

Abstract
Previously we reported that annexin VI is required for the budding of clathrin-coated pits from human fibroblast plasma membranes in vitro. Here we show that annexin VI bound to the NH2-terminal 28-kD portion of membrane spectrin is as effective as cytosolic annexin VI in supporting coated pit budding. Annexin VI–dependent budding is accompanied by the loss of ∼50% of the spectrin from the membrane and is blocked by the cysteine protease inhibitor N-acetyl-leucyl-leucyl-norleucinal (ALLN). Incubation of fibroblasts in the presence of ALLN initially blocks the uptake of low density lipoprotein (LDL), but the cells recover after 1 h and internalize LDL with normal kinetics. The LDL internalized under these conditions, however, fails to migrate to the center of the cell and is not degraded. ALLN-treated cells have twice as many coated pits and twofold more membrane clathrin, suggesting that new coated pits have assembled. Annexin VI is not required for the budding of these new coated pits and ALLN does not inhibit. Finally, microinjection of a truncated annexin VI that inhibits budding in vitro has the same effect on LDL internalization as ALLN. These findings suggest that fibroblasts are able to make at least two types of coated pits, one of which requires the annexin VI–dependent activation of a cysteine protease to disconnect the clathrin lattice from the spectrin membrane cytoskeleton during the final stages of budding.
Keywords: annexin VI, coated pit, spectrin, endocytosis
Receptor-mediated endocytosis of macromolecules depends on the coordinate regulation of receptor sequestration and plasma membrane budding by clathrin-coated pits (3, 51). Coated vesicle formation begins with the assembly of the clathrin lattice and ends with the release of the nascent vesicle after membrane scission. Budding reactions like this are common during intracellular membrane traffic (48, 50) and most appear to be mediated by a coat protein complex. These coats are thought to actively shape the membrane into a vesicle. They may also remodel the lipid and cytoskeletal components of the membrane in preparation for the scission reaction.
Two in vitro systems that reconstitute clathrin-coated pit budding have provided new insights into how coated vesicles form. One system uses perforated A431 cells, which lack cytoplasm but retain many of the regulatory elements required to control receptor clustering and coated pit budding (51). The other system uses purified human fibroblast plasma membranes containing LDL receptors clustered in coated pits. When these membranes are attached to a solid substratum at 4°C, invagination of pits occurs spontaneously upon warming to 37°C. The scission step, however, requires ATP, Ca+2, and cytosol (39). One of the cytosolic proteins essential for scission in this in vitro system is annexin VI (40).
Annexin VI belongs to a family of Ca+2-dependent, phospholipid-binding proteins (16). Each annexin has a variable NH2-terminal region and a conserved COOH-terminal domain called the core (47). The core usually contains four 70-amino-acid long calcium-binding repeats. Annexin VI, however, has eight of these repeats. Several studies indicate annexins play a role in membrane traffic. Annexin VII (synexin) mediates membrane fusion between chromaffin secretory vesicles in vitro (16). Annexin II appears to be involved in membrane fusion between isolated endosomal vesicles (42), is transferred between endosomes during fusion in vitro (19), and rapidly redistributes from the basolateral surface to the apical surface during the transcytosis of glycolithocholate in hepatocytes (60). Annexin II also translocates to the plasma membrane during exocytosis (13). Annexin I is localized to multivesicular bodies and it may be involved in inward vesiculation of these membranes (21). Finally, annexin XIIIb has been implicated in vesicle targeting to the apical surface of MDCK cells (20).
Several annexins have been found to bind actin and spectrin in vitro, raising the possibility they participate in remodeling of the spectrin cytoskeleton (47). Annexin VI, for example, binds to the NH2-terminal 28-kD portion of β-spectrin and this interaction inhibits the ability of spectrin to cross-link actin (59). Spectrin, which is an extended, rod-shaped tetramer composed of two α and two β subunits, forms a cytoskeletal meshwork with actin and other accessory proteins that lines the inner surface of most metazoan cells (10, 24). Several studies suggest that remodeling of this meshwork precedes exocytosis. Stimulation of exocytosis in chromaffin cells causes the depolymerization of membrane-associated F-actin and the reorganization of spectrin into discrete patches on the plasma membrane (7, 56). Antibodies to α-spectrin inhibit exocytosis in these cells, suggesting spectrin must be displaced from sites where vesicles dock and fuse (46). The removal of spectrin may be mediated by the cysteine protease calpain I. Calpain I cleaves both the α and β subunits of brain spectrin, producing subunits that are no longer able to form tetramers or interact with actin (31). Limited proteolysis by calpain also inhibits spectrin from associating with membrane-binding sites (34).
The remodeling of the spectrin–actin cytoskeleton may also be required for endocytosis. For example, endocytosis in erythrocyte ghosts, which is not clathrin mediated, is preceded by the formation of spectrin-free zones at sites of membrane budding (29, 30). It is not known if there is a similar requirement during coated pit budding. However, the recent localization of calpain I to isolated coated vesicles (49) together with the finding that annexin VI binds spectrin (59) and is required for clathrin-coated vesicle budding in vitro (40), suggests a reorganization of the spectrin cytoskeleton might be part of the clathrin budding reaction. Here we report that the binding of annexin VI to spectrin causes the cysteine protease-dependent loss of membrane spectrin. Reagents that block spectrin loss prevent budding both in vivo and in vitro. Cells adapt to the presence of this class of inhibitors by forming a new set of coated pits that are able to bud independently of annexin VI and spectrin removal. The endosomes that form from these pits, however, are unable to carry low density lipoprotein (LDL)1 to the center of the cell and LDL is not degraded.
Materials and Methods
Materials
Coverslips (12-mm round) were obtained from Fisher Scientific Co. (Pittsburgh, PA). 125I-labeled streptavidin (IM 236) was purchased from Amersham Pharmacia Biotech., Inc. (Piscataway, NJ). The following tissue culture agents were obtained from Gibco Laboratories (Grand Island, NY): Dulbecco's modified Eagle's medium (DME, 320–885), Eagle's minimal essential medium (EMEM; 330–1,435), fetal calf serum (FCS; 200–6,140), and penicillin-streptomycin (600–5,145). Human lipoprotein-deficient serum (H-LPPS) was prepared by ultracentrifugation of human plasma (22). SlowFade™ light antifade kit was obtained from Molecular Probes, Inc. (Eugene, OR). Isopropyl-β-d-thiogalactopyranoside was obtained from Stratagene (La Jolla, CA). The following reagents were obtained from Sigma Chemical Co. (St. Louis, MO): poly-l-lysine (P-1524), Hepes (H-3372), PMSF (P-7626), BSA fraction V (A-7906), crystalline BSA (c-BSA; A-7638), ATP (A-5394), thrombin (T-7513), and glutathione-agarose (G-4510). Leupeptin (62070), dithiothreitol (DTT; 43819) and paraformaldehyde (76240) were obtained from Fluka Chemika-Biochemika (Ronkonkoma, NY). Human recombinant calpastatin (208900) and calpain I inhibitor (208719, N-acetyl-leucyl-leucyl-norleucinal [ALLN]) was purchased from Calbiochem-Novabiochem (La Jolla, CA). Biotinylated horse anti–mouse IgG (BA-2000) was purchased from Vector Labs, Inc. (Burlingame, CA). The mouse mAbs used in these experiments were: anticlathrin heavy chain (X-22), provided by F. Brodsky [University of California, San Francisco, CA]; anti–annexin VI IgG from Zymed Labs, Inc. (03-4900; South San Francisco, CA) and antispectrin IgG from Sigma Chemical Co. (S-3396). Rabbit pAb to spectrin was from Sigma Chemical Co. (S-1515). The 125I-labeled LDL was prepared as previously described (22). Fluorescent LDL (pMCA-oleate LDL) was prepared as previously described (2).
Methods
Media and Buffers.
Medium A: DME containing 10% FCS, 20 mM Hepes, pH 7.4, 100 U/ml penicillin, and 100 μg/ml streptomycin. Medium B: DME containing 10% FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Medium C: EMEM containing 20 mM Hepes, pH 7.4. Buffer A: 50 mM Hepes-NaOH, pH 7.4, 100 mM NaCl. Buffer B: 25 mM Hepes-KOH, pH 7.0, 25 mM KCl, 2.5 mM magnesium acetate, 0.2 mM DTT. Buffer C: 2.3 mM NaH2PO4·H2O, 7.7 mM Na2HPO4·7 H2O, 150 mM NaCl, 2 mM MgCl2, pH 7.4.
Cell Culture.
All the in vitro experiments used SV-40–transformed human fibroblasts (designated SV589) as previously described. All in vivo experiments used normal human fibroblasts. SV589 cells were grown as monolayers in medium A and set up for experiments according to a standard format (39). Normal human fibroblasts were grown in medium B and induced to express LDL receptors by replacing the fetal calf serum with human lipoprotein deficient serum as previously described (22).
Recombinant Proteins.
Plasmids encoding COOH-terminal deletions of annexin VI fused with glutathione-S-transferase (GST) were constructed by selective PCR amplification of the corresponding cDNA sequences using oligonucleotides containing flanking restriction sites. The resulting PCR fragments were first cloned into the pCR II vector (Stratagene, La Jolla, CA) and then into the GST expression vector pGEX-KG (28). Oligonucleotide primers used for PCR were: AnxVIwt (residues 1–673), oligonucleotides A (TCTAGACATGGCCAAACCAGCACAGGG) and B (CGCGTCGACCTAGTCCTCACCACCACAGAGAGC); AnxVIΔ379 (residues 1–379), oligonucleotide A and C (CGCGTCGACCTAGAGTCCCTTCATGGCTTTCCG); and AnxVIΔ192 (residues 1–192), oligonucleotide A and D (CGCGTCGACCTACCAT TTCAGTTCCCCTGCCTC). PCR fragments were digested with XbaI and SalI and cloned into pGEX-KG. Recombinant proteins were purified by affinity chromatography on glutathione-agarose beads as previously described (58), with the modification that the purified fusion protein was released from glutathione beads by digesting with thrombin (16 U/ml of beads) for 1 h at room temperature. The released protein was dialyzed in buffer B overnight at 4°C and stored at −80°C.
Plasmid encoding residues 1–272 of β-spectrin fused with GST was constructed by PCR amplification from an NH2-terminal fragment of human β-spectrin (β 5.1) generously provided by V. Bennett (Duke University, Durham, NC) (34). The oligonucleotides used to clone β-spec1–272 were: CGGGATCCCGTGAGCTGA AGCAGGGCA and CCCAAGCTTCTATTGTCTTCAGTTTCC. PCR fragments were digested with BamHI and HindIII and then cloned into pGEX-KG. The proteins were then purified by affinity chromatography as described above.
In Vitro Budding Assay.
Membranes were prepared as previously described (39). In brief, SV589 cells were attached to poly-l-lysine–coated coverslips by centrifugation (1,800 g) at 4°C, incubated with ice-cold medium C containing 2% BSA for 1 h, washed quickly with buffer A and buffer B at 4°C, and then sonicated in buffer B on ice. The attached membranes were washed three times with ice-cold buffer B and either fixed immediately or treated as indicated. Previously we used annexin VI–depleted bovine cytosol containing 1 mM ATP and 150 μM calcium as the starting material for all budding reactions (40). We noted while doing those studies, however, that often during the preparation of cytosol the annexin VI budding activity was lost. We took advantage of this property in the current study and always used as our starting material bovine brain cytosol lacking annexin VI budding activity. To initiate budding, membranes were incubated for 10 min at 37°C in the presence of the inactive bovine brain cytosol (we refer to this as cytosol throughout the paper) that had been dialyzed against buffer B (final concentration in buffer B, 10 mg/ml) plus 1 mM ATP, 150 μM Ca+2 and the indicated additions. The membranes were then washed three times with ice-cold buffer B and fixed with 3% paraformaldehyde in buffer B lacking DTT for 15 min at 4°C, rinsed twice in buffer C, and then held in buffer C at 4°C until assayed.
Indirect 125I-labeled Streptavidin-binding Assays.
Coated pit budding was monitored by measuring the amount of clathrin on the membrane as previously described (39). The same indirect method was used to measure the amount of spectrin and annexin VI on membranes. The primary mAb antibodies were X-22 (1 μg/ml), anti-annexin VI (1 μg/ml) and antispectrin (4.2 μg/ml). Bound antibodies were detected using biotinylated horse anti–mouse IgG (1 μg/ml), and 125I-labeled streptavidin (1 μCi/ml).
LDL Uptake Assay.
The uptake of 125I-labeled LDL (22) and fluorescent LDL (2) were measured using standard methods. All measurements were made on normal human fibroblasts induced to express LDL receptors for 2 d. For fluorescence detection, the cells were fixed with 3% formaldehyde in buffer C, mounted in SlowFade™, and then visualized with a Zeiss photomicroscope III.
Microinjection.
Normal human fibroblasts were induced to express LDL receptors as described (22). The cells were placed in medium C and then comicroinjected with FITC-dextran (3 mg/ml) and the indicated protein using an Eppendorf automated microinjection system (model 5171; Eppendorf Scientific, Inc., Hamburg, Germany). The cells were allowed to recover for 1 h at 37°C in medium B before the uptake of fluorescent-LDL was measured as described above.
Electron Microscopy.
Normal human fibroblasts induced to express LDL receptors were incubated in the presence or absence of 500 μM of ALLN for 1 h, fixed, and then prepared for thin-section EM. The number of coated pits was quantified as previously described (61).
Other Methods.
Protein concentrations were determined using the Bradford assay (Bio-Rad Laboratories, Hercules, CA).
Results
Annexin VI Binds to Inner Membrane Surface
Coated pit budding from membranes attached to a solid substratum at 4°C is detected using an indirect radioimmune assay that measures the relative amount of clathrin on the membrane (43). Previously, we documented that clathrin loss is an accurate gauge of coated vesicle formation (39). Although annexin VI appears to be essential for budding in vitro, other cytosolic factors are also needed. In an effort to identify these factors, we set up a standard reaction mixture consisting of cytosol deficient in active annexin VI (refer to Materials and Methods for details), various recombinant forms of annexin VI, Ca+2, and ATP (Fig. 1 A). No loss of clathrin was detected when membranes were incubated at 37°C in the presence of buffer (Fig. 1 A, bar 1) and only a modest loss occurred in the presence of inactive cytosol (Fig. 1 A, bar 2). By contrast, when 1 nM recombinant annexin VI (AnxVIwt) was added to the cytosol, membrane clathrin declined by 75% (Fig. 1 A, bar 3). Annexin VI lacking the last four calcium binding repeats (AnxVIΔ379) supported budding to the same extent (Fig. 1 A, bar 4). A recombinant annexin VI missing the last six repeats (AnxVIΔ192), however, lacked budding activity (Fig. 1 A, bar 5).
Figure 1.
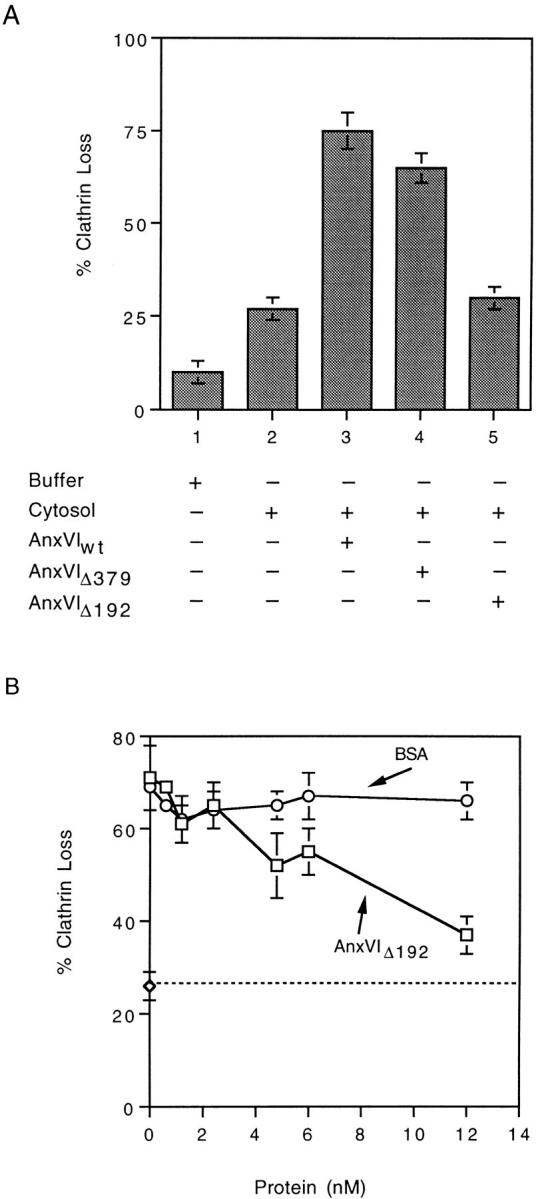
Effects of recombinant wild-type annexin VI (A) and truncated annexin VI (A and B) on budding of clathrin-coated pits. (A) Fibroblast membranes were attached at 4°C and then warmed to 37°C for 10 min in the presence of either buffer alone or cytosol containing 1 nM of the indicated recombinant protein. At the end of the incubation, the amount of clathrin loss was measured as described. Maximum clathrin value was 27,185 cpm/ well of 125I-labeled streptavidin bound with a background of 731 cpm/well. (B) Fibroblast membranes were warmed in cytosol containing 1 nM wild-type annexin VI (Anx VIwt) plus the indicated concentration of AnxVIΔ192 (□) or BSA (○). Maximum clathrin value was 32,475 cpm/well with a background of 1,961 cpm/well. All values are the average of triplicate measurements ± the standard deviation.
In contrast to its inability to support budding, AnxVIΔ192 inhibited the activity of AnxVIwt (Fig. 1 B). Normal clathrin loss occurred in the presence of AnxVIwt alone but with increasing concentrations of AnxVIΔ192 in the mixture, budding was progressively inhibited. At a molar ratio of 12 AnxVIΔ192:1 AnxVIwt, budding was inhibited ∼75% compared with inactive cytosol alone (⋄, dotted line).
The site(s) of action for annexin VI may be in the cytosol, at the plasma membrane, or both. To distinguish between these possibilities, we measured the ability of prebound annexin VI to support budding (Fig. 2 A). Membranes were incubated in the presence (Fig. 2 A, bars 4–6) or absence (bars 1–3) of buffer B containing annexin VI for 15 min at 4°C. The membranes were washed and either not warmed (Fig. 2 A, bar 4) or warmed to 37°C in the presence of either inactive cytosol (Fig. 2 A, bars 2 and 6), buffer alone (Fig. 2 A, bars 1 and 5) or inactive cytosol containing AnxVIwt (Fig. 2 A, bar 3). Just as much clathrin loss occurred from membranes preincubated in the presence of AnxVIwt as from untreated membranes that were warmed in cytosol containing AnxVIwt (Fig. 2 A, compare bar 3 with 6). Buffer alone did not support budding under any condition (Fig. 2 A, bars 1 and 5), nor did cytosol in the absence of AnxVIwt (Fig. 2 A, bar 2). These results suggest prebound annexin VI can activate cytosolic factors necessary for the budding reaction.
Figure 2.
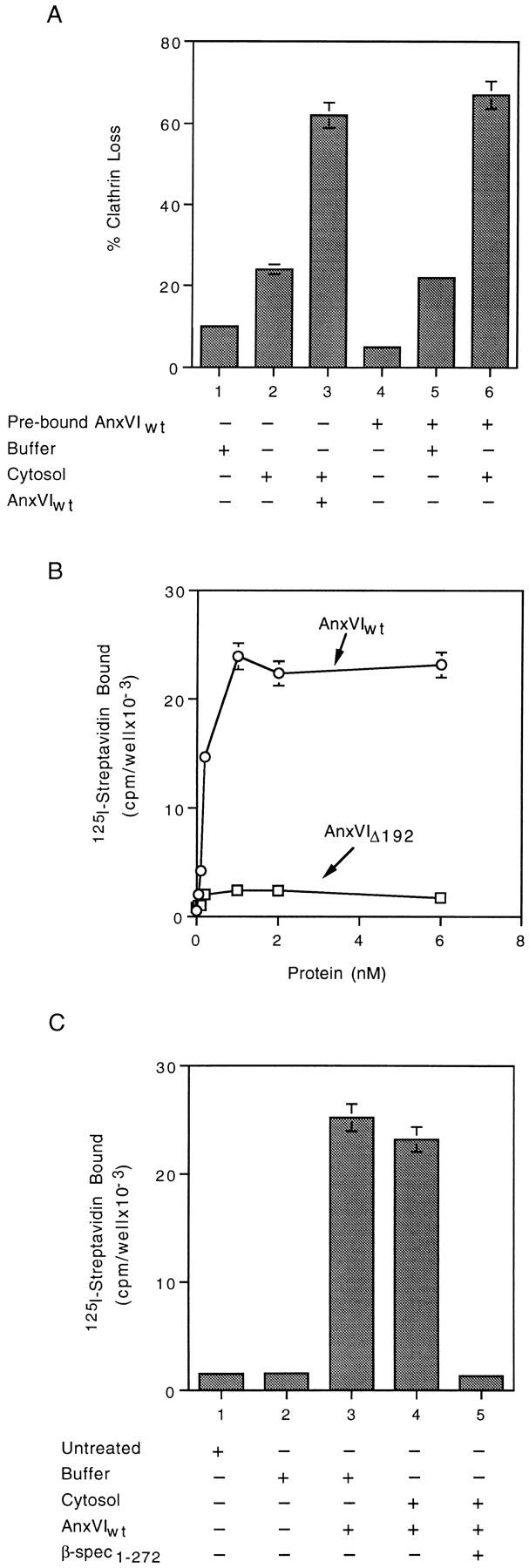
Annexin VI binding to attached membranes (B and C) supports coated pit budding (A). (A) Attached membranes were incubated for 15 min at 4°C in the presence of either buffer alone (bars 1–3) or buffer containing 1 nM AnxVIwt (bars 4–6). Membranes were either assayed immediately (bar 4) or warmed to 37°C for 10 min in the presence of either buffer (bars 1 and 5), cytosol alone (bars 2 and 6), or cytosol containing AnxVIwt (bar 3). Maximum clathrin value was 36,902 cpm/well with a background of 1,182 cpm/well. (B) Attached membranes were incubated in the presence of cytosol containing the indicated concentration of either AnxVIwt (○) or AnxVIΔ192 (□) for 15 min at 4°C. The membranes were washed and assayed for the amount of bound annexin VI as described. Background binding of BSA ranged between 3,056 cpm/well and 8,654 cpm/well and was subtracted from all values. (C) Attached membranes were either not treated (bar 1) or incubated at 4°C for 15 min in the presence of either buffer alone (bar 2), buffer plus 1 nM AnxVIwt (bar 3), cytosol plus 1 nM AnxVIwt (bar 4) or cytosol plus 1 nM AnxVIwt and in 3 nM β-spectrin1–272 (bar 5). Membranes were then assayed for the amount of annexin VI bound. Background binding to BSA has been subtracted from all values. All values are the average of triplicate measurements ± the standard deviation.
We used a radioimmune assay to directly measure membrane binding of annexin VI (Fig. 2 B). Membranes attached by standard methods were incubated for 15 min at 4°C in the presence of buffer B containing cytosol, Ca+2, ATP, and various concentrations of AnxVIwt (○). As the concentration was increased, there was a corresponding increase in the amount of AnxVIwt associated with the membrane. The level of bound AnxVIwt plateaued when the concentration reached 1 nM, suggesting a high-affinity interaction. If we substituted AnxVIΔ192 (□) for AnxVIwt, no binding was detected. AnxVIΔ379 bound as well as wild-type (data not shown). Finally, binding occurred in the presence of buffer alone, so cytosolic factors are not required (Fig. 2 C, bar 3).
The binding characteristics of annexin VI suggested it might be interacting with a protein on the inner surface, rather than a phospholipid. Previously, β-spectrin was identified as an annexin VI–binding protein and the binding site localized to a 28-kD fragment from the NH2-terminal portion of the molecule (59). We expressed in bacteria a cDNA coding for the first 272 amino acids of human β spectrin (β-Spec1–272), which contains the putative annexin VI–binding site, and purified the peptide. The presence of a threefold excess of this peptide completely blocked the binding of AnxVIwt to membranes (Fig. 2 C, compare bar 4 with 5). These results indicate that spectrin is a component of a high-affinity membrane-binding site for annexin VI.
Annexin VI Mediates Remodeling of Membrane Cytoskeleton
The binding of annexin VI to spectrin may be required for remodeling of the membrane cytoskeleton before budding of coated pits. A radioimmune assay was used to measure the relative amount of spectrin on the membrane before and after the completion of the budding reaction (Fig. 3 A). Freshly isolated membranes had ∼6 × 103 cpm/well of antispectrin IgG-binding activity (Fig. 3 A, compare bar 1 with 2). Antispectrin IgG binding activity did not change when the membranes were warmed to 37°C in the presence of buffer (Fig. 3 A, bar 3) or buffer plus AnxVIwt (Fig. 3 A, bar 6), indicating no loss of spectrin under these conditions. Binding activity declined by ∼50%, however, when the buffer was replaced with the standard budding mixture containing AnxVIwt (Fig. 3 A, bar 4). Substitution of AnxVIwt with AnxVIΔ192, which does not affect budding (refer to Fig. 1 A), did not stimulate the loss of spectrin (Fig. 3 A, bar 5). Therefore, conditions that support budding of coated pits reduce the level of spectrin that can be detected on the membrane with this radioimmune assay.
Figure 3.
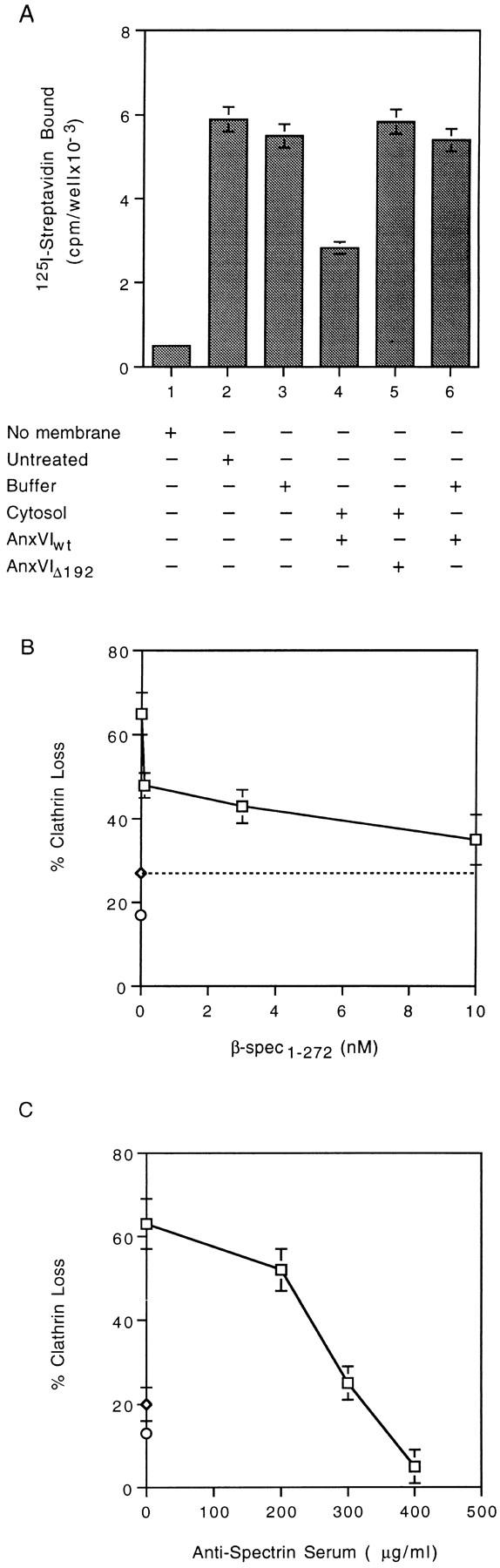
Spectrin loss from membranes (A) accompanies budding (B and C) of coated pits. (A) Attached membranes were warmed to 37°C for 10 min in the presence of buffer alone (bar 3), buffer plus AnxVIwt (bar 6), cytosol plus AnxVIwt (bar 4), or cytosol plus AnxVIΔ192 (bar 5). All wells were then assayed for the presence of spectrin as described. Background was 500 cpm/ well. (B) Attached membranes were incubated for 10 min at 37°C in the presence of cytosol alone (⋄, dashed line) or cytosol containing 1 nM AnxVIwt plus the indicated concentration of β-spectrin peptide (□). At the end of the incubation, the amount of clathrin loss was measured as described. Maximum clathrin value was 30,216 cpm/well with a background of 1,859 cpm/well. (C) Attached membranes were incubated for 10 min at 37°C in the presence of cytosol (⋄, on the ordinate) or cytosol containing 1 nM AnxVIwt plus the indicated concentration of rabbit antihuman spectrin serum (□). At the end of the incubation, the amount of clathrin loss was measured as described. Maximum clathrin value was 28,125 cpm/well with a background of 1,407 cpm/well. All values are the average of triplicate measurements ± the standard deviation.
If the binding of annexin VI to spectrin is linked to spectrin removal during budding, then blocking this interaction should inhibit budding. We tested the effects of different concentrations of β-Spec1–272 (Fig. 3 B), which inhibits annexin VI binding (refer to Fig. 2 C). We saw a 15% clathrin loss when membranes were warmed to 37°C in buffer alone (○, on the ordinate) and ∼27% loss when cytosol lacking AnxVIwt activity was used (⋄, horizontal dotted line). The inclusion of AnxVIwt in the cytosol caused a 65% loss of clathrin, but the loss was progressively suppressed by the addition of increasing amounts of β-Spec1– 272 to the mixture (□). At the highest concentration of β-Spec1–272, we observed an ∼90% inhibition of AnxVIwt – induced budding activity relative to cytosol alone.
Another potential way to interfere with spectrin removal is to use a specific cross-linking agent such as a polyclonal antispectrin antibody (Fig. 3 C). Inactive cytosol caused an ∼20% loss of clathrin (⋄, on the ordinate) but the addition of AnxVIwt stimulated a 62% loss. With increasing concentrations of antispectrin serum added to the mixture, however, there was a progressive decline in the loss of clathrin from the membrane. At the highest concentration (400 μg/ml), clathrin loss was reduced below the amount that occurred in the presence of buffer alone (Fig. 3 C, compare with ○ on the ordinate). Since annexin VI binding was not affected by the presence of the spectrin antiserum (data not shown), the antibody appears to block budding by physically preventing spectrin remodeling.
Spectrin-bound annexin VI could regulate the remodeling of membrane spectrin by activating a cytosolic protease. Since calpain I has been implicated in removing spectrin from the membrane (31, 34), we used the cysteine protease inhibitor ALLN to see if it would block annexin VI–dependent removal of spectrin (Fig. 4 A). Compared with untreated membranes (Fig. 4 A, bar 1), annexin VI caused a significant decrease in the amount of spectrin on the membrane (Fig. 4 A, bar 2). This loss of spectrin was markedly inhibited by adding 5 (Fig. 4 A, bar 3) and 10 μM of ALLN (Fig. 4 A, bar 4) to the incubation. The DMSO carrier used to solubilize the ALLN had no effect (Fig. 4 A, bar 5).
Figure 4.
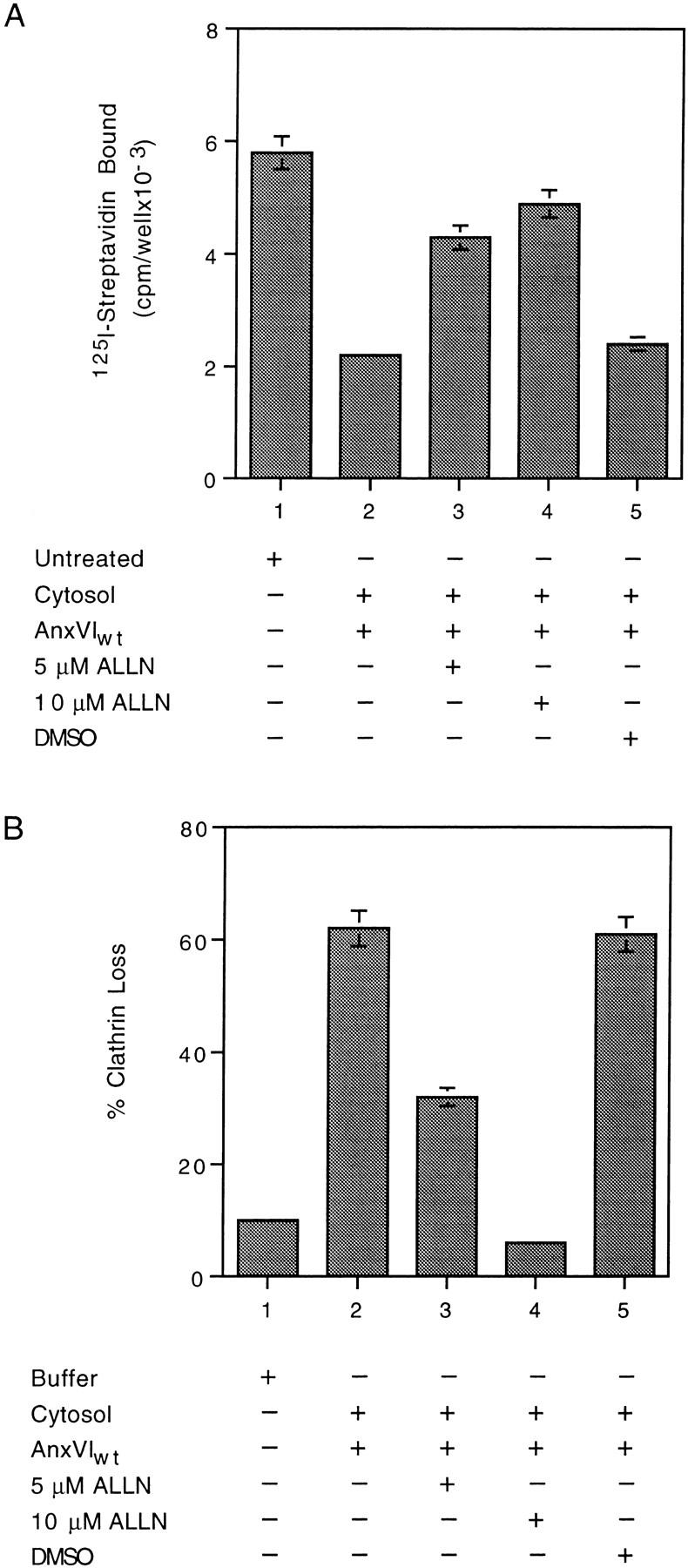
ALLN inhibits loss of spectrin (A) and coated pit budding (B) in vitro. (A) Attached membranes were either not treated (bar 1) or incubated at 37°C for 10 min in the presence of cytosol (bars 2–5) containing the indicated additions. At the end of the incubation, the membranes were assayed for the amount of spectrin as described. (B) Attached membranes were incubated at 37°C in the presence of either buffer (bar 1) or cytosol (bars 2–5) containing the indicated additions. At the end of the incubation, the percent loss of clathrin was measured as described. Maximum clathrin value was 29,098 cpm/well with a background of 1,114 cpm/well. All values are the average of triplicate measurements ± the standard deviation.
ALLN also inhibited coated pit budding (Fig. 4 B). Incubation of membranes in the presence of cytosol plus AnxVIwt caused an ∼60% loss of clathrin. The presence of 5 μM ALLN inhibited loss by 50% (Bar 3), but raising the concentration to 10 μM (Fig. 4 B, bar 4) completely inhibited budding. The endogenous calpain inhibitor, calpastatin, (7 μM) caused a 50% inhibition whereas serine protease inhibitors such as pepstatin and PMSF had no effect (data not shown). These in vitro results suggest the function of annexin VI during endocytosis is to link the activity of a calpain I–like protease to the remodeling of the spectrin cytoskeleton.
Annexin VI Required for Normal Budding In Vivo
If annexin VI is involved in receptor-mediated endocytosis, the results of the in vitro analysis suggest ALLN should inhibit LDL internalization (Fig. 5). Human fibroblasts were incubated for different times in the presence of media containing 125I-labeled LDL, plus or minus the addition of 500 μM of ALLN. Control cells internalized LDL at a constant rate (□) for the first 30–60 min of the incubation, after which the rate began to plateau. The presence of ALLN in the media totally blocked LDL uptake for the first 30 min of incubation (○). Surprisingly, the cells then began to take up LDL, which continued for the duration of the incubation. These results suggested ALLN initially blocks coated pit budding in vivo, but the cells overcome the inhibition by activating an alternative pathway.
Figure 5.

ALLN transiently inhibits uptake of 125I-labeled LDL. Expression of LDL receptors was induced in normal human fibroblasts by standard methods. The cells were preincubated in the presence (○) or absence (□) of 500 μM ALLN for 1 h before the addition of 15 μg/ml of 125I-labeled LDL ± 50-fold excess of cold LDL. The cells were then incubated further for the indicated time before the amount of internalized [125I]LDL was measured as described. Each value is the average of duplicate measurements.
Several studies have shown that when the coated pit pathway is inhibited, cells activate an alternative internalization pathway (38). Cells that have recovered from ALLN were able to internalize LDL, a process thought to require receptor clustering by clathrin coats (5). To better understand the mechanism of recovery from ALLN, we used electron microscopy to examine cells that had been incubated in the presence or absence of ALLN for 60 min (Table I). Both sets of cells had normal-appearing clathrin-coated pits (data not shown), but the ALLN-treated cells contained nearly twice as many pits (0.18 verses 0.32/ μM). The increase in the number of coated pits was confirmed by immunofluorescence (data not shown). In addition, the anti-clathrin IgG radioimmune assay showed that attached plasma membranes from ALLN pretreated cells contained over twofold more clathrin than membranes from control cells (2.3 × 104 cpm of 125I-labeled streptavidin binding (control) compared with 5.8 × 104 [ALLN]). Therefore, ALLN causes an increase in the number of coated pits at the cell surface.
Table I.
Effect of ALLN on Number of Coated Pits
| Treatment | Number of cells evaluated | μM of membrane | Total coated pits | Coated pits/μM | ||||
|---|---|---|---|---|---|---|---|---|
| Control | 106 | 130 | 24 | 0.18 | ||||
| ALLN | 114 | 138 | 44 | 0.32 |
Human fibroblasts were incubated in the presence or absence of 500 μM of ALLN for 1 h at 37°C, fixed, and then prepared for EM thin-section analysis as described.
Coated pits that assembled in the presence of ALLN did not require annexin VI for budding in vitro (Fig. 6 A). We incubated cells in the presence of ALLN for 1 h and prepared membranes. There was little loss of clathrin when these membranes were warmed to 37°C in the presence of buffer (Fig. 6 A, bar 1). Replacing the buffer with inactive cytosol, however, resulted in a 60% loss of clathrin (Fig. 6 A, bar 2). The same amount of clathrin loss occurred regardless of whether annexin VI was added to the cytosol (Fig. 6 A, compare bar 3 with 2) or if ALLN was present in the complete budding mixture (Fig. 6 A, compare bar 4 with 3). Consistent with these results, membranes from ALLN-treated cells did not lose any spectrin during budding (Fig. 6 B, compare bars 3 and 4 with 2). Apparently cells overcome the effects of ALLN by assembling a new population of pits that are not functionally or structurally linked to the spectrin cytoskeleton.
Figure 6.
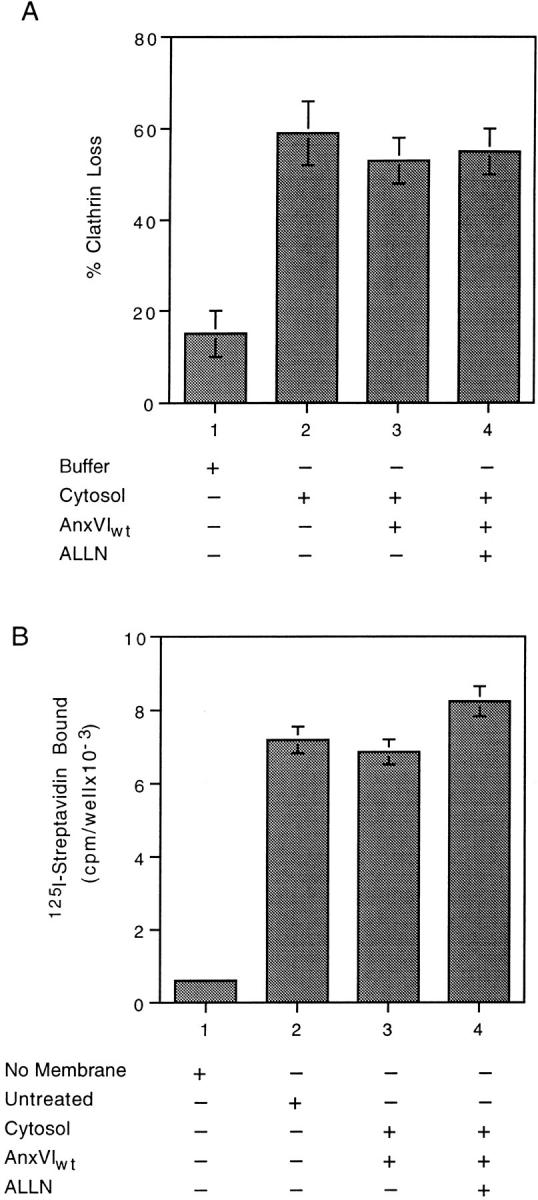
Budding (A) is no longer linked to spectrin loss (B) in membranes from ALLN-treated cells. (A) Attached membranes obtained from cells incubated in the presence of ALLN for 1 h were warmed to 37°C in the presence of buffer alone (bar 1) or warmed in the presence of cytosol (bars 2–4) containing the indicated additions. At the end of the incubation, the percent loss of clathrin was measured as described. Maximum clathrin value was 58,321 cpm/well with a background of 2,536. (B) Attached membranes obtained from cells incubated in the presence of ALLN for 1 h were either not treated (bar 2) or warmed to 37°C for 10 min in the presence of cytosol (bars 3 and 4) containing the indicated additions. At the end of the incubation, the membranes were assayed for the amount of spectrin as described. Background binding is shown in bar 1. In both experiments, each value is the average of triplicate measurements ± the standard deviation.
We next looked at the fate of the LDL internalized by ALLN-treated cells (Fig 7, A and B). Cells were preincubated in the presence (Fig. 7 B) or absence (Fig. 7 A) of ALLN for 60 min before adding 25 μg/ml of fluorescent LDL to each dish and continuing the incubation an additional 90 min. Untreated cells had numerous large fluorescent vesicles aggregated near the center of the cell (Fig. 7 A). By contrast, the fluorescent vesicles in ALLN-treated cells were smaller, more uniform in size and scattered throughout the cell. We also found that LDL degradation was markedly inhibited in ALLN-treated cells (Table II). Using a standard 5-h uptake assay, ∼50% of the internalized 125I-labeled LDL was degraded in control cells, whereas very little degradation was detected in ALLN-treated cells. Instead, these cells contained twofold more nondegraded 125I-labeled LDL than control cells.
Figure 7.

Endosomes are mislocalized (A and B) in cells incubated in the presence of ALLN. Human fibroblasts were incubated in the presence (B) or absence (A) of 500 μM ALLN for 1 h at 37°C before the addition of 25 μg/ml of fluorescent LDL. The cells were incubated an additional 90 min before direct visualization.
Table II.
Effect of ALLN on Degradation of LDL
| Treatment | Receptor-bound protein | Internalized protein | Degraded protein | |||
|---|---|---|---|---|---|---|
| ng/mg | ng/mg | ng/mg | ||||
| Control | 50 | 910 | 1200 | |||
| ALLN | 160 | 2900 | 80 |
Cells were incubated in the presence of 500 μM of ALLN for 1 h before the addition of 15 μg/ml of [125I]LDL ± 50-fold excess cold LDL. Cells were incubated an additional 5 h before the amount of surface, internalized, and then degraded [125I]LDL was measured as described. All values are the average of duplicate measurements.
The dominant-negative–acting AnxVIΔ192 had the same effect on coated pit budding and spectrin loss in vitro as ALLN (refer to Fig. 1 B and Fig. 3 A). Therefore, we microinjected AnxVIwt and AnxVIΔ192 into normal human fibroblasts and compared the effect of these proteins on the uptake of fluorescent LDL (Fig. 8, A–D and Table III). Cells were injected with the indicated recombinant protein mixed with FITC-labeled dextran to mark the cell, allowed to recover for 1 h, and then incubated an additional 1 h in the presence of fluorescent LDL. Cells injected with the AnxVIwt (Fig. 8 A) took up normal amounts of pyrene- labeled LDL, compared with nearby uninjected cells, and delivered it to the perinuclear area of the cell (Fig. 8 B). Internalization of LDL in cells injected with AnxVIΔ192 (Fig. 8 C), by contrast, was markedly reduced (Fig. 8 D) compared with neighboring uninjected cells. In addition, the LDL-positive vesicles in these cells tended to be more randomly distributed. Quantification of these results (Table III) showed that 83% of the cells injected with AnxVIΔ192 had a reduced uptake of LDL and 17% did not internalize any detectable LDL.
Figure 8.

Microinjection of AnxVIΔ192 or antispectrin IgG inhibits LDL uptake. Human fibroblasts were microinjected with 3 mg/ml FITC-dextran and 100 μM AnxVIwt (A and B) or 100 μM AnxVIΔ192 (C and D) or 300 μg/ml anti-rabbit IgG (E and F) or 300 μg/ml antispectrin IgG (G and H). The cells were allowed to recover for 1 h at 37°C and then the uptake of fluorescent LDL (25 μg/ml) for 1 h at 37°C was measured. B, D, F, and H show LDL uptake and A, C, E, and G show FITC-dextran staining of the same cells.
Table III.
Effect of AnxVIΔ192 and Antispectrin on LDL Uptake
| Number of cells evaluated |
Percentage of cells | |||||||
|---|---|---|---|---|---|---|---|---|
| Protein microinjected | ++ Uptake | + Uptake | No uptake | |||||
| AnxVIwt | 48 | 94 | 6 | 0 | ||||
| AnxVIΔ192 | 42 | 0 | 83 | 17 | ||||
| Control IgG | 45 | 91 | 9 | 0 | ||||
| Antispectrin IgG | 39 | 0 | 77 | 23 | ||||
Human fibroblasts were microinjected with the indicated protein mixed with FITC- labeled dextran to mark the injected cell and then incubated for 1 h. The cells were incubated for an additional 1 h in the presence of fluorescent LDL as described. Each value is the average of duplicated experiments.
Like AnxVIΔ192, microinjection of antispectrin IgG also inhibited the uptake of fluorescent LDL (Fig. 8 E–H and Table III). Control IgG did not affect the uptake of the LDL (Fig. 8, E and F), whereas antispectrin IgG markedly reduced uptake (Fig. 8, G and H). Table III shows that the number of affected cells injected with antispectrin IgG was similar to what was seen with AnxVIΔ192. Therefore, three different agents that prevent budding of coated pits in vitro (ALLN, AnxVIΔ192, and antispectrin IgG) also block internalization of LDL in vivo.
Discussion
We have discovered a new set of protein–protein interactions that operate during normal receptor-mediated endocytosis in human fibroblasts. A combination of in vitro and in vivo experiments support the conclusion that annexin VI couples the proteolytic activity of a calpain-like protease to the remodeling of plasma membrane spectrin during budding in human fibroblasts. Three different reagents (ALLN, AnxVIΔ192, and antispectrin IgG) that interfere with the removal of spectrin from plasma membranes block coated pit budding both in vitro and in vivo. Cells adapt to blocking conditions by assembling a new set of pits that are able to bud both in vitro and in vivo without annexin VI–dependent spectrin remodeling. The endosomes that form under these conditions, however, tend to be randomly distributed in the cell rather than migrate to the centrosome region. Therefore, annexin VI is not part of the machinery that physically mediates invagination and scission, but rather is necessary for the formation of endosomes that can move to the correct location in the cell.
These results extend the list of molecular interactions known to be required for coated pit budding in fibroblasts. Clathrin lattice assembly is the end product of interactions between AP2 and its high-affinity membrane binding site (41, 62), AP2 and clathrin (35), and clathrin with itself (35). Invagination of the membrane is driven by the regulated (36) rearrangement of the lattice without the participation of additional cytoplasmic factors (39). At the end of the invagination stage, the pit is a nascent vesicle held onto the membrane by a slender membrane stalk. The scission of this stalk, which may be physically mediated by dynamin (8, 17, 32), apparently cannot release the vesicle unless spectrin remodeling takes place. Remodeling, in turn, requires the binding of annexin VI to spectrin and the activity of a calpain-like, cytoplasmic protease. Only 50% of the spectrin is lost during normal budding (refer to Fig. 3) and some of this probably leaves the membrane with the new coated vesicle. Therefore, annexin VI targets the action of the calpain protease to regions of cytoskeleton that must be freed so scission can release the vesicle.
A connection between the spectrin cytoskeleton and budding of coated pits has not been detected before. Spectrin, actin, band 4.1, and ankyrin form an oligomeric complex (11) that can assemble into a two-dimensional lattice on the cytoplasmic surface of both internal (9) and surface membranes (11). Ankyrin and band 4.1 are both able to bind multiple membrane proteins and tether them to spectrin–actin strands (15). Bound proteins have a restricted lateral mobility in the plane of the membrane (45), which presumably causes them to collect in spectrin-rich regions. Thus, the spectrin lattice shares with the clathrin lattice the ability to create membrane domains capable of carrying out specific functions for the cell. The function of the clathrin lattice is to prepare a portion of membrane for transport to other locations whereas the spectrin lattice seems adapted for retaining the properties of the originating membrane. The function of annexin VI in these cells, therefore, is to mobilize the elements needed to disconnect clathrin domains from spectrin during budding. In this capacity, annexin VI provides a vital communication link between retained and departing portions of a specific region of plasma membrane.
The importance of this link was revealed in cells that were exposed to ALLN for greater than 30 min. After the resumption of receptor-mediated endocytosis there were nearly twofold more coated pits and twice as much clathrin on the membrane. This suggests that in the absence of spectrin remodeling, coated pits that are linked to spectrin become trapped at the surface. The cell compensates by assembling a new population of pits that are not associated with spectrin and, therefore, do not require annexin VI for budding. The endosomes generated by these new pits did not migrate to the center of the cell.
The intracellular movement of membrane vesicles requires both microfilament (44) and microtubule (23, 25) components of the cytoskeleton. Motor proteins such as myosin, kinesin, and dynein bound to vesicle membranes are able to propel the vesicle along the appropriate cytoskeletal track (25). Microtubules and the centrosome-directed motor, dynein, appear to be necessary for the fusion of early with late endosomes during endocytosis (6, 12, 27). Dynein binds to dynactin, a 20 S macromolecular complex involved in maintaining the centrosomal organization of membranes like the Golgi apparatus (1, 57). Another protein in the dynactin complex is centractin (14). Centractin is an actin- related protein that appears to associate with spectrin (33). The two colocalize in cells overexpressing centractin, and spectrin coprecipitates with the dynactin complex. Dynactin has been proposed to be the physical linkage between dynein bound to microtubules and spectrin bound to vesicle membranes (33). If so, then early endosomes lacking spectrin would not link with the dynein–microtubule system and, therefore, be unable to migrate. This may be what happens when annexin VI activity is blocked in fibroblasts. The endosomes that form lack the spectrin necessary to track on microtubules and, as a consequence, fusion with late endosomes containing hydrolytic enzymes (18) is impaired.
Contrary to our initial thinking (40), annexin VI is not an essential component of the coated pit budding machinery. As far as we can tell, it is only needed to release clathrin-coated pits from spectrin. Not all portions of the plasma membrane are covered with a spectrin cytoskeleton. The apical surface of polarized epithelial cells, for example, is relatively deficient in spectrin (45). Annexin VI may not be needed to release coated vesicles from this membrane. Other cytoskeletal factors appear to come into play at this surface because drugs that depolymerize actin microfilaments inhibit coated pit budding from apical membranes, but not from spectrin-rich basolateral membranes (26). Therefore, not all coated pits at the cell surface are equivalent. Superimposed on the basic budding machinery are molecular interactions that must take place to facilitate the release of the budding pit from the particular membrane of origin.
These results suggest that coated pit budding may be normal in cells lacking annexin VI. Consistent with this conclusion, human A431 squamous carcinoma cells lacking annexin VI are able to internalize and recycle transferrin receptors (52). Receptor-mediated endocytosis in these cells is inhibited after disruption of actin filaments (37), suggesting that budding is primarily occurring from membrane domains that are apical-like in character, so annexin VI should not be needed. Interestingly, the ligand-induced degradation of EGF and its receptor is markedly slower in these cells (54) compared with human fibroblasts (55), owing to the diversion of the receptor–EGF complex into a recycling pathway (53). The proportion of internalized LDL that gets degraded in 5 h is also significantly less in these cells compared with matched sets of CHO cells (refer to Table II) (4).
In conclusion, endosomes appear to acquire a set of information during budding that determines where they will migrate in the cell. This information also specifies the membrane of origin for the vesicle. The control of this aspect of membrane traffic is distinctly different in character and molecular mechanism than the control of membrane– membrane recognition and fusion (48, 50). The major function of the annexin family of proteins in membrane traffic (16), therefore, may be to regulate the placement and removal of tags that imprint vesicles with the information necessary for them to assume the correct spatial distribution in the cell.
Acknowledgments
We would like to thank W. Donzell and A. Horton for their valuable technical assistance and S. Baldock for administrative assistance (all three from University of Texas Southwestern Medical Center, Dallas, TX).
This work was supported by a grant from the National Institutes of Health (HL 20948) and the Perot Family Foundation.
Abbreviations used in this paper
- ALLN
N-acetyl-leucyl-leucyl-norleucinal
- GST
glutathione-S-transferase
- LDL
low density lipoprotein
Footnotes
Address all correspondence to Richard G.W. Anderson, Department of Cell Biology and Neuroscience, University of Texas Southwestern Medical Center, Dallas, TX 75235-9039. Tel.: (214) 648-2346. Fax: (214) 648-7577. E-mail: anders06@utsw.swmed.edu
References
- 1.Allan V. Organelle movement. Dynactin: portrait of a dynein regulator. Curr Biol. 1994;4:1000–1002. doi: 10.1016/s0960-9822(00)00225-6. [DOI] [PubMed] [Google Scholar]
- 2.Anderson RGW. Methods for visualization of the LDL pathway in cultured human fibroblasts. Methods Enzymol. 1986;129:201–216. doi: 10.1016/0076-6879(86)29070-9. [DOI] [PubMed] [Google Scholar]
- 3.Anderson, R.G.W. 1991. Molecular motors that shape endocytic membrane. In Intracellular Trafficking of Proteins. C.J. Steer and J.A. Hanover, editors. Cambridge University Press, London, UK. 13–47.
- 4.Anderson RGW, Brown MS, Goldstein JL. Inefficient internalization of receptor-bound low density lipoprotein in human carcinoma A-431 cells. J Cell Biol. 1981;88:44–52. doi: 10.1083/jcb.88.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson RGW, Goldstein JL, Brown MS. A mutation that impairs the ability of lipoprotein receptors to localize in coated pits on the cell surface of human fibroblasts. Nature. 1976;270:695–699. doi: 10.1038/270695a0. [DOI] [PubMed] [Google Scholar]
- 6.Aniento F, Emans N, Griffiths G, Gruenberg J. Cytoplasmic dynein-dependent vesicular transport from early to late endosomes. J Cell Biol. 1993;123:1373–1387. doi: 10.1083/jcb.123.6.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aunis D, Bader MF. The cytoskeleton as a barrier to exocytosis in secretory cells. J Exp Biol. 1988;139:253–266. doi: 10.1242/jeb.139.1.253. [DOI] [PubMed] [Google Scholar]
- 8.Baba T, Damke H, Hinshaw JE, Ikeda K, Schmid SL, Warnock DE. Role of dynamin in clathrin-coated vesicle formation. Cold Spr Harb Symp Quant Biol. 1995;60:235–242. doi: 10.1101/sqb.1995.060.01.027. [DOI] [PubMed] [Google Scholar]
- 9.Beck, K.A., and W.J. Nelson. 1996. The spectrin-based membrane skeleton as a membrane protein-sorting machine. Am. J. Physiol. 270:C1263– 7C1270. [DOI] [PubMed]
- 10.Bennett V. Spectrin-based membrane skeleton: a multipotential adaptor between plasma membrane and cytoplasm (published errata appeared in Physiol. Rev.1991. 71[1]:frontmatter and 71[4]:after 1193) Physiol Rev. 1990;70:1029–1065. doi: 10.1152/physrev.1990.70.4.1029. [DOI] [PubMed] [Google Scholar]
- 11.Bennett V, Gilligan DM. The spectrin-based membrane skeleton and the micron-scale organization of the plama membrane domain. Annu Rev Cell Biol. 1993;9:27–66. doi: 10.1146/annurev.cb.09.110193.000331. [DOI] [PubMed] [Google Scholar]
- 12.Bomsel M, Parton R, Kuznetsov SA, Schroer TA, Gruenberg J. Microtubule- and motor-dependent fusion in vitro between apical and basolateral endocytic vesicles from MDCK cells. Cell. 1990;62:719–731. doi: 10.1016/0092-8674(90)90117-w. [DOI] [PubMed] [Google Scholar]
- 13.Chasserot-Golaz S, Vitale N, Sagot I, Delouche B, Dirrig S, Pradel LA, Henry JP, Aunis D, Bader MF. Annexin II in exocytosis: catecholamine secretion requires the translocation of p36 to the subplasmalemmal region in chromaffin cells. J Cell Biol. 1996;133:1217–1236. doi: 10.1083/jcb.133.6.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clark SW, Meyer DI. Centractin is an actin homologue associated with the centrosome. Nature. 1992;359:246–250. doi: 10.1038/359246a0. [DOI] [PubMed] [Google Scholar]
- 15.Conboy JG. Structure, function and molecular genetics of erythroid membrane skeletal protein 4.1 in normal and abnormal red blood cells. Sem Hematol. 1993;30:58–73. [PubMed] [Google Scholar]
- 16.Creutz CE. The annexins and exocytosis. Science. 1992;258:924–931. doi: 10.1126/science.1439804. [DOI] [PubMed] [Google Scholar]
- 17.Damke H, Gossen M, Freundlieb S, Bujard H, Schmid SL. Tightly regulated and inducible expression of dominant interfering dynamin mutant in stably transformed HeLa cells. Methods Enzymol. 1995;257:209–220. doi: 10.1016/s0076-6879(95)57026-8. [DOI] [PubMed] [Google Scholar]
- 18.Diment S, Stahl P. Macrophage endosomes contain proteases which degrade endocytosed protein ligands. J Biol Chem. 1985;260:15311–15317. [PubMed] [Google Scholar]
- 19.Emans N, Gorvel JP, Walter C, Gerke V, Kellner R, Griffiths G, Gruenberg J. Annexin II is a major component of fusogenic endosomal vesicles. J Cell Biol. 1993;120:1357–1369. doi: 10.1083/jcb.120.6.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fiedler K, Lafont F, Parton RG, Simons K. Annexin XIIIb: a novel epithelial specific annexin is implicated in vesicular traffic to the apical plasma membrane. J Cell Biol. 1995;128:1043–1053. doi: 10.1083/jcb.128.6.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Futter CE, Felder S, Schlessinger J, Ullrich A, Hopkins CR. Annexin I is phosphorylated in the multivesicular body during the processing of the epidermal growth factor receptor. J Cell Biol. 1993;120:77–83. doi: 10.1083/jcb.120.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goldstein JL, Basu SK, Brown MS. Receptor-mediated endocytosis of LDL in cultured cells. Methods Enzymol. 1983;98:241–260. doi: 10.1016/0076-6879(83)98152-1. [DOI] [PubMed] [Google Scholar]
- 23.Goltz JS, Wolkoff AW, Novikoff PM, Stockert RJ, Satir P. A role for microtubules in sorting endocytic vesicles in rat hepatocytes. Proc Natl Acad Sci USA. 1992;89:7026–7030. doi: 10.1073/pnas.89.15.7026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goodman SR, Zimmer WE, Clark MB, Zagon IS, Barker JE, Bloom ML. Brain spectrin: of mice and men. Brain Res Bull. 1995;36:593–606. doi: 10.1016/0361-9230(94)00264-2. [DOI] [PubMed] [Google Scholar]
- 25.Goodson HV, Valetti C, Kreis TE. Motors and membrane traffic. Curr Opin Cell Biol. 1997;9:18–28. doi: 10.1016/s0955-0674(97)80147-0. [DOI] [PubMed] [Google Scholar]
- 26.Gottlieb TA, Ivanov IE, Adesnik M, Sabatini DD. Actin microfilaments play a critical role in endocytosis at the apical but not the basolateral surface of polarized epithelial cells. J Cell Biol. 1993;120:695–710. doi: 10.1083/jcb.120.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gruenberg J, Griffiths G, Howell KE. Characterization of the early endosome and putative endocytic carrier vesicles in vivo and with an assay of vesicle fusion in vitro. J Cell Biol. 1989;108:1301–1316. doi: 10.1083/jcb.108.4.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guan K-L, Dixon JE. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- 29.Hardy B, Bensch KG, Schrier SL. Spectrin rearrangement early in erythrocyte ghost endocytosis. J Cell Biol. 1979;82:654–663. doi: 10.1083/jcb.82.3.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hardy B, Schrier SL. The role of spectrin in erythrocyte ghost endocytosis. Biochem Biophys Res Commun. 1978;81:1153–1161. doi: 10.1016/0006-291x(78)91257-3. [DOI] [PubMed] [Google Scholar]
- 31.Harris AS, Morrow JS. Calmodulin and calcium-dependent protease I coordinately regulate the interaction of fodrin with actin. Proc Natl Acad Sci USA. 1990;87:3009–3013. doi: 10.1073/pnas.87.8.3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hinshaw JE, Schmid SL. Dynamin self-assembles into rings suggesting a mechanism for coated vesicle budding. Nature. 1995;374:190–192. doi: 10.1038/374190a0. [DOI] [PubMed] [Google Scholar]
- 33.Holleran EA, Tokito MK, Karki S, Holzbaur EL. Centractin (ARP1) associates with spectrin revealing a potential mechanism to link dynactin to intracellular organelles. J Cell Biol. 1996;135:1815–1829. doi: 10.1083/jcb.135.6.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu RJ, Bennett V. In vitro proteolysis of brain spectrin by calpain I inhibits association of spectrin with ankyrin-independent membrane binding site(s) J Biol Chem. 1991;266:18200–18205. [PubMed] [Google Scholar]
- 35.Keen JH. Clathrin and associated assembly and disassembly proteins. Annu Rev Biochem. 1990;59:415–438. doi: 10.1146/annurev.bi.59.070190.002215. [DOI] [PubMed] [Google Scholar]
- 36.Lamaze C, Chuang TH, Terlecky LJ, Bokoch GM, Schmid SL. Regulation of receptor-mediated endocytosis by Rho and Rac. Nature. 1996;382:177–179. doi: 10.1038/382177a0. [DOI] [PubMed] [Google Scholar]
- 37.Lamaze C, Fujimoto LM, Yin HL, Schmid SL. The actin cytoskeleton is required for receptor-mediated endocytosis in mammalian cells. J Biol Chem. 1997;272:20332–20335. doi: 10.1074/jbc.272.33.20332. [DOI] [PubMed] [Google Scholar]
- 38.Lamaze C, Schmid SL. The emergence of clathrin-independent pinocytic pathways. Curr Opin Cell Biol. 1995;7:573–580. doi: 10.1016/0955-0674(95)80015-8. [DOI] [PubMed] [Google Scholar]
- 39.Lin HC, Moore MS, Sanan DA, Anderson RGW. Reconstitution of coated pit budding from plasma membranes. J Cell Biol. 1991;114:881–891. doi: 10.1083/jcb.114.5.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin HC, Südhof TC, Anderson RGW. Annexin VI is required for budding of clathrin-coated pits. Cell. 1992;70:283–291. doi: 10.1016/0092-8674(92)90102-i. [DOI] [PubMed] [Google Scholar]
- 41.Mahaffey DT, Peeler JS, Brodsky FM, Anderson RGW. Clathrin-coated pits contain an integral membrane protein that binds the AP-2 subunit with high affinity. J Biol Chem. 1990;265:16514–16520. [PubMed] [Google Scholar]
- 42.Mayorga LS, Beron W, Sarrouf MN, Colombo MI, Creutz C, Stahl PD. Calcium-dependent fusion among endosomes. J Biol Chem. 1994;269:30927–30934. [PubMed] [Google Scholar]
- 43.Moore MS, Mahaffey DT, Brodsky FM, Anderson RGW. Assembly of clathrin-coated pits onto purified plasma membranes. Science. 1987;263:558–563. doi: 10.1126/science.2883727. [DOI] [PubMed] [Google Scholar]
- 44.Murphy C, Saffrich R, Grummt M, Gournier H, Rybin V, Rubino M, Auvinen P, Lutcke A, Parton RG, Zerial M. Endosome dynamics regulated by a Rho protein. Nature. 1996;384:427–432. doi: 10.1038/384427a0. [DOI] [PubMed] [Google Scholar]
- 45.Nelson WJ. Cytoskeleton functions in membrane traffic in polarized epithelial cells. Semin Cell Biol. 1991;2:375–385. [PubMed] [Google Scholar]
- 46.Perrin D, Langley OK, Aunis D. Anti-alpha-fodrin inhibits secretion from permeabilized chromaffin cells. Nature. 1987;326:498–501. doi: 10.1038/326498a0. [DOI] [PubMed] [Google Scholar]
- 47.Raynal P, Pollard HB. Annexins: the problem of assessing the biological role for a gene family of multifunctional calcium- and phospholipid-binding proteins. Biochim Biophys Acta. 1994;1197:63–93. doi: 10.1016/0304-4157(94)90019-1. [DOI] [PubMed] [Google Scholar]
- 48.Rothman JE, Wieland FT. Protein sorting by transport vesicles. Science. 1996;272:227–234. doi: 10.1126/science.272.5259.227. [DOI] [PubMed] [Google Scholar]
- 49.Sato K, Saito Y, Kawashima S. Identification and characterization of membrane-bound calpains in clathrin-coated vesicles from bovine brain. Eur J Biochem. 1995;230:25–31. doi: 10.1111/j.1432-1033.1995.tb20529.x. [DOI] [PubMed] [Google Scholar]
- 50.Schekman R, Orci L. Coat proteins and vesicle budding. Science. 1996;271:1526–1533. doi: 10.1126/science.271.5255.1526. [DOI] [PubMed] [Google Scholar]
- 51.Schmid S. Clathrin-coated vesicle formation and protein sorting: an integrated process. Annu Rev Biochem. 1997;66:511–548. doi: 10.1146/annurev.biochem.66.1.511. [DOI] [PubMed] [Google Scholar]
- 52.Smythe E, Smith PD, Jacob SM, Theobald J, Moss SE. Endocytosis occurs independently of annexin VI in human A431 cells. J Cell Biol. 1994;124:301–306. doi: 10.1083/jcb.124.3.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sorkin A, Krolenko S, Kudrjavtceva N, Lazebnik J, Teslenko L, Soderquist AM, Nikolsky N. Recycling of epidermal growth factor-receptor complexes in A431 cells: identification of dual pathways. J Cell Biol. 1991;112:55–63. doi: 10.1083/jcb.112.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stoscheck CM, Carpenter G. Characterization of the metabolic turnover of epidermal growth factor receptor protein in A-431 cells. J Cell Physiol. 1984;120:296–302. doi: 10.1002/jcp.1041200306. [DOI] [PubMed] [Google Scholar]
- 55.Stoscheck CM, Carpenter G. Downregulation of epidermal growth factor receptors: direct demonstration of receptor degradation in human fibroblasts. J Cell Biol. 1984;98:1048–1053. doi: 10.1083/jcb.98.3.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trifaro JM, Vitale ML. Cytoskeleton dynamics during neurotransmitter release. Trends Neurosci. 1993;16:466–472. doi: 10.1016/0166-2236(93)90079-2. [DOI] [PubMed] [Google Scholar]
- 57.Vallee RB, Sheetz MP. Targeting of motor proteins. Science. 1996;271:1539–1544. doi: 10.1126/science.271.5255.1539. [DOI] [PubMed] [Google Scholar]
- 58.Wang LH, Sudhof TC, Anderson RG. The appendage domain of alpha-adaptin is a high affinity binding site for dynamin. J Biol Chem. 1995;270:10079–10083. doi: 10.1074/jbc.270.17.10079. [DOI] [PubMed] [Google Scholar]
- 59.Watanabe T, Inui M, Chen BY, Iga M, Sobue K. Annexin VI-binding proteins in brain. Interaction of annexin VI with a membrane skeletal protein, calspectin (brain spectrin or fodrin) J Biol Chem. 1994;269:17656–17662. [PubMed] [Google Scholar]
- 60.Wilton JC, Matthews GM, Burgoyne RD, Mills CO, Chipman JK, Coleman R. Fluorescent choleretic and cholestatic bile salts take different paths across the hepatocyte: transcytosis of glycolithocholate leads to an extensive redistribution of annexin II. J Cell Biol. 1994;127:401–410. doi: 10.1083/jcb.127.2.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ying Y-S, Anderson RGW, Rothberg KG. Each caveola contains multiple glycosyl-phosphatidylinositol anchored membrane proteins. Cold Spr Harb Symp Quant Biol. 1992;57:593–604. doi: 10.1101/sqb.1992.057.01.065. [DOI] [PubMed] [Google Scholar]
- 62.Zhang JZ, Davletov BA, Südhof TC, Anderson RGW. Synaptotagmin is a high affinity receptor for clathrin AP-2: implications for membrane recycling. Cell. 1994;78:751–760. doi: 10.1016/s0092-8674(94)90442-1. [DOI] [PubMed] [Google Scholar]