kroemer et al., 1997; Bossy-Wetzel et al., 1998) and possibly other mitochondrial proteins such as apoptosis-inducing factor (AIF) (Zamzami et al., 1996). In the cytosol, cytochrome C forms a complex with Apaf1, a homologue of the Caenorhabditis elegans protein ced-4, and caspase 9, which triggers caspase activation and cell death (Li et al., 1997).
It has been hypothesized that the leakage of cytochrome C from the mitochondria into the cytosol, results from the opening of a mitochondrial channel termed the permeability transition pore (PTP)1 (Zamzami et al., 1995, 1996; Marchetti et al., 1996; Kroemer, 1997a ,b). The PTP functions as a calcium-, voltage-, pH-, and redox-gated channel with several levels of conductance (up to 1.3 nS), poor ion selectivity, and allows the release of solutes with molecular masses up to 1,500 Da (Zoratti and Szabo, 1995; Bernardi and Petronilli, 1996; Ichas et al., 1997). The PTP is thought to be composed of several proteins including hexokinase, the voltage-dependent anion channel present on the outer membrane, the inner membrane adenine nucleotide translocator (ANT), and the matrix cyclophilin D (Zoratti and Szabo, 1995; Nicolli et al., 1996; Bernardi and Petronilli, 1996; Beutner et al., 1996; Brustovetsky and Klingenberg, 1996; Halestrap et al., 1997). Importantly, Bax but not Bcl-2 or Bcl-x, was recently found to copurify with this complex (Marzo et al., 1998). Moreover, Bax triggers the release of cytochrome C from isolated mitochondria after overexpression in many cell types; however, how Bax triggers the release of cytochrome C is still unknown (Xiang et al., 1996; Manon et al., 1997; Juergensmeier et al., 1998; Pastorino et al., 1998; Rosse et al., 1998).
Here, we reconfirm that Bax can trigger the release of cytochrome C after overexpression in different cell types and directly when added to isolated mitochondria. The Bax-induced release of cytochrome C is potentiated by Mg2+ ions and, in contrast to earlier findings, occurs independently of the PTP both in intact cells and on isolated mitochondria.
Materials and Methods
Immunocytochemistry
HeLa cells were transfected with an expression vector (pCI; Promega Corp., Madison, WI) containing His-Bax cDNA under the control of cytomegalovirus promoter using the Fugene-6 method (Boeringer Mannheim Biochemicals, Indianapolis, IN). Cells were fixed in 4% paraformaldehyde in PBS for 10 min and permeabilized in 0.2% Triton X-100 for 10 min at room temperature. After washing, the cells were incubated with the appropriate antibodies in 5% normal goat serum. His tag was revealed with a rabbit polyclonal antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA) used at a 1:100 dilution. Cytochrome C was detected with a mouse monoclonal antibody (PharMingen, San Diego, CA) used at a 1:20 dilution. Anti HSP-70 monoclonal antibody was used at 1:200 from Affinity Bioagents Inc. (Golden, CO).
Mitochondrial Isolation
Before preparation the mice (female, strain 0F1, 10–25 wk) were starved for 16 h. The liver was flushed with PBS and homogenized by hand in 60 ml mitochondrial buffer (MB) (210 mM mannitol, 70 mM sucrose, 10 mM Hepes, pH 7.5, 1 mM EDTA) containing 0.45% BSA. The homogenate was centrifuged for 6 min at 13,000 g. The pellet was dissolved in 30 ml MB containing 0.45% BSA and centrifuged for 3 min at 1,400 g and then the supernatant was recentrifuged for 2 min at 13,000 g. The pellet was resuspended in 8 ml MB and 2 ml was layered on top of a discontinuous sucrose gradient formed by layering 21 ml 1.2 M sucrose, 10 mM Hepes, pH 7.5, 1 mM EDTA, 0.1% BSA on top of 15 ml 1.6 M sucrose, 10 mM Hepes, pH 7.5, 1 mM EDTA, 0.1% BSA. The samples were centrifuged at 27,000 rpm for 2 h in a Beckman SW28 rotor (Fullerton, CA) at 4°C. Mitochondria were recovered at the interface of 1.6 and 1.2 M sucrose, washed, and then resuspended in MB containing 0.5 instead of 1 mM EDTA.
Cytochrome C Release Assay
His-tagged full-length human Bax was expressed in Escherichia coli and purified from the soluble cell fraction on Ni-NTA–agarose followed by heparin and Mono Q FPLC sepharose. The purified protein was stored at −80°C in 25 mM Tris-HCl, 30% glycerol, 0.1 mM DTT, and 1% octyl glucoside, pH 7.5. This sample was diluted 100-fold in the assay buffer to give a final concentration of 170 nM Bax. The mitochondria (100 μg protein) were incubated for 1 h at 30°C in 200 μl KCl buffer (125 mM KCl, 0.5 mM EGTA, 5 mM succinate, 10 mM Hepes-KOH, pH 7.4, 4 mM MgCl2, 5 mM Na2HPO4, 5 μM Rotenon) or 200 ml MS buffer (210 mM mannitol, 70 mM sucrose, 10 mM Hepes-NaOH, pH 7.4, 0.5 mM EGTA, 5 mM succinate, 5 μM Rotenon). The reaction mixtures were centrifuged at 13,000 g for 10 min at 4°C. Mitochondrial pellets corresponding to 5 μg mitochondrial proteins and corresponding supernatant fractions were subjected to 4–20% SDS-PAGE gel electrophoresis and analyzed by Western blotting using a rabbit anti–cytochrome C serum. Equal loading of the mitochondrial pellet was controlled with a anti–Cox-VI antibody (Molecular Probes Inc., Eugene, OR).
Results
Bax Triggers the Release of Cytochrome C After Overexpression in COS Cells
Immunofluorescence studies were designed to test whether overexpression of Bax in HeLa cells could lead to the release of cytochrome C from mitochondria into the cytosol. HeLa cells were transiently transfected with a DNA encoding a His-tagged Bax and immunostained with an anti-His antibody 15 h later. Bax immunostaining appeared as a punctuated staining (Fig. 1 B). A double immunostaining using both an anti-Bax antibody and an antibody to HSP-70, a mitochondrial marker (Fig. 1 A) revealed an overlay of the two immunostainings demonstrating that, after overexpression, Bax is strictly associated with mitochondria. Interestingly, although mitochondria appeared filamentous in nontransfected cells, they were small, round, and aggregated around the nucleus in Bax-overexpressing cells, suggesting that Bax modifies the shape as well as the cellular location of mitochondria. A double immunostaining using anti-Bax and anti–cytochrome C antibodies revealed that in 100% of the Bax-positive cells, a diffuse cytosolic cytochrome C staining was observed whereas Bax-immunonegative cells displayed a filamentous mitochondrial staining (observed in more than 10 experiments; Fig. 2, A and B). These data confirm previous results demonstrating that after Bax overexpression, cytochrome C is released from mitochondria into the cytosol (Manon et al., 1998; Partorino et al., 1998; Rosse et al., 1998). Most of these cells displayed a condensed apoptotic nucleus as indicated by Hoechst staining (Fig. 2 C). Addition of the caspase inhibitor z-VAD-fmk was able to prevent nuclear condensation but could not inhibit Bax-induced cytochrome C release, confirming that caspases are activated downstream of cytochrome C release (Fig. 2, D–F).
Figure 1.

Mitochondrial localization of Bax. HeLa cells were transiently transformed with cDNA encoding a His-tagged Bax and immunostained 15 h later for both the mitochondrial marker HSP-70 (A) and Bax (B). Note that the shape as well as the distribution of the mitochondria has change in the Bax-transfected cell (arrow).
Figure 2.

Immunofluorescence studies of cytochrome C release from mitochondria after overexpression of Bax in HeLa cells. HeLa cells were transiently transfected with a cDNA encoding a His-tagged human Bax and cultured for 15 h in the presence (D–F) or absence (A–C) of z-VAD-fmk before immunostaining for Bax (A and D) and cytochrome C (B and E). C and F are nuclear Hoechst stainings. Arrows, transfected cells.
Bax Induces the Release of Cytochrome C from Isolated Mitochondria
To establish whether the release of cytochrome C was a direct or an indirect effect of Bax on the mitochondria, we tested whether pure recombinant Bax alone was sufficient to trigger the release of cytochrome C from freshly isolated mouse liver mitochondria. Mitochondria energized through complex II with succinate were incubated with 170 nM full-length Bax, 5 μM Bcl-2ΔTM, or 5 μM BaxΔTM for 1 h (Bcl-2ΔTM and BaxΔTM, both proteins lacking the COOH-terminal hydrophilic domain, had been shown to form ion channels in synthetic lipid membranes [Antonsson et al., 1997; Schendel et al., 1997]). The cytochrome C release was subsequently analyzed by Western blotting of both the mitochondrial pellet and the supernatant (Fig. 3 A). Compared with the total amount of cytochrome C contained within mitochondria recovered from the sucrose gradient, we estimated that less than 5% of the total amount of cytochrome C was released from untreated mitochondria after 1 h of incubation under the conditions described above. In contrast, both Bax and BaxΔTM-treated mitochondria had lost 80–95% cytochrome C. Such a release was obtained in 10 independent experiments using three different Bax preparations. Full-length Bax was estimated to be at least ten times more efficient than BaxΔTM (data not shown). Similar results were obtained when pyruvate and malate, instead of succinate, were used as a respiratory substrate (data not shown).
Figure 3.
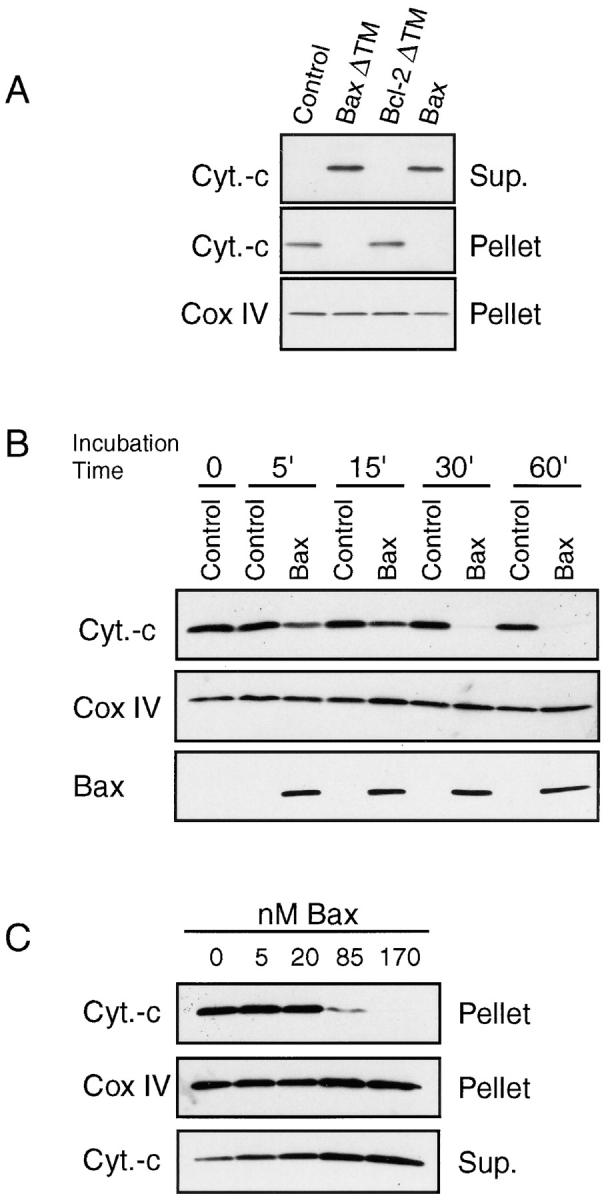
Bax-induced cytochrome C release from isolated mitochondria. (A) Mitochondria isolated from mouse liver were incubated for 1 h at 30°C with 5 μM COOH-terminal truncated Bax (BaxΔTM), 5 μM COOH-terminal truncated Bcl-2 or 170 nM full-length Bax in 200 μl KCl buffer. After 1 h, the reaction mixture was centrifuged at 13,000 g for 5 min. Supernatant and mitochondrial pellets corresponding to 5 μg mitochondrial proteins were subjected to 4–20% SDS-PAGE and analyzed by Western blot. Loading for the mitochondrial pellet was controlled with a Cox IV antibody. (B) Kinetics of Bax-induced cytochrome C release. Mitochondria were incubated with 170 nM Bax or buffer control at 30°C and the mitochondria pellet was analysed over time as in A. In addition to cytochrome C and Cox IV, Bax levels were measured in the mitochondrial pellets. (C) Mitochondria were incubated with different amounts of Bax for 1 h at 30°C and cytochrome C release was measured as in A. Data shown are representative of 3–12 independent experiments performed with at least three different preparations of Bax protein.
Bax Triggers the Release of Cytochrome C from Mitochondria in a Dose- and Time-dependent Manner
The release of cytochrome C was dependent on the concentration of Bax protein, increasing from 5 to 170 nM (Fig. 3 C). Kinetic experiments performed with 170 nM Bax showed that the release of cytochrome C was already detectable after 5 min of incubation and was almost complete after 30 min (Fig. 3 B). Importantly, we noticed during these experiments that the amount of Bax associated with mitochondria remained constant between 5 and 60 min, suggesting that the binding of Bax to mitochondria is a rapid and saturable process. It is of note that levels of endogenous Bax in mouse liver mitochondria were low and only weakly detectable by Western blotting. The amount of Bax associated to mitochondria after adding 170 nM during the different experiments was similar to the level of endogenous Bax found on mitochondria isolated from HeLa cells (data not shown). This finding indicates that the Bax levels used in these studies were within the physiological range.
Bax Effect Is Highly Potentiated by Mg2+ Ions
Several ions were tested for their ability to influence Bax-induced release of cytochrome C from mitochondria (Fig. 4). Sucrose and mannose were used to adjust the osmolarity of all incubation buffers to 300–330 mOsm. Without the addition of any salt, Bax failed to induce the release of cytochrome C (Fig. 4, A and B). However, under these conditions the mitochondria are not respiring due to the lack of ATP production that needs magnesium and phosphate. Therefore, we added 4 mM MgCl2 and 5 mM Na2HPO4, and now Bax was able to trigger >90% cytochrome C release from the mitochondria (Fig. 4 A). Addition of MgCl2 or Na2HPO4 separately showed that MgCl2 induced a strong Bax effect (Fig. 4 B), indicating that the ions and not the respiration were needed for the Bax effect. This was further confirmed as Bax was not inhibited by antimycin, a complex 3 inhibitor (data not shown). NaCl was ineffective demonstrating a key role for Mg2+ ions in this effect (data not shown). As Mg2+ ions have been reported to inhibit the PTP (Zoratti and Szabo, 1995), these results suggested that Bax and the PTP were two separate entities and we therefore further tested this hypothesis using PTP blockers.
Figure 4.

Bax effect is ion dependent. Mitochondria isolated from mouse liver cells were incubated with 170 nM Bax at 30°C in sucrose-mannose buffer with or without addition of salts as indicated in the figure. The salt concentrations were 4 mM MgCl2 and 5 mM Na2HPO4. Cytochrome C release was measured after 1 h as described in Fig. 3.
Bax Triggers the Release of Cytochrome C from Mitochondria Independently of Several Components of the PTP
Opening of the PTP has been proposed to be involved in the release of cytochrome C during apoptosis (Zamzami et al., 1995,1996; Marchetti et al., 1996; Kroemer, 1997a ). Here, we confirmed that calcium, a known PTP opener, triggers cytochrome C release from isolated mitochondria as efficiently as Bax (Fig. 5 A) (Kantrow and Piantadosi, 1997). We then tested whether the Bax effect was mediated through opening of the PTP by using inhibitors.
Figure 5.
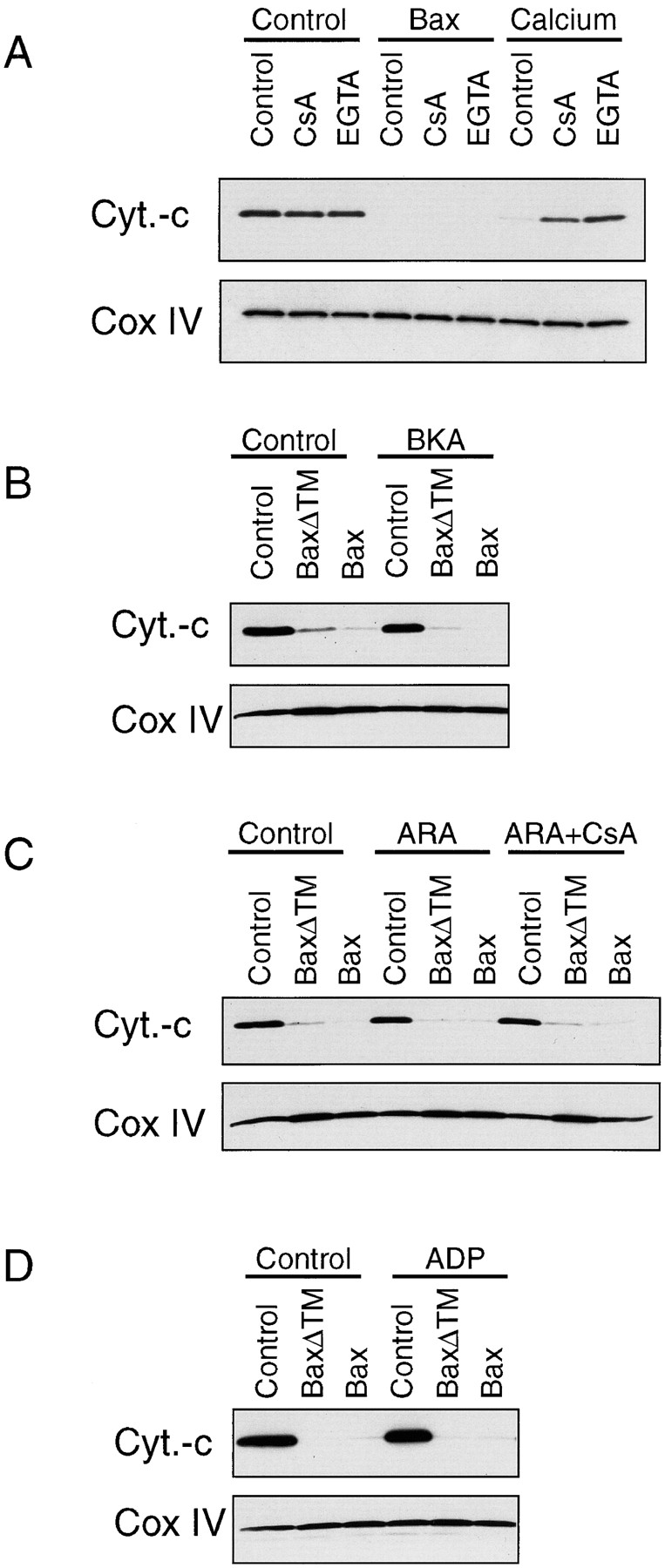
Bax-induced release of cytochrome C from isolated mitochondria is not inhibited by PTP blockers. Several PTP blockers have been tested for their ability to inhibit the release of cytochrome C from isolated mitochondria induced by 170 nM Bax. (A) We compared Bax effect with that of calcium, a PTP opener, and their sensitivity to 10 μM CsA and 0.5 mM EGTA. Cytochrome C and Cox IV in the mitochondrial pellet were measured after a 1-h incubation in KCl buffer at 30°C by Western blotting. The PTP inhibitors BKA (100 μM) (B), a combination of CsA (10 μM) and ARA (50 μM) (C) and ADP (10 μM) (D) were also tested. Mitochondria were preincubated with these compounds for 5 min before addition of Bax. Note that although CsA and EGTA were capable of inhibiting the calcium effect, they failed to block Bax-induced cytochrome C release.
Blockers of the PTP including bongkrekic acid (BKA), CsA, ADP, and EGTA were tested for their ability to inhibit Bax-induced release of cytochrome C from isolated mitochondria. As shown in Fig. 5, none of these compounds was able to block this Bax action. The combination of CsA with the phospholipase A2 inhibitor arachistolic acid (ARA), reported to act as a super-PTP inhibitor (Pastorino et al., 1998), also failed to inhibit the Bax effect. It is of note that all these compounds were able to inhibit the release of cytochrome C induced by the known PTP-opener calcium (Fig. 5 A and data not shown).
PTP blockers were also tested for their ability to inhibit the release of cytochrome C from mitochondria after overexpression of Bax in COS cells (Fig. 6). COS cells cultured in the presence of 10 μM cyclosporin A or 100 μM BKA were transfected with Bax, and cytochrome C release was analyzed 15 h later. These experiments were performed in the presence of z-VAD-fmk to inhibit apoptosis induced by cyclosporin A itself or by Bax. In three separate experiments we found that 100% of the Bax-positive cells displayed a diffuse cytosolic cytochrome C staining. Therefore, as found with isolated mitochondria, neither CsA nor BKA were able to inhibit Bax-induced release of cytochrome C in intact cells (Fig. 6).
Figure 6.

Both Cyclosporin A and BKA fail to inhibit Bax-induced release of cytochrome C in COS cells. COS cells were transfected with a cDNA encoding His-Bax and cultured for 15 h in the presence of 10 μM CsA and 100 μM z-VAD-fmk (A and B) or 100 μM BKA and 100 μM z-VAD-fmk (C and D) before immunostaining for Bax (A and C) and cytochrome C (B and D). Note that all cells that overexpress Bax display a diffuse cytosolic cytochrome C immunostaining. Arrows, transfected cells.
Discussion
During apoptosis of many cell types, cytochrome C has been shown to be released from mitochondria into the cytosol, an event that leads to caspase activation (Kluck et al., 1997; Yang et al., 1997; Li et al., 1997). Although the mechanisms by which cytochrome C is released are not yet understood, more and more evidence suggest that Bax, a channel-forming protein localized on mitochondria, could play a key role in this event. Here, we confirm that both overexpressed Bax or purified Bax added to isolated mitochondria is sufficient to induce release of cytochrome C (Vander Heiden et al., 1997; Rosse et al., 1998), suggesting that Bax could be the component required for cytochrome C release during apoptosis. The release of cytochrome C has been attributed to opening of the PTP, a hypothesis based on many data reporting that opening of the PTP results in Δψm collapse and leakage of many mitochondrial proteins including cytochrome C (Kroemer, 1997a ,b; Kroemer et al., 1997; Marzo et al., 1998; Pastorino et al., 1998). Furthermore Juergensmeier et al. (1998) claimed that cyclosporin A was able to inhibit Bax-induced cytochrome C release from isolated mitochondria. Our results are not in agreement with such a hypothesis as we found that Bax-induced release of cytochrome C from isolated mitochondria could not be prevented by several PTP blockers, including ADP, Mg2+, cyclosporin A, and BKA. Differences in the experimental procedures could explain these conflicting data. In the study of Juergensmeier et al. (1998), mitochondria were incubated for 1 h in a buffer devoid of calcium chelator whereas our experiments were performed in a buffer containing 0.5 mM EGTA. EGTA was used to prevent opening of the PTP by free calcium present as traces in the used buffers. Indeed, we noticed that in the absence of a calcium chelator, mitochondria had a tendency to release spontaneously high levels of cytochrome C (data not shown), an effect that was completely inhibited by EGTA. Interestingly, cyclosporin A had a similar effect as EGTA. Therefore, we strongly believe that in the study of Juergensmeier et al. (1998), CsA is not inhibiting Bax but rather the calcium-activated PTP. We hypothesize that the Bax-induced release of cytochrome C is accompanied by a leakage of calcium from the mitochondria which, in absence of EGTA leads to calcium cycling (Richter, 1993). This calcium cycling would result in PTP opening of the least resistant mitochondria, since mitochondria are not a homogeneous population (Ichas et al., 1997). As these mitochondria undergo permeability transition (PT) they release cytochrome C and calcium into the buffer thereby further increasing the calcium concentration. This would induce more mitochondria to undergo PT starting a positive feedback loop in a similar way as described by Ichas et al. (1997). Thus by adding EGTA to our incubation buffer, we were able to separate the Bax effect from the PTP/calcium effect and therefore, this strongly suggests that Bax acts on the mitochondria independently of several components of the PTP.
Supporting our results obtained with isolated mitochondria, we found that neither cyclosporin A nor BKA, a long-term inhibitor of the PTP, were able to inhibit Bax-induced cytochrome C release after overexpression in COS cells. These results suggest that Bax triggers cytochrome C release independently of at least two major components of the PTP, cyclophilin D (cyclosporin A target), and the ANT (BKA target). These results, however, do not eliminate the possibility that Bax could interact with other components of the PTP as it was recently shown that Bax copurifies with proteins participating in the PTP complex (Marzo et al., 1998).
We have also demonstrated that Bax effect is highly dependent on Mg2+ ions. Among several functions, Mg2+ ions stimulate ATP synthase activity and inhibit the F1-ATPase in the presence of ADP (Boyer, 1997). Thus, it is possible that Bax is active only when the ATP synthase is stimulated, a hypothesis consistent with recent results by Matsuyama et al. (1998) demonstrating a requirement for the mitochondrial F0F1-ATPase proton pump for Bax function in yeast and mammalian cells. How the mitochondrial proton pump activity could contribute to cytochrome C release from mitochondria and Bax-killing activity is, however, unknown. Recently, Bcl-2 was shown to prevent loss of mitochondrial membrane potential, an early event during apoptosis, by stimulating proton efflux (Shimizu et al., 1998). One can hypothesize that both Bax and Bcl-2 act as proton channels: Bcl-2 allowing proton efflux whereas Bax would stimulate proton influx, either directly, being itself a proton channel, or indirectly, through the ATP synthase.
In conclusion, we have reported that Bax can trigger the release of cytochrome C an event occurring early during apoptosis, independently of the activity of the PTP. As a working hypothesis, we propose that during apoptosis the permeability of the mitochondrial membrane is not severely affected, thereby allowing a specific release of a small number of proteins as cytochrome C and possibly AIF into the cytosol. In clear contrast, during necrosis, the permeability of the mitochondrial membrane is severely altered leading to the nonspecific release of proteins into the cytosol, an event consistent with mitochondrial membrane disruption (Vander Heiden et al., 1997).
Acknowledgments
We thank S. Arkinstall for critical reading of the manuscript, T. Wells for encouraging support, and C. Herbert for artwork (all three from Serono Pharmaceutical Research Institute).
Abbreviations used in this paper
- BKA
bongkrekic acid
- CsA
cyclosporin A
- MB
mitochondrial buffer
- PTP
permeability transition pore
- PT
permeability
Footnotes
Address all correspondence to J.-C. Martinou, Serono Pharmaceutical Research Institute, 14 Chemin des Aux, 1228 Plan les Ouates, Geneva, Switzerland. Tel.: (41) 22 706 9822. Fax: (41) 22 794 6965. E-mail: jean-claude.martinou.ch_gva03@serono.com
References
- Antonson B, Conti F, Ciavatta AM, Montesuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod J-J, Mazzei G, Maundrell K, Gambale F, Sadoul R, Martinou J-C. Inhibition of Bax channel forming activity by Bcl-2. Science. 1997;277:370–372. doi: 10.1126/science.277.5324.370. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Petronilli V. The permeability transition pore as a mitochondrial calcium release channel; a critical appraisal. J Bioenerg Biomembr. 1996;28:129–136. doi: 10.1007/BF02110643. [DOI] [PubMed] [Google Scholar]
- Beutner G, Rueck A, Riede B, Welte W, Brdiczka D. Complexes between kinases, mitochondrial porin, and adenylate tranlocator in rat brain resemble the permeability transition pore. FEBS (Fed Eur Biochem Soc) Lett. 1996;396:189–195. doi: 10.1016/0014-5793(96)01092-7. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Newmeyer DD, Green DR. Mitochondrial cytochrome C release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO (Eur Mol Biol Organ) J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer PD. The ATP synthase—a splendid molecular machine. Annu Rev Biochem. 1997;66:717–749. doi: 10.1146/annurev.biochem.66.1.717. [DOI] [PubMed] [Google Scholar]
- Brustovetsky N, Klingenberg M. Mitochondrial ADP/ATP carrier can be reversible converted into a large channel by Ca2+ . Biochemistry. 1996;35:8483–8488. doi: 10.1021/bi960833v. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Woodfield K-Y, Connern CP. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocator. J Biol Chem. 1997;272:3346–3354. doi: 10.1074/jbc.272.6.3346. [DOI] [PubMed] [Google Scholar]
- Hsu Y-T, Wolter KG, Youle RJ. Cytosol-to-membrane redistribution of Bax and Bcl-XLduring apoptosis. Proc Natl Acad Sci USA. 1997;94:3668–3672. doi: 10.1073/pnas.94.8.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichas F, Jouaville LS, Matzat J-P. Mitochondria are excitable organelles capable of generating and conveying electric and calcium currents. Cell. 1997;89:1145–1153. doi: 10.1016/s0092-8674(00)80301-3. [DOI] [PubMed] [Google Scholar]
- Jacobson MD. Bcl-2-related proteins get connected. Curr Biol. 1997;7:R277–R281. doi: 10.1016/s0960-9822(06)00136-9. [DOI] [PubMed] [Google Scholar]
- Juergensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome C from isolated mitochondria. Proc Natl Acad Sci USA. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantrow SP, Piantadosi CA. Release of cytochrome C from liver mitochondria during permeability transition. Biochem Biophys Res Comm. 1997;232:669–671. doi: 10.1006/bbrc.1997.6353. [DOI] [PubMed] [Google Scholar]
- Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome C from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Knudson CM, Korsmeyer SJ. Bcl-2 and Bax function independently to regulate death. Nat Genet. 1997;16:358–363. doi: 10.1038/ng0897-358. [DOI] [PubMed] [Google Scholar]
- Kroemer G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med. 1997a;3:614–620. doi: 10.1038/nm0697-614. [DOI] [PubMed] [Google Scholar]
- Kroemer G. Mitochondrial implication in apoptosis. Towards an endosymbiont hypothesis of apoptosis evolution. Cell Death Diff. 1997b;4:443–456. doi: 10.1038/sj.cdd.4400266. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Zamzami N, Susin SA. Mitochondrial control of apoptosis. Immunol Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome C and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Manon S, Chaudhuri B, Guerin M. Release of cytochrome C and the decrease of cytochrome C oxidase in Bax-expression yeast cells, and prevention of these effects by coexpression of Bcl-xL . FEBS (Fed Eur Biochem Soc) Lett. 1997;415:29–32. doi: 10.1016/s0014-5793(97)01087-9. [DOI] [PubMed] [Google Scholar]
- Marchetti P, Castedo M, Susin SA, Zamzami N, Hirsch T, Haeffner A, Hirsch F, Geuskens M, Kroemer G. Mitochondrial permeability transition is a central coordinating event of apoptosis. J Exp Med. 1996;184:1155–1160. doi: 10.1084/jem.184.3.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo I, Brenner C, Zamzami N, Susin SA, Beutner G, Brdiczka D, Remy R, Xie Z-H, Reed JC, Kroemer G. The permeability transition pore complex: a target for apoptosis regulation by caspases and Bcl-2-related proteins. J Exp Med. 1998;187:1261–1271. doi: 10.1084/jem.187.8.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S, Xu Q, Velours J, Reed JC. The mitochondrial F0F1-ATPase proton pump is required for function of the proapoptotic protein Bax in yeast and mammalian cells. Mol Cell. 1998;1:327–336. doi: 10.1016/s1097-2765(00)80033-7. [DOI] [PubMed] [Google Scholar]
- Minn AJ, Velez P, Schendal SL, Liang H, Muchmore SW, Fesik SW, Fill M, Thompson CB. Bcl-xLforms an ion channel in synthetic lipid membranes. Nature. 1997;385:353–357. doi: 10.1038/385353a0. [DOI] [PubMed] [Google Scholar]
- Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, Nettesheim D, Chang BS, Thompson CB, Wong S-L, et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 1996;381:335–341. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- Newmeyer DD, Farschon DM, Reed JC. Cell-free apoptosis in Xenopusegg extracts: inhibition by Bcl-2 and requirement for an organelle fraction enriched in mitochondria. Cell. 1994;79:353–364. doi: 10.1016/0092-8674(94)90203-8. [DOI] [PubMed] [Google Scholar]
- Nicolli A, Basso E, Petronilli V, Wenger RM, Bernardi P. Interactions of cyclophilin with mitochondrial inner membrane and regulation of the permeability transition pore, a cyclosporin A-sensitive channel. J Biol Chem. 1996;271:2185–2192. doi: 10.1074/jbc.271.4.2185. [DOI] [PubMed] [Google Scholar]
- Pastorino JG, Chen S-T, Tafani M, Snyder JW, Farber JL. The over expression of Bax produces cell death upon induction of the mitochondrial permeability transition. J Biol Chem. 1998;273:7770–7775. doi: 10.1074/jbc.273.13.7770. [DOI] [PubMed] [Google Scholar]
- Reed JC. Double identity for proteins of the Bcl-2 family. Nature. 1997;387:773–776. doi: 10.1038/42867. [DOI] [PubMed] [Google Scholar]
- Richter C. Pro-oxidants and mitochondrial Ca2+: their relationship to apoptosis and oncogenesis. FEBS (Fed Eur Biochem Soc) Lett. 1993;325:104–107. doi: 10.1016/0014-5793(93)81423-w. [DOI] [PubMed] [Google Scholar]
- Rosse T, Olivier R, Monney L, Rager M, Conus S, Fellay I, Jansen B, Borner C. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome C. Nature. 1998;391:496–499. doi: 10.1038/35160. [DOI] [PubMed] [Google Scholar]
- Schendel SL, Xie Z, Montal MO, Matsuyama S, Montal M, Reed J. Channel formation by antiapoptotic protein Bcl-2. Proc Natl Acad Sci USA. 1997;94:5113–5118. doi: 10.1073/pnas.94.10.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger PH, Gross A, Yin X-M, Yamamoto K, Siato M, Waksman G, Korsmeyer SJ. Comparison of the ion channel characteristics of proapoptotic BAX and antiapoptotic BCL-2. Proc Natl Acad Sci USA. 1997;94:11357–11362. doi: 10.1073/pnas.94.21.11357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S, Eguchi Y, Kamiike W, Funahashi Y, Mignon A, Lacronique V, Matsuda H, Tsujimoto Y. Bcl-2 prevents apoptotic mitochondrial dysfunction by regulating proton flux. Proc Natl Acad Sci USA. 1998;95:1455–1459. doi: 10.1073/pnas.95.4.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden, M.G., N.S. Chandel, E.K. Williamson, P.T. Schumacker, and C.B. Thompson. Bcl-xlregulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- Wolter KG, Hsu Y-T, Smith CL, Nechushtan A, Xi X-G, Youle RJ. Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang J, Chao DT, Korsmeyer ST. Bax-induced cell death may not require interleukin 1β-converting enzyme-like proteases. Proc Natl Acad Sci USA. 1996;93:14559–14563. doi: 10.1073/pnas.93.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cia J, Peng T-I, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: Release of cytochrome C from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Migotte B, Kroemer G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med. 1995;182:367–377. doi: 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamzami N, Susin SA, Marchetti P, Hirsch T, Gomez-Monterrey I, Castedo M, Kroemer G. Mitochondrial control of nuclear apoptosis. J Exp Med. 1996;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha H, Fisk HA, Yaffe MP, Mahajan N, Herman B, Reed JC. Structure function comparisons of the proapoptotic protein Bax in Yeast and mammalian cells. Mol Cell Biol. 1996;16:6494–6508. doi: 10.1128/mcb.16.11.6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoratti M, Szabo I. The mitochondrial permeability transition, . Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]