

Abstract
We have investigated the axonal transport of neurofilament protein in cultured neurons by constricting single axons with fine glass fibers. We observed a rapid accumulation of anterogradely and retrogradely transported membranous organelles on both sides of the constrictions and a more gradual accumulation of neurofilament protein proximal to the constrictions. Neurofilament protein accumulation was dependent on the presence of metabolic substrates and was blocked by iodoacetate, which is an inhibitor of glycolysis. These data indicate that neurofilament protein moves anterogradely in these axons by a mechanism that is directly or indirectly dependent on nucleoside triphosphates. The average transport rate was estimated to be at least 130 μm/h (3.1 mm/d), and ∼90% of the accumulated neurofilament protein remained in the axon after detergent extraction, suggesting that it was present in a polymerized form. Electron microscopy demonstrated that there were an abnormally large number of neurofilament polymers proximal to the constrictions. These data suggest that the neurofilament proteins were transported either as assembled polymers or in a nonpolymeric form that assembled locally at the site of accumulation. This study represents the first demonstration of the axonal transport of neurofilament protein in cultured neurons.
Keywords: neurofilament, cytoskeleton, slow axonal transport, axon, neuron
Axonal proteins are synthesized in the cell bodies of neurons and transported along axons by two distinct mechanisms known as fast and slow axonal transport (Tytell et al., 1981; Vallee and Bloom, 1991). Fast axonal transport represents the movement of proteins associated with membranous organelles at modal rates of ∼50–400 mm/d, whereas slow axonal transport represents the movement of cytoskeletal and cytosolic proteins at modal rates of ∼0.2–8 mm/d (Lasek et al., 1984).
The molecular mechanism of slow axonal transport is not known and has been the subject of much controversy in recent years. According to one hypothesis, cytoskeletal polymers are the transport vehicle and cytosolic proteins move by association with the moving polymers (Lasek, 1986; Vallee and Bloom, 1991; Baas and Brown, 1997). According to another hypothesis, cytoskeletal polymers in axons are stationary and cytoskeletal and cytosolic proteins are transported along axons in a nonpolymeric form (Weisenberg et al., 1987; Hollenbeck, 1989; Nixon, 1991; Hirokawa et al., 1997). At present both hypotheses are based on indirect evidence, due primarily to the lack of an experimentally accessible system in which the axonal transport of cytoskeletal proteins can be visualized directly in living cells.
Two approaches that have held great promise for studies on slow axonal transport are fluorescence photobleaching and photoactivation. In the first study of this kind, Keith (1987) reported anterograde movement of tubulin in the neurites of cultured PC12 cells. However, more recent studies on tubulin in cultured PC12 cells (Lim et al., 1989), cultured chick sensory neurons (Lim et al., 1990), and developing neurons in zebrafish and grasshopper embryos (Sabry et al., 1995; Takeda et al., 1995), as well as on tubulin, actin, and neurofilament proteins in cultured mouse sensory neurons (Okabe and Hirokawa, 1990, 1992; Okabe et al., 1993; Takeda et al., 1994), have all reported no detectable movement. The only system in which these techniques have demonstrated movement is in cultured embryonic frog neurons (Okabe and Hirokawa, 1993; Reinsch et al., 1991), but Okabe et al. (1992) have argued that this movement is an artifact of the rapid growth of these neurons in culture and that it reflects passive dragging of the axonal cytoskeleton by the growth cone rather than bona fide slow axonal transport. More recently, Chang et al. (1998) have shown that the movement of tubulin in cultured frog embryonic neurons is dependent on the adhesiveness of the culture substratum and is not observed when the axons are induced to grow at comparable rates on more highly adhesive substrates. The explanation for these observations is presently unclear, but they have cast doubt on the significance of the observed movement. Thus the photobleaching and photoactivation approaches have yet to provide a clear and unequivocal demonstration of slow axonal transport in cultured neurons.
In spite of the negative results obtained with the photobleaching and photoactivation approaches, it is clear that active transport of cytoskeletal proteins must occur in culture for axon growth to be sustained. Soluble proteins can diffuse freely in axons but the efficiency of diffusion as a mechanism for net transport of molecules along an axon declines with the square of the distance (e.g., Popov and Poo, 1992). Sabry et al. (1995) have shown that diffusion of proteins from the cell body becomes limiting for axon growth at a distance that is inversely related to the rate of growth. For the fastest growing axons, this distance was calculated to be <10 μm, but even in the slowest growing axons diffusion could not support the growth of axons longer than 200 μm. Further support for the active transport of cytoskeletal proteins in cultured neurons has come from the work of Campenot et al. (1996) who adapted the radioisotopic pulse-labeling paradigm to cell culture using a compartmentalized culture dish assembly. Using this approach, the authors demonstrated the anterograde movement of a wave of pulse-labeled tubulin at a modal rate of ∼1 mm/d, which is consistent with the rates that have been obtained using radioisotopic pulse labeling in whole animals. However, if axonal transport of cytoskeletal proteins does occur in cultured neurons, why has it not been revealed by the photobleaching and photoactivation approaches? There are several possible explanations (see Discussion), but the issue presently remains unresolved. Meanwhile, these data clearly indicate that there is a need for alternative experimental approaches.
For many decades, it has been known that physical constriction of axons can impede the movement of axonally transported materials. This was first observed by Weiss and Hiscoe (1948) in their seminal study on axoplasmic transport in regenerating peripheral nerves. Constriction of these nerves led to an accumulation of axoplasm proximal (upstream) to the site of compression, which caused the individual axons (and therefore the entire nerve) to become locally distended. More recent ultrastructural analyses have shown that this axonal enlargement is caused by an accumulation of neurofilaments and membranous organelles proximal to the site of constriction (e.g., Schmidt and Plurad, 1985; LeBeau et al., 1988). These observations suggest that axonally transported materials, especially neurofilaments and membranous organelles, are susceptible to an increase in the resistance to their movement such as that encountered at an axonal constriction. In this paper we demonstrate that the nerve constriction paradigm of Weiss and Hiscoe can also be applied to cultured neurons by constricting single axons with fine glass fibers. Using quantitative immunofluorescence microscopy in conjunction with digital image processing techniques, we show that neurofilament protein accumulates proximal to these constrictions in an energy dependent manner and at a rate consistent with that of slow axonal transport in whole animals. To the best of our knowledge, this is the first demonstration of the axonal transport of neurofilament protein in cultured neurons.
Materials and Methods
Cell Culture
Neurons dissociated from the dorsal root ganglia of E16.5 rat embryos were plated onto glass coverslips coated with polylysine and laminin at ∼24–30 cells/cm2 as described by Brown (1997). Cultures were maintained at 37°C in Leibovitz's L-15 medium (phenol red free; GIBCO BRL) supplemented with 10% adult rat serum (prepared as described by Hawrot and Patterson, 1979), 0.5% hydroxypropylmethylcellulose (Methocel™; Dow Corning), 50 ng/ml 2.5S nerve growth factor (Collaborative Research), 0.6% glucose, and 2 mM l-glutamine (Bray, 1991). For experiments on axons in the presence and absence of metabolic substrates, the above medium was rinsed and replaced with Dulbecco's PBS (GIBCO BRL) supplemented with 0.5% Methocel™ and 50 ng/ml 2.5S nerve growth factor, with or without 0.6% glucose (Sigma Chemical Co.) and 0.055% sodium pyruvate (Sigma Chemical Co.). All studies were performed between 14 and 71 h after the time of plating.
Constriction of Axons
Glass fibers for constriction were pulled to a diameter of 0.7–1.8 μm from 1-mm-diam borosilicate glass rods (World Precision Instruments) using a Sutter P-87 Flaming-Brown pipette puller. The cultures were maintained at ∼37°C on the stage of a Nikon Diaphot microscope using a Nicholson ASI-400 Air Stream Incubator and the medium was covered with a thin layer of silicone fluid (dimethylpolysiloxane; Sigma Chemical Co.; viscosity = 5 centistokes) to prevent evaporation. The microscope was situated on a vibration isolation table and the air stream was directed onto the culture dish from beneath the microscope stage to avoid vibration of the glass fiber.
To constrict an axon, a glass fiber was attached to a Narashige 3-axis hydraulic micromanipulator at a 45° angle to the coverslip and then oriented perpendicular to the axon by rotating the culture dish (see Fig. 1). All subsequent manipulations were performed while observing the axon using a Zeiss 63×/1.4 NA Phase Apochromat oil immersion objective. The fine flexible fiber was positioned with its tip touching the glass coverslip ∼40 μm to one side of the axon and then advanced downward, causing it to bend and flatten out across the axon. The length of the glass fiber in contact with the coverslip ranged from ∼80–140 μm. Forces generated during bending of the fiber sometimes caused it to slip along the substrate, shearing the axon. When axons were damaged during constriction, this was almost always the cause. To avoid this problem, downward movements in the Z dimension were countered by fine adjustments in the X dimension to keep the tip of the fiber in place. Because of slight drift of the microscope stage (<0.5 μm/h), it was often necessary to make fine adjustments to the micromanipulator in the X and Y dimensions during the course of our observations to prevent movement of the fiber relative to the axon. To release the constriction, essentially the same movements were performed as during application of the constriction, but in reverse. Once out of contact with the axon, the fiber could be moved away rapidly without further incident.
Figure 1.

Method for constricting single axons in cell culture. A fine flexible glass fiber is oriented perpendicular to the axon and at a 45° angle to the horizontal with its tip touching the substrate to one side of the intended constriction site (A). The fiber is then advanced downward against the coverslip, causing it to bend and flatten out across the axon (B).
All constrictions were performed on axons that had branched by bifurcation of the growth cone to give rise to two sister axons. This allowed us to compare the neurofilament distribution along the constricted axon with its nonconstricted sister (see Results). In choosing which of the two sister axons to constrict, we selected the axon that was closest to the desired orientation and then rotated the dish to bring that axon into precise orientation. Constrictions were applied approximately midway between the branch point and the growth cone, at least 80 μm from each. Axons were not constricted if either of the sister axons were fasciculated with each other or with other axons, or if they intersected other axons within 80 μm of the constriction site.
Fixation and Immunostaining
For immunofluorescence microscopy, cultures were rinsed twice with PBS (138 mM NaCl, 2.7 mM KCl, and 10 mM sodium phosphate, pH 7.4) and then fixed with 4% formaldehyde in PBS containing 1% sucrose for 30 min. After fixation the cells were extracted with 1% Triton X-100 in PBS containing 0.3 M NaCl and then stained using a rabbit polyclonal antibody specific for low molecular weight neurofilament triplet polypeptide (NF-L)1 and a secondary antibody conjugated to lissamine rhodamine (Jackson ImmunoResearch Labs) as described by Brown (1997). In earlier experiments the constriction was released immediately before fixation to avoid damaging the axon due to movement of the glass fiber during the PBS rinses and the addition of the fixative solution. In some of these experiments, constricted axons were extracted before fixation by rinsing the cells once with PBS, once with PHEM (60 mM NaPipes, 25 mM NaHepes, 10 mM NaEGTA, and 2 mM MgCl2, pH 6.9), and then treating them with 0.02% saponin in a solution composed of PHEM and 0.19 M NaCl (Black et al., 1986). The interval between release of the constriction and addition of the extraction or fixation solution ranged from 1 to 3 min. In later experiments, we found that we were able to fix constricted axons directly on the microscope stage without removal of the glass fiber and without damage to the axons, though ∼50% of the axons fixed in this manner had to be discarded because they adhered to the glass fiber as it was removed, causing them to detach from the substrate. For these experiments, two bent syringe needles were mounted on opposite sides of the culture dish before constriction and each was connected to a syringe using flexible plastic tubing. The tubing functioned to prevent transmission of vibration to the dish when suction or pressure was applied to the syringes and during syringe exchange. Immediately before fixation, ∼1 ml of medium was withdrawn using one syringe, leaving ∼0.5 ml in the dish, and then ∼3.5 ml fixative solution containing 1 mM EGTA was added using the other syringe. During the rinses, the cells remained undisturbed because they lay within the circular well in the center of the culture dish assembly. After fixation for 15 min, the glass fiber was removed and then the cells were fixed for another 15 min in fresh fixative.
Microscopy, Image Acquisition, and Analysis
Cells were observed using a Nikon Diaphot 300 microscope and digital images were acquired using a Photometrics CCD camera equipped with a Kodak KAF 1400 chip. Images of constrictions were acquired under phase contrast using a Zeiss 63×/1.4 NA Phase Apochromat oil immersion objective. Fluorescence images were acquired using a Zeiss 25×/0.8 NA Phase Plan Neofluar multi-immersion objective or a Nikon 100×/1.3 NA Plan Fluor Phase oil immersion objective. Fluorescence intensity was analyzed along individual axons using the segmented mask method developed by Brown et al. (1992). In brief, digital image processing techniques were performed on flat-field corrected images to create a mask of the axon that was subdivided into contiguous segments of equal length. In this study, we used a segment length of 4.1 μm for all analyses. The pixel intensities were then corrected for background using a complementary background mask and the total fluorescence intensity for each segment was calculated by summing the corrected intensities of the individual pixels in that segment. Note that this method of analysis measures the total fluorescence intensity per unit length of axon regardless of axon diameter, so no correction is required for axonal volume. All image processing and analysis procedures were performed on a Macintosh computer using Oncor-Image software (Oncor Inc.). Digital images were prepared for publication using Adobe Photoshop software.
Electron Microscopy
For electron microscopy, constricted axons were fixed on the microscope stage with the glass fiber in place by gently withdrawing most of the medium from the culture dish and then gently adding an excess volume of fixative using the syringe assembly described above. Fixation in the presence of Methocel™ was found to produce sheets of flocculent material which tended to peel off the substrate during subsequent processing, resulting in frequent loss or damage to the cells of interest. For this reason, we replaced the L-15-based culture medium with the same medium lacking Methocel™ immediately before constriction for electron microscopy. The fixative solution was composed of 2% glutaraldehyde (EM grade; Polysciences), 0.2% tannic acid, and 150 mM sodium phosphate, pH 7.4 (320 mOs). The fixed cells were treated with osmium tetroxide and uranyl acetate, dehydrated with a graded series of ethanol solutions, and embedded in Poly/Bed 812 epoxy resin, essentially as described by Baas and Ahmad (1993). The glass coverslip was dissolved by exposing the coverslip to 48% hydrofluoric acid for 10 min (Yu et al., 1996) and the plastic dish was removed as described by Whitlon and Baas (1992). The embedded cells and their axons were located under phase contrast and the axons of interest were marked by scoring a circle in the block using a Leitz diamond-scoring object marker. Ribbons of silver/gold sections were cut using a Reichert-Jung Ultracut E microtome, retrieved on hexagonal mesh copper grids, stained with uranyl acetate and lead citrate, and examined with a Jeol JEM-1010 transmission electron microscope at 80 kV. Images were acquired using a Gatan Model 791 cooled CCD camera with a 16 bit 1024 × 1024 chip mounted on the 35-mm camera port of the electron microscope column, using a Macintosh computer and Gatan Digital Micrograph software. Digital images and montages were prepared for publication using Adobe Photoshop software.
Microinjection and Extraction of Fluorescent Dextran
Cell bodies of cultured neurons were injected with 10 mg/ml tetramethyl rhodamine isothiocyanate dextran (Sigma Chemical Co.; average Mw = 76,000) in 50 mM potassium glutamate, pH 7.0, using a Medical Systems Corporation PLI-100 pressure injection apparatus with a compressed nitrogen gas pressure source. Micropipettes were pulled using a Sutter P-87 Flaming-Brown pipette puller and maneuvered using a Narashige 3-axis hydraulic micromanipulator. After injection, the dishes were returned to the incubator for a few hours and then placed on the microscope stage and photographed under epifluorescence illumination immediately before extraction and then after extraction for 2, 5, and 10 min. The rinsing and extraction protocol was the same as described above, except that it was performed directly on the microscope stage. The fluorescence intensity in the cell bodies was quantified using Oncor-Image software (Oncor Inc.).
Results
Observations on Constricted Axons
Fig. 1 shows the method that we used for constricting axons, which is described in Materials and Methods. Fig. 2, A–D shows an axon before constriction and then after constriction for 1, 30, and 120 min. In most cases, constricted axons showed no sign of injury; growth cones continued to exhibit filopodial and/or lamellipodial activity, large membranous organelles visible by phase contrast microscopy continued to move on both sides of the constriction, and the axon retained its normal refractivity under phase contrast optics. When damage did occur, it generally resulted from sudden movement of the glass fiber and was apparent due to an immediate change in refractivity followed by beading and subsequent fragmentation of the axon.
Figure 2.

Morphological changes at the site of constriction. Phase contrast images of an axon immediately before constriction (A) and after constriction for 1, 30, and 120 min (B–D). The cell was fixed after constriction for 2 h and then the glass fiber was removed (E) and the cell was stained for NF-L by immunofluorescence microscopy (F). Bar, 5 μm.
Within 15 min after application of a constriction the axons began to enlarge on both sides of the glass fiber (Fig. 2 C). The swellings generally appeared to enlarge at comparable rates, though at later times the proximal swelling often became more elongated, exhibiting a slightly more gradual taper than the distal swelling (Fig. 2 D). Both the proximal and distal swellings exhibited continual changes in shape throughout the duration of the constriction, implying extensive motile activity within them. Large membrane-bound organelles, which appear as round or elongated bright or dark structures by phase contrast microscopy (Overly et al., 1996), were observed to accumulate in the swellings as they enlarged. This suggests that the axonal swellings were formed, at least in part, by the accumulation of axonally transported membrane-bound organelles whose movement was impeded by the constriction.
During the course of these studies we have constricted a total of 191 axons under various conditions and for various periods of time. 161 of these axons survived, which represents a survival rate of 84%. In 10 cases, the axons were observed to extend a short collateral branch from the proximal swelling which ran alongside the glass fiber for a distance of <40 μm. In eight of these axons, the branch extended towards the tip of the glass fiber and in two cases it extended away from the tip. Such collateral branches were never observed to extend from distal swellings. In seven cases, the distal swelling was observed to pull away from the glass fiber in a distal direction, remaining connected to the proximal swelling by a very thin strand of axon, but this was never observed proximally (these cells were excluded from our analyses). In 18 cases, the axon was observed to form sinusoidal bends reminiscent of those observed during the shortening of axons detached from the substrate or severed from their cell bodies (Shaw and Bray, 1977; George et al., 1988). In 13 of these axons the sinusoids were located proximal to the constriction and in the other 5 they were located distally. Of the 30 axons that did not survive constriction, 29 died due to severing of the axon during initial application of the constriction and one died of unknown causes at a later point during the experiment.
Quantification of Neurofilament Protein Accumulation
To determine whether neurofilament protein accumulated at the axonal constrictions, constricted axons were fixed and then stained for NF-L by immunofluorescence microscopy (Fig. 2, E and F). In all cases, the proximal swelling stained more intensely than elsewhere along the axon but the distal swelling did not (Fig. 2 F). To quantify the relative amount of neurofilament protein proximal and distal to the constriction, we acquired digital images of constricted and nonconstricted axons and then analyzed the fluorescence intensity along their length using the segmented mask method (see Materials and Methods).
All the experiments described in this paper were performed on cultures obtained from dorsal root ganglia. Though these ganglia contain exclusively sensory neurons, these cells are not a homogeneous population and they do vary in their size and neurofilament protein content (Lawson, 1992). This makes it difficult to accurately compare the effect of constriction on the neurofilament protein distribution in different cells. To control for this variation, we took advantage of the fact that axons in these cultures typically branch by bifurcation of the growth cone, giving rise to two sister axons that have similar length, diameter, and growth behavior (Bray et al., 1987). By restricting our analyses to axons that branched in this manner we were able to constrict one of the two sister axons and then compare the neurofilament protein distribution with its nonconstricted sister control (Fig. 3).
Figure 3.

Method of analysis of constricted and control sister axons. This schematic diagram depicts a cultured neuron with a bifurcating axon. One of the sister branches is constricted and the other serves as a nonconstricted control. After constriction for a certain period of time, the cell is fixed, neurofilament protein is visualized by immunofluorescence microscopy, and then the fluorescence intensity is quantified along both axons using the segmented mask method (see Materials and Methods). The rectangles drawn with dashed lines demarcate the measurement windows located proximal and distal to the constriction (P1 and D1), and at corresponding distances along the nonconstricted sister axon (P2 and D2). The total fluorescence intensity in each measurement window is a relative measure of the amount of neurofilament protein in that portion of the axon. To calculate the proximal and distal accumulation ratios, the total fluorescence intensities in the proximal and distal measurement windows of the constricted axon (P1 and D1, respectively) are divided by the total fluorescence intensities in the corresponding measurement windows of the nonconstricted sister axon (P2 and D2, respectively), i.e., P1/P2 and D1/D2. Thus, the proximal and distal accumulation ratios are each a measure of the accumulation or depletion of neurofilament protein in the constricted axon relative to its nonconstricted sister.
Fig. 4 shows the neurofilament protein distribution along three axons that were constricted for different lengths of time, and along their corresponding nonconstricted sister axons. Constriction for 5 s had no apparent effect; both the constricted and control sister axons exhibited a fairly uniform distribution of neurofilament protein along their entire length, which is typical for axons in these cultures. In contrast, constriction for 30 min resulted in a pronounced increase in the amount of neurofilament protein proximal to the constriction and a smaller increase distally. Constriction for 2 h resulted in a further increase in the amount of neurofilament protein proximal to the constriction, but no further increase distal to the constriction. Proximal to the constriction, the amount of neurofilament protein typically declined over a distance of 5–10 segments (∼20–40 μm) whereas distal to the constriction the decline was more abrupt.
Figure 4.

Neurofilament protein distribution along constricted and control sister axons. Neurons were fixed and stained for neurofilament protein by immunofluorescence microscopy using a rabbit polyclonal antibody to NF-L and the fluorescence intensity was quantified along constricted and control sister axons by the segmented mask method (see Materials and Methods). The three profiles on the left represent axons that were constricted for 5 s, 30 min, and 2 h. The three profiles on the right represent the corresponding control sister axons, which were not constricted. Each graph represents the fluorescence intensity profile along a 300-μm length of axon. The total axon lengths were 739, 500, and 611 μm for the constricted axons and 700, 435, and 776 μm for the control axons, respectively. The arrow on each constricted axon profile indicates the segment that contained the constriction. Each point in the graphs represents the fluorescence intensity for a single 4.1-μm segment along the axon, which is a relative measure of the amount of neurofilament protein in that segment. Proximal is left and distal is right in all cases. The horizontal bars along the abscissa of each plot represent the locations of the 41-μm (10 segment) measurement windows that were used for calculation of the proximal and distal accumulation ratios.
To allow quantitative comparison of the neurofilament protein accumulation in different cells, we defined two axonal measurement windows proximal and distal to the constriction site. Each window measured 41 μm in length, which corresponded to 10 segments in the segmented mask analysis. This window length was selected because it was the minimum size necessary to include the entire neurofilament protein accumulation in all the axons that we analyzed. The segment containing the glass fiber was excluded from the windows because it contained portions of both the proximal and distal swellings. The total fluorescence intensity in each window was divided by the corresponding intensity in an identically sized measurement window at an equivalent distance along the control sister axon to obtain a ratio which we call the accumulation ratio. The accumulation ratio is a measure of the amount of neurofilament protein proximal or distal to the constriction site relative to the corresponding nonconstricted sister axon (Fig. 3). An accumulation ratio of >1 means that there is more neurofilament protein in the constricted axon than in the sister control, and a ratio of <1 indicates that there is less.
Fig. 5 A shows the mean proximal and distal accumulation ratios after constriction for different durations. To establish the extent of variation in neurofilament protein content between sister axons, we performed sham experiments in which neither sister axon was constricted. This yielded mean accumulation ratios of 1.0 (range = 0.7–2.1, n = 7) for the proximal measurement window and 0.9 (range = 0.4–1.4, n = 7) for the distal measurement window, indicating that sister axons were not significantly different from each other in their neurofilament content (P = 0.6, t test). These data confirm the validity of using sister axon comparisons in our quantitative analyses. After constriction of axons for 5 s, there was no detectable change in the amount of neurofilament protein either proximal to the constriction (mean accumulation ratio = 1.0, range = 0.5–1.4, n = 5) or distal to the constriction (mean accumulation ratio = 0.9, range = 0.6–1.5, n = 5). This confirms that there was no redistribution of neurofilament protein as an immediate consequence of compression of the axon with the glass fiber. After constriction for 30 min, the average amount of neurofilament protein was 110% higher proximally (mean accumulation ratio = 2.1, range = 0.6– 4.3, n = 4) and 60% higher distally (mean accumulation ratio = 1.6, range = 1.0–2.6, n = 4) but these values were not significantly different from axons constricted for 5 s (P = 0.3 and 0.1 respectively, t test). After constriction for 2 h, the average amount of neurofilament protein was 630% higher proximally (mean accumulation ratio = 7.3, range = 2.0–12.4, n = 6), which represented a statistically significant increase compared with axons constricted for 5 s (P = 0.01, t test) and 30 min (P = 0.04, t test). In contrast, the average amount of neurofilament protein distal to the constriction increased by only 30% (mean accumulation ratio = 1.3, range = 0.4–2.3, n = 6), and was not significantly different from axons constricted for 5 s (P = 0.3, t test) and 30 min (P = 0.6, t test). These data indicate that there was a marked time-dependent accumulation of neurofilament protein proximal to the constrictions, which suggests that neurofilament protein is transported anterogradely in these axons.
Figure 5.

Quantitative analysis of neurofilament protein accumulation proximal and distal to the constriction site after constriction for various durations and under various experimental conditions. Each column represents the mean accumulation ratio for the number of cells indicated and the error bars represent the standard deviation about the mean. An accumulation ratio >1 indicates an accumulation of neurofilament protein in the constricted axon relative to the control sister axon and an accumulation ratio <1 indicates a depletion. (A) Time course of accumulation. For the sham experiment, no constriction was performed so as to allow comparison of the normal variability between sister axons. For the other experiments, the axons were constricted for either 5 s, 30 min, or 2 h. The mean proximal accumulation ratio after 2 h was significantly greater than the corresponding distal accumulation ratio (P = 0.01, t test), and significantly greater than the proximal accumulation ratios after 5 s (P = 0.01, t test) and 30 min (P = 0.04, t test). All other accumulation ratios were not significantly different from each other. (B) Constriction in the presence of an inhibitor of glycolysis. Cells were preincubated in medium containing 2 mM sodium iodoacetate for 1 h and then constricted for 2 h in the same medium. The mean proximal accumulation ratio was significantly less than for axons constricted for 2 h in the absence of inhibitor (P = 0.02, t test). (C) Permeabilization of constricted axons with detergent to extract soluble neurofilament protein. Cells were constricted for 2 h and then extracted with 0.02% saponin before fixation as described in Materials and Methods. The mean proximal accumulation ratio was significantly greater than the corresponding distal accumulation ratio (P = 0.01, t test) and significantly greater than the proximal accumulation ratio in unextracted axons after constriction for 5 s (P = 0.01, t test).
To confirm that the accumulation of neurofilament protein proximal to the constrictions was not due to axonal shortening, we measured the average distance of the constriction site from the cell body for six different axons. The average distance was 370 μm at the time of constriction (minimum = 263 μm, maximum = 496 μm) and 375 μm after constriction for 2 h (minimum = 270 μm, maximum = 504 μm). The average change in axon length was 1.2 ± 2.4% (mean ± standard deviation; minimum = −3.5%, maximum = +2.9%). These measurements demonstrate that there was no significant change in axon length proximal to the constrictions during these experiments and thus the accumulation of neurofilament protein cannot be accounted for by shortening of the proximal axon.
Metabolic Requirements of Neurofilament Protein Accumulation
To examine the metabolic requirements of neurofilament protein accumulation, we investigated the effects of iodoacetate, which is an inhibitor of the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (Sabri and Ochs, 1971). Axons constricted for 2 h in the presence of 2 mM iodoacetate did not develop swellings either proximal or distal to the constriction and exhibited greatly reduced organelle movement, which is consistent with the known dependence of organelle translocation on ATP (Adams, 1982; Brady et al., 1982). In addition, our quantitative analyses revealed no significant accumulation of neurofilament protein proximal to the constriction compared with axons constricted for 5 s (P = 0.2, t test; Fig. 5 B), which suggests that neurofilament protein accumulation was dependent on glycolysis.
To confirm the dependence of neurofilament protein accumulation on glycolysis, we also constricted axons in a medium that lacked metabolic substrates. For these experiments, we replaced the L-15-based culture medium with a simpler Dulbecco's PBS-based medium, either without any metabolic substrates or with glucose and pyruvate as the sole metabolic substrates. In the presence of glucose and pyruvate (fed), axons exhibited apparently normal swellings proximal and distal to the constriction and neurofilament protein was observed to accumulate proximally (Fig. 6). However, the magnitude of the accumulation was lower than for axons constricted in the L-15-based medium and this suggests that the physiology of these cells was compromised somewhat in the simpler PBS-based medium, which lacked serum as well as numerous other components that are present in the L-15 formulation (see Materials and Methods). In the absence of glucose and pyruvate (starved) the axons did not swell either proximal or distal to the constriction and the accumulation of neurofilament protein was reduced significantly (P = 0.01, t test). Collectively, these data indicate that the anterograde axonal transport of neurofilament protein is an active process that is dependent, directly or indirectly, on nucleoside triphosphates.
Figure 6.
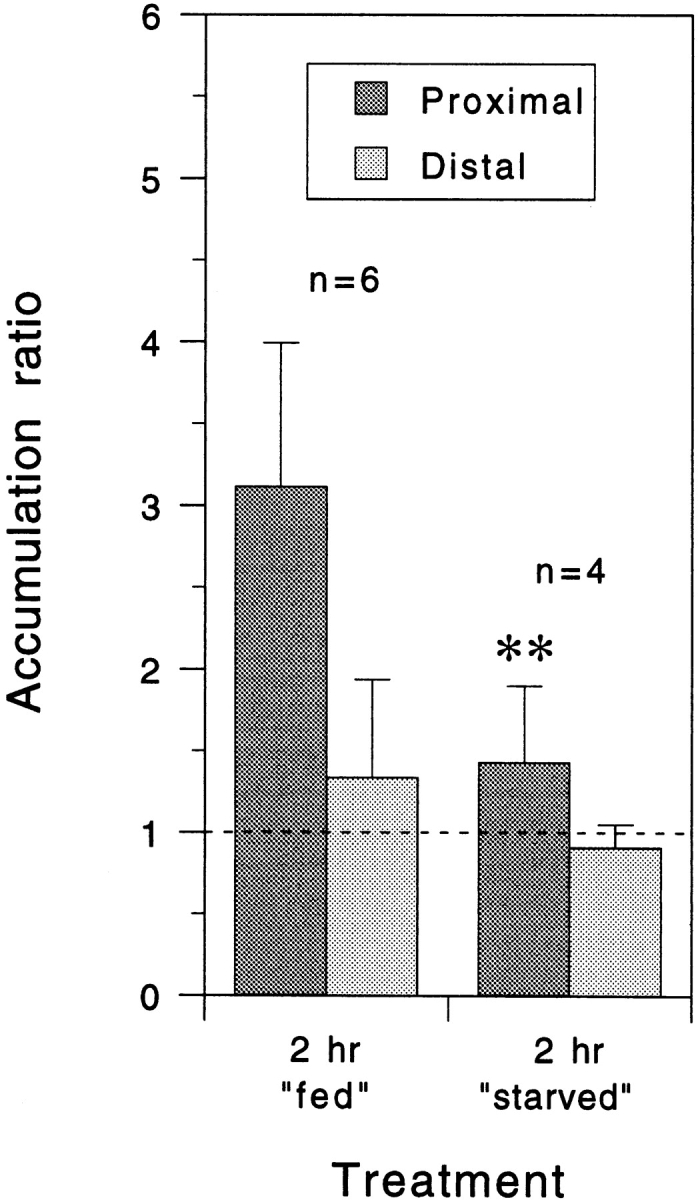
Quantitative analysis of neurofilament protein accumulation proximal and distal to the constriction site after constriction for 2 h in the presence or absence of metabolic substrates. Each column represents the mean accumulation ratio for the number of cells indicated and the error bars represent the standard deviation about the mean as described in the legend to Fig. 5. For these experiments, the L-15-based culture medium was replaced with a simpler Dulbecco's PBS-based medium with or without 0.6% glucose and 0.055% sodium pyruvate (fed and starved, respectively). The mean proximal accumulation ratio in the presence of glucose and pyruvate was lower than for axons constricted for the same period of time in L-15-based medium (Fig. 5 A) but it was, nevertheless, reduced significantly when these two substrates were omitted (P = 0.01, t test).
Detergent Extraction of Constricted Axons
To address the form in which the neurofilament protein accumulated, we constricted axons for 2 h and then permeabilized them with detergent under conditions that stabilize neurofilament polymers in order to extract neurofilament protein subunits and diffusible oligomers. The cells were then fixed, immunostained and analyzed as described above. Previous studies in our laboratory have shown that neurofilament polymers in cultured neurons splay apart from each other when treated with the concentrations of detergent that are normally used to extract cultured cells (Brown, 1997, 1998). This splaying phenomenon presented a problem for our studies because we found that it interfered with creation of the segmented mask that we used for our quantitative analyses. However, we found that cells could be extracted without inducing splaying by using lower concentrations of detergent such as 0.02% saponin, which has been used to extract soluble proteins from cells by Nakata et al. (1987) and Okabe et al. (1993). To confirm that 0.02% saponin was sufficient to permeabilize our cells, we microinjected neurons with 70,000 mol wt fluorescent dextran and then quantified the fluorescence intensity within the cell bodies before and after detergent treatment (see Materials and Methods). Fig. 7 shows that 96% of the fluorescent dextran diffused out of the cells within 2 min after addition of 0.02% saponin. After 10 min, the proportion extracted had increased to 97%. Subsequent treatment with 1% Triton X-100 for 10 min (data not shown) increased the proportion extracted to 98%. Thus treatment with 0.02% saponin for 10 min extracted 99% of the Triton X-100 soluble fluorescence in these neurons.
Figure 7.


Extraction of neurons with 0.02% saponin. (A) Cell body of a cultured neuron that was microinjected with tetramethyl rhodamine isothiocyanate dextran (average mol wt = 76,000). (B) The same cell body after extraction with 0.02% saponin in PHEM + 0.19 M NaCl for 10 min as described in Materials and Methods. Bar, 15 μm. (C) Quantitative data for five cells imaged before extraction and after extraction for 2, 5, and 10 min. For each cell, the total fluorescence intensity in the cell body after extraction was expressed as a percentage of the total fluorescence intensity in the cell body before extraction. The error bars represent the standard deviation about the mean for each time point. 97% of the fluorescent dextran was extracted from the cells within 10 min.
Fig. 5 C shows the proximal and distal accumulation ratios for five axons that were constricted for 2 h and then extracted with 0.02% saponin before fixation and immunostaining. The average proximal accumulation ratio was significantly greater than the average distal accumulation ratio (P = 0.01, t test), and significantly greater than the average proximal accumulation ratio in unextracted axons after constriction for 5 s (P = 0.01, t test). The magnitude of the average proximal accumulation ratio was 6.5, which is 89% of the average accumulation ratio for unextracted cells after constriction for 2 h (Fig. 5 A). This suggests that 89% of the accumulated neurofilament protein was present in a polymerized form, though this must be considered an approximate value given the large variability encountered among different cells in these experiments (see Discussion).
Electron Microscopy of Constricted Axons
To examine the ultrastructure of constricted axons, we fixed cells after constriction for 2 h and then processed them for electron microscopy. Fig. 8 A shows a longitudinal section of an axon taken parallel to the glass coverslip and passing through the center of the proximal and distal swellings. The swellings contained numerous membrane-bound organelles including mitochondria, small vesicles with light or dark lumens, large heterogeneous multilamellar organelles, and small tubules that resembled smooth endoplasmic reticulum. Distally, large multilamellar organelles predominated, though smaller vesicles and tubules were also observed (Fig. 8, D and E). Proximally, small vesicles and tubules predominated and there were very few multilamellar organelles (Fig. 8, B and C). Mitochondria were observed to accumulate on both sides of the constrictions. The distinct size and appearance of the membranous organelles in the proximal and distal swellings is consistent with previous descriptions of anterogradely and retrogradely moving organelles (e.g., Smith, 1980; Tsukita and Ishikawa, 1980; Fahim et al., 1985; Hirokawa et al., 1990), and this indicates that the constrictions impeded both anterograde and retrograde fast axonal transport.
Figure 8.
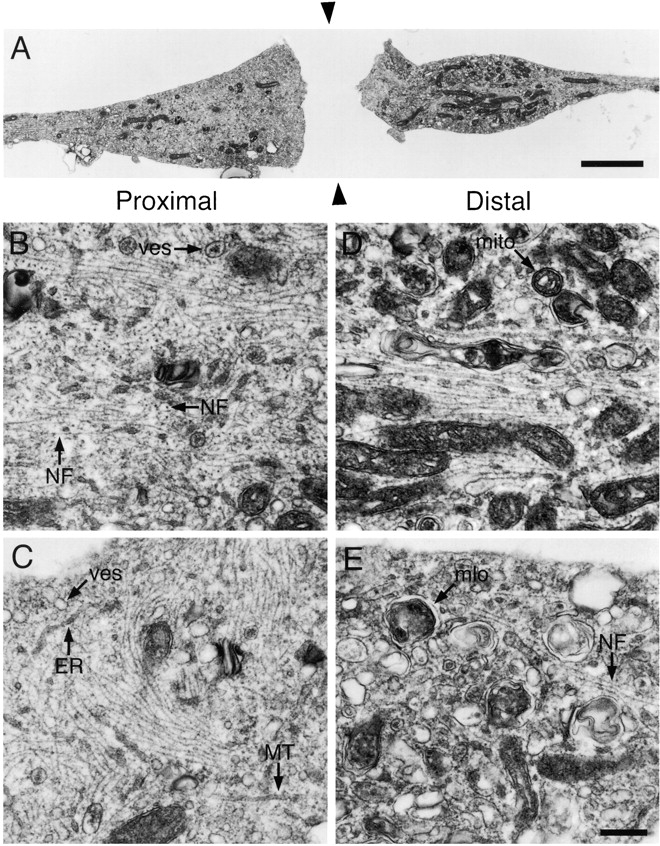
Electron micrographs of constricted axons. Axons were constricted for 2 h and then fixed and processed for electron microscopy as described in Materials and Methods. Sections were cut parallel to the glass coverslip and longitudinal with respect to the axis of the axon. Proximal is left and distal is right. (A) Low magnification montage showing the axonal swellings on either side of the constriction. This section passed through the approximate center of the proximal and distal swellings, which was ∼1.2 μm from the surface of the coverslip. The gap between the swellings represents the space occupied by the glass fiber (black arrowheads), which was removed after fixation. (B and C) High magnification views of proximal swellings. Note the numerous and highly disorganized neurofilaments arranged singly or in clusters and oriented longitudinally, obliquely, and transversely within the axon. (D and E) High magnification views of distal swellings. Note the presence of relatively few neurofilaments and numerous large multilamellar membranous organelles. The images in B and D correspond to regions of the swellings shown in A. ER, endoplasmic reticulum; MT, microtubule; mito, mitochondrion; mlo, multilamellar organelle; NF, neurofilament; ves, vesicle. Bars: (A) 2 μm; (B–E) 0.25 μm.
The proximal and distal swellings also contained neurofilaments and microtubules. Distally there were relatively few neurofilaments and they were generally oriented parallel or oblique to the longitudinal axis of the axon (Fig. 8, D and E). Sometimes we observed a core bundle of neurofilaments within the center of the distal swelling (Fig. 8 D). In contrast to the distal swellings, the proximal swellings contained many more neurofilaments and these polymers were highly disorganized, coursing longitudinally, obliquely, and transversely throughout the swelling (Fig. 8, B and C). The presence of large numbers of transversely and obliquely oriented neurofilaments is a dramatic departure from the normal longitudinal alignment of these polymers in axons, and the frequent occurrence of longitudinally and transversely oriented neurofilaments adjacent to each other suggests that they were entangled. Similar accumulations of neurofilaments have been observed proximal to long-term constrictions of peripheral nerves in laboratory animals (LeBeau et al., 1988; Schmidt and Plurad, 1985) and in human diseases that are thought to involve an impairment of slow axonal transport, such as giant axonal neuropathy (Donaghy et al., 1988) and amyotrophic lateral sclerosis (Hirano et al., 1984).
Serial longitudinal sections taken parallel to the glass coverslip revealed that the constricted axons were continuous and undamaged beneath the glass fiber (Fig. 9). By noting the interference colors of the sections as they came off the diamond knife and subsequently examining each section in the electron microscope it was possible to estimate the thickness of the axon at the constriction site. For the two axons that we analyzed in this way, the thicknesses were estimated to be ∼90 and 290 nm, which indicates that the axons were only partially constricted and that there may have been considerable variability in the extent of constriction for different axons (see Discussion). Within the region of continuity beneath the glass fiber, we observed neurofilaments, microtubules, and a variety of membranous organelles (Fig. 9 B). Some of the neurofilaments and microtubules were oriented longitudinally and passed directly under the fiber, whereas others were oriented transversely. These observations indicate that constriction did not completely disrupt the continuity of the axonal cytoskeleton and thus it is possible that the movement of axonally transported materials, including neurofilament proteins, may have been only partially impaired in our experiments (see Discussion).
Figure 9.

The axon is continuous across the constriction site. Axons were constricted for 2 h and then fixed and processed for electron microscopy as described in Materials and Methods. Sections were cut parallel to the glass coverslip and longitudinal with respect to the axis of the axon. Proximal is left and distal is right. (A) Low magnification montage showing continuity of the axon beneath the glass fiber. This section was located ∼75–165 nm from the surface of the coverslip. (B) High magnification view of the region of continuity beneath the glass fiber in A. Note the presence of numerous neurofilaments and microtubules passing beneath the glass fiber. The black arrowheads indicate the location of the glass fiber, which was removed after fixation. See legend to Fig. 8 for key to abbreviations. Bars: (A) 2 μm; (B) 0.25 μm.
Discussion
We have adapted the classic constriction paradigm of Weiss and Hiscoe (1948) to single cells by constricting axons in culture with fine glass fibers. The compressive force exerted by the glass fiber caused the axon to become constricted locally but electron microscopy confirmed that it remained continuous and undamaged. 84% of the axons survived this constriction procedure which is an indication of the remarkable resilience of these slender processes.
Within minutes of applying the constriction, anterogradely and retrogradely transported organelles began to accumulate causing the axon to swell proximal and distal to the glass fiber. This is consistent with the observations of many different laboratories that the movement of axonally transported membranous organelles can be retarded by locally increasing the physical resistance to movement. For example, similar accumulations have been observed proximal and distal to experimentally applied constrictions of peripheral nerves in vivo (e.g., Kapeller and Mayor, 1969a,b; Matthews, 1973) and isolated myelinated frog axons ex vivo (Smith, 1980), and at naturally occurring nodal constrictions along myelinated axons in vivo (Berthold et al., 1993; Fabricius et al., 1993; Zimmermann, 1996). Anterogradely and retrogradely transported organelles have also been induced to accumulate in the axons of cultured neurons by optical trapping of large organelles to create an intra-axonal obstruction (Martenson et al., 1993).
In addition to the accumulation of bidirectionally transported membranous organelles, our quantitative analyses also revealed a significant accumulation of neurofilament protein proximal to the constriction (average = 630%) that was significantly reduced in the presence of an inhibitor of glycolysis and in the absence of metabolic substrates. We also observed a much smaller increase in the average amount of neurofilament protein distal to the constriction (average = 30%), but this increase was not statistically significant and did not increase between 30 min and 2 h. These observations indicate that neurofilament protein is transported anterogradely in these axons in an energy-dependent manner and that the movement of this cytoskeletal protein, like membranous organelles, is also susceptible to an increase in the resistance to movement caused by local narrowing of the axon.
If neurofilament protein is transported anterogradely in these axons then we might expect to observe a depletion distal to the constriction in addition to an accumulation proximally. One possible explanation for the absence of a distal depletion in our studies could be that only a small proportion of the total neurofilament protein in these axons is transported, in which case loss of the moving fraction of protein distal to the constriction might not result in a significant change in neurofilament content. Another possible explanation is that neurofilament protein transport was only partially impaired and that a significant fraction of the transported neurofilament protein may have been able to pass across the constriction. The latter possibility is supported by our electron microscopic observations that have shown that the glass fibers caused only partial constriction of the axons. In theory, it is also possible that a proportion of the axonal neurofilament protein is transported retrogradely, as proposed by Glass et al. (1994). However, even if retrograde movement does occur, our data suggest that it would have to represent a small proportion of the total transported protein.
One of the principle issues in the controversy surrounding slow axonal transport has been the form in which the cytoskeletal proteins move (Baas and Brown, 1997; Hirokawa et al., 1997). In the simplest scenario, we might expect that the form in which a transported protein accumulates at an axonal constriction might represent the form in which it is transported. To examine quantitatively the proportion of the accumulated neurofilament protein that was polymerized, we extracted constricted axons with detergent before fixation and immunostaining. We found that, on average, 89% of the neurofilament protein in a 41-μm window of axon proximal to the glass fiber was not extracted by the detergent. Since the only detergent-insoluble form in which neurofilament proteins are known to exist is the neurofilament polymer, this suggests that 89% of the accumulated neurofilament protein was polymerized, though this percentage must be considered a rough approximation because of the large variability in the accumulation ratios that we have encountered in these experiments (see below for further discussion). The proposal that the transported neurofilament protein accumulated in the form of polymer is supported by our electron microscopic observations that demonstrated the presence of an abnormally large number of neurofilament polymers in the axonal swellings proximal to the constrictions. However, simply the fact that a transported protein accumulates in a polymerized form does not prove that polymers actually move. Thus, even though our data are consistent with the hypothesis that axonal neurofilament protein moves in the form of assembled polymers, we cannot exclude the possibility that it is transported in a nonpolymeric form that assembled locally at the site of accumulation.
One of the challenges that we have faced in these studies has been the large variability in neurofilament protein accumulation among different cells. For example, the magnitude of the neurofilament accumulation ranged from 100% (approximately twofold) to 1,140% (∼11-fold) for six axons constricted for 2 h (Fig. 5 A). It is possible that some of this variability was due to intrinsic differences in the rate of neurofilament protein transport in different neurons. Such differences are possible given the heterogeneity of neuronal types present in dorsal root ganglia (Lawson, 1992). However, we believe that a more important factor was variability in the extent of constriction for different axons, and this is supported by our electron microscopic observations on the thickness of constricted axons beneath the glass fiber. Among the factors that may determine the extent of constriction, the most important are likely to be the diameter, stiffness, and uniformity of the glass fibers and the uniformity of the glass coverslip. We made every effort to use glass fibers of similar dimensions but in practice this was difficult to control precisely. In addition, we observed that particulate material in the culture medium tended to adhere to the glass surfaces which may also have contributed to variability in the extent of constriction. Given the narrow diameter of these axons, differences in the extent of constriction of just hundreds of nanometers could have had significant effects on the extent of retardation of neurofilament protein transport.
It is possible to estimate the rate of transport in this experimental paradigm based on the magnitude of the neurofilament protein accumulation. For example, we obtained an average accumulation ratio of 7.3 for six axons constricted for 2 h (Fig. 5 A). This corresponds to a 6.3-fold increase in the neurofilament content of the 41-μm-long proximal measurement window. Since the neurofilament content is fairly constant along these axons before constriction (for example, see analyses of control sister axons in Fig. 5 A), this amount of accumulated neurofilament protein can be considered to be equivalent to the amount contained in a 258-μm (6.3 × 41 μm) length of axon. If we assume that 100% of the axonal neurofilament protein is transported and that no neurofilament protein can pass through the constriction, then we obtain an average transport rate of 130 μm/h (3.1 mm/d). This transport rate should probably be considered a minimum estimate because it would be proportionately higher if a portion of the transported neurofilament protein was able to pass through the constriction or if <100% of the protein was moving. In addition, the segment containing the constriction was excluded from our analyses because it contained portions of both the proximal and distal swellings, but we calculate that including this segment would have increased our estimate of the transport rate by <15%. The modal transport rate for neurofilament proteins in adult neurons in vivo ranges from 0.25 to 3 mm/d (Lasek et al., 1993), but the transport rate of individual neurofilament proteins can range from <0.005 mm/d to >72 mm/day (Lasek et al., 1992; Lasek et al., 1993). Thus, the average transport rate in these embryonic neurons ex vivo is higher than the modal transport rate in most adult neurons in vivo (Lasek et al., 1993) but well within the broad range of rates at which individual neurofilament proteins are capable of moving.
All previous attempts to demonstrate the movement of neurofilament protein in cultured neurons have employed the technique of fluorescence photobleaching using fluorescently labeled NF-L or NF-H microinjected into cultured mouse dorsal root ganglion neurons (Okabe et al., 1993; Takeda et al., 1994). These studies reported turnover of the neurofilament protein within the bleached zone, implicating exchange between the polymer and subunit pool, but the zone itself did not move. Microinjection of biotinylated neurofilament protein resulted in its incorporation into neurofilaments >200 μm from the cell body within 3 h but this is also not indicative of axonal transport because it is known that cytoskeletal proteins can diffuse over such distances along axons in this period of time (e.g., Popov and Poo, 1992). Similar results have been obtained on tubulin and actin in a variety of systems using photobleaching and photoactivation (see Introduction). But if transport is occurring, why have these techniques not revealed movement? At present, the answer is unclear. One possible explanation is that there is photodamage to the slow transport mechanisms in these cells during the photobleaching or photoactivation process (e.g., Keith and Farmer, 1993; McIntosh et al., 1990). However, these techniques have been used successfully to visualize cytoskeletal polymer movement in nonneuronal cells so this explanation is not compelling (e.g., Keating et al., 1997; Waterman-Storer and Salmon, 1997). Another possibility is that the proportion of the axonal neurofilament protein that is transported is too small to be detected by these techniques. For example, Sabry et al. (1995) have estimated that the photoactivation technique would not be capable of detecting movement if 10% or less of the protein was moving. Nixon and colleagues have proposed that there are distinct stationary and moving populations of neurofilament proteins in axons, but this argument is based entirely on one study in optic nerve axons that did not account for the presence of comigrating SCa and SCb proteins on one-dimensional polyacrylamide gels (Nixon and Logvinenko, 1986; Nixon, 1998). In fact, analyses of neurofilament protein transport in optic nerve axons using two dimensional PAGE, which allows separation of comigrating SCa and SCb proteins, has shown that there is a single population of neurofilament proteins that is transported at a broad range of rates, with no evidence for distinct stationary and moving populations (Lasek et al., 1992, 1993). Furthermore, since slow axonal transport is required to support the elongation of long axons (Sabry et al., 1995), if <10% of the axonal proteins were moving then they would have to be transported at >10 times the rate of axon growth in order to support axon elongation. Since the modal rate of slow axonal transport in whole animals is broadly comparable to the rate of axonal growth it seems likely that a large fraction of the axonal protein must be transported.
The axonal transport of neurofilament proteins has attracted considerable interest in recent years because an impairment in this movement has been implicated in the etiology of certain neurodegenerative diseases, most notably amyotrophic lateral sclerosis (Williamson et al., 1996; Julien, 1997). Cultured neurons offer a special opportunity for studies on the transport mechanisms because of their accessibility to direct observation and experimental manipulation. The data in this paper demonstrate for the first time that neurofilament proteins are indeed transported in the axons of cultured neurons, in spite of the failure of photobleaching approaches to detect such movement. Future progress on the mechanisms of movement, including the form in which the proteins move, will depend on the development of live cell models in which the movement of neurofilament proteins or polymers can be visualized directly in living cells.
Acknowledgments
We thank Virginia Lee for providing the NF-L antibody and Peter Baas and Bob Hikida for their advice on electron microscopy.
Abbreviations used in this paper
- NF-L
low molecular weight neurofilament triplet polypeptide
- PHEM
buffer containing Pipes, Hepes, EGTA, and MgCl2
Footnotes
Address correspondence to Anthony Brown, Department of Biological Sciences, Ohio University, Athens, OH 45701. Tel.: (740) 593-2330. Fax: (740) 593-0300. E-mail: browna1@ohiou.edu
Funded by a grant from the National Institute of Neurological Disorders and Stroke.
References
- Adams RJ. Organelle movement in axons depends on ATP. Nature. 1982;297:327–329. doi: 10.1038/297327a0. [DOI] [PubMed] [Google Scholar]
- Baas PW, Ahmad FJ. The transport properties of axonal microtubules establish their polarity orientation. J Cell Biol. 1993;120:1427–1437. doi: 10.1083/jcb.120.6.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, Brown A. Slow axonal transport: the polymer transport model. Trends Cell Biol. 1997;7:380–384. doi: 10.1016/S0962-8924(97)01148-3. [DOI] [PubMed] [Google Scholar]
- Berthold C-H, Fabricius C, Rydmark M, Andersén B. Axoplasmic organelles at nodes of Ranvier. I. Occurrence and distribution in large myelinated spinal root axons of the adult cat. J Neurocytol. 1993;22:925–940. doi: 10.1007/BF01218351. [DOI] [PubMed] [Google Scholar]
- Black MM, Keyser P, Sobel E. Interval between the synthesis and assembly of cytoskeletal proteins in cultured neurons. J Neurosci. 1986;6:1004–1012. doi: 10.1523/JNEUROSCI.06-04-01004.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady ST, Lasek RJ, Allen RD. Fast axonal transport in extruded axoplasm from squid giant axon. Science. 1982;218:1129–1131. doi: 10.1126/science.6183745. [DOI] [PubMed] [Google Scholar]
- Bray, D. 1991. Isolated chick neurons for the study of axonal growth. In Culturing Nerve Cells. G. Banker and K. Goslin, editors. MIT Press, Cambridge, MA. 119–135.
- Bray D, Bunge MB, Chapman K. Geometry of isolated sensory neurons in culture. Effects of embryonic age and culture substratum. Exp Cell Res. 1987;168:127–137. doi: 10.1016/0014-4827(87)90422-8. [DOI] [PubMed] [Google Scholar]
- Brown A. Visualization of single neurofilaments by immunofluorescence microscopy of splayed axonal cytoskeletons. Cell Motil Cytoskelet. 1997;38:133–145. doi: 10.1002/(SICI)1097-0169(1997)38:2<133::AID-CM3>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Brown A. Contiguous phosphorylated and non-phosphorylated domains along axonal neurofilaments. J Cell Sci. 1998;111:455–467. doi: 10.1242/jcs.111.4.455. [DOI] [PubMed] [Google Scholar]
- Brown A, Slaughter T, Black MM. Newly assembled microtubules are concentrated in the proximal and distal regions of growing axons. J Cell Biol. 1992;119:867–882. doi: 10.1083/jcb.119.4.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campenot RB, Lund K, Senger DL. Delivery of newly synthesized tubulin to rapidly growing distal axons of rat sympathetic neurons in compartmented cultures. J Cell Biol. 1996;135:701–709. doi: 10.1083/jcb.135.3.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SH, Rodionov VI, Borisy GG, Popov SV. Transport and turnover of microtubules in frog neurons depend on the pattern of axonal growth. J Neurosci. 1998;18:821–829. doi: 10.1523/JNEUROSCI.18-03-00821.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaghy M, King RHM, Thomas PK, Workman JM. Abnormalities of the axonal cytoskeleton in giant axonal neuropathy. J Neurocytol. 1988;17:197–208. doi: 10.1007/BF01674207. [DOI] [PubMed] [Google Scholar]
- Fabricius C, Berthold C-H, Rydmark M. Axoplasmic organelles at nodes of Ranvier. II. Occurrence and distribution in large myelinated spinal cord axons of the adult cat. J Neurocytol. 1993;22:941–954. doi: 10.1007/BF01218352. [DOI] [PubMed] [Google Scholar]
- Fahim MA, Lasek RJ, Brady ST, Hodge AJ. AVEC-DIC and electron microscopic analyses of axonally transported particles in cold-blocked squid giant axons. J Neurocytol. 1985;14:689–704. doi: 10.1007/BF01170822. [DOI] [PubMed] [Google Scholar]
- George EB, Schneider BF, Lasek RJ, Katz MJ. Axonal shortening and the mechanisms of axonal motility. Cell Motil Cytoskelet. 1988;9:48–59. doi: 10.1002/cm.970090106. [DOI] [PubMed] [Google Scholar]
- Glass JD, Griffin JW. Retrograde transport of radiolabeled cytoskeletal proteins in transected nerves. J Neurosci. 1994;14:3915–3921. doi: 10.1523/JNEUROSCI.14-06-03915.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawrot E, Patterson PH. Long-term culture of dissociated sympathetic neurons. Methods Enzymol. 1979;58:574–584. doi: 10.1016/s0076-6879(79)58174-9. [DOI] [PubMed] [Google Scholar]
- Hirano A, Donnenfeld H, Sasaki S, Nakano I. Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1984;43:461–470. doi: 10.1097/00005072-198409000-00001. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Terada S, Funakoshi T, Takeda S. Slow axonal transport: the subunit transport model. Trends Cell Biol. 1997;7:384–388. doi: 10.1016/S0962-8924(97)01133-1. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Yoshida Y, Sato-Yoshitake R, Kawashima T. Brain dynein localizes on both anterogradely and retrogradely transported membranous organelles. J Cell Biol. 1990;108:111–126. doi: 10.1083/jcb.111.3.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ. The transport and assembly of the axonal cytoskeleton. J Cell Biol. 1989;108:223–227. doi: 10.1083/jcb.108.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien JP. Neurofilaments and motor neuron disease. Trends Cell Biol. 1997;7:243–249. doi: 10.1016/S0962-8924(97)01049-0. [DOI] [PubMed] [Google Scholar]
- Kapeller K, Mayor D. An electron microscopic study of the early changes distal to a constriction in sympathetic nerves. Proc R Soc Lond B Biol Sci. 1969a;172:53–63. doi: 10.1098/rspb.1969.0011. [DOI] [PubMed] [Google Scholar]
- Kapeller K, Mayor D. An electron microscopic study of the early changes proximal to a constriction in sympathetic nerves. Proc R Soc Lond B Biol Sci. 1969b;172:39–51. doi: 10.1098/rspb.1969.0010. [DOI] [PubMed] [Google Scholar]
- Keating TJ, Peloquin JG, Rodionov VI, Momcilovic D, Borisy GG. Microtubule release from the centrosome. Proc Natl Acad Sci USA. 1997;94:5078–5083. doi: 10.1073/pnas.94.10.5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith CH. Slow transport of tubulin in the neurites of differentiated PC12 cells. Science. 1987;235:337–339. doi: 10.1126/science.2432662. [DOI] [PubMed] [Google Scholar]
- Keith CH, Farmer MA. Microtubule behavior in PC12 neurites: variable results obtained with photobleach technology. Cell Motil Cytoskelet. 1993;25:345–357. doi: 10.1002/cm.970250405. [DOI] [PubMed] [Google Scholar]
- Lasek RJ. Polymer sliding in axons. J Cell Sci Suppl. 1986;5:161–179. doi: 10.1242/jcs.1986.supplement_5.10. [DOI] [PubMed] [Google Scholar]
- Lasek RJ, Garner JA, Brady ST. Axonal transport of the cytoplasmic matrix. J Cell Biol. 1984;99:212–221. doi: 10.1083/jcb.99.1.212s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasek RJ, Paggi P, Katz MJ. Slow axonal transport mechanisms move neurofilaments relentlessly in mouse optic axons. J Cell Biol. 1992;117:607–616. doi: 10.1083/jcb.117.3.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasek RJ, Paggi P, Katz MJ. The maximum rate of neurofilament transport in axons: a view of molecular transport mechanisms continuously engaged. Brain Res. 1993;616:58–64. doi: 10.1016/0006-8993(93)90192-p. [DOI] [PubMed] [Google Scholar]
- Lawson, S.N. 1992. Morphological and biochemical cell types of sensory neurones. In Sensory Neurons. S.A. Scott, editor. Oxford University Press, New York, NY. 27–59.
- LeBeau JM, Ellisman MH, Powell HC. Ultrastructural and morphometric analysis of long term peripheral nerve regeneration through silicone tubes. J Neurocytol. 1988;17:161–172. doi: 10.1007/BF01674203. [DOI] [PubMed] [Google Scholar]
- Lim S-S, Edson KJ, Letourneau PC, Borisy GG. A test of microtubule translocation during neurite elongation. J Cell Biol. 1990;111:123–130. doi: 10.1083/jcb.111.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S-S, Sammak PJ, Borisy GG. Progressive and spatially differentiated stability of microtubules in developing neuronal cells. J Cell Biol. 1989;109:253–263. doi: 10.1083/jcb.109.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martenson C, Stone K, Reedy M, Sheetz M. Fast axonal transport is required for growth cone advance. Nature. 1993;366:665–679. doi: 10.1038/366066a0. [DOI] [PubMed] [Google Scholar]
- Matthews MR. An ultrastructural study of axonal changes following constriction of post-ganglionic branches of the superior cervical ganglion in the rat. Philos Trans R Soc Lond Ser B Biol Sci. 1973;264:480–505. doi: 10.1098/rstb.1973.0002. [DOI] [PubMed] [Google Scholar]
- McIntosh, J.R., M. Coue, and G.P.A. Vigers. 1990. Fluorescent microtubules are light sensitive. In Optical Microscopy for Biology: Proceedings of the International Conference on Video Microscopy. June 4–7, 1989. Chapel Hill, North Carolina. B. Herman and K. Jacobson, editors. Wiley-Liss, NY. 173– 181.
- Nakata T, Hirokawa N. Cytoskeletal reorganization of human platelets after stimulation revealed by the quick-freeze deep-etch technique. J Cell Biol. 1987;105:1771–1780. doi: 10.1083/jcb.105.4.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon, R.A. 1991. Axonal transport of cytoskeletal proteins. In The Neuronal Cytoskeleton. R.D. Burgoyne, editor. Wiley-Liss, Inc., New York, NY. 283–307.
- Nixon RA. The slow axonal transport of cytoskeletal proteins. Curr Opin Cell Biol. 1998;10:87–92. doi: 10.1016/s0955-0674(98)80090-2. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Logvinenko KB. Multiple fates of newly synthesized neurofilament proteins: evidence for a stationary neurofilament network distributed nonuniformly along axons of retinal ganglion cells. J Cell Biol. 1986;102:647–659. doi: 10.1083/jcb.102.2.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe S, Hirokawa N. Turnover of fluorescently labelled tubulin and actin in the axon. Nature. 1990;343:479–482. doi: 10.1038/343479a0. [DOI] [PubMed] [Google Scholar]
- Okabe S, Hirokawa N. Differential behavior of photoactivated microtubules in growing axons of mouse and frog neurons. J Cell Biol. 1992;117:105–120. doi: 10.1083/jcb.117.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe S, Hirokawa N. Do photobleached fluorescent microtubules move? Re-evaluation of fluorescence laser photobleaching both in vitro and in growing Xenopusaxons. J Cell Biol. 1993;120:1177–1186. doi: 10.1083/jcb.120.5.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe S, Miyasaka H, Hirokawa N. Dynamics of the neuronal intermediate filaments. J Cell Biol. 1993;121:375–386. doi: 10.1083/jcb.121.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overly CC, Rieff HI, Hollenbeck PJ. Organelle motility and metabolism in axons vs dendrites of cultured hippocampal neurons. J Cell Sci. 1996;109:971–980. doi: 10.1242/jcs.109.5.971. [DOI] [PubMed] [Google Scholar]
- Popov S, Poo MM. Diffusional transport of macromolecules in developing nerve processes. J Neurosci. 1992;12:77–85. doi: 10.1523/JNEUROSCI.12-01-00077.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinsch SS, Mitchison TJ, Kirschner MW. Microtubule polymer assembly and transport during axonal elongation. J Cell Biol. 1991;115:365–379. doi: 10.1083/jcb.115.2.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabri MI, Ochs S. Inhibition of glyceraldehyde-3-phosphate dehydrogenase in mammalian nerve by iodoacetic acid. J Neurochem. 1971;18:1509–1514. doi: 10.1111/j.1471-4159.1971.tb00013.x. [DOI] [PubMed] [Google Scholar]
- Sabry J, O'Connor TP, Kirschner MW. Axonal transport of tubulin in Ti1 pioneer neurons in situ. Neuron. 1995;14:1247–1256. doi: 10.1016/0896-6273(95)90271-6. [DOI] [PubMed] [Google Scholar]
- Schmidt RE, Plurad SB. Ultrastructural appearance of intentionally frustrated axonal regeneration in rat sciatic nerve. J Neuropathol Exp Neurol. 1985;44:130–146. doi: 10.1097/00005072-198503000-00002. [DOI] [PubMed] [Google Scholar]
- Shaw G, Bray D. Movement and extension of isolated growth cones. Exp Cell Res. 1977;104:55–62. doi: 10.1016/0014-4827(77)90068-4. [DOI] [PubMed] [Google Scholar]
- Smith RS. The short term accumulation of axonally transported organelles in the region of localized lesions of single myelinated axons. J Neurocytol. 1980;9:39–65. doi: 10.1007/BF01205226. [DOI] [PubMed] [Google Scholar]
- Takeda S, Funakoshi T, Hirokawa N. Tubulin dynamics in neuronal axons of living zebrafish embryos. Neuron. 1995;14:1257–1264. doi: 10.1016/0896-6273(95)90272-4. [DOI] [PubMed] [Google Scholar]
- Takeda S, Okabe S, Funakoshi T, Hirokawa N. Differential dynamics of neurofilament-H protein and neurofilament-L protein in neurons. J Cell Biol. 1994;127:173–185. doi: 10.1083/jcb.127.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita S, Ishikawa H. The movement of membranous organelles in axons: electron microscopic identification of anterogradely and retrogradely transported organelles. J Cell Biol. 1980;84:513–530. doi: 10.1083/jcb.84.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tytell M, Black MM, Garner JA, Lasek RJ. Axonal transport: each major component reflects the movement of distinct macromolecular complexes. Science. 1981;214:179–181. doi: 10.1126/science.6169148. [DOI] [PubMed] [Google Scholar]
- Vallee RB, Bloom GS. Mechanisms of fast and slow axonal transport. Annu Rev Neurosci. 1991;14:59–92. doi: 10.1146/annurev.ne.14.030191.000423. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer CM, Salmon ED. Actomyosin-based retrograde flow of microtubules in lamella of migrating epithelial cells influences microtubule dynamic instability and turnover and is associated with microtubule breakage and treadmilling. J Cell Biol. 1997;139:417–434. doi: 10.1083/jcb.139.2.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenberg RC, Flynn J, Gao B, Awodi S, Skee F, Goodman SR, Riederer BM. Microtubule gelation-contraction: essential components and relation to slow axonal transport. Science. 1987;238:1119–1122. doi: 10.1126/science.2446388. [DOI] [PubMed] [Google Scholar]
- Weiss PA, Hiscoe HB. Experiments on the mechanism of nerve growth. J Exp Zool. 1948;107:315–396. doi: 10.1002/jez.1401070302. [DOI] [PubMed] [Google Scholar]
- Whitlon DS, Baas PW. Improved methods for using glass coverslips in cell culture and electron microscopy. J Histochem Cytochem. 1992;40:875–877. doi: 10.1177/40.6.1588032. [DOI] [PubMed] [Google Scholar]
- Williamson TL, Marszalek JR, Vechio JD, Bruijn LI, Lee MK, Xu Z, Brown RH, Jr, Cleveland DW. Neurofilaments, radial growth of axons, and mechanisms of motor neuron disease. Cold Spring Harbor Symp Quant Biol. 1996;61:709–723. [PubMed] [Google Scholar]
- Yu W, Schwei MJ, Baas PW. Microtubule transport and assembly during axon growth. J Cell Biol. 1996;133:151–157. doi: 10.1083/jcb.133.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann H. Accumulation of synaptic vesicle proteins and cytoskeletal specializations at the peripheral node of Ranvier. Microsc Res Technique. 1996;34:462–473. doi: 10.1002/(SICI)1097-0029(19960801)34:5<462::AID-JEMT6>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]